


Abstract
Antibiotic resistance is a serious problem which may be caused by bacterial dormancy. It has been suggested that bacterial toxin–antitoxin systems induce dormancy. We analyzed the genome-wide role of Staphylococcus aureus endoribonuclease toxin MazF using RNA-Seq, Ribo-Seq and quantitative proteomics. We characterized changes in transcriptome, translatome and proteome caused by MazF, and proposed that MazF decreases translation directly by cleaving mRNAs, and indirectly, by decreasing translation factors and by promoting ribosome hibernation. Important pathways affected during the early stage of MazF induction were identified: MazF increases cell wall thickness and decreases cell division; MazF activates SsrA-system which rescues stalled ribosomes, appearing as a result of MazF mRNA cleavage. These pathways may be promising targets for new antibacterial drugs that prevent bacteria dormancy. Finally, we described the overall impact of MazF on S. aureus cell physiology, and propose one of the mechanisms by which MazF might regulate cellular changes leading to dormancy.
Graphical Abstract
Graphical Abstract.
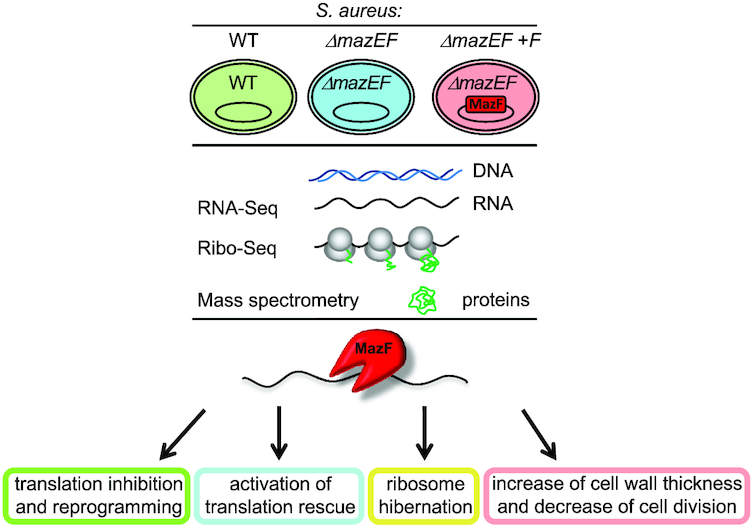
MazF toxin causes alterations in Staphylococcus aureus transcriptome, translatome and proteome that underlie bacterial dormancy.
INTRODUCTION
Staphylococcus aureus is a Gram-positive human pathogen, colonizing about one third of the world's population and causing a variety of diseases, from minor skin infections to life-threatening diseases (1). Antibiotic treatment is one of the main approaches of modern medicine used to combat bacterial infections; however, the effective use of antibiotics is constantly undermined due to the emergence of resistant pathogens, posing a serious public health problem (2).
Antibiotic resistance typically evolves via natural selection through random mutation or horizontal gene transfer, generating resistant cells that survive in the presence of antibiotics. It is now widely appreciated that bacterial cells may escape the bactericidal activity of antibiotics without undergoing genetic change (3). These cells are genetically identical to susceptible bacteria but have entered a non-growing quiescent or dormant state in which cells are metabolically inactive, and, thus, recalcitrant to antibiotics. Dormancy, latency, stasis, quiescence and persistence are conceptually related terms used to describe the propensity of bacteria to arrest their growth in response to environmental or host-imposed stress (3–6). The molecular mechanisms underlying bacterial dormancy are not well understood.
Bacterial toxin–antitoxin systems (TASs) have been proposed as factors primarily responsible for the formation of cell dormancy; however, their role in this process is still unclear. TAS is a two-component ubiquitous module found in almost all bacteria, which is composed of a toxin and an antitoxin counteracting the toxin activity (7–9). By decreasing cell growth, toxins may help to maintain a population of dormant cells, which are not killed by antibiotics in an otherwise sensitive culture. In S. aureus MazEF is a type II TAS that consists of two proteins: a stable toxin MazF, and unstable antitoxin MazE (10,11). MazF is an endoribonuclease that preferentially cleaves mRNAs at UACAU sites (12–14). In our recent work, MazF cleavage preference has been studied, extending the previously reported cleavage motif (15). The endonucleolytic activity of MazF is neutralized when it binds stoichiometrically to antitoxin MazE (16). Preferential digestion of MazE by stress-inducible ATP-dependent ClpCP proteolytic unit frees MazF to exert its toxicity (17,18).
MazF was extensively investigated in E. coli, where it was shown to block protein synthesis (19,20) and is involved in reversible growth inhibition, drug tolerance and persistence generation (21). Later works demonstrated that the role of E. coli MazF in cell dormancy, however, is not yet fully clear (reviewed in (22)). Much less is known about S. aureus MazF, where its properties and function could differ from its E. coli homolog. Not only the MazF cleavage motifs do not entirely coincide, but also the total number of TASs is vastly different: unlike E. coli with numerous TASs, S. aureus has only three type II TASs. In S. aureus, MazF may be activated by stress (23), the activation of MazF leads to bacteriostasis (15,24) and alters susceptibility to antibiotics (14). Recently, it has been demonstrated that MazF promotes S. aureus biofilm antibiotic tolerance leading to transition from an acute to chronic infection that cannot be eradicated with antibiotics and is less virulent (25). It has been suggested that MazF not only made the bacteria more tolerant to antibiotics but also made the bacteria more tolerant to the host (25). Thus, the MazEF system may conceivably be one of the important players in S. aureus dormancy and antibiotic tolerance.
Endoribonuclease toxins, when activated, would predictably substantially change gene expression, which can be measured genome-wide by high throughput RNA sequencing (RNA-Seq). However, diverse studies have shown that mRNA levels only partially explain protein levels in the cell because of the complexity of protein translation, which is the most common toxin target. The development of ribosome sequencing (Ribo-Seq) has allowed the global analysis of translation in vivo (26–28). Ribo-Seq is based on sequencing of ribosome-protected mRNA fragments (RPFs). In contrast to RNA-Seq, Ribo-Seq only captures those mRNAs that are being actively translated. However, although Ribo-Seq provides information on which proteins are being translated in the cell and at first approximation on their translation levels, it remains an indirect method to estimate protein abundance. Only a combination of all three techniques, RNA-Seq, Ribo-Seq and quantitative proteomics can provide definite information about gene expression.
In S. aureus, the genome-wide role of MazEF TAS has not been investigated, although RNA- and Ribo-Seq have been applied to explore MazF toxin in Eschrichia coli (29). Herein, we used RNA-Seq, Ribo-Seq and quantitative proteomics to analyze the effect of MazF toxin on the transcriptome, translatome and proteome of S. aureus. We characterized the correlation between transcription, translation, and protein levels and demonstrated that MazF decreases translation directly by cleaving mRNA, and indirectly, by decreasing levels of translation factors, like rrf/frr, levels of genes responsible for amino acid biosynthesis, tRNA aminoacylation and modifications and by promoting ribosome hibernation. Our results suggest a plausible model where MazF not only suppresses transcription and translation but rather changes the translational program in a way that the cell is prepared for dormancy, for instance, promoting ribosome hibernation, increasing cell wall thickness, and decreasing cell division.
MATERIALS AND METHODS
Bacteria strains and plasmids
In this study we use HG003 strain, either wild-type (30), or deleted for mazEF (14). The mazEF deleted strain was constructed as described in (14), without affecting the sigB locus located downstream of the mazEF genes. Briefly, the mazEF knockout mutant was generated by replacing the mazE and mazF open reading frames with a lox flanked erythromycin resistance cassette, without affecting the upstream transcriptional promoter and the downstream rho independent transcriptional terminator. The resistant cassette was further removed, generating a markerless HG003 mazEF deleted mutant (14). Strains were transformed either with an empty pRAB11 vector (31), or a vector carrying HG003 mazF gene under control of inducible anhydrotetracycline (ATc) promoter (pRAB11-mazF) (15,23). Cultures were grown on Mueller Hinton Broth media in the presence of 15 μg/ml of chloramphenicol (Cm) until OD600 of 0.5–0.7 at 37°C with shaking. To induce mazF expression, anhydrotetracycline (ATc) was added up to a final concentration 0.2 μM and cultures were incubated 10–180 min at 37°C with shaking.
RNA-Seq
Cultures with either the empty vector or the vector containing mazF were grown untill OD600 of 0.5 at 37°C with shaking. ATc induction was done for 10 min. For total RNA isolation 8 ml of culture were immediately mixed with 40 ml of –80°C cold ethanol–acetone mixture (1:1). Cells were collected by centrifugation for 10 min at 4000 g, 4°C. Pellets were washed with 1 ml of cold TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0), resuspended in 100 μl of TE with 1 mg/ml of lysostaphin (AMBI) and 2 μl of RNasinePlus (Promega), and incubated for 5 min at 37°C. Total RNAs were purified with the ReliaPrep RNA kit (Promega) in biological duplicates. Ribosomal RNAs were depleted with RiboZero rRNA Removal Kit for Bacteria (Illumina). Libraries were created using the Illumina TruSeq stranded mRNA kit. First strand cDNAs were synthesized with random hexamer primer. Libraries were sequenced at Fasteris SA (Geneva, Switzerland) on an Illumina HiSeq 3000/4000, single-reads, 1 × 50 bp.
Analysis of RNA-Seq
Results of RNA-Seq were aligned to RefSeq assembly GCF_000013425.1 (NCTC 8325) combined with pRAB11 sequence (accession JN635500.1) using STAR aligner. The aligned reads were associated with the RefSeq gene annotations (the tmRNA region was added to the annotations manually using the location from AureoWiki). A typical library contained 24–34 million of reads per sample aligned to genome, with a high correlation between biological replicates (Supplementary Figure S1B, Table S1). Only genes with more than 40 reads per sample (average over samples) were retained for further analysis. Read counts were normalized by the total number of reads excluding reads aligned to ribosomal RNA, and differential gene fold changes and P-values were obtained by limma/voom (32).
RNA-Seq results were validated using TaqMan reverse transcription real-time quantitative polymerase chain reaction (RTqPCR) with MGB Double-Dye probes (Supplementary Table S4) as described in supplementary methods.
Detection of MazF cleavage sites
The potentially cleaved genes were identified in the genome by the dips in RNA-Seq coverage similar to published methods (29). First, RNA-Seq coverage was constructed from single end sequencing assuming length of fragments to be equal to 200 nt. Gene coverage was analyzed including 50 nt upstream. Second, cleavage ratio was constructed as ratio of the normalized RNA-Seq coverage of ΔmazEF+F to ΔmazEF samples. The genes were defined as potential MazF cleavage targets if the difference between minimum and maximum of cleavage ratio along the gene was >8-fold, creating the ‘dip’ (Supplementary Figure S2B). The genes with RNA-Seq coverage in ΔmazEF sample below 10 near the cleavage minimum were ignored to evade false positives in low expressed genes. The method described here differs from that of (29), where only the cleavage ratio in the minimum along the gene was used. This difference makes our approach less biased toward detecting down regulated, but not cleaved, genes.
To verify that this method detects mRNA cleavage, we checked that the detected cleavage minima predominantly had a UACAU cleavage motif in the 150 nt vicinity (Supplementary Figure S2C). With the uncertainty on the length of the mRNA fragments in the library, we cannot expect a more precise association of the cleavage minima with cleavage site in each particular gene.
Ribo-Seq
To obtain S. aureus polysomes 1 ml of an overnight culture was diluted in 250 ml of media and grown until an OD600 of 0.5 at 37°C. ATc induction was done for 10 min. To block translation Cm was added up to a final concentration of 0.3 mM, and culture was incubated for 2 min at 37°C and then quickly chilled. After centrifugation pellets were washed in 2 ml of cold Resuspension Buffer (RB) (20 mM Tris (pH 8.0), 10 mM MgCl2, 5 mM CaCl2, 100 mM NH4Cl, 1 mM Cm), and resuspended in 0.8 ml of cold Lysis Buffer [RB plus 0.1% IGEPAL, 0.4% Triton X-100, 100 U/ml RNase-free DNase I (Roche), 0.5 U/μl SUPERaseIn (Ambion), Protease Inhibitors (Roche)]. Suspension was incubated on ice for 5 min and frozen in drops in liquid nitrogen. Drops were milled five times with 1-min cycles at 30 Hz in CryoMill MM400 (Retsch) in a 10 ml grinding jar with 15 mm grinding ball.
For cell extract preparation, cell powder was thawed in 50 ml tube and centrifuged at 4000 g, 4°C for 1 min. Supernatant (SN) was clarified at 16 000 g, 4°C for 1 min and centrifuged in a new tube at 16 000 g, 4°C for 10 min.
To obtain ribosome footprints 1000 U of S7 Micrococcal Nuclease S7 (Roche) was added per 1 mg of nucleic acids. Samples were incubated for 1 h at 25°C with rotation at 190 rpm. Reactions were quenched by the addition of EGTA up to a final concentration 6 mM. Nucleic acids (0.5 mg) were loaded onto a linear 10–50% sucrose gradients prepared on Gradient Buffer [20 mM Tris (pH 8.0), 10 mM MgCl2, 100 mM NH4Cl]. Tubes were centrifuged at 217 000 g (35 000 rpm, SW41 Ti rotor, Beckman Coulter) for 3 h at 4°C. Fractions were collected using a UA/6 detector (ISCO). After centrifugation fractions (750 μl) were incubated with 10 μl of proteinase K (NEB, 800 U/ml) and 10 μl of SDS 10% overnight at 4°C. RNAs were isolated with phenol–chloroform extraction. Libraries were prepared as described (26,33) and sequenced on an Illumina HiSeq 3000/4000, single-reads, 1 × 50 bp.
Analysis of Ribo-Seq
Reads were aligned by STAR, with the adaptor sequence removed using ‘--clip3pAdapterSeq CTGTAGGCACCATCAATAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC’ option. The position of the P-site was determined by the study of the distribution of the positions of the fragments around the beginning of the ORFs, as described (26) using only ORFs without other annotated features 50 nt upstream in the genome (to exclude possible complications of translation of genes within operons). Alignment of the first significant peak of the distribution with the AUG codon corresponded to 3′-end of the fragment being in the nucleotide position 16 of the open reading frame, so the P-site was further defined as the 3′-position of the read minus 15, and only RPF with the length 25–31 were used. The translational signal was defined by the number of RPF with the P-site within gene CDS, excluding first 30 and last 6 nucleotides, to remove the effects of translational initiation and termination. Only genes with >40 reads on average were retained for analysis. The signal was normalized by the total number of reads aligned to the genome (excluding plasmid and rRNA reads). Fold changes were computed by limma/voom (32). Typical number of Ribo-Seq reads corresponding to coding genes was about 4 million per sample with very good reproducibility between biological replicates (Supplementary Figure S7B, Table S1). Translational efficiency (TE) was defined as the ratio of Ribo-Seq and RNA-Seq signals in RPKM. TE and its fold change was calculated using Xtail (34) allowing to identify the statistical significance of the result.
TMT-MS2 quantitative proteomics
ATc induction was done for 10 min. For total protein extraction 25 ml of cultures with OD600 of 0.7 were collected and washed three times with 1 ml of phosphate-buffered saline (PBS) buffer. Cells were lysed in the presence of 400 μl of lysis buffer (LB) [PBS, 200 μg/ml lysostaphin (AMBI), 200 μg/ml DNAse I, protease inhibitors (Roche)] for 20 min at 37°C, chilled on ice, and sonicated 10 times with 30-s cycles using Cell Disrupter B-30 (Branson). Extracts were clarified by centrifugation for 10 min at 14 000 g, 4°C. Total protein concentration was measured in supernatants by the Bradford protein assay. This method permitted to obtain of about 1 mg of total protein with a concentration of about 2.5–3.0 mg/ml. Samples were mixed with Laemmli Sample Buffer (SB) and analyzed by SDS polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Blue staining or western blot.
Tandem Mass Tags Mass Spectrometry (TMT-MS2) was done at the Proteome Sciences R&D GmbH&Co KG. Samples were digested with trypsin, TMT-labeled, and subjected to Strong Cation Exchange (SCX) chromatography. Each fraction was analyzed by nano LC–MS/MS and MS2 (Thermo LTQ-Orbitrap Velos mass spectrometer). Data were analyzed with the MASCOT and SEQUEST databases. Filtering, normalization and quantification was achieved using Proteome Discoverer (Thermo Scientific). P-values and the log-fold change was further obtained using limma package.
Transmission electron microscopy (TEM)
ΔmazEF cells carrying either empty vector or vector containing mazF were grown at 37°C until an OD600 of 0.5–0.6. After ATc induction cultures were incubated for 0, 10, 60 and 180 min at 37°C with shaking. 2 ml of cell cultures were collected by centrifugation for 1 min, 10 000 g, 4°C. Pellets were washed with 1 ml of PBS and then resuspended in 1 ml of fresh 4% (v/v) glutaraldehyde in PBS. After 15 min incubation samples were spun for 1 min, 10 000 g, 4°C and washed with 1 ml of PBS. Samples obtained in two independent experiments were analyzed on Morgagni TEM (FEI) at the Microscopy PFMU core platform, CMU, University of Geneva. Cell wall thickness measurements were done using TEM image software: six measurements per cell in 10 different cells of each strain with nearly equatorial cut surfaces were averaged. The P-values were computed using two-sided Wilcoxon signed-rank test. Cell size was analyzed using ImageJ software. For analysis of cell division the Fisher's exact test was used to compare the number of cells with and without division septum.
Antibodies
Polyclonal rabbit anti-MazF antibodies were described in (15) and used in dilution 1:500. Polyclonal rabbit anti-Hpf antibodies were described in (35) and used in dilution 1:4000. Polyclonal rabbit anti-Pbp4 antibodies were used in dilution 1:5000. Goat anti-rabbit IgG–HRP conjugate (BioRad) were used in dilution 1:5000.
RESULTS
Transcriptome analysis after MazF induction
We expected that MazF modifies S. aureus physiology directly by cleavage of mRNA at a specific motif, and/or globally by indirect regulation. This global effect is expected to modify the activation of pathways relevant to dormancy. Using conditional expression of mazF (Supplementary Figure S1A) we explored in detail how MazF toxin affects cellular physiology (Figure 1A).
Figure 1.

Analysis of transcriptome by RNA-Seq after mazF induction. (A) Experimental design. S. aureus wild type (WT) cells or cells deleted for mazEF (ΔmazEF), were transformed with an empty vector or a vector carrying inducible mazF (ΔmazEF+F). Cells were collected after 10 min of mazF induction when MazF protein had been produced, but cell growth was not yet inhibited (15,23), and analyzed by RNA-Seq, Ribo-Seq and quantitative mass spectrometry. (B) MazF expression leads to mRNA cleavage on UACAU on genome-wide level. Comparison of RNA-Seq coverage 500 nt before and after UACAU sequences: WT to ΔmazEF (green) and ΔmazEF+F to ΔmazEF (red). (C) log2-fold change (log-FC) in RNA levels of WT compared to ΔmazEF (left) and of ΔmazEF+F compared to ΔmazEF (right). RNA-Seq is expressed in counts per million reads (CPM). Upregulated genes (P < 0.05), downregulated genes (P < 0.05) and non-differentially expressed genes (non-DE) (P > 0.05) are marked in red, blue and grey, respectively. Only genes with more than 40 reads per gene averaged per sample (above the grey highlighted region) were retained for the further analysis. (D) Distribution of the log-FC of RNA-Seq between WT and ΔmazEF (left) and ΔmazEF+F and ΔmazEF (right). The genes with identified MazF cleavage site are indicated in cyan.
First, we analyzed the effect of mazF induction on the S. aureus transcriptome by RNA-Seq. The RNA-Seq libraries showed high correlation between both biological replicates (Supplementary Figure S1B, Table S1). In the S. aureus genome (NCTC 8325) there are 2843 genes annotated as open reading frames (ORFs). 1684 (60%) of them contain in total 3406 UACAU sequences that are potentially cleaved by MazF. The distribution of this motif over coding gene sequences (CDS) does not have any preferential position bias (Supplementary Figure S2A). In WT both MazE and MazF proteins are present, and even though MazF is neutralized by MazE we cannot exclude some level of endogenous MazF activity, as reported (14). We analyzed the RNA-Seq read coverage 500 nucleotides (nt) before and after each of UACAU sequences (Figure 1B), and observed a decrease of reads around the potential MazF cleavage sites when MazF expression was induced, while no global changes were found between WT and ΔmazEF strain. This result provides evidence that in WT MazF activity is generally neutralized by MazE, while, in the absence of MazE, MazF leads to mRNA cleavage on UACAU on a genome-wide level.
Recently, we identified MazF cleavage motifs in vivo using the nEMOTE (non-phosphorylated exact mapping of transcriptome ends) technique (15). This method allows detection of the exact cleavage sites with nucleotide precision. However, it has limitations, and it was possible to identify cleavages only in highly expressed genes. In our analysis herein, we applied a different approach similar to (29,36). We identified MazF cleavages by the pronounced dips in the ratio between ΔmazEF+F and ΔmazEF RNA-Seq coverage (Supplementary Figure S2B and C). By this method 339 genes that may be directly cut by MazF were detected (Supplementary Table S2). These cleavages mostly corresponded to the significantly down-regulated genes and have only 10% overlap with the genes identified by nEMOTE in (15) (Supplementary Figure S2D).
In previous studies, it was proposed that E. coli MazF cleaves closely upstream of the AUG start codon and generates leaderless mRNAs, which are translated by ‘stress-ribosomes’ lacking anti-Shine-Dalgarno sequence also created by MazF (36,37). Later, this finding was argued (29). We analyzed whether S. aureus MazF created such transcripts, and found only one gene (SAOUHSC_01137), which was cleaved by MazF in the proximity of the start codon and which was translated slightly better after MazF induction. We concluded that MazF does not create translated leaderless transcripts in S. aureus.
Principal Component Analysis (PCA) allows to summarize the systematic patterns of variations in the data. PCA demonstrated close similarity of biological duplicates (Supplementary Figure S3A). ΔmazEF cluster is closer to WT cluster, while ΔmazEF+F is notably different from the WT and ΔmazEF groups, indicating that mazF induction leads to larger gene expression changes than deletion of mazEF. Clustering was similar for PCA limited to only high (RPKM > 25) or low (RPKM < 25) expressed genes, and even limited to only transcripts with or without UACAU motifs, suggesting that MazF changed expression not only of cleaved genes, but rather impacted the entire transcriptome.
We compared changes in transcriptomes when mazEF was deleted (WT versus ΔmazEF) and when mazF was induced in a ΔmazEF background (ΔmazEF+F versus ΔmazEF) (Figure 1C, Supplementary Table S1). Similar number of genes were up- and down-regulated (569 and 573 genes, respectively) in WT compared to ΔmazEF, while transcription of 1685 genes was unchanged. In contrast, we observed that MazF caused considerable perturbation in the transcriptome. Upon mazF induction only 867 genes were not changed, while 488 and 1474 genes were up- and down-regulated, respectively, corresponding to about 17% and 52% of analyzed genes. Importantly, upon mazF induction, many genes were significantly down-regulated (with log2-fold change (log-FC)<-2). 17% of up-regulated genes indicates that, even if MazF decreased transcription and inhibited cell growth, the cell remained biologically active. RNA-Seq data were validated by RTqPCR in independent experiments for 18 transcripts (vraS, lytR, rsbW, htrA1, frr/rrf, murA1, tagA, purR, pbp2, pbp4, stp1, yjbH, fusA, hup, trfA, spx, trxA, hpf) differently affected by MazF (Supplementary Figure S4, Table S4).
The distribution of the RNA-Seq log-FC for the WT to ΔmazEF comparison showed one peak with the average of zero (Figure 1D, left). In the comparison of ΔmazEF+F to ΔmazEF, however, the histogram of the RNA-Seq log-FC was bimodal with two pronounced peaks, indicating that there are two families of genes (Figure 1D, right). One group – with the average log-FC of zero, and a second group of significantly down regulated genes with average log-FC –2.5. This distribution suggests that there are two mechanisms of gene regulation by MazF. The genes in the first group did not display an overall decrease of expression, but had a variance in expression change, indicating an indirect effect of MazF on them. For comparison, the distribution of the RNA-Seq log-FC between replicates showed a very narrow peak around zero (Supplementary Figure S3B). The transcripts in the second group probably correspond to those directly cleaved by MazF, leading to strongly reduced expression. Indeed, distribution of the genes that we identified as cut by MazF (Supplementary Table S2), explicitly correlate with the second peak supporting the idea of two mechanisms of gene regulation by MazF (Figure 1D, right). Not all genes from the second group were identified as cleaved, due to the strict criteria of our analysis, chosen to avoid false positive artifacts. It is certainly possible that more genes, if not all, from the second group are MazF targets.
To examine if MazF may generally affect the stability of mRNAs besides it direct cleavage targets we analyzed if the change in mRNA levels upon mazF induction can be attributable to the change of mRNA stability. We performed an RNA stability assay by adding rifampicin to halt de novo mRNA synthesis in ΔmazEF cells expressing either empty vector or inducible MazF after 10 min of ATc induction. The decay of the mRNAs was monitored by RTqPCR at different time points of rifampicin treatment (Supplementary Figure S5). For transcripts that were not affected by MazF (gyrB), and for transcripts that were decreased upon mazF induction (frr/rrf, hup, fusA), we observed slightly increased mRNA degradation. For the transcripts that were increased upon mazF induction (spx, hpf) we observed slight mRNA stabilization. Although individual decay rates varied for each gene tested, we found evidence of both increased and decreased decay. Thus, there appears to be no global non-specific effect on RNA decay, but rather a gene specific effect.
Analysis of S. aureus translatome
Global impact of MazF on translation was studied by Ribo-Seq (Figure 2A). Ribosome profiles from ΔmazEF cells slightly differed from the WT. Ribosome profiles after MazF expression in ΔmazEF background showed decreased polysomes and the appearance of a pronounced 100S peak, representing ribosome dimers (38). The 100S ribosomes are translationally inactive and correspond to the resting form in the ribosome cycle, hence, the process of 100S formation has been named ‘ribosomal hibernation’. 100S peak was not affected by RNase treatment and was not observed in Δhpf strain (Supplementary Figure S6). Thus we concluded that MazF expression led to appearance of hibernating ribosomes that are sequestrated from active translation and are preserved in a form of dimers.
Figure 2.

Analysis of translatome after mazF induction by Ribo-Seq. (A) 10–15% sucrose density gradient analysis of polysomes from the WT and ΔmazEF cells, harboring either empty plasmid or plasmid with mazF gene (F). Positions of 30S, 50S, monosomes (70S), ribosome dimers (100S), and polysomes are indicated. (B) The frequency distribution of RPFs between 21 and 38 nt obtained from nuclease digestion in the WT replicate 1. Codon periodicity for each length fragment is indicated in green, violet, and orange for the 1st, 2nd and 3rd nt position in codon, respectively. (C) Reads attributed to the 1st, 2nd and 3rd nt of a codon in WT (green), ΔmazEF (blue) and ΔmazEF+F (red) samples in duplicates. (D) Ribosomes stall at the end of the transcripts cleaved by MazF. Mean change in RPFs between ΔmazEF+F and ΔmazEF (red) at the position -1 relative to U(A)CAU sequences. For comparison, mean change in RPFs between WT and ΔmazEF (blue) is shown in the same region. (E) MazF induction leads to the activation of SsrA-tagging system, which consists of tmRNA and protein SmpB, and to the decrease of RPFs at the 3′ end of transcripts. ΔmazEF (blue) and ΔmazEF+F (red) strains were compared for tmRNA levels, ribosome density on MLD part of tmRNA, protein levels of SmpB, and ribosome density at the 5′ and 3′ ends of transcripts. SmpB protein levels were evaluated by quantitative mass spectrometry (Supplementary Table S1) and have arbitrary normalization. P-values are indicated above.
The quality of Ribo-Seq analysis depends on the size and homogeneity of ribosome protected fragments (RPFs) obtained after nuclease digestion. Optimal digestion was obtained with 1000 U of S7 nuclease per 1 mg of total RNA (Supplementary Figure S7A). Libraries were prepared from digested RPFs and, after quality control, subtraction of rRNA and plasmid reads, showed good reproducibility between biological replicates (Supplementary Figure S7B, Table S1). The length distribution of RPFs ranged from 22 to 40 nt and showed a single peak with a maximum at 27 nt and well-defined three-nucleotide periodicity which is a characteristic property of protein coding ORFs (Figure 2B and Supplementary Figure S7C). This was larger than E. coli (24 nt) and smaller than eukaryotic (28–32 nt) RPFs. RPFs of length 25–31 nt, demonstrating good correlation with the 1st nt of codons, were selected for subsequent analysis (Figure 2C).
When MazF cleaves mRNA it creates stop-less transcripts, where ribosomes reaching the end of prematurely truncated mRNA stall because of the absence of a complete codon in the A-site, as was reported for E. coli (29). We analyzed changes in RPFs near the MazF cleavage site, and indeed observed accumulation of ribosomes at the position preceding UACAU sequences (Figure 2D).
In bacteria the stop-less transcripts are rescued by the SsrA-tagging system, consisting of transfer-messenger RNA (tmRNA) and protein SmpB, in the process called trans-translation. tmRNA is a bifunctional molecule that has tRNA-like domain (TLD) acting as a tRNA, and messenger-like domain (MLD) acting as a mRNA. Elongation factor Tu with the help of SmpB delivers tmRNA to the A-site of the stalled ribosome, where mRNA template is changed with MLD, and translation continues. Most of the knowledge about the bacterial translation rescue systems derives from E. coli (39,40). In S. aureus, there is only one translation rescue system, which is essential. It terminates translation by a normal stop codon and contributes to degradation of partially synthesized proteins by proteases (17,41). We observed increase of tmRNA transcription, increase of RPFs on MLD of tmRNA and increase of SmpB protein production after MazF induction (Figure 2E). We concluded that the SsrA-tagging system, playing an important role in the rescue of stalled ribosomes and in the resumption of protein synthesis, was activated by MazF in S. aureus. MazF induction resulted in about 50% decrease of ribosome footprints at the 3′ end of transcripts, due to the ribosomes running off from the 3′ end of cleaved transcripts, but did not significantly change the ribosome occupancy at the 5′ end of transcripts and within the transcripts (Figure 2E, right). The PCA for Ribo-Seq (Supplementary Figure S8) was similar to those of RNA-Seq, confirming the conclusion that MazF causes indirect regulation of translation of many genes.
Comparison of the Ribo-Seq of WT versus ΔmazEF and ΔmazEF+F versus ΔmazEF (Figure 3, Supplementary Table S1) led to the results similar to RNA-Seq. We found similar numbers of up- and down-regulated genes, 443 and 543, respectively in WT compared to ΔmazEF, while translation of 1781 genes was not changed. MazF led to substantial decrease of RPFs. Only 250 genes had more RPFs, while 1187 genes had less RPFs when MazF was induced, corresponding to 9.0% and 43% of analyzed genes. 1330 genes were unchanged. Many genes were significantly down-regulated with log-FC<-2, indicating the substantial reduction of translation.
Figure 3.

Changes in translatome after mazF induction. log-FC in RPFs in WT compared to ΔmazEF (left) and in ΔmazEF+F compared to ΔmazEF (right). Genes with increased RPFs (P < 0.05), decreased RPFs (P < 0.05) and genes without significant change in RPFs (P > 0.05) are marked in red, blue and grey. Only genes with more than 40 reads per gene averaged per sample (above the grey highlighted region) were retained for the further analysis.
Correlation between transcriptome, translatome and proteome
At the first approximation, the change in number of RPFs in Ribo-Seq corresponds to the change in translation and is more directly related to the change in protein levels, than RNA-Seq. However, translation does not always occur with the same speed and outcome. Translation is further modified by additional factors, which may influence protein production, as, e.g. the presence of co-translational factors or chaperones assisting folding of nascent chains. Thus, changes in translation alone also do not necessarily imply changes in the abundance of the translated proteins. By this reasoning, the relation between RNA-Seq, Ribo-Seq and quantitative proteomics data should be analyzed.
Protein abundance was estimated by Tandem Mass Tags Mass Spectrometry (TMT-MS2) under the same conditions of mazF induction as before (Supplementary Figure S9A). PCA showed that WT, ΔmazEF and ΔmazEF+F duplicates form separated clusters (Supplementary Figure S9B).
To study the relation between RNA-Seq, Ribo-Seq, and proteomics we compared the changes of these parameters between either WT and ΔmazEF (Figure 4, left), or between ΔmazEF+F and ΔmazEF (Figure 4, right). Use of the differential expression allowed to avoid the possible biases associated with overall gene expression or CDS length, and is the only possible approach for proteomics data, which does not provide reliable absolute measurements.
Figure 4.

Correlation between changes in RNA-Seq, Ribo-Seq and proteomics. log-FC in RNA-Seq, Ribo-Seq, and proteomics in WT compared to ΔmazEF (left) and ΔmazEF+F compared to ΔmazEF (right). Comparison of Ribo-Seq versus RNA-Seq, RNA-Seq versus proteomics, and Ribo-Seq versus proteomics, are indicated. The scales are the same for the graphs on the right and left side of the picture for the clear comparison. Pearson correlation is indicated above each panel. The numbers of the genes in each group are shown. The genes are defined as differentially expressed for log-FC larger than 0.3 for RNA- and Ribo-Seq, and larger than 0.2 for proteins and P < 0.05. Colors indicate the homodirectional change (red), opposite change (green), change only in x-axis (blue), change only in y-axis (violet), no change (gray). Genes which were identified as a directly cleaved by MazF are indicated by black dots.
First, we studied the relation between transcriptome and translatome (Figure 4, top). For the case of WT vs ΔmazEF the Pearson correlation was rather small (0.29) and overall shape of the distribution was round. This indicates, that the difference caused by removal of mazEF is not pronounced, and the correlation is mostly dominated by random noise. We can still see, that the number of genes that changed homodirectionally in a statistically significant way (up or down regulated in both RNA- and Ribo-Seq, in red) was larger, than of those that changed in opposite directions (up in RNA- and down in Ribo-Seq, or vice versa, in green), indicating that the changes were indeed related.
The effect of mazF induction, on the contrary, caused significant modification of both translatome and transcriptome, with a large number of downregulated genes. With the large changes, the Pearson correlation was higher (0.63), confirming the strong relation between RNA- and Ribo-Seq. The subset of genes showing opposite changes such as increased mRNA but decreased Ribo-Seq, may be explained by mRNA preservation in a translationally inactive, but stable form. Genes showing decreased mRNA levels but increased Ribo-Seq, may be translated more efficiently. More complex scenarios could involve combination of transcriptional and translational regulatory events.
Middle and bottom panels of Figure 4 demonstrated the relation between RNA- and Ribo-Seq and protein levels. Both RNA- and Ribo-Seq clearly correlate with the protein levels (Pearson correlation 0.45 and 0.4) in ΔmazEF+F vs ΔmazEF comparison, even if this correlation was, as expected, weaker than between RNA- and Ribo-Seq (0.63). Quite a lot of genes were significantly changed only in RNA- and Ribo-Seq but not in proteome (genes marked in violet). A possible explanation for these genes is the experimental timing. Probably 10 min of mazF induction caused changes in RNA levels, but was not enough for the proteome to fully respond. Also, we did not examine protein stability, only steady-state protein levels.
An important observation is that the transcripts identified as cleaved by MazF (marked by black dots) mainly correspond to the genes downregulated at all stages of gene expression: transcription, translation and protein production. However, we noticed that not all transcripts which were decreased upon mazF induction were cleaved by MazF. To demonstrate the spectrum of possible relations between RNA-Seq, Ribo-Seq and MazF cleavage activity we compiled the plots of RNA-Seq and Ribo-Seq density maps and MazF cleavage map in ΔmazEF and ΔmazEF+F samples for several representative situations: transcripts which we identified as cleaved by MazF (lytR, pyrR); transcripts which have both RNA- and Ribo-Seq signals decreased but no cleavage detected (vraS, frr/rrf); transcripts which have RNA-Seq signal increased and Ribo-Seq signal increased or not changed (epiE, hpf); transcripts which have RNA-Seq increased and Ribo-Seq decreased (ilvC), and, finally transcripts which have RNA-Seq decreased and Ribo-Seq increased (SAOUHSC_02842) (Supplementary Figure S10).
Quantitative data for both transcript abundances (RNA-Seq) and protein synthesis rates (Ribo-Seq) enabled us to infer the relative translation efficiency (TE). To explain better the crosstalk of transcription and translation, it was proposed to calculate TE as the ratio of Ribo- and RNA-Seq signals (26), which corresponds to the number of ribosomes per length of mRNA, or ribosome density. In our data set changes in TE had very low or even anti-correlation with proteins (0.091 for WT versus ΔmazEF and -0.22 for ΔmazEF+F versus ΔmazEF) (Supplementary Figure S11). Thus, TE, calculated in this way, cannot fully explain protein abundance. Indeed, TE depends on many factors such as codon usage, availability of the tRNAs, sequence-dependent determinants (like Shine-Dalgarno sequences) and mRNA secondary structures (42). TE can also change over time and conditions for a given mRNA, reflecting dynamic regulation at the level of translation. However, in some particular cases in our data set (see below) TE correlated with protein levels and may explain increased protein even when transcription was decreased.
MazF affects expression of many specific groups of genes, in particular, genes involved in cell wall biosynthesis and cell division
We identified several functional groups of genes where transcription, translation or protein level were changed upon MazF expression (Figure 5, Supplementary Table S3). MazF cleaves mRNA and reduces polysomes, indicating that translation and protein synthesis are reduced. The decreased translation can be seen, in particular, for the groups connected to protein synthesis, tRNA aminoacylation, signal transduction, transport and binding proteins, energy metabolism, cell envelope and cell division (Figure 5A, Supplementary Table S3). Translation of many others groups of genes, such as amino acid biosynthesis, translation and ribosome recycling factors, tRNA and rRNA modification was reduced (Supplementary Table S3), that is also in agreement with the general translation inhibition. We observed, however, that the translation of some groups of genes was increased upon mazF induction: e.g. groups of genes of the arginine deiminase (ADI) pathway (Figure 5A), ATP dependent chaperones, TCA cycle, glycolysis, gluconeogenesis and the pentose phosphate pathway (Supplementary Table S3), indicating that particular pathways were activated upon MazF induction. Interestingly, upregulation of the ADI pathway was associated with cell wall biosynthesis and increased cell wall thickness (43,44), observed in vancomycin-intermediate S. aureus strains (45).
Figure 5.

MazF affects specific pathways. (A) Example of groups of genes with decreased and increased translation (log2-FC) represented as a box plot. Comparison of WT to ΔmazEF (blue) and ΔmazEF+F to ΔmazEF (red). The other groups of genes are present in Supplementary Table S3. (B) Examples of groups of genes with decreased translation, but increased protein levels. Changes in transcription (RNA-Seq), translation (Ribo-Seq), translational efficiency (TE), and protein levels for WT compared to ΔmazEF (blue) and for ΔmazEF+F compared to ΔmazEF (red). Genes of ribosomal proteins, purine and pyrimidine biosynthesis and ATP synthase are presented. The P-values for the group of genes being overall differentially expressed are indicated above each group, and were computed using two-sided Wilcoxon test.
While overall transcription and translation correlated well with protein levels we observed several examples of the opposite behavior. For example, genes of ribosomal proteins, pyrimidine and purine ribonucleotide biosynthesis, and genes of ATP synthase (Figure 5B, Supplementary Table S3) had decreased translation but increased protein level. In some cases, this may be explained by increased TE, as observed, e.g. for ATP synthase and pyrimidine and purine biosynthesis genes. However, for genes encoding ribosomal proteins the protein levels were increased even without increase in TE.
We found that many genes involved in cell wall biosynthesis, cell division, and fatty acid metabolism were affected upon MazF expression. Some of these genes were cleaved by MazF (Supplementary Table S2) and (15). To address the role of MazF in cell wall structure we examined genes in the S. aureus cell wall biosynthetic pathway (Supplementary Figure S12) (44,46–49). Translation and/or protein levels of many key enzymes involved in cell wall biosynthesis and several side pathways connected with cell wall biosynthesis, such as ADI pathway, pyrimidine biosynthesis, TCA cycle, glycolysis were increased by MazF (Supplementary Figure S12, Table S3). Simultaneously MazF suppressed expression of many genes involved in cell division (Supplementary Table S3).
To further verify the effect of MazF on cell wall and cell division we examined cell morphology changes after different times of mazF induction by transmission electron microscopy (TEM) (Figure 6 and Supplementary Figure S13). Without induction no visible difference was detected in ΔmazEF cells carrying either the empty vector or the plasmid expressing MazF. After 180 min of MazF induction, we observed an increase of cell wall thickness and a decrease in the number of dividing cells, as determined by the number of cells with a division septum. These effects were also visible after 60 min of induction (Figure 6B, C and Supplementary Figure S13). We concluded that MazF increased cell wall thickness and decreased the number of dividing cells, in agreement with the inhibition of the cell growth and bacteriostasis (15).
Figure 6.

MazF increases thickness of cell wall and decreases cell division. (A) ΔmazEF cell carrying either empty vector or plasmid expressing MazF (ΔmazEF+F) were treated with ATc for 0, 10, 60 and 180 min to induce mazF expression and analyzed by transmission electron microscopy (TEM). Electron microscopy images are presented at 2 different magnifications (upper and lower panels), and the scale bars indicate 1 μm or 200 nm, respectively. Images at 0 and 180 min of induction are shown. More images are shown on the Supplementary Figure S13. (B and C) Analysis of cell wall thickness (B) and cell size (C) after 0, 60 and 180 min of mazF induction. ΔmazEF and ΔmazEF+F samples are indicated in blue and red, respectively. The error bars indicate standard deviations (SDs). The P-values were computed using two-sided Wilcoxon signed-rank test. (D) Analysis of cell division after 0, 60 and 180 min of mazF induction. The Fisher's exact test was used to compare the number of cells with and without division septum. P-values are indicated.
DISCUSSION
In this work, we have used multiple techniques to probe the effects of MazF toxin on S. aureus physiology in an effort to understand how this toxin might regulate cellular changes leading to dormancy. Understanding the causes of dormancy is critical for the management of antibiotic resistance. Presently, it is not known how many distinct mechanisms culminate in S. aureus dormancy, and it is certainly possible that there exists more than one pathway, or that there are redundancies among multiple pathways. Our study now contributes a more comprehensive understanding of the link between MazF toxin and its relation to dormancy. We have summarized our collective observations in a working model (Figure 7) demonstrating examples of the pathways that may be regulated by MazF and lead to bacterial dormancy.
Figure 7.

Model of MazF impact on cell physiology and its implication on cell survival and dormancy. MazE degradation unleashes MazF endoribonuclease activity. MazF cleaves many RNAs and suppresses transcription and translation of many genes rather than a particular set of genes. MazF changes the translational program and specifically affects several pathways: co-translational quality control, ribosome hibernation and recycling, cell division and cell wall thickness. Activation of these pathways may contribute to reversible bacteria dormancy, which allow cells to survive unfavorable growth conditions. Impact of MazF is indicated in red arrows. Activation of the other pathways by MazF is also possible.
Role of MazF in translation inhibition
Whereas we identified only 339 cleaved genes, MazF expression caused a decrease of about 50% of transcripts according to both RNA- and Ribo-Seq. The cleaved transcripts belonged to many different gene families without any statistically significant enrichment. We concluded that MazF cleaves a wide range of mRNAs as it was described for E. coli (29), affecting the entire transcriptome and translatome and causing global changes in cell metabolism.
Even if MazF inhibits cell growth, we did not observe complete drop of polysomes (Figure 2A), showing that the cell remained active, similar to what was reported in E. coli (50). We found that about 17% of transcripts were increased upon MazF induction, indicating that some pathways were upregulated. Notably, in E. coli MazF selectively enabled the synthesis of about 10% of proteins that either promoted cell death or ensured the survival of small subpopulation under stress conditions (20). We conclude that in S. aureus MazF activity does not lead to a complete inhibition but rather to an alteration of the translation program, thereby potentially preparing the cell for long-term survival. In this work, we demonstrated three examples of such pathways that may be activated by MazF, namely translation rescue and co-translational quality control, ribosome hibernation and recycling, and finally, cell division and cell wall thickness (Figure 7).
Role of MazF in translation rescue and co-translational quality control
Cleavage by MazF results in the formation of stop-less transcripts, where stalled ribosomes are accumulated. We observed that MazF activated the SsrA system that rescues stalled ribosomes and resolves non-stop translation. MazF also increased the levels of the ATP-dependent chaperones, which deal with partially synthesized peptides appearing during translation rescue. Both the SsrA system and ATP-dependent chaperones are important players in co-translational quality control mechanisms. Thus, we concluded that in S. aureus MazF induction leads to activation of co-translational quality control mechanisms that resume protein synthesis to maintain cell survival upon dormancy and possibly to facilitate the exit from dormancy state later (Figure 7).
Role of MazF in ribosome hibernation and recycling
A role for MazF in translation inhibition was discussed in several studies mostly performed on E. coli, and several targets of MazF were proposed. It was suggested that E. coli MazF reduces translation through cleavage of both mRNAs and rRNAs (19,29,36,37). In particular, it cleaves rRNA and mRNAs encoding ribosomal proteins, leading to disruption of rRNA maturation and ribosome biogenesis, and, thus, decreases cell growth (29). Earlier we also found that MazF cleaves mRNAs encoding ribosomal proteins in S. aureus (15). In this study, we observed reduced translation of these mRNAs, but, surprisingly, without reduction of the protein levels. We observed, however, a significant decrease of factors responsible for translation, tRNA aminoacylation, ribosome disassembly, and increase of hibernation-promoting factor, Hpf (Supplementary Figure S14).
In bacteria the majority of non-translating ribosomes hibernate in the form of 100S dimers, which sequester ribosomes away from active translation, and prevent ribosome degradation by an unknown pathway. In S. aureus Hpf mediates ribosome dimerization and is required for a long-term survival (51). On the one hand, Hpf suppresses translation through titrating active ribosomes. On the other hand, HpfSa decreases ribosome turnover and preserves them, thereby the dimers can be dissociated and reassembled into active ribosomes upon the exit of quiescence (38). To resume translation ribosome hibernation is reversed by 100S dissociation. Recently, it was shown that in S. aureus ribosome recycling factor (Rrf, also known as Frr) and elongation factor G (EF-G, also known as FusA) synergistically split 100S dimers and, in certain conditions, this process may include activity of HflX GTPase (52). Notably, we observed that MazF decreased levels of rrf (Supplementary Figure S14A). Levels of EF-G and hflX were also decreased but to a lesser extent (Supplementary Table S3), and both these mRNAs were cleaved by MazF (15). Increased levels of hibernation factor and decreased levels of dimer disassembly factors favors ribosome hibernation. These observations are consistent with a model that in S. aureus MazF suppresses translation mainly acting at the level of translation and ribosome hibernation factors, rather than ribosomal proteins (Figure 7). Such a mechanism allows the cell to rapidly decrease translation but also rapidly restart it if needed, e.g. to exit from the dormant state.
Role of MazF in cell wall thickness
MazF affected translation and protein levels of many genes involved in cell wall biosynthesis as illustrated schematically (Supplementary Figure S12). MazF also altered several side metabolic pathways involved in the synthesis of the cell wall. Namely, translation of ADI pathway genes, arcRCDBA, where ArcR is the transcription activator of arc operon, were significantly increased. Upregulated ADI pathway is associated with elevation of ammonia and activation of Glm pathway, which produces UDP-N-acetyl-glucosamine (GlcNAc), the cardinal precursor metabolite for cell wall peptidoglycan (43), and with elevation of ATP, a substrate necessary for Mur ligases in cell wall synthesis (44). Both pathways culminate in increased cell wall thickness. Accordingly, increased cell wall thickness, observed in vancomycin-intermediate S. aureus strains (VISA), was linked to arcB upregulation and elevation of ammonia and ATP (45).
Another example of pathways altered upon MazF expression are purine and pyrimidine biosynthesis and pathways connected to it. Upon MazF expression glycolysis, TCA cycle and pentose phosphate pathway were mostly upregulated in both RNA- and Ribo-Seq, providing substrates for de novo synthesis of nucleotides. We observed that many genes involved in purine and pyrimidine biosynthesis were cleaved by MazF and decreased in both RNA- and Ribo-Seq when mazF was expressed. However, TE of those genes was increased, and accordingly, protein abundance also. Increased purine biosynthesis might be needed to maintain at certain level ATP production for the needs of cell wall synthesis, as it was observed for VISA strains where genes involved in purine biosynthesis were upregulated (53), or for S. aureus persistence to rifampicin (54). Increased pyrimidine biosynthesis provides UMP, which is an important substrate for UDP-GlcNAc, required for the cell wall synthesis. Consistent with our observation, metabolomic analysis revealed that MazF in E. coli increases NMP and NDP levels (50).
Several other observations also connect MazF activity to the cell wall thickness. First, increase of cell wall thickness was observed in a ΔyjbH mutant (55), where MazF was activated (23). Second, deletion of trfA, which stabilized MazE (17), restored sensitivity of cells resistant to cell wall antibiotics (56). Moreover, in other bacteria a possible role of MazF in cell wall biogenesis was also noticed. For example, in E. coli mRNAs isolated from polysomal fractions and responsible for ‘cell structure’ were upregulated upon MazF induction (36).
Possible role of MazF in bacterial dormancy/persistence
The terms of dormancy and persistence are often confusing in the literature. Persister cells are non-growing subpopulations tolerant to antibiotics, and often significantly present in biofilms (5). Persisters arise due to dormancy, defined as a state in which cells decrease metabolism and arrest growth (3,5). Dormant bacteria are also often tolerant to antibiotics.
It has been suggested that MazF in E. coli is involved in drug tolerance and persistence generation (21). In S. aureus activation of MazF leads to quiescence (24) and dormancy (15) and alters susceptibility to antibiotics (14). In this study we induced MazF only for 10 min, thus the changes we observed possibly correspond to a very early stage leading to the entrance to dormancy. In this regard, our observations concerning ribosome hibernation and co-translational quality control systems are very interesting. Both mechanisms promote cell survival and, possibly, reversible cell dormancy instead of cell death (Figure 7).
Ribosome hibernation has emerged as one of the pivotal cellular processes for entry into dormancy and subsequent resuscitation. MazF could favor ribosome hibernation by increasing levels of Hpf, and decreasing ribosome recycling factors. Mechanisms that cause dissociation of the ribosome dimers might decrease cell viability and prevent bacterial dormancy. Accordingly, antibiotic tolerance has been mediated by ribosome dimerization in the absence of Mycobacterial HflX (57). Disruption of 100S dimers, either by decreasing activity of Hpf, or by increasing activity of EF-G/Rrf/HflX, makes these factors possible and promising targets for future antibacterial drug discovery.
Another important connection to dormancy uncovered in our study is the increased transcription and translation of ADI and TCA cycle genes. Upregulation of ADI pathway had been connected to vancomycin resistance (45), while the increased activity of TCA cycle had been connected to persistence in S. aureus (58). Genes encoding enzymes of TCA cycle, ribosomal proteins and ADI pathway were also upregulated in S. aureus biofilms (59) that may be linked to persistence.
Increased cell wall thickness and changes in expression of cell wall related genes caused by MazF, described in this study, may be connected to bacterial dormancy (15) and persistence. Accordingly, activation of the cell wall stress stimulon and peptidoglycan biosynthesis was observed in intracellular S. aureus persisters upon antibiotic exposure (60). Taken together, our observations are important in the regard that approaches leading to MazF inactivation may synergize bactericidal effects of cell wall antibiotics.
MazEF regulation by stress
MazF endoribonuclease activity is neutralized by antitoxin MazE. MazE protein level is controlled by the ATP-dependent ClpCP protease and requires an adaptor protein TrfA (17,18). Transcription of TrfA is regulated in part by activator Spx (61). Spx protein level is, in turn, controlled by ClpXP protease and requires an adaptor protein YjbH (62,63). One can imagine, that stress such as antibiotics, that causes inhibition of transcription and/or translation may inhibit mazEF expression, and prevent de novo synthesis of MazE and MazF. The unstable MazE is degraded and nuclease activity of MazF is unleashed. It has been suggested that in E. coli stress induces transcription of TASs without activating toxins and that transcriptional induction results from antitoxin degradation and relief of transcriptional autoregulation (64). However, the S. aureus MazEF system is unique because it does not repress its own promoter (65). Accordingly, recently we demonstrated that YjbH aggregates in response to stress, thereby initiating YjbH-Spx-TrfA cascade, which results in MazE degradation and, consequently, MazF toxin activation (23). These observations were confirmed by the fact that inhibition of cell growth caused by MazF induction was reverted by trfA deletion (15). Therefore, it is very possible that in S. aureus environmental stress, such as antibiotics, may modulate MazEF function and unleash MazF endoribonuclease activity.
CONCLUSION
We characterized changes and correlations in transcriptome, translatome, and proteome caused by MazF. Important pathways affected during the early stage following MazF induction were identified. These pathways may be promising targets for new antibacterial drugs that prevent bacteria dormancy and synergize the efficacy of conventional antibiotics.
DATA AVAILABILITY
All RNA- and Ribo-Seq data from this study are available at NCBI Gene Expression Omnibus (GSE155036). Mass spectrometry raw data are available at ProteomeXchange (PXD021129).
Supplementary Material
ACKNOWLEDGEMENTS
We kindly thank Friedrich Götz (The University Tübingen, Germany) for supplying the HG003 and the HG003 mazEF deleted strains, Mee-Ngan Yap (Saint Louis University, USA) for anti-Hpf antibody, and William L. Kelley (University of Geneva, Switzerland) for invaluable help and critical reading of the manuscript.
Contributor Information
Fedor Bezrukov, Department of Physics and Astronomy, The University of Manchester, Manchester M13 9PL, UK.
Julien Prados, Department of Microbiology and Molecular Medicine, Faculty of Medicine, University of Geneva, Geneva 1211, Switzerland.
Adriana Renzoni, Department of Microbiology and Molecular Medicine, Faculty of Medicine, University of Geneva, Geneva 1211, Switzerland; Division of Infectious Diseases, Department of Medicine, Geneva University Hospitals and Medical School, Geneva 1211, Switzerland.
Olesya O Panasenko, Department of Microbiology and Molecular Medicine, Faculty of Medicine, University of Geneva, Geneva 1211, Switzerland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Helmut Horten Stiftung (to O.O.P.); Ernst and Lucie Schmidheiny Foundation (to O.O.P.); Swiss National Science Foundation [310030-169404 to A.R.]. Funding for open access charge: Swiss National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998; 339:520–532. [DOI] [PubMed] [Google Scholar]
- 2. Aslam B., Wang W., Arshad M.I., Khurshid M., Muzammil S., Rasool M.H., Nisar M.A., Alvi R.F., Aslam M.A., Qamar M.U.et al.. Antibiotic resistance: a rundown of a global crisis. Infect. Drug Resist. 2018; 11:1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balaban N.Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D.I., Brynildsen M.P., Bumann D., Camilli A., Collins J.J.et al.. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019; 17:441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewis K. Persister cells. Annu. Rev. Microbiol. 2010; 64:357–372. [DOI] [PubMed] [Google Scholar]
- 5. Wood T.K., Knabel S.J., Kwan B.W.. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013; 79:7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rittershaus E.S., Baek S.H., Sassetti C.M.. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe. 2013; 13:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masuda H., Inouye M.. Toxins of prokaryotic toxin-antitoxin systems with sequence-specific endoribonuclease activity. Toxins (Basel). 2017; 9:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otsuka Y. Prokaryotic toxin-antitoxin systems: novel regulations of the toxins. Curr. Genet. 2016; 62:379–382. [DOI] [PubMed] [Google Scholar]
- 9. Yamaguchi Y., Park J.H., Inouye M.. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011; 45:61–79. [DOI] [PubMed] [Google Scholar]
- 10. Schuster C.F., Bertram R.. Toxin-antitoxin systems of Staphylococcus aureus. Toxins (Basel). 2016; 8:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sierra R., Viollier P., Renzoni A.. Linking toxin-antitoxin systems with phenotypes: a Staphylococcus aureus viewpoint. Biochim. Biophys. Acta Gene Regul. Mech. 2019; 1862:742–751. [DOI] [PubMed] [Google Scholar]
- 12. van Rensburg J.J., Hergenrother P.J.. Detection of endogenous MazF enzymatic activity in Staphylococcus aureus. Anal. Biochem. 2013; 443:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu L., Inoue K., Yoshizumi S., Kobayashi H., Zhang Y., Ouyang M., Kato F., Sugai M., Inouye M.. Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 2009; 191:3248–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuster C.F., Mechler L., Nolle N., Krismer B., Zelder M.E., Gotz F., Bertram R.. The MazEF toxin-antitoxin system alters the beta-lactam susceptibility of Staphylococcus aureus. PLoS One. 2015; 10:e0126118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sierra R., Prados J., Panasenko O.O., Andrey D.O., Fleuchot B., Redder P., Kelley W.L., Viollier P.H., Renzoni A.. Insights into the global effect on Staphylococcus aureus growth arrest by induction of the endoribonuclease MazF toxin. Nucleic Acids Res. 2020; 48:8545–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu Z., Donegan N.P., Memmi G., Cheung A.L.. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 2007; 189:8871–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donegan N.P., Marvin J.S., Cheung A.L.. Role of adaptor TrfA and ClpPC in controlling levels of SsrA-tagged proteins and antitoxins in Staphylococcus aureus. J. Bacteriol. 2014; 196:4140–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donegan N.P., Thompson E.T., Fu Z., Cheung A.L.. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010; 192:1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y., Zhang J., Hoeflich K.P., Ikura M., Qing G., Inouye M.. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell. 2003; 12:913–923. [DOI] [PubMed] [Google Scholar]
- 20. Amitai S., Kolodkin-Gal I., Hananya-Meltabashi M., Sacher A., Engelberg-Kulka H.. Escherichia coli MazF leads to the simultaneous selective synthesis of both “death proteins” and “survival proteins. PLoS Genet. 2009; 5:e1000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tripathi A., Dewan P.C., Siddique S.A., Varadarajan R.. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 2014; 289:4191–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fraikin N., Goormaghtigh F., Van Melderen L.. Type II toxin-antitoxin systems: evolution and revolutions. J. Bacteriol. 2020; 202:e00763-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Panasenko O.O., Bezrukov F., Komarynets O., Renzoni A.. YjbH solubility controls Spx in Staphylococcus aureus: implication for MazEF toxin-antitoxin system regulation. Front. Microbiol. 2020; 11:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fu Z., Tamber S., Memmi G., Donegan N.P., Cheung A.L.. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J. Bacteriol. 2009; 191:2051–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma D., Mandell J.B., Donegan N.P., Cheung A.L., Ma W., Rothenberger S., Shanks R.M.Q., Richardson A.R., Urish K.L. The toxin-antitoxin MazEF drives staphylococcus aureus biofilm formation, antibiotic tolerance, and chronic infection. mBio. 2019; 10:e01658-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014; 15:205–213. [DOI] [PubMed] [Google Scholar]
- 28. Brar G.A., Weissman J.S.. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015; 16:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Culviner P.H., Laub M.T.. Global analysis of the E. coli toxin mazf reveals widespread cleavage of mRNA and the inhibition of rRNA maturation and ribosome biogenesis. Mol. Cell. 2018; 70:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herbert S., Ziebandt A.K., Ohlsen K., Schafer T., Hecker M., Albrecht D., Novick R., Gotz F.. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 2010; 78:2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helle L., Kull M., Mayer S., Marincola G., Zelder M.E., Goerke C., Wolz C., Bertram R.. Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology. 2011; 157:3314–3323. [DOI] [PubMed] [Google Scholar]
- 32. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGlincy N.J., Ingolia N.T.. Transcriptome-wide measurement of translation by ribosome profiling. Methods. 2017; 126:112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiao Z., Zou Q., Liu Y., Yang X.. Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun. 2016; 7:11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basu A., Yap M.N.. Disassembly of the Staphylococcus aureus hibernating 100S ribosome by an evolutionarily conserved GTPase. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E8165–E8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sauert M., Wolfinger M.T., Vesper O., Muller C., Byrgazov K., Moll I.. The MazF-regulon: a toolbox for the post-transcriptional stress response in Escherichia coli. Nucleic. Acids. Res. 2016; 44:6660–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vesper O., Amitai S., Belitsky M., Byrgazov K., Kaberdina A.C., Engelberg-Kulka H., Moll I.. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell. 2011; 147:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Basu A., Yap M.N.. Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic. Acids. Res. 2016; 44:4881–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neubauer C., Gillet R., Kelley A.C., Ramakrishnan V.. Decoding in the absence of a codon by tmRNA and SmpB in the ribosome. Science. 2012; 335:1366–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huter P., Muller C., Arenz S., Beckert B., Wilson D.N.. Structural basis for ribosome rescue in bacteria. Trends Biochem. Sci. 2017; 42:669–680. [DOI] [PubMed] [Google Scholar]
- 41. Flynn J.M., Levchenko I., Seidel M., Wickner S.H., Sauer R.T., Baker T.A.. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gingold H., Pilpel Y.. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011; 7:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cui L., Murakami H., Kuwahara-Arai K., Hanaki H., Hiramatsu K.. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 2000; 44:2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lovering A.L., Safadi S.S., Strynadka N.C.. Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 2012; 81:451–478. [DOI] [PubMed] [Google Scholar]
- 45. Tan X.E., Neoh H.M., Looi M.L., Chin S.F., Cui L., Hiramatsu K., Hussin S., Jamal R.. Activated ADI pathway: the initiator of intermediate vancomycin resistance in Staphylococcus aureus. Can. J. Microbiol. 2017; 63:260–264. [DOI] [PubMed] [Google Scholar]
- 46. Barreteau H., Kovac A., Boniface A., Sova M., Gobec S., Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008; 32:168–207. [DOI] [PubMed] [Google Scholar]
- 47. Swoboda J.G., Campbell J., Meredith T.C., Walker S.. Wall teichoic acid function, biosynthesis, and inhibition. ChemBioChem. 2010; 11:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown S., Santa Maria J.P. Jr., Walker S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013; 67:313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sobral R., Tomasz A.. The staphylococcal cell wall. Microbiol Spectr. 2019; 7:doi:10.1128/microbiolspec.GPP3-0068-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mok W.W., Park J.O., Rabinowitz J.D., Brynildsen M.P.. RNA futile cycling in model persisters derived from MazF accumulation. MBio. 2015; 6:e01588-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ueta M., Wada C., Wada A.. Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells. 2010; 15:43–58. [DOI] [PubMed] [Google Scholar]
- 52. Basu A., Shields K.E., Yap M.F.. The hibernating 100S complex is a target of ribosome-recycling factor and elongation factor G in Staphylococcus aureus. J. Biol. Chem. 2020; 295:6053–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mongodin E., Finan J., Climo M.W., Rosato A., Gill S., Archer G.L.. Microarray transcription analysis of clinical Staphylococcus aureus isolates resistant to vancomycin. J. Bacteriol. 2003; 185:4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yee R., Cui P., Shi W., Feng J., Zhang Y.. Genetic screen reveals the role of purine metabolism in Staphylococcus aureus persistence to rifampicin. Antibiotics (Basel). 2015; 4:627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Renzoni A., Andrey D.O., Jousselin A., Barras C., Monod A., Vaudaux P., Lew D., Kelley W.L.. Whole genome sequencing and complete genetic analysis reveals novel pathways to glycopeptide resistance in Staphylococcus aureus. PLoS One. 2011; 6:e21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Renzoni A., Kelley W.L., Barras C., Monod A., Huggler E., Francois P., Schrenzel J., Studer R., Vaudaux P., Lew D.P.. Identification by genomic and genetic analysis of two new genes playing a key role in intermediate glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2009; 53:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rudra P., Hurst-Hess K.R., Cotten K.L., Partida-Miranda A., Ghosh P.. Mycobacterial HflX is a ribosome splitting factor that mediates antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang W., Chen J., Chen G., Du X., Cui P., Wu J., Zhao J., Wu N., Zhang W., Li M.et al.. Transposon mutagenesis identifies novel genes associated with staphylococcus aureus persister formation. Front. Microbiol. 2015; 6:1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Resch A., Rosenstein R., Nerz C., Gotz F.. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl. Environ. Microbiol. 2005; 71:2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Peyrusson F., Varet H., Nguyen T.K., Legendre R., Sismeiro O., Coppee J.Y., Wolz C., Tenson T., Van Bambeke F.. Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat. Commun. 2020; 11:2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jousselin A., Kelley W.L., Barras C., Lew D.P., Renzoni A.. The Staphylococcus aureus thiol/oxidative stress global regulator Spx controls trfA, a gene implicated in cell wall antibiotic resistance. Antimicrob. Agents Chemother. 2013; 57:3283–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Engman J., von Wachenfeldt C.. Regulated protein aggregation: a mechanism to control the activity of the ClpXP adaptor protein YjbH. Mol. Microbiol. 2015; 95:51–63. [DOI] [PubMed] [Google Scholar]
- 63. Engman J., Rogstam A., Frees D., Ingmer H., von Wachenfeldt C.. The YjbH adaptor protein enhances proteolysis of the transcriptional regulator Spx in Staphylococcus aureus. J. Bacteriol. 2012; 194:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. LeRoux M., Culviner P.H., Liu Y.J., Littlehale M.L., Laub M.T.. Stress can induce transcription of toxin-antitoxin systems without activating toxin. Mol. Cell. 2020; 79:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Donegan N.P., Cheung A.L.. Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 2009; 191:2795–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA- and Ribo-Seq data from this study are available at NCBI Gene Expression Omnibus (GSE155036). Mass spectrometry raw data are available at ProteomeXchange (PXD021129).