


Abstract
During homologous recombination, Dbl2 protein is required for localisation of Fbh1, an F-box helicase that efficiently dismantles Rad51–DNA filaments. RNA-seq analysis of dbl2Δ transcriptome showed that the dbl2 deletion results in upregulation of more than 500 loci in Schizosaccharomyces pombe. Compared with the loci with no change in expression, the misregulated loci in dbl2Δ are closer to long terminal and long tandem repeats. Furthermore, the misregulated loci overlap with antisense transcripts, retrotransposons, meiotic genes and genes located in subtelomeric regions. A comparison of the expression profiles revealed that Dbl2 represses the same type of genes as the HIRA histone chaperone complex. Although dbl2 deletion does not alleviate centromeric or telomeric silencing, it suppresses the silencing defect at the outer centromere caused by deletion of hip1 and slm9 genes encoding subunits of the HIRA complex. Moreover, our analyses revealed that cells lacking dbl2 show a slight increase of nucleosomes at transcription start sites and increased levels of methylated histone H3 (H3K9me2) at centromeres, subtelomeres, rDNA regions and long terminal repeats. Finally, we show that other proteins involved in homologous recombination, such as Fbh1, Rad51, Mus81 and Rad54, participate in the same gene repression pathway.
INTRODUCTION
DNA double-strand breaks (DSBs) occur intrinsically during normal cell growth or are caused by exogenous factors. In addition, DSBs are essential intermediates during programmed recombination events, such as meiosis and mating-type switching in yeast (1). Eukaryotic cells have evolved two mechanistically distinct pathways to repair DSBs during mitosis: non-homologous end joining (NHEJ) and homologous recombination (HR) (2). In the NHEJ pathway, which can function throughout the entire cell cycle, two ends of DSBs are ligated together with little or no requirement for homology (3). On the other hand, HR is restricted to the late S and G2 phases, where it uses homologous double-stranded DNA (dsDNA) to mediate error-free DSB repair. HR repair is initiated by the 5′→3′ resection of DSB ends to expose 3′ single-stranded DNA (ssDNA) for replication protein A (RPA). The subsequent displacement of RPA by Rad51 requires HR mediators, such as Rad52 and the Rad55–Rad57 complex in yeast (4–8). The resulting Rad51–ssDNA presynaptic filament is responsible for locating a homologous DNA sequence and then catalysing strand invasion, which pairs the ssDNA overhang with the homologous duplex (9,10). Other proteins, such as the F-box DNA helicase Fbh1 in Schizosaccharomyces pombe and the Srs2 helicase in Saccharomyces cerevisiae, are negative regulators of Rad51 (11–14). The recruitment of Fbh1 to DSBs is further regulated by the Dbl2 protein (15), which was first identified in a screening as a protein that localises to DNA DSBs (16). The Swi2/Snf2-related Rad54 protein plays multiple roles in regulating Rad51. Based on results from yeast, it serves as a positive regulator at early stages of recombination by enhancing the homologous DNA pairing reaction (17–20), promoting Rad51-mediated strand invasion (19–21), enabling D-loop formation (22–24), and mediating the migration of the nascent Holliday structure (25). However, it also works as a negative regulator of Rad51 at later stages of recombination by preventing non-specific binding of Rad51 to dsDNA or by removing Rad51 from dsDNA to expose a free 3′-OH primer terminus for DNA synthesis (17,26,27). Once DNA synthesis is initiated, two different routes can be used. In the first, the second end of DSB can be engaged to stabilise the D-loop structure, generating two Holliday junctions (28). In the second, the invading strand can be dismantled from the D-loop by helicases such as Fml1 in S. pombe and anneal with the complementary strand and associate with the other end of the DSB (29,30). The Holliday junctions are further processed by resolvases, such as Mus81–Eme1 (31). The second mechanism is preferred during mitosis to avoid potentially harmful events such as loss of heterozygosity.
More recently, HR proteins have been shown to play critical roles in maintaining genome integrity during DNA replication (32–34). Rad51, Rad52 and BRCA2 in mammalian cells safeguard arrested forks from becoming dysfunctional and protect dysfunctional forks from excessive resection, thus allowing their successful merger with converging forks (34–36). When forks lose replicative competence, HR proteins restart forks that likely result in the construction of a new replisome (33,37–40).
The mechanism by which chromatin is disassembled and reassembled during DSB repair has also been intensively studied. In budding yeast, chromatin disassembly in the vicinity of a DSB requires the remodelling complex INO80 and MRX (41,42), which is essential for timely DNA resection (43–45). Nucleosome assembly is regulated by histone chaperones, which bind specific histones to mediate their deposition into chromatin. In human cells, chromatin reassembly after DSB repair requires both the HIRA-mediated replication-independent pathway and the CAF-1-mediated replication-dependent pathway, suggesting that they act in a coordinated manner to accurately re-establish the chromatin structure after DNA repair (46). A previous study in budding yeast showed that the histone chaperone Asf1 is required for chromatin assembly after DSB repair by promoting acetylation of histone H3 on lysine 56 via the histone acetyltransferase Rtt109 (41). Interestingly, a recent study reported specific recruitment of both histone H3 chaperones, Hir and CAF-1, to meiotic DSBs in budding yeast (47). In fission yeast, the loss of the HIRA complex leads to an increased susceptibility to DNA damaging agents; this finding supports the idea that the HIRA complex plays a role in protecting cells against DSBs (48).
In addition to DSB repair, the HIRA complex has been implicated in multiple aspects of transcriptional regulation. In some contexts, HIRA is necessary for transcriptional activation. For example, the induction of specific genes in fission yeast in response to environmental stress is HIRA-dependent (49). HIRA is also required for transcriptional repression. S. cerevisiae Hir1 and Hir2 were initially identified as repressors of histone gene expression (50). In fission yeast, HIRA represses expression of heterochromatic regions, subtelomeric genes, Tf2 long terminal repeat (LTR) retrotransposons and their remnants, and it also limits the levels of cryptic intragenic transcripts (48,51–53).
However, the interplay between HIRA and HR in the repression of gene expression has not been reported. Here, we show for the first time that the Dbl2 protein functions in repression of regions located closer to long tandem and long terminal repeats compared with the loci with no change in expression. We find that dbl2 deletion upregulates a large number of genes, including Tf2 LTR retrotransposons, subtelomeric genes, meiotic genes and antisense transcripts. Interestingly, Dbl2 targets overlap with HIRA targets, and simultaneous deletion of dbl2 and hip1 or slm9 does not lead to cumulative increase in expression from most tested loci. Furthermore, genetic interactions between dbl2 and HIRA extend to histone modifications, growth at 37°C, and sensitivity to the thiabendazole. Consistently, dbl2 deletion completely restored the silencing defect of hip1Δ and slm9Δ at the outer centromere, indicating that Dbl2 might act upstream of HIRA. Finally, we show that other proteins involved in HR, including Fbh1, Rad51, Mus81 and Rad54, participate in repression of the tested Dbl2-regulated genes.
MATERIALS AND METHODS
Strains, growth media and general methods
The genotypes of the strains used in this study are listed in Supplementary Table S1. Strains carrying a deletion have been either constructed as described previously (54) or purchased from Bioneer (55) and from the National BioResource Project (NBRP, Japan). Rich YES, minimal EMM2 and sporulation PMG-N media were used to grow and mate S. pombe strains (56). If necessary, 0.15 g/l G418, 0.1 g/l nourseothricin, 0.2 g/l hygromycin B, 40 μM camptothecin (CPT) or 15 μg/ml thiabendazole (TBZ) were added. For spot assays, 10-fold serial dilutions of exponential phase cultures were spotted onto media in the presence or absence of drug and incubated for 3 days. S. pombe was transformed using the lithium acetate method (54). Yeast two-hybrid analysis was performed as previously described (15).
β-galactosidase assay
All lacZ reporters previously constructed (48) were introduced into the dbl2Δ background by using standard genetic crosses. Approximately 2.2–2.5 × 108 cells were harvested from cultures grown to OD595 = 0.75–0.8 in YES at 30°C, washed with H2O and stored at –80°C. The β-galactosidase assay was performed as previously described with modifications in the cell lysis protocol (57). Frozen samples were resuspended in 250 μL of Breaking buffer (100 mM Tris-HCl pH 8.0, 1 mM DTT, 20% glycerol) supplemented with 12.5 μl of 0.1 M PMSF and disrupted using glass beads (3 cycles of 2 min vortexing and 2 min chilling on ice). Additional 250 μl of Breaking buffer was added, the cell extracts were transferred to a new tube and centrifuged at 20 000 g for 15 min at 4°C. The clarified supernatants were stored at –20°C. 200 μl of the supernatants were mixed with 800 μl of Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, 10% glycerol) and pre-incubated for 5 min at 30°C. To initiate the reaction, 200 μl of o-nitrophenol-β-d-galactoside (ONPG) in Z-buffer (4 mg/ml) was added to the consecutive tubes in 30 s interval to keep the incubation time strictly 90 min at 30°C. The reactions were terminated by addition of 500 μl of 1M Na2CO3 and the absorbance of the reaction mix was measured at 420 nm (Thermo Scientific Multiskan GO). The Bradford assay was used to determine the protein concentration (58). β-galactosidase activity was measured in extracts of three parallel cultures for each strain and is reported as nmol of o-nitrophenol/min/mg of total protein.
Heterochromatin assay
Silencing in the outer domain of centromeric region was assessed using a strain containing ade6+ marker gene inserted into otr1R of centromere 1 (59). A marker gene inserted into this region is subjected to strong silencing, which gives a red colour to colonies grown with limited adenine in the medium. Defective silencing in this region leads to whiter colonies.
Total RNA isolation and RNA-seq library preparation
Total RNA of wt, dbl2Δ, wt + CPT, dbl2Δ + CPT, mus81Δ and rad51Δ was isolated as previously described (60). Four biological replicates grown to the exponential phase (OD595 = 0.5–0.55) in YES at 30°C were used for each strain. Total RNA was quantified with a Qubit™ 3.0 Fluorometer using the Qubit RNA HS assay (Thermo Fisher Scientific) and the RNA quality was verified by NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific). Recommended input of 1 μg of total RNA was processed to DNA library preparation according to the Illumina TruSeq Stranded Total RNA protocol. Ribosomal RNA depletion and reverse transcription of the cleaved RNA fragments were performed as described in TruSeq Stranded Total RNA Human/Mouse/Rat preparation kit (Illumina). The quantification of the DNA library was performed with a Qubit™ 3.0 Fluorometer using Qubit DNA HS assay (Thermo Fisher Scientific). Quality of the DNA library was analysed with an Agilent 2100 Bioanalyzer (Agilent Technologies). Sequencing was performed using Illumina NextSeq500 technology with the application of paired-end sequencing (2 × 75 bp reads). Adapters and low-quality ends of sequenced reads were removed using Trimmomatic (version 0.36) (61) based on quality control statistics generated by FastQC (version 0.11.5). After trimming, fragments without sufficient length of both reads (at least 35 bp) were removed from read sets. Filtered reads were used in downstream analyses.
RNA-seq data analysis
Expression of individual transcripts for each sequenced sample was estimated separately using Salmon (version 0.7.2) (62). Count vectors were aggregated into the summary table and normalised for different sequencing depths among samples using edgeR (version 3.12.1) (63). The tool also assessed the statistical significance of expression changes among biological replicates of selected groups (WT and dbl2 mutant, normal and CPT environment). We considered transcripts to be significantly changed if they met three conditions: (i) ≥1.5-fold change between two conditions; (ii) a calculated false discovery rate (FDR) ≤0.05 and (iii expression was observed in each biological replicate. Due to high sequence similarity of the Tf2 LTR retrotransposons, we analysed these together by summing all reads mapped to any of the Tf2 location (13 records in the PomBase gff3 annotation). Finally, we excluded transfer RNA and ribosomal RNA due to sequence similarity between paralogues and great deviations induced by rRNA depletion, respectively. Alternatively, filtered reads were also mapped to the reference yeast genome using Hisat2 (version 2.0.5) (64). Quality of the mapping was assessed using summary reports generated by Qualimap (version 2.2.1) (65). Coverage tracks were extracted from resulting BAM files using the bamCoverage tool from DeepTools (version 3.1.3) (66) and visually inspected with IGV (version 2.4.8) (67). Data analysis processing was automated using pipelines implemented in the SnakeLines framework (manuscript in preparation) running on the Snakemake workflow engine (version 5.2.2) (68). Reference genomic and transcriptomic sequences of S. pombe were downloaded from the PomBase database (version ASM294v2) (69). We used an associated gff3 annotation file to identify genomic regions with low sequential complexity. In particular, we selected all regions annotated as repeats, retrotransposons, polyA, centromeres, gaps or low complexity regions. We also reannotated the genome with Tandem Repeats Finder (version 4.9.0) (70), which identified 1688 tandem repeat regions between 24 and 9368 bp long (median length 46 bp). According to the length of the repeated motif, we divided them into two groups: short tandem repeats (motif length < 7 bp, 266 regions) and long tandem repeats (motif length > 6 bp, 1422 regions). Finally, we used the TetraplexFinder tool from QuadBase2 (other sources 4) to locate G-quadruplex motifs in the genome (71). Functional enrichment analysis was performed using g:Profiler (version e94_eg41_p11_6f51822) with the g:SCS multiple testing correction method applying significance threshold of 0.05 (72,73).
The significance of overlaps between different gene lists was assessed using the hypergeometric test. We considered the definition of the hypergeometric distribution with k successes (upregulated genes in both mutants) in n draws (upregulated genes in mutant 1), without replacement, from a finite population of size N (all genes) that contains exactly K objects with the feature (upregulated genes in mutant 2) that fits our data well. The P-value indicates the probability that the observed overlap happened by chance.
Quantitative PCR (qPCR)
RNA was isolated from yeast strains grown in YES at 30°C either to the exponential phase (OD595 = 0.5–0.55) or to the stationary phase (OD595 = 7–8) using Thermo Fisher Scientific kit. First strand complementary DNA (cDNA) was prepared using Thermo Fisher Scientific components (OligodT, Random hexamer, RiboLock RNase Inhibitor and RevertAid Reverse Transcriptase) according to manufacturer's instructions. For qPCR, FastStart DNA Master SYBR Green master mix (Roche) was used as instructed. Two genes (act1 and tbp1) were used for normalisation. Primers used for measuring gene expression are listed in Supplementary Table S2.
MNase digestion of chromatin, library preparation and analysis
One-hundred millilitres of wt (SP65), wt (SP072), dbl2Δ (SP067) and hip1Δ (SP456) were grown to OD595 = 0.75–0.8 in YES at 30°C. MNase digestion of chromatin, DNA sequencing and bioinformatics analyses were performed as previously described (74) with minor modifications. Three biological replicates were used for each sample. Fragmented DNA libraries were prepared using TruSeq Nano DNA Library Prep Kit (Illumina) according to the manufacturer‘s protocol with some modifications. We prepared libraries with half the volume of reagents and omitting the fragmentation step at the beginning of library preparation and without size selection. After the end repair step, all DNA fragments were captured with a fourfold volume of AMPure XP beads (Beckman Coulter). Next, DNA fragments were 3′-adenylated and ligated to indexed adapters using TruSeq DNA CD Indexes (Illumina). Libraries were amplified with eight cycles of PCR. The final DNA libraries were quantified with a Qubit™ 3.0 Fluorometer using the Qubit DNA HS assay (Thermo Fisher Scientific). The quality of the DNA libraries was analysed by Agilent 2100 Bioanalyzer using a High Sensitivity DNA assay (Agilent Technologies). The libraries were normalised to 4 nM and then pooled, denatured and diluted to a loading concentration of 1.8 pM for clustering at high output Illumina flowcell. Sequencing was performed using Illumina NextSeq500 technology with the application of paired-end sequencing (2 × 75 bp reads).
Chromatin immunoprecipitation and deep sequencing (ChIP-seq)
We performed ChIP-seq analyses with two independent biological replicates for each sample as previously described (75) with the following modifications. Four-hundred millilitres of S. pombe WT and dbl2Δ cells were grown to the exponential phase (OD595 = 0.5) in YES medium at 30°C. The extracted chromatin was sheared with the Bioruptor sonicator (Diagenode) using 30 cycles of 30 s ON, 30 s OFF at high power settings. For each IP, 5 μg of the respective antibody were used (H3: ab1791, H3K9me2: ab1220, both from Abcam). The washed precipitated material and input chromatin extracts were decrosslinked for 6 h at 65°C, treated with RNase A (Thermo Fisher Scientific) for 1 h at 37°C followed by proteinase K (Roche) treatment for 2 h at 55°C. DNA was isolated using phenol–chloroform extraction and sodium acetate/ethanol precipitation. DNA was further purified on SPRIselect beads (Beckman Coulter, B23317) to remove RNA and small DNA fragments.
For library construction, Illumina TruSeq Nano DNA kit with Illumina TruSeq DNA CD Indexes were used according to the manufacturer's instructions, with the following exceptions: no initial DNA fragmentation was performed and DNA was purified after end-repair using 4 volumes of AMPure Beads. Libraries were pooled and sequenced on an Illumina NextSeq 500 instrument (35 nt paired-end mode) using the NextSeq 500/550 High Output Reagent Cartridge v2 75 cycles with the NextSeq 500/550 High OutPut Flow Cell Cartridge v2.5. Adapter sequences were trimmed during export to FASTQ. Reads were then quality-trimmed using Trimmomatic 0.39 (61). The obtained reads were mapped to the fission yeast genome (ftp://ftp.pombase.org/pombe/genome_sequence_and_features/genome_sequence/; downloaded 09/2020) using HISAT 2.1.0 (76) and samtools 1.7 (77). BAM files were further processed using Deeptools 3.3.1 (66). For each sample, a read coverage was determined using bamCoverage. Normalisation of each H3K9me2 coverage dataset to the corresponding total H3 coverage was applied using bigwigCompare. The log2-transformed H3-normalised data from dbl2Δ were further normalised to wt. Coverage files were inspected visually in IGV 2.6.3 (67) and analysed using R 4.0.2 (www.r-project.org/) and RStudio 1.2.5019. The scripts used for ChIP-seq data processing and analyses are available at https://github.com/mprevorovsky/bagelova-polakova-dbl2-histones.
Western blotting analysis
Primary anti-histone H3 (ab176842), anti-histone H3K9ac (ab4441), anti-histone H3K4me3 (ab8580), anti-histone H3K9me2 (ab1220) and anti-histone H3K9me3 (ab8898) antibodies and the secondary goat anti-rabbit IgG H&L (conjugated to horseradish peroxidase, HRP) (ab205718) and goat anti-mouse IgG H&L (HRP) (ab205719) antibodies were purchased from Abcam. All the other chemicals were obtained from Sigma-Aldrich. For each strain, one liter of culture was grown in YES medium to mid-log phase (OD595 = 0.8) and the cells were collected by centrifugation (4000 g for 5 min at 4°C). Yeast cell powders were prepared from frozen cell pellets using SPEX SamplePrep 6770 Freezer/Mill cooled by liquid nitrogen. Proteins were extracted using Buffer II (50 mM Tris–HCl pH 8.0, 300 mM NaCl, 1 mM EDTA, 0.1% NP-40, 1 mM Mg-acetate, 1 mM imidazole, 10% glycerol, complete protease and phosphatase inhibitors and 1 mM PMSF) (78) in a ratio of 1 g of yeast powder to 1 ml of Buffer II for 20 min at 4°C. Extracts were cleared by centrifugation (41 000 g for 10 min at 4°C) and proteins were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting onto a polyvinylidene fluoride membrane (PVDF, 0.45 μm, Millipore). For the immunodetection, the primary and secondary antibodies were used at dilutions of 1:2000 and 1:10 000, respectively. The quantitative analysis was done on digitalized images using ImageJ software (National Institutes of Health). The Student's t-tests for paired comparison were performed on the data from experiments repeated four times.
RESULTS
Dbl2 represses expression of both coding and non-coding genes
Yeast two-hybrid system analysis showed that Dbl2 fused to the GAL4 DNA-binding domain can trigger expression from the reporter gene, suggesting a possible function of Dbl2 in gene expression regulation (Supplementary Figure S1). To address this phenomenon, we analysed the expression profiles of dbl2Δ and wild-type cells grown in YES medium to the exponential phase using strand-specific RNA-seq. We validated RNA-seq data for multiple genes using qPCR (Figure 1A). We observed much smaller differences in the level of transcripts of selected genes between dbl2Δ and wild-type cells grown in minimal EMM2 medium to the exponential phase (Supplementary Figure S2A) or in cells grown in YES medium to the stationary phase (except for SPBPB2B2.08) (Supplementary Figure S2B).
Figure 1.

Deletion of dbl2 leads to significant change in expression of more than 500 genes. A wild-type strain (SP065) and dbl2Δ mutant (SP067) were cultivated in standard YES media and their transcriptomes were analysed using RNA-seq. (A) dbl2Δ RNA-seq data for multiple genes were validated using qPCR. Upregulated genes and genes with no difference in expression were randomly selected from dbl2Δ RNA-seq data. The plotted values are the mean of four independent biological replicates ± standard error of the mean. (B) The majority of differentially expressed genes in the dbl2Δ mutant are upregulated. (C) Upregulated genes in the dbl2Δ mutant (511) are represented mostly by non-coding (295) and protein-coding genes (204), with a few pseudogenes (10), snoRNA (1) and snRNA (1). (D) Venn diagrams showing overlap between genes upregulated in the dbl2Δ mutant and genes upregulated under the indicated condition. Only genes included in both analyses were compared. The P-value indicates the probability that the observed overlap happened by chance.
There were 530 significantly altered RNA levels in the dbl2Δ mutant grown in YES medium to the exponential phase (FDR < 0.05; fold change > 1.5), indicating that Dbl2 influences the expression of a large number of loci (Supplementary Table S3). The majority of these genes (96%), including both protein-coding and non-coding genes, showed increased transcript levels in the dbl2Δ mutant (Figure 1B). Deletion of dbl2 increased the expression of at least 4% of fission yeast protein-coding genes. Interestingly, 58% of the upregulated genes belong to non-coding RNA, mainly antisense RNA (Figure 1C). Antisense RNA can arise in the cells either via read-through transcription at convergent genes or via initiation of antisense transcription from cryptic promoters. The antisense RNAs in the dbl2Δ mutant did not map preferentially to convergent genes (49%), suggesting that they are not a result of faulty transcriptional termination (Supplementary Table S3). The transcriptome of the dbl2Δ mutant was further enriched for transcripts from Tf2 LTR retrotransposons (2.1-fold increase), as well as transcripts from subtelomeric genes (Figure 2A), and meiotic genes (P < 2.4 × 10−29) (Figure 1D) (79). We analysed expression levels of individual Tf2 LTR retrotransposons in strains with an integrated lacZ reporter adjacent to an LTR (48). Reporter gene analysis showed that the suppression of only a few Tf2 mRNA levels is dependent upon Dbl2 (Supplementary Figure S2C).
Figure 2.

Genome-wide localisation of genes misregulated in dbl2Δ for the three S. pombe chromosomes. (A) Illustration of S. pombe chromosomes highlighting the subtelomeres (subtel)—box in grey and black adjacent to telomeres. The black box of the subtelomere represents telomere-adjacent SH region, which contain heterochromatin and the grey box of the subtelomere represents a highly condensed knob structure generated by Sgo2 (83). The term ‘subtelomere’ refers to the 100-kb-long region from the telomere end of chromosomes 1 and 2 (83). The ends of chromosome 3 contain rDNA repeats marked in red close to the telomeres; rRNA was depleted in the RNA-seq experiment. The length of the three S. pombe centromeres cen1, cen2 and cen3, is about 40 kb, 65 kb and 110 kb, respectively (153). Differentially expressed (DE) genes between dbl2Δ (SP067) and wt (SP065) on forward and reverse strands are represented as vertical lines on the upper and lower part of the chromosomal plot, respectively. DE genes in the dbl2Δ mutant were significantly enriched at the subtelomeric regions of chromosomes I and II (78 from 530 DE genes, chi-squared test, P < 2.12 × 10−49) but not at the centromeric region (4 from 530 DE genes, chi-squared test, P < 0.97). The statistics were performed using chi2 contingency test implemented in the scipy Python package (154). (B) Distance between Dbl2-affected genes and the closest specific genomic element. DE genes between wild-type and the dbl2Δ mutant (grey colour) are significantly closer to long terminal repeats and long tandem repeats than genes without significant change in expression (white colour). The lines across the boxes represent the median values of distance for NDE versus DE genes from long terminal repeats—26 577 bp vs 16568 bp; long tandem repeats—6000 bp versus 4114 bp; short tandem repeats—23 987 bp versus 23 030 bp; and G-quadruplexes—153 155 bp versus 139 204 bp. The boxes show the middle 50% of scores while the whiskers represent the maximum and minimum values, except for points determined as outliers outside of the 1.5 × interquartile range. (C) Venn diagram showing overlap between genes upregulated (>1.5-fold) in the dbl2Δ mutant with genes covered with γH2A. Only genes included in both analyses were compared. The P-value indicates the probability that the observed overlap happened by chance.
Furthermore, many of the genes upregulated in the dbl2Δ mutant overlap with the core environmental stress response genes (CESR) (P < 1.5 × 10−20) (80) and less with irradiation-induced (P < 3.1 × 10−12) (81), heat-shock-specific (P < 1 × 10−6), oxidative-stress-specific (P < 5.4 × 10−5), cadmium-specific (P < 8 × 10−3) and osmotic stress-specific (P < 7 × 10−3) genes (Figure 1D) (80). The dbl2Δ mutant is sensitive to the topoisomerase poison CPT (15), which prevents normal DNA re-ligation and therefore causes DSBs. To determine whether the changes in RNA levels in dbl2Δ are due to increased DNA damage, we analysed the expression profile of wild-type cells grown in YES medium supplemented with CPT using RNA-seq (Supplementary Table S3). When we compared the expression response to CPT with the response to dbl2 deletion, we detected only a very small overlap between the two profiles (P < 0.02) (Figure 1D). These results do not support the notion that the altered expression profile in dbl2Δ is due to increased DNA damage, although we cannot exclude this possibility. To further analyse the protein-coding genes influenced by Dbl2, we performed functional enrichment analysis. The Gene Ontology (GO) terms that are overrepresented in the list of upregulated protein-coding genes are related to meiosis, detoxification and transport (Supplementary Figure S3).
S. pombe possesses three chromosomes, of which chromosomes I and II contain a distinct form of heterochromatin at their ends, whereas chromosome III contains arrays of ribosomal DNA repeats next to the telomeres (82,83). Our analysis of the localisation of misregulated genes in the dbl2Δ mutant showed that, while there were many misregulated genes throughout the genome, they were significantly enriched at the subtelomeric regions of chromosomes I and II (chi-squared test, P < 2.12 × 10−49) (Figure 2A). We did not identify similar enrichment of misregulated genes at the centromeric region (chi-squared test, P < 0.97) (Figure 2A). We next compared the distribution of the misregulated loci in dbl2Δ with the distribution of repetitive DNA elements such as LTRs (84), motifs of seven or more (long tandem repeats), motifs of six or fewer (short tandem repeats) (70) and G4 structures (71). We found that the localisation of the misregulated genes significantly correlated with the localisation of long terminal and long tandem repeats (P < 6.7 × 10−11 and P < 2 × 10−6, respectively) (Figure 2B, Supplementary Figure S4). Nevertheless, we observed only a very small correlation between the transcript fold changes and the distance from a long terminal or a long tandem repeat (Supplementary Figure S5). Furthermore, we did not find any correlation with the localisation of short tandem repeats and G4 structures (Figure 2B).
Closer inspection of the localisation of the misregulated genes revealed that they significantly overlapped with genomic loci decorated by phosphorylated histone H2A (γH2A) (P < 2 × 10−15) (Figure 2C). γH2A marks are formed at natural replication fork barriers, retrotransposons, heterochromatin region and at the region repressed by Clr3/Clr6-mediated histone deacetylation (85). Overall, these data suggest that Dbl2 represses the expression of meiotic genes, subtelomeric genes, Tf2 LTR retrotransposons and non-coding RNA genes. Compared with the loci with no change in expression, the misregulated loci are closer to long terminal and long tandem repeats.
Dbl2 represses loci targeted by HIRA
In S. pombe, the main pathways that have been implicated in the repression of gene expression include RNA interference (RNAi) machinery (dcr1, ago1, rdp1) (86–88), silencing machinery (clr1, clr3, clr4 and clr6) (89–94) and histone chaperone HIRA (hip1 and slm9) (48,95). To determine whether Dbl2 fits into one of these pathways, we compared our dbl2Δ RNA-seq data with tiling array data from previous studies (48,92). We observed the most significant overlap between genes upregulated in the dbl2Δ mutant and genes upregulated in the hip1Δ and slm9Δ mutants (P < 2.3 × 10−69 and P < 3.5 × 10−51, respectively) and, to a lesser degree, with the clr6–1 mutant (P < 8.9 × 10−27) (Figure 3). We identified less significant overlap between genes upregulated in the dbl2Δ mutant and genes upregulated in mutants involved in silencing machinery – clr3Δ (P < 2.6 × 10−15), clr1Δ (P < 4 × 10−14) and clr4Δ (P < 1.2 × 10−10) – and the RNAi pathway – dcr1Δ (P < 3.3 × 10−9), rdp1Δ (P < 2.5 × 10−8) and ago1Δ (P < 4 × 10−6). The greater overlap with the HIRA complex and Clr6 indicates that Dbl2 might participate within the same biological pathway. This overlap cannot be simply explained by downregulation of genes encoding the HIRA complex or Clr6 in dbl2Δ because the loss of dbl2 did not lead to a change in their expression. HIRA histone chaperone mediates replication-independent histones H3-H4 deposition (96–100) and has been implicated in assembly of heterochromatin and silencing in various organisms (101–104). Clr6 functions as the catalytic core of the class I histone deacetylase (HDAC) complex, which regulates gene expression from numerous loci (93). Of note, HIRA plays a role in inhibiting expression of many genes, which are normally repressed in growing vegetative cells (48). These genes include those located in subtelomeric regions, Tf2 LTR retrotransposons, meiotic genes and antisense transcripts (48,51).
Figure 3.

Comparison of upregulated genes in the dbl2Δ and selected mutants that are known to affect gene expression. Venn diagrams showing overlap between genes upregulated (>1.5-fold) in the dbl2Δ mutant with genes upregulated (>1.5-fold) in the indicated deletion mutant. Only genes included in both analyses were compared. The P-values indicate the probability that the observed overlap happened by chance.
Genetic interactions between dbl2 and genes associated with gene repression
Significant overlap between genes upregulated in the hip1Δ, slm9Δ, clr6–1 and dbl2Δ mutants prompted us to analyse the genetic interactions between the HIRA and Clr6 complexes and the dbl2 gene. For comparison, we also included a mutant in the clr4 methyltransferase (105), required for transcriptional silencing, and a mutant in the dcr1 gene involved in RNAi (86,87). For epistasis analyses, we first selected several genes that were upregulated in both mutants—dbl2Δ and another mutant involved in the analysis. Then, we homogenized the set of genes used in these experiments. We assayed the effects of single and double mutants on the transcript levels from several loci using qPCR. Introduction of the dbl2Δ allele into the hip1Δ and slm9Δ background did not result in an additive increase in transcript levels compared with the dbl2Δ single mutant, except for SPBC947.05c and SPAC869.07c in dbl2Δhip1Δ (Figure 4A, B). Otherwise, the level of transcripts in the double mutants resembled that of the dbl2Δ single mutant; except for SPBC3E7.02c, SPBC1773.06c and SPAC27D7.09c whose transcript levels were reduced in dbl2Δslm9Δ. It was surprising that combining the mutation in hip3Δ with dbl2Δ led to a slight additive increase in transcript levels from half of the selected loci (SPCC965.11c, SPCC663.06c, SPAC3G9.11c, SPBC947.05c, SPAC11D3.01c, SPAC869.07c, SPAC27D7.09c and SPCC737.04) compared with any of the single mutants (Figure 4C). In addition, at three loci (SPAC15E1.02c, SPBPB21E7.07 and SPAC212.08c) the double mutant showed the transcript levels similar to hip3Δ and at two loci (SPBC3E7.02c and SPBPB2B2.08) similar to dbl2Δ. Hip3 is another subunit of the HIRA complex, which consists of four subunits (Hip1, Slm9, Hip3 and Hip4). Our data suggest that Hip3 may have a cellular function that is independent of other subunits of the HIRA complex. This is also supported by the fact that genes encoding the HIRA complex exhibit different genetic interactions in S. pombe (48,95,101,106–108). When clr6–1 was combined with dbl2Δ, the double mutant showed additive increase in the transcript levels at three measured loci (SPCC663.06c, SPBPB21E7.07 and SPCC737.04) compared with any of the single mutants (Figure 4D). Moreover, at four loci (SPBC16E9.16c, SPCC965.11c, SPAC11D3.01c and SPAC212.08c), the transcript levels in dbl2Δclr6–1 resembled that of dbl2Δ and equally at four loci (SPAC4H3.08, SPAC3G9.11c, SPBC947.05c and SPAC869.07c) resembled that of clr6–1. Comparing expression of the dbl2Δ and dcr1Δ mutants, the transcript levels in the double mutant were at seven loci (SPBC16E9.16c, SPAC4H3.08, SPCC663.06c, SPAC15E1.02c, SPBC1773.06c, SPBPB21E7.07 and SPCC737.04) similar to dbl2Δ and at three loci (SPBC3E7.02c, SPBPB2B2.08 and SPAC27D7.09c) similar to dcr1Δ. In addition, at one locus (SPCC965.11c) the level of transcripts was increased compared with any of the single mutants (Figure 4E). Finally, combining the clr4Δ and dbl2Δ mutations resulted in an additive effect on transcript levels from half of the tested loci (SPBC16E9.16c, SPAC4H3.08, SPCC965.11c, SPCC663.06c, SPBC947.05c, SPBC1773.06c and SPCC737.04) compared with any of the single mutants (Figure 4F). In addition, at five loci (SPBC3E7.02c, SPAC15E1.02c, SPAC11D3.01c, SPAC212.08c and SPAC27D7.09c) the double mutant showed transcript levels similar to the clr4Δ mutant and at two loci (SPBPB21E7.07 and SPBPB2B2.08) similar to the dbl2Δ mutant. We would like to stress that not all genes behaved the same way in each mutant background. Therefore, the complex pattern of results obtained from epistasis analyses makes it difficult to draw a general conclusion about the relationship between dbl2 and genes involved in gene repression pathways.
Figure 4.

Gene expression in the dbl2Δ single and double mutants in combination with mutations of genes involved in gene expression. RNA was isolated from cells in the exponential phase (OD595 = 0.5–0.55), and gene expression was analysed using qPCR. The data represent transcript levels relative to wild-type after normalisation to act1 and tbp1. The plotted values are the mean of three independent biological replicates ± standard error of the mean; asterisks denote P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) from two-tailed Student's t-tests, which was used to assess the significance of difference between the single mutants and the double mutant. (A) Gene expression in the dbl2Δ (SP067), hip1Δ (SP456) and dbl2Δhip1Δ (SP467) mutants compared with wt (SP065). (B) Gene expression in the dbl2Δ (SP067), slm9Δ (SP462) and dbl2Δslm9Δ (SP471) mutants compared with wt (SP065). (C) Gene expression in the dbl2Δ (SP067), hip3Δ (SP458) and dbl2Δhip3Δ (SP468) mutants compared with wt (SP065). (D) Gene expression in the dbl2Δ (SP067), clr6–1 (SP415) and dbl2Δclr6–1 (SP829) mutants compared with wt (SP065). (E) Gene expression in the dbl2Δ (SP067), dcr1Δ (SP435) and dbl2Δdcr1Δ (SP501) mutants compared with wt (SP065). (F) Gene expression in the dbl2Δ (SP067), clr4Δ (SP434) and dbl2Δclr4Δ (SP503) mutants compared with wt (SP065).
Given that protein–DNA interactions depend on the temperature (109), we tested the growth of single and double mutants at 37°C to further explore whether the analysed proteins function in a common or a parallel pathway. A previous study demonstrated that deletion of any subunit of the HIRA complex (hip1Δ, hip3Δ, hip4Δ and slm9Δ) results in temperature sensitivity (51). Combining mutations in hip1Δ, hip4Δ or slm9Δ with dbl2Δ rescued the growth of HIRA mutants at 37°C, suggesting that Dbl2 could act in the same pathway as the HIRA complex (Figure 5). The dbl2Δhip3Δ double mutant did not grow at 37°C, confirming that Hip3 behaves differently from the other members of the HIRA complex. The Clr6 complex is present in S. pombe in four distinct complexes (complex I, I’, I’’ and II) that share a common catalytic subunit, Clr6 (93). Clr6-1 and the pst2Δ and alp13Δ mutants (subunits of complex II) were sensitive to higher temperature. Combining mutations in clr6-1, pst2Δ and alp13Δ with dbl2Δ did not improve growth at 37°C (Figure 5). Moreover, introduction of the dbl2Δ mutation into the dcr1Δ background resulted in temperature sensitivity, indicating some functional redundancy between these two proteins.
Figure 5.
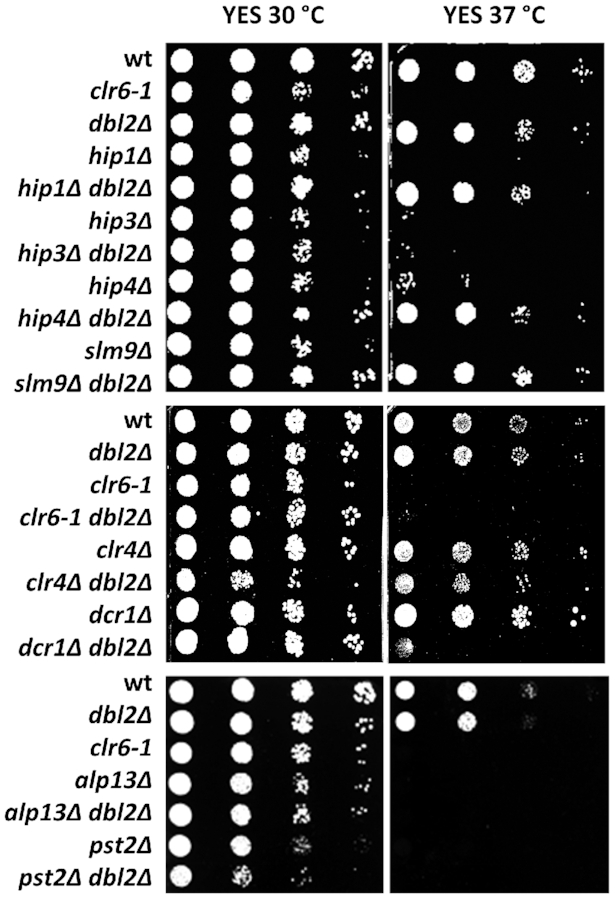
Deletion of dbl2 suppresses the thermosensitivity of three out of four HIRA mutants, but not Clr6 complex mutants. Wild-type (SP065) and mutant strains clr6–1 (SP415), dbl2Δ (SP067), hip1Δ (SP456), hip1Δdbl2Δ (SP467), hip3Δ (SP458), hip3Δdbl2Δ (SP468), hip4Δ (SP460), hip4Δdbl2Δ (SP470), slm9Δ (SP462), slm9Δdbl2Δ (SP471), clr6–1dbl2Δ (SP829), alp13Δ (SP527), dbl2Δalp13Δ (SP553), pst2Δ (SP535), dbl2Δpst2Δ (SP561), dcr1Δ (SP435), dbl2Δdcr1Δ (SP501), clr4Δ (SP434) and clr4Δdbl2Δ (SP503) were cultivated until the exponential phase in YES medium. Tenfold serial dilutions of cell suspensions were spotted on the indicated plates. Images were taken after 3-day cultivation at 30 or 37°C (as indicated).
Dbl2 antagonises the silencing activity of Hip1 and Slm9 at outer centromeres
HIRA proteins have also been implicated in heterochromatin assembly and silencing in a range of organisms. Loss of HIRA protein in fission yeast leads to defective pericentric heterochromatin formation (101). To investigate whether Dbl2 affects pericentric heterochromatin, we tested the dbl2Δ mutant in a colour-based silencing assay (59). For comparison, we included deletion mutants of genes encoding HIRA subunits. As expected, HIRA null mutants alleviated silencing of the ade6+inserted at the outer centromeric repeat region (otr1R::ade6+) (Figure 6A, B). The dbl2Δ mutant did not show defects in the pericentric silencing (Figure 6B). We next evaluated our results by measuring expression of heterochromatic repeats at centromeres (dg/dh) in the single and double mutants by qPCR (Figure 6C). Consistent with our results from the colour-based assay, the hip1Δ and slm9Δ mutants displayed increased levels of pericentric transcripts, while the dbl2Δ mutant did not show any defect in silencing. Notably, the dbl2Δ mutation restored pericentric silencing in the hip1Δ and slm9Δ mutants, but not in the hip3Δ mutant. In addition, qPCR revealed that dbl2, hip1 or slm9 deletion had no effect on the expression of subtelomeric repeats (Figure 6C). We noticed an increased level of subtelomeric transcripts only in the hip3Δ mutant and in the corresponding double mutants with dbl2Δ. Furthermore, loss of pericentric heterochromatin causes impaired centromere cohesion (110,111), resulting in chromosome missegregation and sensitivity to the microtubule destabilizing drug TBZ. To test whether dbl2 deletion in HIRA mutant cells restores pericentric heterochromatin, we analysed the sensitivity of single and double mutants to TBZ. As expected, hip1Δ or slm9Δ showed severe sensitivity to TBZ (101) (Figure 6D). Combining dbl2Δ with hip1Δ or slm9Δ suppressed the TBZ sensitivity of HIRA mutants (Figure 6D). In contrast, the dbl2Δhip3Δ double mutant showed increased sensitivity to TBZ compared with single mutants (Figure 6D), consistent with our previous qPCR results of pericentric transcripts. In conclusion, these data suggest that Dbl2 is involved in suppression of expression within the specific subnuclear domains, but in contrast to HIRA, its absence does not alleviate silencing at centromeres. However, dbl2 deletion completely restored the silencing defect of hip1Δ and slm9Δ mutants at the outer centromere, indicating that Dbl2 may act in an antagonistic manner to Hip1 and Slm9. Notably, this phenomenon does not apply to hip3Δ.
Figure 6.

Deletion of dbl2 suppresses the silencing defect of hip1Δ and slm9Δ mutants at the outer centromeric region. (A) Schematic representation of the otr1R::ade6+ reporter and dg/dh repeats in the centromeric region. (B) Growth of strains on YES with limited adenine. The test strain with ade6+gene insert (SP392) with additional deletion of dbl2 (SP485, SP486), hip1 (SP489) and slm9 (SP487, SP488) were cultivated until the exponential phase, and tenfold serial dilutions of cell suspensions were spotted on the indicated plates. Images were taken after 3-day cultivation at 30°C. (C) qPCR analysis of dg/dh repeats and subtelomeric transcripts. RNA was isolated from wild-type (SP065); single mutants dbl2Δ (SP067), hip1Δ (SP456), slm9Δ (SP462) and hip3Δ (SP458); and double mutants dbl2Δhip1Δ (SP467), dbl2Δslm9Δ (SP471) and dbl2Δhip3Δ (SP468) grown to the exponential phase (OD595 = 0.5–0.55). The data represent transcript levels relative to wild-type after normalisation to act1 and tbp1. The plotted values are the mean of three independent biological replicates ± standard error of the mean; asterisks denote P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) from two-tailed Student's t-tests, which was used to assess the significance of difference between the single mutants and the double mutant. (D) Growth of strains on YES and YES supplemented with thiabendazole (TBZ) (15 μg/ml). The strains—wt (SP065), dbl2Δ (SP067), hip1Δ (SP456), dbl2Δhip1Δ (SP467), hip3Δ (SP458), dbl2Δhip3Δ (SP468), hip4Δ (SP460), dbl2Δhip4Δ (SP470), slm9Δ (SP462) and dbl2Δslm9Δ (SP471) – were cultivated until the exponential phase, and tenfold serial dilutions of cell suspensions were spotted on the indicated plates. Images were taken after 3-day cultivation at 30°C.
Dbl2 affects nucleosome occupancy and histone modifications
Loss of HIRA (hip1Δ) leads to a global reduction in nucleosome occupancy, which is most pronounced towards the 3′ end of the genes (74). Reduced levels of specific nucleosome peaks had been detected in hip1Δ over individual ORFs, at Tf2 LTR retrotransposons, at some promoters and at heterochromatic repeats (74). Since our data suggest that Dbl2 and HIRA could act in the same pathway, we wondered whether Dbl2 promotes nucleosome occupancy at similar loci. We addressed this possibility by using a chromatin sequencing technology to measure genome-wide nucleosome occupancy (74,112). Chromatin derived from three biological replicates was digested with MNase to generate a DNA ladder with a highly similar molecular weight distribution between wild-type and a deletion mutant (Supplementary Figure S6A). To validate our results, we compared nucleosome patterns of wild-type and hip1Δ with already published results (74). Chromatin at the 5′ end of eukaryotic gene typically consists of a nucleosome-depleted region (NDR) located immediately upstream of the RNA pol II transcription start site (TSS), which is surrounded by an ordered nucleosomal array that extends into the coding sequence (113,114). As previously described, a comparison of average nucleosome positions surrounding the TSS in the hip1Δ mutant did not alter the NDR or the +1 nucleosome peak, but it showed reduced amplitudes of the nucleosome peaks in coding region (Figure 7A). By contrast, comparison of average nucleosome occupancy from dbl2Δ and wild-type revealed slightly increased amplitudes of the –2, –1 and +1 nucleosome peaks (relative to NDR) (P < 1.35 × 10−4), suggesting that Dbl2 loss could lead to increased nucleosome stability in these regions (Figure 7A). Furthermore, plots of average nucleosome profiles of Tf2 LTR retrotransposons from hip1Δ showed reduced nucleosome peaks downstream of the TSS and marked changes to the nucleosome positioning, mainly at the 3′-end of the genes (Supplementary Figure S6B). Consistent with our previous results, the dbl2Δ mutant showed slightly increased nucleosome peaks around the TSS and did not exhibit any changes to nucleosome positioning (Supplementary Figure S6B). We further confirmed these results by investigating the nucleosome profiles of both mutants at the individual genes (Supplementary Figure S7). Finally, we compared the chromatin organisation of HIRA-repressed genes (48) and Dbl2-repressed genes with the complete set of S. pombe coding genes. In both cases, we found that these genes exhibited lower peaks in the coding sequences and narrower and shallower NDR (Supplementary Figure S7H). This type of nucleosome profiles correlates with genes exhibiting lower median expression levels (114). Taken together, we found that Dbl2 and HIRA suppress expression of genes with similar chromatin organisation, however, unlike the hip1Δ mutant, the dbl2Δ mutant did not show any reduction in the nucleosome occupancy. On the contrary, our data suggest that in the dbl2Δ mutant the –2, –1 and +1 nucleosome peaks are slightly increased, indicating its role in destabilisation of the nucleosomes around the TSS.
Figure 7.

Nucleosome occupancy and histone modifications are altered in the dbl2 and hip1 mutants compared with the wild-type. (A) Average nucleosome occupancy profiles for all genes aligned at the transcription start site (TSS) in wild-type (SP072, SP065), hip1Δ (SP456) and dbl2Δ (SP067) strains. The plotted values are the mean of three independent biological replicates. To compare empirical distribution between wild-type and dbl2Δ, we used the Kolmogorov–Smirnov test from open-source scipy library, which showed statistically significant differences (P < 1.35 × 10−4). (B, C) Western blots were used to measure histone modifications in the indicated strains. The levels of histone modifications were normalised to total histone H3. The plotted values are the mean of four independent biological replicates ± standard error of the mean; asterisks denote P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) from two-tailed Student's t-tests, which was used to assess the significance of difference between wild-type and the mutants. (D) H3K9me2 occupancy at subtelomeres, centromeres and rDNA (marked by black horizontal bars) is increased in the dbl2Δ mutant. H3K9me2 ChIP-seq coverage in each sample was first normalised to the corresponding total H3 ChIP-seq coverage, and the dbl2Δ values were then further normalised to the WT values from the corresponding biological replicate. Final normalised H3K9me2 occupancy was plotted along all three fission yeast chromosomes. Results from two independent experiments are shown. The subtelomeres are here defined as regions of H3K9me2 enrichment in wild-type cells. (E) H3K9me2 occupancy at LTRs is increased in the dbl2Δ mutant. H3K9me2 occupancy normalised as in (D) was plotted for 239 LTR regions (LTR and 1 kb flanking regions) and for matched random control loci as a heatmap (right panel) or average locus profiles (left panel). Results from two independent experiments are shown.
In addition to nucleosome positioning, gene expression can be regulated through changes in histone modification patterns. In general, euchromatin is rich in hyperacetylated histones (e.g. H3K9ac) and methylated histones H3 at lysine 4 (e.g. H3K4me3), whereas heterochromatin is characterised by methylation of histone H3 at lysine 9 (e.g. H3K9me2 and H3K9me3) (115). Previous studies have shown that hip1Δ exhibits a substantial increase in H3K9ac levels (95). To gain further insight into Dbl2 functions, we tested the levels of histone modifications in the dbl2Δ, hip1Δ and hip3Δ single mutants and in the dbl2Δhip1Δ and dbl2Δhip3Δ double mutants using western blotting (Figure 7B, C). Our analysis showed the increased levels of H3K9ac and decreased levels of H3K9me2 and H3K9me3 in the hip1Δ and hip3Δ mutants. On the contrary, the levels of H3K9ac were significantly decreased in the single and double mutants of dbl2Δ and the levels of H3K9me2 and H3K9me3 were significantly increased in the dbl2Δ mutant. These data are in agreement with the previous conclusion that HIRA is required for histone deacetylation via Clr6 (95). Of note, with respect to histone modification, dbl2 is epistatic to both subunits of the HIRA complex (hip1 and hip3). To analyse H3K9me2 further, we performed ChIP-seq analysis (Figure 7D, E). The dbl2Δ mutant showed elevated levels of H3K9me2 at constitutive heterochromatic regions, i.e., centromeres, subtelomeres and rDNA compared with the wild-type cells (Figure 7D, Supplementary Figure S8). Moreover, a closer examination of the data revealed elevated levels of H3K9me2 at LTRs in the dbl2Δ mutant (Figure 7E). Taken together, these results suggest that although gene expression in the dbl2Δ mutant is mostly increased, Dbl2 is required for nucleosome destabilisation and reduction of H3K9me2 at centromeres, subtelomeres, rDNA regions and LTRs.
Proteins involved in HR repress expression from the same genomic regions as Dbl2
In HR, Dbl2 is required for the formation of Fbh1 DNA helicase foci in order to remove Rad51 protein and to process DNA joint molecules (15). To address whether Dbl2 also exerts its function through Fbh1 in the process of the repression of gene expression, we assayed the effects of fbh1Δ and dbl2Δ single and double mutants on transcript levels from several loci using qPCR. The transcript levels in fbh1Δ at measured loci were similar to those in dbl2Δ; combining the dbl2Δ and fbh1Δ mutations did not show any additive increase in transcript levels compared with the single mutants (Figure 8A). To further examine this phenomenon, we assayed the transcript levels in the F-box (fbh1L14A/P15A) fbh1 mutant and in the helicase-defective (fbh1D485N) fbh1 mutant (13). The transcript levels in the fbh1D485N mutant were comparable to those in the fbh1Δ mutant, whereas the fbh1L14A/P15A mutant also showed increased – albeit to a lesser extent – transcript levels, indicating that Fbh1 and mainly its helicase domain function with Dbl2 in the repression of gene expression.
Figure 8.

Cells lacking homologous recombination proteins show expression profiles similar to dbl2Δ. (A) RNA was isolated from wild-type (SP065), dbl2Δ (SP067), fbh1Δ (SP070), fbh1D485N (SP633), fbh1L14A/P15A (SP629), dbl2Δfbh1Δ (SP613), rad51Δ (SP068), dbl2Δrad51Δ (SP735), mus81Δ (SP375), rad54Δ (SP025) and dmc1Δ (SP069) strains in the exponential phase, and gene expression was analysed using qPCR. The data represent transcript levels relative to wild-type after normalisation to act1 and tbp1. The plotted values are the mean of three independent biological replicates ± standard error of the mean; asterisks denote P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) from two-tailed Student's t-tests, which was used to assess the significance of difference between fbh1Δ and fbh1L14A/P15A. (B) Venn diagrams showing overlap between genes upregulated in the dbl2Δ mutant (SP067) and genes upregulated in the rad51Δ (SP068) or mus81Δ (SP375) mutant. Only genes included in both analyses were compared. The P-value indicates the probability that the observed overlap happened by chance. (C) RNA was isolated from wild-type (SP065), dbl2Δ (SP067), ku70Δ (SP700) and lig4Δ (SP842) strains in exponential phase and gene expression was analysed using qPCR. The dbl2Δ strain was used as a positive control. (D, E) RNA was isolated from wild-type (SP065), rad51Δ (SP068), hip1Δ (SP456) and rad51Δhip1Δ (SP793) strains in the exponential phase and gene expression was analysed using qPCR. The plotted values are the mean of three independent biological replicates ± standard error of the mean; asterisks denote P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***) from two-tailed Student's t-tests, which was used to assess the significance of difference between the single mutants and the double mutant. (F) Model of repression of gene expression near hard-to-replicate sites such as repetitive sequences. In wild-type cells, hard-to-replicate sites trigger replication fork stalling or collapse, which is associated with the generation of single-stranded or double-stranded DNA breaks. Homologous recombination proteins along with the HIRA histone chaperone ensure that repressive chromatin is assembled and genome integrity is maintained.
A key Fbh1 function is to disrupt Rad51 nucleoprotein filaments via two activities: DNA unwinding/translocation and ubiquitin ligation (12). We therefore hypothesised that in the dbl2Δ or fbh1Δ mutants the level of transcripts at measured loci will be affected by Rad51 accumulation. We tested this hypothesis by analysing a rad51Δ deletion mutant (15). To our surprise, the rad51Δ mutant showed increased transcript levels similar to the dbl2Δ mutant (except for SPBPB21E7.07), suggesting that the presence of Rad51 protein is necessary to repress gene expression. The corresponding double mutant with dbl2Δ displayed decreased transcript levels of four tested loci and a slight increase (statistically not significant) of SPAC4H3.08 compared with the single mutants (Figure 8A). These unexpected results prompted us to assay the level of transcripts in deletion mutants of other genes encoding further proteins involved in HR. We selected Rad54, which enables D-loop formation (22–24) and promotes DNA strand invasion by the Rad51 presynaptic filament (19–21), and Mus81, which is a subunit of the Mus81–Eme1 Holliday junction resolvase (14,31,116–119). Both rad54Δ and mus81Δ deletion mutants showed increased transcript levels at measured loci. In order to validate the qPCR data, we compared the expression profiles of the rad51Δ and mus81Δ mutants with those of wild-type by using strand-specific RNA-seq (Figure 8B, Supplementary Table S3). As expected, the genes that were upregulated in dbl2Δ were highly correlated with upregulated genes in the rad51Δ and mus81Δ mutants (P < 3.8 × 10−87 and P < 8.5 × 10−177, respectively). To exclude the possibility that the altered expression profiles are due to the presence of the deletion cassettes used to generate corresponding mutants, we analysed the impact of the dmc1 gene deletion. Dmc1 is a meiosis-specific recombinase that mediates homologous DNA pairing (120), so its deletion in mitotically dividing cells should not cause any effect. Using qPCR, we detected no change in expression of measured genes in the dmc1Δ strain. We next asked whether genes involved in NHEJ are also required for gene repression. Using qPCR, we assayed transcript levels in deletion mutants of genes encoding protein Pku70 and the DNA repair ligase Lig4 (Figure 8C). The Ku70/Ku80 complex binds dsDNA ends, inhibits end-resection and allows the ligation of DSBs by Lig4 (1,121,122). Cells lacking Pku70 or Lig4 did not show any increase in transcript levels (Figure 8C), suggesting that the NHEJ pathway is not required for gene repression at measured loci.
The ability of dbl2Δ to suppress the silencing defect of hip1Δ and slm9Δ mutants at pericentric region prompted us to test genetic interactions between rad51 and hip1. The rad51Δ and hip1Δ double mutants showed mostly decreased transcript levels compared with the single mutants at genomic loci (Figure 8D). Although the nature of the decreased transcript levels in the double mutants remains unclear, the non-additive phenotype of the double mutants suggests that they could act in the same pathway. Furthermore, deletion of rad51Δ restored the silencing defect of hip1Δ at the outer centromere region (Figure 8E). In addition, combining mutations in hip1Δ or slm9Δ with rad51Δ partially rescued the growth of all mutants at 37°C (Supplementary Figure S9). In conclusion, these results suggest a coordinated pathway involving proteins of HR and the HIRA complex in the repression of a large number of genes. It is possible that this coordinated action of HR and HIRA also contributes to the regulation of centromeric silencing.
DISCUSSION
We present here that the Dbl2 protein along with other proteins involved in HR or replication fork integrity, such as Fbh1, Rad51, Rad54 and Mus81, are required for repression of gene expression (Figure 8). We linked the Dbl2 protein to the HIRA histone chaperone by genetic interactions (Figures 4–7). Our data suggest a possible role for HR proteins in coordinating nucleosome occupancy to assemble repressive chromatin at a large number of genes, which are close to repeat sequences.
Our results suggest that Dbl2 could function in the same pathway as the HIRA complex regarding repression of gene expression at euchromatic and heterochromatic loci. Notably, cells lacking either Dbl2 or HIRA factors (Hip1 or Slm9) exhibit increased levels of antisense RNA and RNA from the Tf2 LTR retrotransposons, subtelomeric genes and meiotic genes (48,51,95). The transcript levels in the hip1Δdbl2Δ or slm9Δdbl2Δ double mutants are mostly similar to that of dbl2Δ single mutant, indicating that Dbl2 might act upstream of HIRA (Figures 4A, B and 6C). Moreover, introduction of the dbl2Δ mutation into the hip1Δ, hip4Δ and slm9Δ background rescues the growth of HIRA mutants at 37°C (Figure 5) or in the presence of TBZ (Figure 6D). Furthermore, genetic interactions between Dbl2 and HIRA extend to histone modifications, because introduction of dbl2Δ into hip1Δ and hip3Δ reduces H3K9ac to levels found in the dbl2Δ mutant (Figure 7B, C).
There are two main pathways in S. pombe that repress antisense expression at euchromatic loci. The Clr4 complex and RNAi factors (e.g. Dcr1) mediate degradation of read-through antisense RNA via a mechanism involving nuclear exosome (88), and a pathway involving the Clr6, Set2 and the HIRA complex represses initiation of antisense transcripts from cryptic promoters (93,95). At some loci dbl2 acts in a redundant manner with clr4 (Figure 4F) and genes with increased antisense levels in the dbl2Δ mutant do not map preferentially to convergent loci (49%). These findings suggest that Dbl2 rather acts in a pathway involving HIRA. However, the epistasis analyses regarding repression of gene expression at euchromatic loci showed a very complex pattern of interactions between dbl2 and clr4, dcr1, clr6-1 and hip3 (Figure 4). Therefore, these results cannot be used to unambiguously describe genetic pathways involved in these processes.
A previous report revealed that cells lacking the HIRA histone chaperone experience a global reduction in nucleosome occupancy at both euchromatic and heterochromatic loci (74). Our results indicate that nucleosome peaks around the TSS are slightly increased in the dbl2Δ mutant compared with the wild-type (Figure 7A), indicating that Dbl2 could be involved in removing excess nucleosomes. In support of this notion, Rad51 overexpression stimulates the histone eviction at DSBs in bre1Δ mutant (123). In wild-type cells, Bre1 stimulates histone eviction at DSBs by H2B ubiquitination (123). Furthermore, a recent study analysing regulators of heterochromatin spreading in different chromatin contexts demonstrated that the HIRA histone chaperone and genes involved in HR pathway, such as rad50 and sfr1, act in opposite ways (124). The authors detected these actions mainly at the ectopic heterochromatin domain composed of dh element embedded in gene-rich euchromatin. The HIRA complex has been implicated in heterochromatic domains expansion, while rad50 and sfr1 have been found to function in heterochromatin destabilisation (124). Consistently, we show that H3K9me2 nucleosomes are elevated in the dbl2Δ mutant at constitutive heterochromatic regions and LTRs (Figure 7D, E). It remains unclear how these chromatin changes could increase gene expression in the dbl2Δ mutant, although it has been shown that H3K9me2 domains are transcriptionally active and contain modifications associated with euchromatic transcription (125). Additional studies in HR-deficient mutants should be now conducted in order to establish how HR proteins affect chromatin status and stability.
Both the Dbl2 protein and the HIRA complex are involved in repression of Tf2 LTR retrotransposons. Tf2 LTR retrotransposons are organised into Tf bodies, the function of which depends on CENP-B (Abp1, Cbh1 and Cbh2), the Set1 histone methyltransferase (126) and HDAC proteins (Clr6, Clr3, and Hst4) (92,127–129). The RNAi machinery plays only an accessory role to the exosome in this process (82,130,131). Importantly, CENP-B-mediated inhibition of Tf2 LTR transcription is independent of the repressive function of HIRA complex (128). The positive genetic interactions between dbl2 and HIRA suggest that HIRA and Dbl2 might represent a common pathway of Tf2 repression distinct from CENP-B regulation (132). However, it still needs to be experimentally confirmed. In human cells after infection with naked viral DNAs, HIRA co-localises with viral genomes, binds to incoming viral DNAs and deposits H3.3 onto them (133). Given the parallels between Tf2 and viral DNA, it will be interesting to determine whether the human ortholog of Dbl2 (ZGRF1) is also required for repression of viral DNA.
It is surprising that hip3 behaves differently from the other members of the HIRA complex (Figures 4, 5 and 6). The dbl2Δhip3Δ double mutant showed at some loci a slight additive increase in transcript levels compared with the single mutants (Figures 4C and 6C), and introduction of dbl2Δ did not rescue the growth of hip3Δ at 37°C or on TBZ (Figures 5 and 6D). These findings indicate a very different, but currently unknown, function for Hip3 in gene expression. Nevertheless, these results are consistent with a previous report that linked different physical interactions, involving HIRA (ortholog of Hip1 and Slm9), UBN1 (ortholog of Hip4) and ASF1, to the distinct functional properties of active chaperone complexes in HeLa cells (134). Systematic mapping of Hip3 physical and genetic interactions will be required to better understand its function in gene expression and chromatin remodelling.
Our analyses further revealed that other proteins involved in HR, such as Fbh1, Rad51, Rad54 and Mus81, are also required for the repression of gene expression (Figure 8A, B). The role of HR proteins in the regulation of gene expression has also been observed in other organisms. In human cell lines with deficiency in HR repair genes, expression of numerous genes required for DNA damage repair, cell cycle regulation and DNA replication is upregulated or downregulated (135). For example, the expression levels of three genes encoding DSB end resection enzymes, BLM, DNA2 and EXO1, are all reduced in HR-deficient cell lines created by depletion of independent HR genes (135). In plants, HR proteins such as RAD51D, RAD51 and XRCC2 have been shown to regulate expression of pathogen-related genes and genes important for DNA repair, abiotic stress and transcriptional regulation (136–138). Our RNA-seq analysis of the dbl2Δ mutant did not reveal differentially expressed protein-coding genes involved in DNA repair, but there were differentially expressed genes involved in meiosis, detoxication and transport. Moreover, these misregulated loci in the dbl2Δ mutant are near repetitive DNA elements, such as long tandem and long terminal repeats (Figure 2). However, this correlation between the localisation of misregulated genes and the localisation of LTRs may be an indirect consequence of the preference of Tf LTR retrotransposons to insert in the promoters of certain types of genes (139,140). Our findings are also similar to recent studies that have implicated HR proteins in the repression of repeat sequences and transposable elements. Loss of Brca1 in mice results in transcriptional derepression of satellite DNA sequences (141). Furthermore, the loss of RAD51 and ATR checkpoint kinase triggers satellite expression and the loss of SET-25 methyltransferase leads to expression of retrotransposons and tissue-specific genes in Caenorhabditis elegans (142).
How do HR and HIRA orchestrate the assembly of repressive chromatin?
Our data provide evidence that cells lacking Dbl2 exhibit increased levels of transcripts located closer to long terminal repeats and long tandem repeats compared with the loci with no change in expression. LTRs in S. pombe impair DNA replication by blocking fork progression, a property that results from Sap1 binding to LTRs (132). The authors proposed that the function of this block is to control the direction of transposon replication, possibly by coordinating lagging strand synthesis, which prevents single strand annealing from complementary direct repeats (132). CENP-B factors counteract Sap1 activity and promote replication fork progression through the LTRs (132). Blocked replication forks are potential sources of genome instability because they can lead to replisome collapse and DSB formation (143). Consistent with the fact that LTR regions are prone to replication fork stalling, some of the loci misregulated in dbl2Δ overlap with genomic loci covered with γH2A (Figure 2C) (85). Transient formation of γH2A is assumed to result from replication fork arrest or collapse during the S-phase (85). If the LTR loci in question are constantly experiencing stalled replication forks during normal replication, their DNA damage and repair load should be higher, which should result in a different histone landscape compared with other chromosomal regions, which keeps replenishing itself by continuous cell division. This finding is in agreement with our results that the dbl2Δ cells grown in the stationary phase did not show significant changes in gene expression (Supplementary Figure S2). We propose that Dbl2 and other HR proteins are implicated in repression of genes located near hard-to-replicate sites such as LTRs (Figure 8B). The involvement of Mus81–Eme1 and Rad54 in this process suggests that stalled replication forks could be converted into DSBs (144–148) and processed by HR. In the absence of HR proteins, other pathways such as NHEJ should be involved in DSB repair. A study recently reported that several chromatin modifications which occur proximal to DSBs, are indeed pathway-specific (149). Additional studies should now be conducted to establish whether different histone modifications depending on HR or NHEJ repair are established at these regions and how they affect gene expression.
It is notable that in minimal medium we observed much smaller changes in gene expression in the dbl2Δ mutant (Supplementary Figure S2). The specific effect of Dbl2 under YES growth conditions is not clear, however, the data may reflect either more replication–transcription collisions under rapid growth conditions or the environmental control of the epigenetic state. Recent experiments determining the role of TOR2, a major regulator of eukaryotic cellular growth, revealed that epigenetic stability of subtelomeric chromatin responds to cellular growth conditions (150). This is remarkably similar to significant upregulation of genes at the subtelomeric regions of chromosomes I and II in dbl2Δ (chi-squared test, P < 2.12 × 10−49). Whether the effect of Dbl2 and TOR2 is related remains to be investigated.
Our data suggest a link between Dbl2 and the HIRA histone chaperone. A recent study showed that S. pombe cells lacking HIRA experience a global reduction in nucleosome occupancy at gene sequences, although there are some regions of the genome that are more severely perturbed (74). Our observation that HR and HIRA could act in the same pathway would explain why specific regions of the genome show greater dependency upon HIRA than others. Replication protein A (RPA), which is best known for its role in DNA repair and replication (151), has been recently implicated in human cells also in the efficient deposition of newly synthesised H3.3 by the HIRA histone chaperone at promoters and enhancers (152). Although it is presently unclear how this process is elicited at a mechanistic level, it is tempting to speculate that Dbl2 in concert with HR may contribute to the eviction of histones followed by HIRA-mediated nucleosome occupancy to assemble repressive chromatin near long terminal and long tandem repeats (Figure 8F).
DATA AVAILABILITY
RNA-seq data are available at the National Center for Biotechnology Information (NCBI) under accession numbers PRJNA601919 and PRJNA659385. The ChIP-seq data are available at the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under an accession number E-MTAB-9619. The scripts used for ChIP-seq data processing and analyses are available at https://github.com/mprevorovsky/bagelova-polakova-dbl2-histones.
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful to Simon Whitehall (Newcastle University) and Nicholas Kent (Cardiff University) for strains, protocols and helpful discussions; Matthew Whitby (University of Oxford), Li-Lin Du (National Institute of Biological Sciences) and Robin Allshire (University of Edinburg) for strains; Peter Schlogelhofer (University of Vienna), Randy Hyppa (Fred Hutchinson Cancer Research Center), Gerry Smith (Fred Hutchinson Cancer Research Center), Adrianna Skoneczna (Polish Academy of Sciences), Lumir Krejci (Masaryk University) and Jan Palecek (Masaryk University) for helpful discussions and Maria Balazova (Slovak Academy of Sciences), Kristina Hrasnova (Slovak Academy of Sciences), Anetta Bakosova (Slovak Academy of Sciences), Barbora Huraiova (Comenius University) and Katarina Gaplovska-Kysela (Comenius University) for help with experiments.
Author contributions: S.B.P., I.M., A.P., L.C., M.P., J.Gr and T.Sz planned experiments and analysed data. I.M., A.P., S.B.P., T.Se, A.J., Z.B., M.S., N.M., K.P., L.C. and L.S. performed experiments. J.B., J.Ga and M.P. performed bioinformatic analysis. S.B.P., J.Gr and I.M. wrote the paper with contributions from all authors.
Contributor Information
Ivana Misova, Institute of Animal Biochemistry and Genetics, Centre of Biosciences, Slovak Academy of Sciences, 840 05 Bratislava, Slovakia.
Alexandra Pitelova, Institute of Animal Biochemistry and Genetics, Centre of Biosciences, Slovak Academy of Sciences, 840 05 Bratislava, Slovakia.
Jaroslav Budis, Comenius University Science Park, 841 04 Bratislava, Slovakia; Geneton Ltd., 841 04 Bratislava, Slovakia; Slovak Centre of Scientific and Technical Information, 811 04 Bratislava, Slovakia.
Juraj Gazdarica, Geneton Ltd., 841 04 Bratislava, Slovakia; Slovak Centre of Scientific and Technical Information, 811 04 Bratislava, Slovakia; Department of Molecular Biology, Faculty of Natural Sciences, Comenius University in Bratislava, 841 04 Bratislava, Slovakia.
Tatiana Sedlackova, Comenius University Science Park, 841 04 Bratislava, Slovakia; Geneton Ltd., 841 04 Bratislava, Slovakia.
Anna Jordakova, Department of Cell Biology, Faculty of Science, Charles University, 128 00 Praha 2, Czechia.
Zsigmond Benko, Institute of Animal Biochemistry and Genetics, Centre of Biosciences, Slovak Academy of Sciences, 840 05 Bratislava, Slovakia; Department of Molecular Biotechnology and Microbiology, Faculty of Science and Technology, University of Debrecen, H-4010 Debrecen, Hungary.
Maria Smondrkova, Department of Genetics, Faculty of Natural Sciences, Comenius University in Bratislava, 841 04 Bratislava, Slovakia.
Nina Mayerova, Department of Genetics, Faculty of Natural Sciences, Comenius University in Bratislava, 841 04 Bratislava, Slovakia.
Karoline Pichlerova, Institute of Animal Biochemistry and Genetics, Centre of Biosciences, Slovak Academy of Sciences, 840 05 Bratislava, Slovakia.
Lucia Strieskova, Comenius University Science Park, 841 04 Bratislava, Slovakia; Geneton Ltd., 841 04 Bratislava, Slovakia.
Martin Prevorovsky, Department of Cell Biology, Faculty of Science, Charles University, 128 00 Praha 2, Czechia.
Juraj Gregan, Advanced Microscopy Facility, VBCF and Department of Chromosome Biology, Max Perutz Labs, University of Vienna, Vienna Biocenter (VBC), 1030 Vienna, Austria.
Lubos Cipak, Cancer Research Institute, Biomedical Research Center, Slovak Academy of Sciences, 845 05 Bratislava, Slovakia.
Tomas Szemes, Comenius University Science Park, 841 04 Bratislava, Slovakia; Geneton Ltd., 841 04 Bratislava, Slovakia; Slovak Centre of Scientific and Technical Information, 811 04 Bratislava, Slovakia; Department of Molecular Biology, Faculty of Natural Sciences, Comenius University in Bratislava, 841 04 Bratislava, Slovakia.
Silvia Bagelova Polakova, Institute of Animal Biochemistry and Genetics, Centre of Biosciences, Slovak Academy of Sciences, 840 05 Bratislava, Slovakia; Department of Genetics, Faculty of Natural Sciences, Comenius University in Bratislava, 841 04 Bratislava, Slovakia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
People Programme (Marie Curie Actions) European Union's Seventh Framework Programme under REA grant agreement [0070/01/02]; Slovak Research and Development Agency [APVV-18-0219, APVV-17-0130, APVV-16-0120]; Slovak Grant Agency, VEGA [1/0013/2020, 2/0034/19, 1/0048/16, 2/0026/18, 1/0450/18, 2/0039/19]; Grant Agency of Charles University [GAUK248120]; European Cooperation in Science and Technology Action CA16124 Bio-Brillouin; and by the Austrian Science Fund (FWF) [P30516]. Funding for open access charge: [APVV-18-0219].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mimitou E.P., Symington L.S.. DNA end resection—unraveling the tail. DNA Repair (Amst.). 2011; 10:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ceccaldi R., Rondinelli B., D’Andrea A.D.. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016; 26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krogh B.O., Symington L.S.. Recombination proteins in yeast. Annu. Rev. Genet. 2004; 38:233–271. [DOI] [PubMed] [Google Scholar]
- 4. Ranjha L., Howard S.M., Cejka P.. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma. 2018; 127:187–214. [DOI] [PubMed] [Google Scholar]
- 5. Grishchuk A.L., Kohli J.. Five RecA-like proteins of Schizosaccharomyces pombe are involved in meiotic recombination. Genetics. 2003; 165:1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Young J.A., Hyppa R.W., Smith G.R.. Conserved and nonconserved proteins for meiotic DNA breakage and repair in yeasts. Genetics. 2004; 167:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellermeier C., Schmidt H., Smith G.R.. Swi5 acts in meiotic DNA joint molecule formation in Schizosaccharomyces pombe. Genetics. 2004; 168:1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haruta N., Kurokawa Y., Murayama Y., Akamatsu Y., Unzai S., Tsutsui Y., Iwasaki H.. The Swi5-Sfr1 complex stimulates Rhp51/Rad51- and Dmc1-mediated DNA strand exchange in vitro. Nat. Struct. Mol. Biol. 2006; 13:823–830. [DOI] [PubMed] [Google Scholar]
- 9. Bianco P.R., Tracy R.B., Kowalczykowski S.C.. DNA strand exchange proteins: a biochemical and physical comparison. Front. Biosci. 1998; 3:D570–603. [DOI] [PubMed] [Google Scholar]
- 10. Morrical S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015; 7:a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krejci L., Macris M., Li Y., Van Komen S., Villemain J., Ellenberger T., Klein H., Sung P.. Role of ATP hydrolysis in the antirecombinase function of Saccharomyces cerevisiae Srs2 protein. J. Biol. Chem. 2004; 279:23193–23199. [DOI] [PubMed] [Google Scholar]
- 12. Tsutsui Y., Kurokawa Y., Ito K., Siddique M.S.P., Kawano Y., Yamao F., Iwasaki H. Multiple regulation of Rad51-mediated homologous recombination by fission yeast Fbh1. PLos Genet. 2014; 10:e1004542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Osman F., Dixon J., Barr A.R., Whitby M.C.. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 2005; 25:8084–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorenz A., West S.C., Whitby M.C.. The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic. Acids. Res. 2010; 38:1866–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Polakova S., Molnarova L., Hyppa R.W., Benko Z., Misova I., Schleiffer A., Smith G.R., Gregan J.. Dbl2 regulates Rad51 and DNA joint molecule metabolism to ensure proper meiotic chromosome segregation. PLos Genet. 2016; 12:e1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu Y., Ren J.Y., Zhang J.M., Suo F., Fang X.F., Wu F., Du L.L.. A proteome-wide visual screen identifies fission yeast proteins localizing to DNA double-strand breaks. DNA Repair (Amst.). 2013; 12:433–443. [DOI] [PubMed] [Google Scholar]
- 17. Chi P., Kwon Y., Seong C., Epshtein A., Lam I., Sung P., Klein H.L.. Yeast recombination factor Rdh54 functionally interacts with the Rad51 recombinase and catalyzes Rad51 removal from DNA. J. Biol. Chem. 2006; 281:26268–26279. [DOI] [PubMed] [Google Scholar]
- 18. Petukhova G., Stratton S., Sung P.. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature. 1998; 393:91–94. [DOI] [PubMed] [Google Scholar]
- 19. Petukhova G., Sung P., Klein H.. Promotion of Rad51-dependent D-loop formation by yeast recombination factor Rdh54/Tid1. Genes Dev. 2000; 14:2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Komen S., Petukhova G., Sigurdsson S., Stratton S., Sung P.. Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell. 2000; 6:563–572. [DOI] [PubMed] [Google Scholar]
- 21. Tan T.L., Essers J., Citterio E., Swagemakers S.M., de Wit J., Benson F.E., Hoeijmakers J.H., Kanaar R.. Mouse Rad54 affects DNA conformation and DNA-damage-induced Rad51 foci formation. Curr. Biol. 1999; 9:325–328. [DOI] [PubMed] [Google Scholar]
- 22. Jaskelioff M., Van Komen S., Krebs J.E., Sung P., Peterson C.L.. Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J. Biol. Chem. 2003; 278:9212–9218. [DOI] [PubMed] [Google Scholar]
- 23. Kwon Y., Chi P., Roh D.H., Klein H., Sung P.. Synergistic action of the Saccharomyces cerevisiae homologous recombination factors Rad54 and Rad51 in chromatin remodeling. DNA Repair (Amst.). 2007; 6:1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon Y., Seong C., Chi P., Greene E.C., Klein H., Sung P.. ATP-dependent chromatin remodeling by the Saccharomyces cerevisiae homologous recombination factor Rdh54. J. Biol. Chem. 2008; 283:10445–10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bugreev D. V, Mazina O.M., Mazin A.V.. Rad54 protein promotes branch migration of Holliday junctions. Nature. 2006; 442:590–593. [DOI] [PubMed] [Google Scholar]
- 26. Solinger J.A., Kiianitsa K., Heyer W.-D.. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51:dsDNA filaments. Mol. Cell. 2002; 10:1175–1188. [DOI] [PubMed] [Google Scholar]
- 27. Li X., Heyer W.-D.. RAD54 controls access to the invading 3′-OH end after RAD51-mediated DNA strand invasion in homologous recombination in Saccharomyces cerevisiae. Nucleic. Acids. Res. 2009; 37:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szostak J.W., Orr-Weaver T.L., Rothstein R.J., Stahl F.W.. The double-strand-break repair model for recombination. Cell. 1983; 33:25–35. [DOI] [PubMed] [Google Scholar]
- 29. Nassif N., Penney J., Pal S., Engels W.R., Gloor G.B.. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell. Biol. 1994; 14:1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorenz A., Osman F., Sun W., Nandi S., Steinacher R., Whitby M.C.. The fission yeast FANCM ortholog directs non-crossover recombination during meiosis. Science. 2012; 336:1585–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boddy M.N., Gaillard P.H., McDonald W.H., Shanahan P., Yates J.R., Russell P.. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001; 107:537–548. [DOI] [PubMed] [Google Scholar]
- 32. Carr A.M., Lambert S.. Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013; 425:4733–4744. [DOI] [PubMed] [Google Scholar]
- 33. Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T.. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010; 37:492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M.. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V.. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010; 17:1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Higgs M.R., Reynolds J.J., Winczura A., Blackford A.N., Borel V., Miller E.S., Zlatanou A., Nieminuszczy J., Ryan E.L., Davies N.J.et al.. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell. 2015; 59:462–477. [DOI] [PubMed] [Google Scholar]
- 37. Hashimoto Y., Puddu F., Costanzo V.. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2011; 19:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iraqui I., Chekkal Y., Jmari N., Pietrobon V., Fréon K., Costes A., Lambert S.A.E.. Recovery of arrested replication forks by homologous recombination is error-prone. PLos Genet. 2012; 8:e1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyabe I., Mizuno K., Keszthelyi A., Daigaku Y., Skouteri M., Mohebi S., Kunkel T.A., Murray J.M., Carr A.M.. Polymerase δ replicates both strands after homologous recombination–dependent fork restart. Nat. Struct. Mol. Biol. 2015; 22:932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mizuno K., Miyabe I., Schalbetter S.A., Carr A.M., Murray J.M.. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013; 493:246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen C.-C., Carson J.J., Feser J., Tamburini B., Zabaronick S., Linger J., Tyler J.K.. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008; 134:231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsukuda T., Fleming A.B., Nickoloff J.A., Osley M.A.. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005; 438:379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paull T.T., Gellert M.. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell. 1998; 1:969–979. [DOI] [PubMed] [Google Scholar]
- 44. van Attikum H., Fritsch O., Hohn B., Gasser S.M.. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004; 119:777–788. [DOI] [PubMed] [Google Scholar]
- 45. Gospodinov A., Vaissiere T., Krastev D.B., Legube G., Anachkova B., Herceg Z.. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol. Cell. Biol. 2011; 31:4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li X., Tyler J.K.. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. Elife. 2016; 5:e15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brachet E., Béneut C., Serrentino M.-E., Borde V.. The CAF-1 and Hir histone chaperones associate with sites of meiotic double-strand breaks in budding yeast. PLoS One. 2015; 10:e0125965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anderson H.E., Wardle J., Korkut S.V., Murton H.E., Lopez-Maury L., Bahler J., Whitehall S.K.. The fission yeast HIRA histone chaperone is required for promoter silencing and the suppression of cryptic antisense transcripts. Mol. Cell. Biol. 2009; 29:5158–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chujo M., Tarumoto Y., Miyatake K., Nishida E., Ishikawa F.. HIRA, a conserved histone chaperone, plays an essential role in low-dose stress response via transcriptional stimulation in fission yeast. J. Biol. Chem. 2012; 287:23440–23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spector M.S., Raff A., DeSilva H., Lee K., Osley M.A.. Hir1p and Hir2p function as transcriptional corepressors to regulate histone gene transcription in the Saccharomyces cerevisiae cell cycle. Mol. Cell. Biol. 1997; 17:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greenall A., Williams E.S., Martin K.A., Palmer J.M., Gray J., Liu C., Whitehall S.K.. Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. J. Biol. Chem. 2006; 281:8732–8739. [DOI] [PubMed] [Google Scholar]
- 52. Cheung V., Chua G., Batada N.N., Landry C.R., Michnick S.W., Hughes T.R., Winston F.. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 2008; 6:e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nourani A., Robert F., Winston F.. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006; 26:1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gregan J., Rabitsch P.K., Rumpf C., Novatchkova M., Schleiffer A., Nasmyth K.. High-throughput knockout screen in fission yeast. Nat. Protoc. 2006; 1:2457–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim D.U., Hayles J., Kim D., Wood V., Park H.O., Won M., Yoo H.S., Duhig T., Nam M., Palmer G.et al.. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2010; 28:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sabatinos S.A., Forsburg S.L.. Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 2010; 470:759–795. [DOI] [PubMed] [Google Scholar]
- 57. Guarente L. Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol. 1983; 101:181–191. [DOI] [PubMed] [Google Scholar]
- 58. Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976; 72:248–254. [DOI] [PubMed] [Google Scholar]
- 59. Ekwall K., Cranston G., Allshire R.C.. Fission yeast mutants that alleviate transcriptional silencing in centromeric flanking repeats and disrupt chromosome segregation. Genetics. 1999; 153:1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lyne R., Burns G., Mata J., Penkett C.J., Rustici G., Chen D., Langford C., Vetrie D., Bähler J.. Whole-genome microarrays of fission yeast: characteristics, accuracy, reproducibility, and processing of array data. BMC Genomics. 2003; 4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Okonechnikov K., Conesa A., García-Alcalde F.. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2015; 32:btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koster J., Rahmann S.. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 69. Wood V., Harris M.A., McDowall M.D., Rutherford K., Vaughan B.W., Staines D.M., Aslett M., Lock A., Bähler J., Kersey P.J.et al.. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Res. 2012; 40:D695–D699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999; 27:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dhapola P., Chowdhury S.. QuadBase2: web server for multiplexed guanine quadruplex mining and visualization. Nucleic Acids Res. 2016; 44:W277–W283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J.. g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016; 44:W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Reimand J., Kull M., Peterson H., Hansen J., Vilo J.. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic. Acids. Res. 2007; 35:W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gal C., Moore K.M., Paszkiewicz K., Kent N.A., Whitehall S.K.. The impact of the HIRA histone chaperone upon global nucleosome architecture. Cell Cycle. 2015; 14:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Převorovský M., Oravcová M., Tvarůžková J., Zach R., Folk P., Půta F., Bähler J.. Fission yeast CSL transcription factors: Mapping their target genes and biological roles. PLoS One. 2015; 10:e0137820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kizer K.O., Phatnani H.P., Shibata Y., Hall H., Greenleaf A.L., Strahl B.D.. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 2005; 25:3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mata J., Lyne R., Burns G., Bähler J.. The transcriptional program of meiosis and sporulation in fission yeast. Nat. Genet. 2002; 32:143–147. [DOI] [PubMed] [Google Scholar]
- 80. Chen D., Toone W.M., Mata J., Lyne R., Burns G., Kivinen K., Brazma A., Jones N., Bähler J.. Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell. 2003; 14:214–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Watson A., Mata J., Bähler J., Carr A., Humphrey T.. Global gene expression responses of fission yeast to ionizing radiation. Mol. Biol. Cell. 2004; 15:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cam H.P., Sugiyama T., Chen E.S., Chen X., FitzGerald P.C., Grewal S.I.S.. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 2005; 37:809–819. [DOI] [PubMed] [Google Scholar]
- 83. Tashiro S., Nishihara Y., Kugou K., Ohta K., Kanoh J.. Subtelomeres constitute a safeguard for gene expression and chromosome homeostasis. Nucleic Acids Res. 2017; 45:10333–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bowen N.J., Jordan I.K., Epstein J.A., Wood V., Levin H.L.. Retrotransposons and their recognition of pol II promoters: a comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res. 2003; 13:1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rozenzhak S., Mejía-Ramírez E., Williams J.S., Schaffer L., Hammond J.A., Head S.R., Russell P.. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 2010; 6:e1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Reinhart B.J., Bartel D.P.. Small RNAs correspond to centromere heterochromatic repeats. Science (80-.). 2002; 297:1831–1831. [DOI] [PubMed] [Google Scholar]
- 87. Volpe T.A., Kidner C., Hall I.M., Teng G., Grewal S.I.S., Martienssen R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002; 297:1833–1837. [DOI] [PubMed] [Google Scholar]
- 88. Zofall M., Fischer T., Zhang K., Zhou M., Cui B., Veenstra T.D., Grewal S.I.S.. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature. 2009; 461:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ekwall K., Ruusala T.. Mutations in rik1, clr2, clr3 and clr4 genes asymmetrically derepress the silent mating-type loci in fission yeast. Genetics. 1994; 136:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Grewal S.I., Bonaduce M.J., Klar A.J.. Histone deacetylase homologs regulate epigenetic inheritance of transcriptional silencing and chromosome segregation in fission yeast. Genetics. 1998; 150:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thon G., Bjerling K.P., Nielsen I.S.. Localization and properties of a silencing element near the mat3-M mating-type cassette of Schizosaccharomyces pombe. Genetics. 1999; 151:945–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hansen K.R., Burns G., Mata J., Volpe T.A., Martienssen R.A., Bahler J., Thon G.. Global effects on gene expression in fission yeast by silencing and RNA interference machineries. Mol. Cell. Biol. 2005; 25:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nicolas E., Yamada T., Cam H.P., FitzGerald P.C., Kobayashi R., Grewal S.I.S.. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat. Struct. Mol. Biol. 2007; 14:372–380. [DOI] [PubMed] [Google Scholar]
- 94. Zhang K., Fischer T., Porter R.L., Dhakshnamoorthy J., Zofall M., Zhou M., Veenstra T., Grewal S.I.S.. Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science (80-.). 2011; 331:1624–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yamane K., Mizuguchi T., Cui B., Zofall M., Noma K., Grewal S.I.S.. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol. Cell. 2011; 41:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Amin A.D., Vishnoi N., Prochasson P.. A global requirement for the HIR complex in the assembly of chromatin. Biochim. Biophys. Acta. 2012; 1819:264–276. [DOI] [PubMed] [Google Scholar]
- 97. Green E.M., Antczak A.J., Bailey A.O., Franco A.A., Wu K.J., Yates J.R., Kaufman P.D.. Replication-independent histone deposition by the HIR complex and Asf1. Curr. Biol. 2005; 15:2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Prochasson P., Florens L., Swanson S.K., Washburn M.P., Workman J.L.. The HIR corepressor complex binds to nucleosomes generating a distinct protein/DNA complex resistant to remodeling by SWI/SNF. Genes Dev. 2005; 19:2534–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ray-Gallet D., Quivy J.-P., Scamps C., Martini E.M.-D., Lipinski M., Almouzni G.. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell. 2002; 9:1091–1100. [DOI] [PubMed] [Google Scholar]
- 100. Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y.. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004; 116:51–61. [DOI] [PubMed] [Google Scholar]
- 101. Blackwell C., Martin K.A., Greenall A., Pidoux A., Allshire R.C., Whitehall S.K.. The Schizosaccharomyces pombe HIRA-like protein Hip1 is required for the periodic expression of histone genes and contributes to the function of complex centromeres. Mol. Cell. Biol. 2004; 24:4309–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sharp J.A., Fouts E.T., Krawitz D.C., Kaufman P.D.. Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr. Biol. 2001; 11:463–473. [DOI] [PubMed] [Google Scholar]
- 103. Zhang R., Poustovoitov M.V., Ye X., Santos H.A., Chen W., Daganzo S.M., Erzberger J.P., Serebriiskii I.G., Canutescu A.A., Dunbrack R.L.et al.. Formation of MacroH2A-Containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell. 2005; 8:19–30. [DOI] [PubMed] [Google Scholar]
- 104. Phelps-Durr T.L., Thomas J., Vahab P., Timmermans M.C.P.. Maize rough sheath2 and its Arabidopsis orthologue ASYMMETRIC LEAVES1 interact with HIRA, a predicted histone chaperone, to maintain knox gene silencing and determinacy during organogenesis. Plant Cell. 2005; 17:2886–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rea S., Eisenhaber F., O’Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D.et al.. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000; 406:593–599. [DOI] [PubMed] [Google Scholar]
- 106. Roguev A., Bandyopadhyay S., Zofall M., Zhang K., Fischer T., Collins S.R., Qu H., Shales M., Park H.O., Hayles J.et al.. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008; 322:405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ryan C.J., Roguev A., Patrick K., Xu J., Jahari H., Tong Z., Beltrao P., Shales M., Qu H., Collins S.R.et al.. Hierarchical modularity and the evolution of genetic interactomes across species. Mol. Cell. 2012; 46:691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Barrales R.R., Forn M., Georgescu P.R., Sarkadi Z., Braun S.. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 2016; 30:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Driessen R.P.C., Sitters G., Laurens N., Moolenaar G.F., Wuite G.J.L., Goosen N., Dame R.T. Effect of temperature on the intrinsic flexibility of DNA and its interaction with architectural proteins. Biochemistry. 2014; 53:6430–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fischer T., Cui B., Dhakshnamoorthy J., Zhou M., Rubin C., Zofall M., Veenstra T.D., Grewal S.I.S.. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bernard P., Allshire R.C.. Centromeres become unstuck without heterochromatin. Trends Cell Biol. 2002; 12:419–424. [DOI] [PubMed] [Google Scholar]
- 112. Kent N.A., Adams S., Moorhouse A., Paszkiewicz K.. Chromatin particle spectrum analysis: A method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res. 2011; 39:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jiang C., Pugh B.F.. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009; 10:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lantermann A.B., Straub T., Strålfors A., Yuan G.C., Ekwall K., Korber P.. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat. Struct. Mol. Biol. 2010; 17:251–257. [DOI] [PubMed] [Google Scholar]
- 115. Martin C., Zhang Y.. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005; 6:838–849. [DOI] [PubMed] [Google Scholar]
- 116. Cromie G.A., Hyppa R.W., Taylor A.F., Zakharyevich K., Hunter N., Smith G.R.. Single holliday junctions are intermediates of meiotic recombination. Cell. 2006; 127:1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Oh S.D., Lao J.P., Taylor A.F., Smith G.R., Hunter N.. RecQ Helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol. Cell. 2008; 31:324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Jessop L., Lichten M.. Mus81/Mms4 endonuclease and Sgs1 helicase collaborate to ensure proper recombination intermediate metabolism during meiosis. Mol. Cell. 2008; 31:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C.. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011; 147:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. San Filippo J., Sung P., Klein H.. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008; 77:229–257. [DOI] [PubMed] [Google Scholar]
- 121. Shim E.Y., Chung W.H., Nicolette M.L., Zhang Y., Davis M., Zhu Z., Paull T.T., Ira G., Lee S.E.. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010; 29:3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Clerici M., Mantiero D., Guerini I., Lucchini G., Longhese M.P.. The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 2008; 9:810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zheng S., Li D., Lu Z., Liu G., Wang M., Xing P., Wang M., Dong Y., Wang X., Li J.et al.. Bre1-dependent H2B ubiquitination promotes homologous recombination by stimulating histone eviction at DNA breaks. Nucleic. Acids. Res. 2018; 46:11326–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Greenstein R.A., Ng H., Barrales R.R., Tan C., Braun S., Al-Sady B.. Local chromatin context dictates the genetic determinants of the heterochromatin spreading reaction. 2020; bioRxiv doi:31 May 2020, preprint: not peer reviewed 10.1101/2020.05.26.117143. [DOI] [PMC free article] [PubMed]
- 125. Jih G., Iglesias N., Currie M.A., Bhanu N.V., Paulo J.A., Gygi S.P., Garcia B.A., Moazed D.. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. 2017; 547:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lorenz D.R., Mikheyeva I. V, Johansen P., Meyer L., Berg A., Grewal S.I.S., Cam H.P. CENP-B cooperates with Set1 in bidirectional transcriptional silencing and genome organization of retrotransposons. Mol. Cell. Biol. 2012; 32:4215–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sugiyama T., Cam H.P., Sugiyama R., Noma K., Zofall M., Kobayashi R., Grewal S.I.S.. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell. 2007; 128:491–504. [DOI] [PubMed] [Google Scholar]
- 128. Cam H.P., Noma K., Ebina H., Levin H.L., Grewal S.I.S.. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature. 2008; 451:431–436. [DOI] [PubMed] [Google Scholar]
- 129. Durand-Dubief M., Sinha I., Fagerström-Billai F., Bonilla C., Wright A., Grunstein M., Ekwall K.. Specific functions for the fission yeast Sirtuins Hst2 and Hst4 in gene regulation and retrotransposon silencing. EMBO J. 2007; 26:2477–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hansen K.R., Burns G., Mata J., Volpe T.A., Martienssen R.A., Bahler J., Thon G.. Global effects on gene expression in fission yeast by silencing and RNA interference machineries. Mol. Cell. Biol. 2005; 25:590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yamanaka S., Mehta S., Reyes-Turcu F.E., Zhuang F., Fuchs R.T., Rong Y., Robb G.B., Grewal S.I.S.. RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature. 2013; 493:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zaratiegui M., Vaughn M.W., Irvine D.V., Goto D., Watt S., Bähler J., Arcangioli B., Martienssen R.A.. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature. 2011; 469:112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rai T.S., Glass M., Cole J.J., Rather M.I., Marsden M., Neilson M., Brock C., Humphreys I.R., Everett R.D., Adams P.D.. Histone chaperone HIRA deposits histone H3.3 onto foreign viral DNA and contributes to anti-viral intrinsic immunity. Nucleic Acids Res. 2017; 45:11673–11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pchelintsev N.A., McBryan T., Rai T.S., VanTuyn J., Ray-Gallet D., Almouzni G., Adams P.D.. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013; 3:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Peng G., Lin C.C.J., Mo W., Dai H., Park Y.Y., Kim S.M., Peng Y., Mo Q., Siwko S., Hu R.et al.. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014; 5:3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Durrant W.E., Wang S., Dong X.. Arabidopsis SNI1 and RAD51D regulate both gene transcription and DNA recombination during the defense response. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:4223–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wang S., Durrant W.E., Song J., Spivey N.W., Dong X.. Arabidopsis BRCA2 and RAD51 proteins are specifically involved in defense gene transcription during plant immune responses. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:22716–22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wang Y., Xiao R., Wang H., Cheng Z., Li W., Zhu G., Wang Y., Ma H.. The Arabidopsis RAD51 paralogs RAD51B, RAD51D and XRCC2 play partially redundant roles in somatic DNA repair and gene regulation. New Phytol. 2014; 201:292–304. [DOI] [PubMed] [Google Scholar]
- 139. Jeffares D.C., Rallis C., Rieux A., Speed D., Převorovský M., Mourier T., Marsellach F.X., Iqbal Z., Lau W., Cheng T.M.K.et al.. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat. Genet. 2015; 47:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Guo Y., Levin H.L.. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 2010; 20:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zhu Q., Pao G.M., Huynh A.M., Suh H., Tonnu N., Nederlof P.M., Gage F.H., Verma I.M.. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011; 477:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Padeken J., Zeller P., Towbin B., Katic I., Kalck V., Methot S.P., Gasser S.M.. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 2019; 33:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Szilard R.K., Jacques P.T., Laramée L., Cheng B., Galicia S., Bataille A.R., Yeung M., Mendez M., Bergeron M., Robert F.et al.. Systematic identification of fragile sites via genome-wide location analysis of γ-H2AX. Nat. Struct. Mol. Biol. 2010; 17:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kaliraman V., Mullen J.R., Fricke W.M., Bastin-Shanower S.A., Brill S.J.. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001; 15:2730–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Doe C.L., Osman F., Dixon J., Whitby M.C.. DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 2004; 32:5570–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Fricke W.M., Bastin-Shanower S.A., Brill S.J.. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair (Amst.). 2005; 4:243–251. [DOI] [PubMed] [Google Scholar]
- 147. Ehmsen K.T., Heyer W.-D.. Saccharomyces cerevisiae Mus81-Mms4 is a catalytic, DNA structure-selective endonuclease. Nucleic Acids Res. 2008; 36:2182–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Arnaudeau C., Lundin C., Helleday T.. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells11Edited by J. Karn. J. Mol. Biol. 2001; 307:1235–1245. [DOI] [PubMed] [Google Scholar]
- 149. Clouaire T., Rocher V., Lashgari A., Arnould C., Aguirrebengoa M., Biernacka A., Skrzypczak M., Aymard F., Fongang B., Dojer N.et al.. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell. 2018; 72:250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Cohen A., Habib A., Laor D., Yadav S., Kupiec M., Weisman R.. TOR complex 2 in fission yeast is required for chromatin-mediated gene silencing and assembly of heterochromatic domains at subtelomeres. J. Biol. Chem. 2018; 293:8138–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wold M.S. REPLICATION PROTEIN A:A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 152. Zhang H., Gan H., Wang Z., Lee J.-H., Zhou H., Ordog T., Wold M.S., Ljungman M., Zhang Z.. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol. Cell. 2017; 65:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Allshire R.C., Ekwall K.. Epigenetic regulation of chromatin states in Schizosaccharomyces pombe. Cold Spring Harb. Perspect. Biol. 2015; 7:a018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J.et al.. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are available at the National Center for Biotechnology Information (NCBI) under accession numbers PRJNA601919 and PRJNA659385. The ChIP-seq data are available at the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under an accession number E-MTAB-9619. The scripts used for ChIP-seq data processing and analyses are available at https://github.com/mprevorovsky/bagelova-polakova-dbl2-histones.