

Abstract
The proteasome is a key protease in the eukaryotic cells which is responsible for various important cellular processes such as the control of the cell cycle, immune responses, protein homeostasis, inflammation, apoptosis, and the response to proteotoxic stress. Acting as a major molecular machine for protein degradation, proteasome first identifies damaged or obsolete regulatory proteins by attaching ubiquitin chains and subsequently utilizes conserved pore loops of the heterohexameric ring of AAA+ (ATPases associated with diverse cellular activities) to pull and mechanically unfold and translocate the misfolded protein to the active site for proteolysis. A detailed knowledge of the reaction mechanism for this proteasomal proteolysis is of central importance, both for fundamental understanding and for drug discovery. The present study investigates the mechanism of the proteolysis by the proteasome with full consideration of the protein’s flexibility and its impact on the reaction free energy. Major attention is paid to the role of the protein electrostatics in determining the activation barriers. The reaction mechanism is studied by considering a small artificial fluorogenic peptide substrate (Suc-LLVY-AMC) and evaluating the activation barriers and reaction free energies for the acylation and deacylation steps, by using the empirical valence bond method. Our results shed light on the proteolysis mechanism and thus should be important for further studies of the proteasome action.
Graphical Abstract

I. INTRODUCTION
The proteolysis is a major biological function not only in the protein regulation but also in maintaining cellular quality.1 Proteasomes are large self-compartmentalized protein complexes which are primarily responsible for selective proteolysis. They are found in all forms of life, including both prokaryotes and eukaryotes.1–3 A bulk of the protein degradation (up to 90%)4 in the nucleus of eukaryotic cells is achieved by the ubiquitin–proteasome pathway (UPP) through a selective and ATP-dependent degradation of misfolded or short-lived or potentially toxic proteins. In many cellular processes, e.g., oxidative stress and cell cycle, the proteasomal degradation pathway has turned to be very crucial. The role of ubiquitin in proteolytic pathways and the importance of proteolytic degradation inside cells were acknowledged in the award of the 2004 Nobel Prize in Chemistry to Aaron Ciechanover, Avram Hershko, and Irwin Rose.5,6
The canonical form of the proteasome in eukaryotes is known as 26S proteasome, which is a 2.5 MDa protein complex. The 26S proteasome is composed of two main components: a 20S catalytic core particle and a 19S regulatory complex (Figure 1).7 The 20S core particle, which has a molecular weight of ~700 kDa, is capped with 19S regulatory particle at both ends.1,2,8 In terms of shape, the 20S core particle has a cylinder or barrel shape formed with four stacked rings, each of which is made from seven heteromeric subunits.2 It is composed of one or two different α and β subunits, where the α subunit consists of 233 amino acids and the β subunit consists of 211 residues.3 Analysis of several crystal structures from mammalian 20S proteasome1,3 have shown that the cylindrical 20S particle is made of two identical halves composed of 14 unique proteins (having individual molecular weights of approximately 20–30 kDa)9 in an α7β7β7α7 arrangement.10,11 This particle is a hollow barrel with a length and diameter of 15 and 11 nm, respectively. The central chamber inside the core particle is formed by two inner β rings and holds a volume of 84 nm3, which can interestingly be compared with a folded protein with molecular weight of ~70 kDa.12 An inhibitor bound crystal structure reveals that the two innermost β rings and two outermost α rings arranged to form antechambers (formed by α7β7) with a volume of ~60 nm3, which has a potential to allow larger polypeptides to enter the system.12
Figure 1.
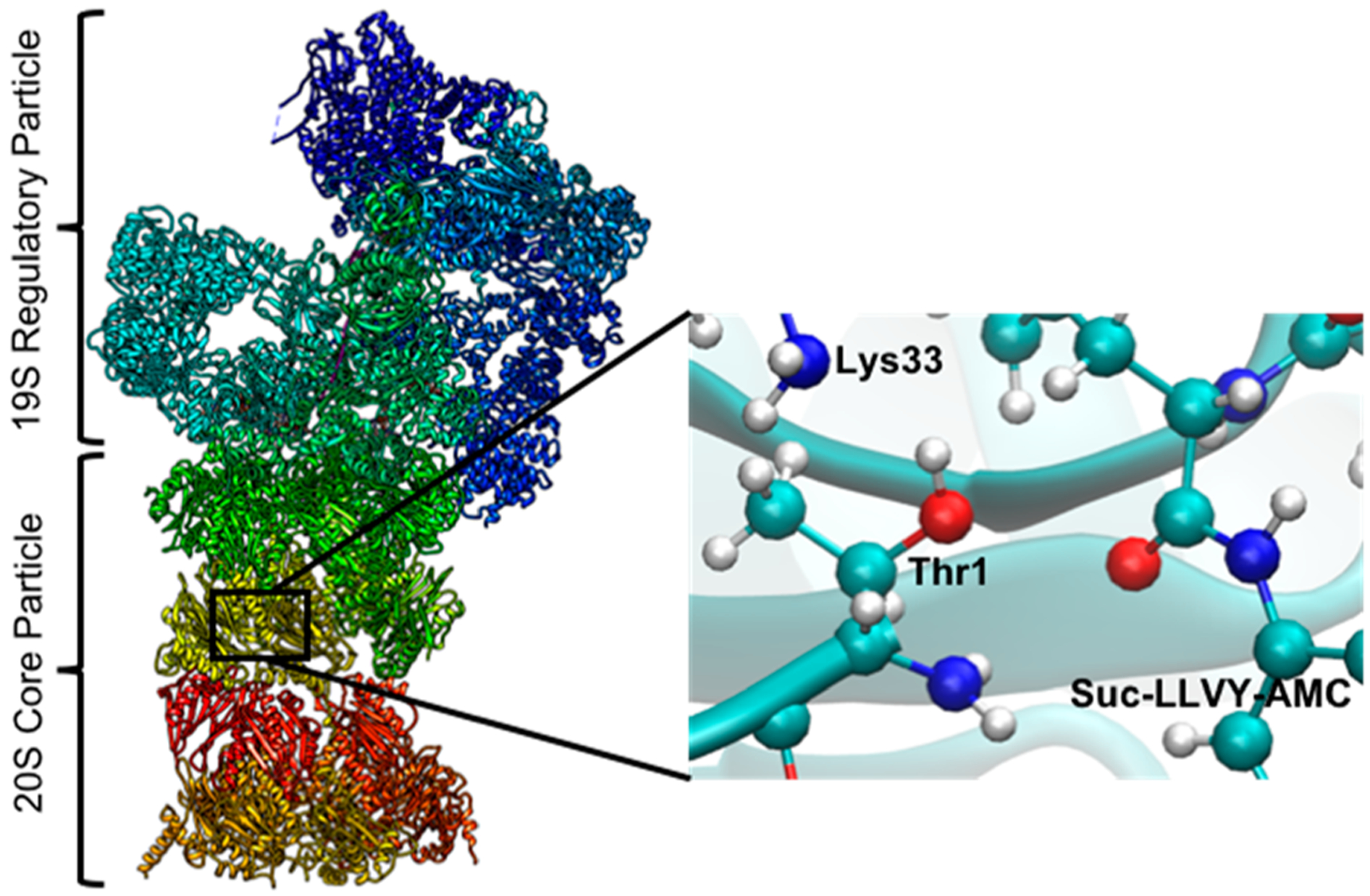
The left-hand panel shows the entire 26S proteasome. The right-hand panel is the zoom-out view of the active site on the β5 subunit with the docked substrate.
In the nucleus of eukaryotic cells, within the ubiquitin/proteasome system (UPS), the proteins are modified by covalent bonds during the course of attachment of a small protein (with a molecular weight of 9 kDa) called “ubiquitin”.13 The degradation mechanism or proteolysis starts with a signal which is initiated by the attachment of certain kind of ubiquitin protein chains to other ubiquitins through lysine residues, the process of which is termed as “polyubiquitination”.1,14–16 The following process is composed of a series of functional steps including identification of polyubiquitinated substrate protein, the course of their unfolding, and finally their translocation to the 20S proteasome’s inner chamber. This process is accomplished by the 19S regulatory particle, which is composed of two main structural sections: lid and base.13,17 While the lid has nine non-ATP dependent subunits (Rpn3, Rpn5, Rpn6, Rpn7, Rpn8, Rpn9, Rpn11, Rpn12, and Rpn15), the base is composed of a ring of six homologous AAA-ATPase subunits (ATPases Associated to a variety of cellular Activities),18–21 which are called Rpt 1 to 6 (Regulatory particle ATPase) as well as the non-ATP-dependent subunits Rpn1, Rpn2, Rpn10, and Rpn13 (Regulatory particle non-ATPase).1 The Rpt units are supposedly responsible for the unfolding process of the client substrate proteins as well as preventing free access of the substrate to the 20S core particle’s inner chamber by acting as an efficient gate keeper.22 On the other hand, the Rpn units serve a variety of functional responsibilities, which include binding polyubiquitinated substrate to Rpn10 and Rpn13 and release of the ubiquitin by cleaving its bond with the client protein.11
In proteasomes, the proteolytic active sites are the β subunits in the innermost two rings. Three forms of β-types exist in proteasomes: β1, β2, and β5 which have caspase-like, trypsin-like, and chymotrypsin-like activities.20,22 All the six active sites (two β1, two β2, and two β5) are functionally independent.3 The inhibitor (Ac-LLnL-al) bound crystal structure reveals that N-terminal threonines of all the β subunits are bound to the inhibitor covalently.23 Early studies1,24,41 including structural and mutational analyses recognized threonine as the most essential component of the active site, establishing the proteasome as a novel type protease.2,3
The proteasome stands as an attractive target for therapeutic agents which can inhibit cell proliferation in the treatments of such diseases as cancer.25 Despite the fact that the discovery of proteasome inhibitors has received intense research focus, development of a specific inhibitor has still remained a challenge, given the complexity of finding a suitable pharmacological application.26–28 Proteasome inhibitors can be classified to many groups based on the warhead pharmacophore and related chemical properties. These groups include peptide aldehyde inhibitors, β-lactone inhibitors, peptide boronic acid inhibitors, peptide epoxyketone inhibitors, and peptide vinyl sulfone inhibitors.29 Three of the inhibitors (bortezomib,30–34 ixazomib,35 and carfilzomib30,34,36–39) are currently approved by the FDA as marketed drugs in the limited treatment of multiple myeloma or mantle-cell lymphoma.40
In the context of the above knowledge, it is important to understand the mechanism of proteolysis by proteasomes. The proteolytic mechanism has remained a focused topic during the past decade,1,2,41–48 and several proposals have been made regarding the activation of the threonine residue in the active site.1,45–48 According to the suggestions of a few reports, the threonine activation happens through a proton transfer (PT) from Thr1–Ox to the terminus nitrogen Thr1–Nz (Figure 2).41 A few reports also predicted the proton transfer to happen between Ox and Nz indirectly via a water-mediated pathway.42,43,45 The possibility that a nearby lysine residue (Lys33) acts as a proton acceptor was also been considered but has been largely dismissed given the assumption that at neutral pH it will likely remain positively charged.1 Following the first proton transfer (regardless of the proton acceptor), the deprotonated oxygen of the threonine residue (Thr1), Ox, will now attack the carbonyl carbon of the scissile peptide bond in the client substrate (as depicted as step 2 in Figure 3). Finally, the group in the P1 pocket (7-amino-4-methylcoumarin, i.e., the AMC group) will leave, probably accepting a proton from the N-terminal of the threonine residue (steps 4 and 5 in Figure 3). The fact that the last proton transfer step may or may not involve additional water molecule has also been debated.47,48
Figure 2.

The first step in the reaction scheme involving a direct proton transfer from Ox to Nz of Thr1. The subscript letters (e.g., x and z) denote the specified atoms (see text).
Figure 3.

A proteolysis mechanism scheme with a proton transfer through Nx followed by nucleophilic attack on the substrate Suc-LLVY-AMC. The subscript letters (e.g., x and z) denote the specified atoms (see text).
Given the above possible mechanisms, we studied here the entire reaction profile for both the acylation and deacylation steps, using the empirical valence bond (EVB) method.49 In our study, we have considered the issue of potential proton transfer routes and examined an alternative sequence of reaction steps. Furthermore, we paid significant attention to the identification of the rate-determining step in the context of previously published proposals.47,48
It should be noted that previous theoretical studies were not based on a very consistent approach. For example, refs 47 and 48 used an approach where the reaction coordinate is determined by energy minimization and then used an uncoupled free energy calculation, where the MM part is allowed to fluctuate around fixed QM region. This uncoupled approach may lead to problems with the evaluation of the crucial electrostatic contributions. For example, in the current case, the presence of several ionizable residues in the reaction center plays a pivotal role. This poses a major challenge in determining the reaction free energy. That is, as pointed out by the authors in refs 47 and 48, the sampling of the MM subsystem was performed with the QM subsystem (which includes these important residues) frozen at each sampling step along the reaction path. However, a proper coupled QM/MM sampling may be crucial for accurate determination of the reaction free energy. Thus, in this report, we treated the system with the EVB method, which involves full configurational averaging and the change of the charges of the reacting (QM) atoms during the sampling process.
Proteasomal peptidase activities are usually measured with fluorogenic peptide substrate.41 It is classified based on the residues at the P1 position, where proteasomes cleave the amide bond of a small artificial oligopeptide substrate to release a fluorogenic leaving group, e.g., 7-amino-4-methylcoumarin (AMC).26–29 The proteasome activity of a well-known fluorogenic compound “succinyl-leucyl-leucyl-valyltyrosyl-7-amino-4-methylcoumarin” (Suc-LLVY-AMC) has been studied experimentally, and the corresponding kcat has been reported.41 Thus, we investigated in this report the reaction profile of Suc-LLVY-AMC with the proteasome, that generates amine AMC and carboxylic acid Suc-LLVY. This was done for the acylation step, given that the release of fluorogenic compound was only measured as a sign of peptide bond breaking in the experimental setup.
II. METHODS
To prepare the enzyme–substrate complex, we considered the proteasome–inhibitor complex (EPX) in PDB ID: 1G65.50 Considering the EPX position at the active site as a reference structure, we docked Suc-LLVY-AMC into the proteasome. The study of the reaction profile was done by using our EVB approach,49,51,52 and the corresponding EVB free energy surfaces were calculated by utilizing the FEP/US sampling technique.53 The MOLARIS-XG software54 with the ENZYMIX force field55,56 was used to generate the surfaces for each step. The surfaces were calibrated using the results of a previous careful ab initio study57 of the relevant reaction in water. A part of this procedure involves recalibration of the proton transfer energies using the corresponding pKa’s in water. The charges and corresponding EVB parameters are given in the Supporting Information. The center of the simulation sphere was set to the geometric center of the EVB reacting atoms. The system was immersed in an 25 Å water sphere by using the surface-constrained all-atom solvent (SCAAS) model.58 This system was further surrounded by a 2 Å surface of Langevin dipoles and then a bulk continuum. All other atoms beyond this sphere were fixed at their initial positions in the X-ray crystal structure, and the electrostatic interaction from outside of the sphere was turned off. The local reaction field (LRF)59 was used to treat the long-range effects. Water molecules in the entire simulation were generated by using MOLARIS-XG.54 The protonation states of the ionizable residues were determined by calculating the pKa’s using the Protein Dipoles Langevin Dipoles (PDLD) method.60 After this preparation of the system, the structure was equilibrated by increasing the temperature from 10 to 300 K for around 200 ps. Three different staring structures were generated from this relaxation and were further used for the FEP simulation, which involved 11 frames where each frame was simulated for 30 ps with a time step of 1 fs.
III. RESULTS AND DISCUSSION
Detailed proposed reaction schemes for the acylation step are provided in Figures 3 and 4 (the end frames of each step in the acylation reaction are shown in Figure 5). The initial step of the reaction mechanism of the proteolysis by proteasomes is the activation of threonine-1 residue. This activation can be achieved by transferring the threonine-OH proton to a possible nearby proton acceptor. As mentioned earlier, the mechanism for this particular proton transfer step received considerable attention during the past two decades.38,40–42 Some workers suggested that the nucleophile Thr1-O is activated by a direct proton transfer to its own N-terminal amino group (Thr1-NH2). Notably, Zhan et al.47 reported that this proton transfer will happen via a water molecule instead of a direct proton transfer. Regardless of the mechanism of the initial proton transfer, Zhan et al.47 concluded that the rate-determining step for the acylation reaction involves the last proton transfer, where the proton is transferring from N-terminal (Thr1-NH3) of the threonine to the NH group of the scissile peptide bond of AMC. At any rate, we are trying here to determine the actual mechanism of the proteolysis reaction.
Figure 4.

A proteolysis mechanism scheme with proton transfer through Nz followed by nucleophilic attack on the substrate Suc-LLVY-AMC. The subscript letters (e.g., x and z) denote the specified atoms (see text).
Figure 5.

Structure of the end frames for each step (as depicted in Figure 4). Step 1 involves the proton transfer from Thr1 to Lys33. Step 2 involves proton transfer from Lys33 to Thr1. Step 3 involves the nucleophilic attack of Thr1 to substrate that leads to the tetrahedral intermediate. Step 4 involves the proton transfer from Thr1 to substrate. Step 5 involves leaving of the AMC group.
As a start, we focused on the evaluation of the pKa’s of the groups that can be involved in the proton transfer. This is crucial since we cannot accept assumptions that groups like Lys33 must be protonated.1 The pKa calculations were performed with our reliable semimacroscopic Protein Dipoles Langevin Dipoles in its linear response approximation (PDLD/S-LRA).61 In the present case, the results are sensitive to the simulation conditions and the ionization conditions of the protein ionizable groups (that were determined by our CG model54) and to whether the inclusion of these groups is done explicitly or implicitly. Our consensus values for the pKa’s of Lys33 and the NH2 end group of Thr1 were 5.4 ± 2 and 3.0 ± 2, respectively. This means that both groups are deprotonated. Note, however, that it is incorrect to assume that if for example Lys33 is protonated, it cannot be used as a PT path. That is, all what is needed is to add to the PT energy the free energy of transferring the proton to the bulk (1.38(pH − pKa)). The simulations explored the two general reaction paths in Figures 3 and 4, where the energetics of the reference reactions in water is given in Figure 6. The two reaction paths (e.g., through Lys and through Thr) involve similar reaction steps (steps 1, 4, and 5) but an alternate sequence of steps.
Figure 6.

Calculated energetics of the reference acylation reaction in water for the two competing paths. The blue and red lines correspond to the paths of the reactions in Figures 3 and 4, respectively.
The calculated reaction profiles of the acylation reaction in the protein are summarized in Figure 7. The figure considers two feasible paths for the initial PT, finding that a nucleophilic attack appears to have a lower transition state for the path where the NH3 of Thr1 is protonated. For both possible paths we found that the nucleophilic attack is the rate-determining step. In contrast to the results of Zhan et al.,47 we found that the last step of the acylation process cannot be rate determining. The first and last frames of the nucleophilic attack step are provided in Figure 8 (only Lys33, Thr1, and Suc-LLVY-AMC are shown). The figure indicates that the system is getting structurally very well-prepared at this stage, to undergo relatively smoothly step 4 of the reaction.
Figure 7.

Calculated free energy profile for the acylation reaction in the protein in the paths of Figures 3 and 4, respectively. The error range of the calculations is ±2 kcal/mol.
Figure 8.

End frame structures of step 3 of Figure 4. The distances are shown in blue, and the dihedral angles of the scissile peptide bond are shown in violet.
At this stage, we decided to examine the deacylation step, which can be rate-limiting. The reaction scheme of the deacylation step is depicted in steps 6–9 of Figure 9, where the reference reaction in water is given in Figure 10. The energetics of the entire reaction (including deacylation steps) in protein is described in Figure 11.
Figure 9.

Entire mechanism scheme. Deacylation process (steps 6–9). The subscript letters (e.g., x and z) denote the specified atoms (see text).
Figure 10.

Energetics of the acylation and deacylation steps in water.
Figure 11.

Energetics of the acylation and deacylation steps in the protein.
The overall calculations identified the nucleophilic attack step in the acylation and the deacylation barriers as the rate-determining steps. The calculated barriers that correspond to kcat are 14.5 ± 2 and 21.0 ± 2 kcal/mol for the acylation and deacylation steps, respectively, while the observed barrier that corresponds to kcat is 18.6 kcal/mol.41 On the other hand, the calculated barriers that correspond to kcat/Km is 14.5 kcal/mol − ΔG bind and 15.0 kcal/mol − ΔG bind for the acylation and deacylation steps, respectively. However, since kcat is smaller in the deacylation step than in the acylation step, then the complex kinetics can account for the underestimate.
IV. CONCLUDING REMARKS
In this report, we have studied the mechanism of the proteolysis by the proteasome, considering small artificial fluorogenic peptide substrate (Suc-LLVY-AMC). We evaluated the activation barriers and reaction free energies of the entire reaction profile for both the acylation and deacylation steps using the EVB method. Our study indicated that two pathways are feasible for the initial PT, but that a nucleophilic attack step appears to have a lower transition state for the path where the NH3 of Thr1 is being protonated. For both possible paths, we found that the nucleophilic attack is the rate-determining step, in contrast to the conclusions that emerged from the study of Zhan et al.,47 where it was suggested that the last proton transfer in the acylation step is the rate-determining step. Our calculations identified the nucleophilic attack steps in the acylation and the deacylation barriers as the origin for the value of kcat for each step, where the barrier for the deacylation (21 ± 2 kcal/mol) is the higher barrier.
The current results are quite sensitive to the ionization states of the ionizable groups near the active site and the effective dielectric assumed for their electrostatic effect. This can lead, for example, to a shift in the pKa of Lys33, but the energetic for the proton transfer step will tend to compensate for the change in pKa.
The understanding of the mechanism of the proteasome should be useful for several advances, in addition to the basic understanding of this important reaction. One direction can involve the development and refinement of effective inhibitors. Another direction (which is now explored by our group) is the overall process of the protein insertion and cleavage. At any rate, the analysis of the proteolysis reaction is an important building stone in the understanding of the action of the proteasome.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health R35 GM122472 and the National Science Foundation Grant MCB 1707167. We thank the University of Southern California High Performance Computing and Communication Center for computational resources.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jpcb.0c04435
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.0c04435.
All EVB parameters and other details (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Marques AJ; Palanimurugan R; Matias AC; Ramos PC; Dohmen RJ Catalytic mechanism and assembly of the proteasome. Chem. Rev 2009, 109, 1509–1536. [DOI] [PubMed] [Google Scholar]
- (2).Finley D Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem 2009, 78, 477–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Förster F; Unverdorben P; Śledz P; Baumeister W Unveiling the Long-Held Secrets of the 26S Proteasome. Structure 2013, 21, 1551–1562. [DOI] [PubMed] [Google Scholar]
- (4).Serrano-Aparicio N; Świderek K; Moliner V Theoretical Study of the Inhibition Mechanism of Human 20S Proteasome by Dihydroeponemycin. Eur. J. Med. Chem 2019, 164, 399–407. [DOI] [PubMed] [Google Scholar]
- (5).Ciechanover A; Heller H; Elias S; Haas AL; Hershko A ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc. Natl. Acad. Sci. U. S. A 1980, 77, 1365–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hershko A; Ciechanover A; Heller H; Haas AL; Rose IA Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. U. S. A 1980, 77, 1783–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ding Z; Xu C; Sahu I; Wang Y; Fu Z; Huang M; Wong CC; Glickman MH; Cong Y Structural Snapshots of 26S Proteasome Reveal Tetraubiquitin-Induced Conformations. Mol. Cell 2019, 73, 1150–1161. [DOI] [PubMed] [Google Scholar]
- (8).Xie SC; et al. Te structure of the PA28–20S proteasome complex from Plasmodium falciparum and implications for proteostasis. Nat. Microbiol 2019, 4, 1990–2000. [DOI] [PubMed] [Google Scholar]
- (9).Tanaka K The proteasome: Overview of structure and functions. Proc. Jpn. Acad., Ser. B 2009, 85, 12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).de la Peña AH; Goodall EA; Gates SN; Lander GC; Martin A Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 2018, 362, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dong Y; Zhang S; Wu Z; Li X; Wang WL; Zhu Y; Stoilova-Mcphie S; Lu Y; Finley D; Mao Y Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lowe J; Stock D; Jap B; Zwickl P; Baumeister W; Huber R Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995, 268, 533–539. [DOI] [PubMed] [Google Scholar]
- (13).Rousseau A; Bertolotti A Regulation of Proteasome Assembly and Activity in Health and Disease. Nat. Rev. Mol. Cell Biol 2018, 19, 697. [DOI] [PubMed] [Google Scholar]
- (14).Zhang Y; Vukovic L; Rudack T; Han W; Schulten K Recognition of Poly-Ubiquitins by the Proteasome through Protein Refolding Guided by Electrostatic and Hydrophobic Interactions. J. Phys. Chem. B 2016, 120, 8137–8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wagner ND; Russell DH Defining Noncovalent Ubiquitin Homodimer Interfacial Interactions through Comparisons with Covalently Linked Diubiquitin. J. Am. Chem. Soc 2016, 138, 16588–16591. [DOI] [PubMed] [Google Scholar]
- (16).Hicke L; Schubert HL; Hill CP Ubiquitin-Binding Domains. Nat. Rev. Mol. Cell Biol 2005, 6, 610–621. [DOI] [PubMed] [Google Scholar]
- (17).Ye Y; Blaser G; Horrocks MH; Ruedas-Rama MJ; Ibrahim S; Zhukov AA; Orte A; Klenerman D; Jackson SE; Komander D Ubiquitin Chain Conformation Regulates Recognition and Activity of Interacting Proteins. Nature 2012, 492, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Majumder P; et al. Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Javidialesaadi A; Stan G Asymmetric Conformational Transitions in AAA+ Biological Nanomachines Modulate Direction-Dependent Substrate Protein Unfolding Mechanisms. J. Phys. Chem. B 2017, 121, 7108–7121. [DOI] [PubMed] [Google Scholar]
- (20).Javidialesaadi A; Flournoy S; Stan G Role of Diffusion in Unfolding and Translocation of Multidomain Titin I27 Substrates by a Clp ATPase Nanomachine. J. Phys. Chem. B 2019, 123, 2623–2635. [DOI] [PubMed] [Google Scholar]
- (21).Zhu Y; et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat. Commun 2018, 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Matyskiela ME; Lander GC; Martin A Conformational Switching of the 26S Proteasome Enables Substrate Degradation. Nat. Struct. Mol. Biol 2013, 20, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vinitsky A; Cardozo C; Sepp-Lorenzino L; Michaud C; Orlowski M Biochemical Properties of Insect and Crustacean Proteasomes. J. Biol. Chem 1994, 269, 29860–29866. [PubMed] [Google Scholar]
- (24).Groll M; Ditzel L; Lowe J; Stock D; Bochtler M; Bartunik HD; Huber R Structure of 20S Proteasome from Yeast at 2.4Å Resolution. Nature 1997, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- (25).Dou QP; Zonder JA Overview of Proteasome Inhibitor-Based Anti-cancer Therapies: Perspective on Bortezomib and Second-Generation Proteasome Inhibitors versus Future Generation Inhibitors of Ubiquitin-Proteasome System. Curr. Cancer Drug Targets 2014, 14, 517–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kisselev AF; van der Linden WA; Overkleeft HS Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol 2012, 19, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dorsey BD; Iqbal M; Chatterjee S; Menta E; Bernardini R; Bernareggi A; Cassara PG; D’Arasmo G; Ferretti E; De Munari S; Oliva A; Pezzoni G; Allievi C; Strepponi I; Ruggeri B; Ator MA; Williams M; Mallamo JP Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J. Med. Chem 2008, 51, 1068–1072. [DOI] [PubMed] [Google Scholar]
- (28).Zhou H-J; Aujay MA; Bennett MK; Dajee M; Demo SD; Fang Y; Ho MN; Jiang J; Kirk CJ; Laidig GJ; Lewis ER; Lu Y; Muchamuel T; Parlati F; Ring E; Shenk KD; Shields J; Shwonek PJ; Stanton T; Sun CM; Sylvain C; Woo TM; Yang J Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem 2009, 52, 3028–3038. [DOI] [PubMed] [Google Scholar]
- (29).Beck P; Dubiella C; Groll M Covalent and non-covalent reversible proteasome inhibition. Biol. Chem 2012, 393, 1101–1120. [DOI] [PubMed] [Google Scholar]
- (30).Demo SD; Kirk CJ; Aujay MA; Buchholz TJ; Dajee M; Ho MN; Jiang J; Laidig GJ; Lewis ER; Parlati F; Shenk KD; Smyth MS; Sun CM; Vallone MK; Woo TM; Molineaux CJ; Bennett MK Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [DOI] [PubMed] [Google Scholar]
- (31).Adams J; Palombella VJ; Sausville EA; Johnson J; Destree A; Lazarus DD; Maas J; Pien CS; Prakash S; Elliott PJ Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [PubMed] [Google Scholar]
- (32).Adams J; Behnke M; Chen S; Cruickshank AA; Dick LR; Grenier L; Klunder JM; Ma Y-T; Plamondon L; Stein RL Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett 1998, 8, 333–338. [DOI] [PubMed] [Google Scholar]
- (33).Adams J The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 2004, 4, 349–360. [DOI] [PubMed] [Google Scholar]
- (34).Mirabella AC; Pletnev AA; Downey SL; Florea BI; Shabaneh TB; Britton M; Verdoes M; Filippov DV; Overkleeft HS; Kisselev AF Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem. Biol 2011, 18, 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kupperman E; Lee EC; Cao Y; Bannerman B; Fitzgerald M; Berger A; Yu J; Yang Y; Hales P; Bruzzese F; Liu J; Blank J; Garcia K; Tsu C; Dick L; Fleming P; Yu L; Manfredi M; Rolfe M; Bolen J Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [DOI] [PubMed] [Google Scholar]
- (36).O’Connor OA; Stewart AK; Vallone M; Molineaux CJ; Kunkel LA; Gerecitano JF; Orlowski RZ A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res 2009, 15, 7085–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Elofsson M; Splittgerber U; Myung J; Mohan R; Crews CM Towards subunit-specific proteasome inhibitors: synthesis and evaluation of peptide α′, β′-epoxyketones. Chem. Biol 1999, 6, 811–822. [DOI] [PubMed] [Google Scholar]
- (38).Kuhn DJ; Chen Q; Voorhees PM; Strader JS; Shenk KD; Sun CM; Demo SD; Bennett MK; van Leeuwen FWB; Chanan-Khan AA; Orlowski RZ Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Parlati F; Lee SJ; Aujay M; Suzuki E; Levitsky K; Lorens JB; Micklem DR; Ruurs P; Sylvain C; Lu Y; Shenk KD; Bennett MK Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [DOI] [PubMed] [Google Scholar]
- (40).Kuhn DJ; Hunsucker SA; Chen Q; Voorhees PM; Orlowski M; Orlowski RZ Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009, 113, 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kisselev AF; Songyang Z; Goldberg AL Why Does Threonine, and Not Serine, Function as the Active Site Nucleophile in Proteasomes? J. Biol. Chem 2000, 275, 14831–14837. [DOI] [PubMed] [Google Scholar]
- (42).Genin E; Reboud-Ravaux M; Vidal J Proteasome Inhibitors: Recent Advances and New Perspectives in Medicinal Chemistry. Curr. Top. Med. Chem 2010, 10, 232–256. [DOI] [PubMed] [Google Scholar]
- (43).Myung J; Kim KB; Crews CM The Ubiquitin-Proteasome Pathway and Proteasome Inhibitors. Med. Res. Rev 2001, 21, 245–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Duggleby HJ; Tolley SP; Hill CP; Dodson EJ; Dodson G; Moody PC Penicillin Acylase Has A Single-amino-acid Catalytic Centre. Nature 1995, 373, 264–268. [DOI] [PubMed] [Google Scholar]
- (45).Groll M; Nazif T; Huber R; Bogyo M Probing Structural Determinants Distal to the Site of Hydrolysis That Control Substrate Specificity of the 20S Proteasome. Chem. Biol 2002, 9, 655–662. [DOI] [PubMed] [Google Scholar]
- (46).Groll M; Huber R; Potts BC Crystal Structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in Complex with the 20S Proteasome Reveal Important Consequences of beta-Lactone Ring Opening and A Mechanism for Irreversible Binding. J. Am. Chem. Soc 2006, 128, 5136–5141. [DOI] [PubMed] [Google Scholar]
- (47).Wei DH; Fang L; Tang MS; Zhan CG Fundamental Reaction Pathway for Peptide Metabolism by Proteasome: Insights from First-Principles Quantum Mechanical/Molecular Mechanical Free Energy Calculations. J. Phys. Chem. B 2013, 117, 13418–13434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wei DH; Lei BL; Tang MS; Zhan CG Fundamental Reaction Pathway and Free Energy Profile for Inhibition of Proteasome by Epoxomicin. J. Am. Chem. Soc 2012, 134, 10436–10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Warshel A; Weiss RM An empirical valence bond approach for comparing reactions in solutions and in enzymes. J. Am. Chem. Soc 1980, 102, 6218–6226. [Google Scholar]
- (50).Groll M; Kim KB; Kairies N; Huber R; Crews CM Crystal Structure of Epoxomicin: 20S Proteasome Reveals a Molecular Basis for Selectivity of alpha ‘, beta ‘-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc 2000, 122, 1237–1238. [Google Scholar]
- (51).Kamerlin SCL; Warshel A The empirical valence bond model: theory and applications. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2011, 1, 30–45. [Google Scholar]
- (52).Kamerlin SCL; Warshel A The EVB as a quantitative tool for formulating simulations and analyzing biological and chemical reactions. Faraday Discuss. 2010, 145, 71–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zwanzig RW High Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys 1954, 22, 1420–1426. [Google Scholar]
- (54).Feliks M. https://github.com/mfx9/MolarisTools (accessed September 2016).
- (55).Lee FS; Chu ZT; Warshel A Oscillations of the energy gap for the initial electron-transfer step in bacterial reaction centers. J. Comput. Chem 1993, 14, 161–185. [Google Scholar]
- (56).Warshel A, Chu ZT; Villa J; Strajbl M; Schutz CN; Shurki A; Vicatos S; Plotnikov NV; Schopf P Molaris-XG, v 9.15 University of Southern California, Los Angeles, 2012. [Google Scholar]
- (57).Strajbl M; Florian J; Warshel A Ab Initio Evaluation of the Potential Surface for General Base Catalyzed Methanolysis of Formamide: A Reference Solution Reaction for Studies of Serine Proteases. J. Am. Chem. Soc 2000, 122, 5354–5366. [Google Scholar]
- (58).King G; Warshel A Investigation of the Free Energy Functions for Electron Transfer Reactions. J. Chem. Phys 1990, 93, 8682–8692. [Google Scholar]
- (59).Lee FS; Warshel A A Local Reaction Field Method for Fast Evaluation of Long-Range Electrostatic Interactions in Molecular Simulations. J. Chem. Phys 1992, 97, 3100–3107. [Google Scholar]
- (60).Lee FS; Chu ZT; Warshel A Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem 1993, 14, 161–185. [Google Scholar]
- (61).Warshel A; Sharma PK; Kato M; Parson WW Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1647–1676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.