


Abstract
The tRNA modification N6-threonylcarbamoyladenosine (t6A) is universally conserved in all organisms. In bacteria, the biosynthesis of t6A requires four proteins (TsaBCDE) that catalyze the formation of t6A via the unstable intermediate l-threonylcarbamoyl-adenylate (TC-AMP). While the formation and stability of this intermediate has been studied in detail, the mechanism of its transfer to A37 in tRNA is poorly understood. To investigate this step, the structure of the TsaBD heterodimer from Escherichia coli has been solved bound to a stable phosphonate isosteric mimic of TC-AMP. The phosphonate inhibits t6A synthesis in vitro with an IC50 value of 1.3 μM in the presence of millimolar ATP and L-threonine. The inhibitor binds to TsaBD by coordination to the active site Zn atom via an oxygen atom from both the phosphonate and the carboxylate moieties. The bound conformation of the inhibitor suggests that the catalysis exploits a putative oxyanion hole created by a conserved active site loop of TsaD and that the metal essentially serves as a binding scaffold for the intermediate. The phosphonate bound crystal structure should be useful for the rational design of potent, drug-like small molecule inhibitors as mechanistic probes or potentially novel antibiotics.
INTRODUCTION
Transfer RNAs from all forms of life are subject to a large number of enzyme-catalysed post-transcriptional chemical modifications that fine tune both its structure and function in the cell (1–4). The N6-threonylcarbamoyladenosine (t6A) modification (5) is found at position 37 adjacent to the anticodon in tRNAs that decode 5′-ANN codons (6,7), and is universally conserved throughout life (8). Structurally, t6A enhances codon recognition by disrupting base pairing with U33 in the anticodon loop and by interacting with both anticodon bases as well as the first base of the mRNA codon (9). Mutants in both bacteria (10,11) and eukaryotes (11,12) that lack t6A typically have severe growth defects or, as in the case of Escherichia coli, are inviable (13). In many organisms t6A is further modified to the hydrolytically labile cyclized hydantoin ct6A, which is formed by ATP-catalysed cyclization of t6A (14,15) (Figure 1). This has prompted a call for re-evaluation of the structural explanation for its role in codon recognition. The catalytic machinery for the biosynthesis of t6A differs significantly between bacteria and eukaryotes/archaea. In eukaryotes, t6A is catalysed by the Tcs1 (SUA5) protein (10) in conjunction with the KEOPS complex (16–19), which contains five proteins. KEOPS was originally identified as a transcription complex (20) involved in telomere regulation (21). In addition to its requirement for t6A tRNA modification, KEOPS proteins in yeast have t6A-independent roles in telomere replication via SUA5p (22) and as an auxiliary sensor of DNA double strand breaks (23). KEOPS mutations have been recently implicated in neurodegeneration with nephrotic disease in humans (24–26). In mitochondria, a minimal t6A system operates, consisting of Tcs1 and the mitochondrial protein Qri7 (27). Expression of Qri7 in the cytoplasm can complement loss of KEOPS proteins in t6A formation. This result has implicated the KEOPS Qri7 homolog OSGEP (Tcs3) as the main catalytic component responsible for t6A synthesis. In human mitochondria, t6A formation was recently found to function as a CO2 sensor, in that low CO2 levels result in low t6A formation and reduced translation of mitochondrial proteins (28). Based on the dependence of t6A in bacteria for normal growth, and the apparent divergence of the protein machinery between bacterial and eukaryotic t6A synthesis, this modification has been identified as a potential antibiotic target.
Figure 1.

Biosynthesis of t6A in bacteria. In most bacterial tRNAs, t6A is further cyclized to the hydantoin ct6A, which is hydrolytically unstable to commonly used isolation procedures.
As shown in Figure 1, the biosynthesis of t6A in bacteria involves four distinct proteins (29). The first step in the biosynthesis is carried out by TsaC1(YrdC)/TsaC2 (SUA5), which are short/long bacterial homologs, respectively, of eukaryotic Tcs1/SUA5. These enzymes catalyse an l-threonine-dependent adenylation reaction, using bicarbonate (possibly via CO2), to form a putative l-threonine carbamate that is adenylated with ATP to give the unstable product TC-AMP (30). This mechanism was also shown to operate in vitro for both eukaryotes and archaea (31). There is evidence that although TsaC1 and TsaC2 catalyse the same reaction, they may operate by different mechanisms due to the involvement in TsaC2 of additional hinge and SUA5 domains (32). The TsaBDE proteins together in the presence of ATP can catalyse the addition reaction of exogenously supplied TC-AMP with the tRNA substrate to give t6A and AMP as products (30). Similarly, in yeast it was shown that SUA5p and Qri7 can catalyse t6A formation via TC-AMP diffusion without direct protein-protein interaction (33). The bacterial TsaD protein is a homolog to the ancestral (COG0533) family of carbamoyltransferase proteins that include KAE1/OSGEP/TobZ that utilize carbamoyl adenylate substrates or intermediates to carbamoylate nucleophiles (34,35). Previous structural and mechanistic studies in Salmonella tymphimurium (36), E. coli (37) and Thermotoga maritima (38–41) have shown a functional requirement for the formation of a TsaBD heterodimer that binds tRNA (38) and likely binds TC-AMP. The role of the small TsaE protein in t6A formation is more complicated. It was characterized initially as an ATPase with low intrinsic activity (42–44) that is stimulated by the presence of TsaBD (29,37). As shown in Figure 1, the chemical transformation from TC-AMP to t6A does not formally require ATP. Indeed, bacterial t6A formation is observed in vitro in the absence of this protein (30); however, as explicitly shown in T. maritima (38), TsaE catalysed ATP-hydrolysis is required for multiple turnover.
A comparison of structural studies in bacteria show that the quaternary structure for the TsaBDE complex is species dependent. In E. coli and S. typhimurium, small angle X-ray scattering (SAXS) and isothermal calorimetry (ITC), respectively, were used to determine the ternary complex is a TsaB1D1E1 heterotrimer, the formation of which is ATP-dependent (36,37). In E. coli, there is a novel additional ADP binding site at the TsaB–TsaD interface (37). The T. maritima proteins form a hexameric TsaB2D2E2 complex as a dimer of heterotrimers with TmTsaB as the dimerization domain (38,39). The role of the TsaE protein in both systems appears to be regulatory in nature, in that TmTsaE binding to the TmTsaB2D2E1 complex has been shown to be competitive with tRNA (38). This is consistent with the observation in E. coli that EcTsaBDE does not bind tRNA (37), while EcTsaBD does (38). TsaE-catalysed ATP hydrolysis is required for additional turnover, possibly via a resetting of the active conformation of TsaBD (38). Missoury et al. (39,40) observed that TmTsaE binding to TmTsaBD induces a partial denaturing of the TsaD active site. Both studies are in agreement that TmTsaE binding precludes the binding of tRNA. Questions remain as to the exact mechanism and timing of the conformational effects elicited by TsaE during the catalytic cycle, as well as the timing of tRNA binding relative to TC-AMP loading onto TsaD and the potential role of TsaC1/C2 in the latter. Each of these steps may be specifically tailored in mesophiles versus thermophiles, due in part to the instability of TC-AMP and the need to avoid wasteful ATP utilization.
While a structure of TC-AMP bound to TsaC2 from Sulfolobus tokodaii has been reported (34,45), there are as yet no structures of TC-AMP bound to the TsaBD heterodimer. Luthra et al. (41) have recently reported the structure of T. maritima TsaB2D2E2 with the unstable ligand carboxy-AMP bound in the TsaD active site. This structure was used to model TC-AMP binding to the complex. Efforts at solving a structure of TsaBD with TC-AMP itself have been hampered by the known instability of this substrate (30). In an effort to circumvent this barrier, we report here the preparation of chemically stable analogs of TC-AMP and an assessment of their ability to inhibit the formation of t6A in the E. coli system. The most potent inhibitor is an isosteric phosphonate analog, in which the adenylate ester oxygen of TC-AMP is replaced by a methylene group. We show here that this stable analog binds to the EcTsaBD heterodimer with affinity in the low micromolar range and we report the crystal structure of the bound complex. Interestingly, the active site Zn ion interacts with the inhibitor by chelating oxygens from both the carboxylate of the l-threonine moiety as well as one of the oxygen atoms of the phosphonate. The structure suggests that TsaBD binds TC-AMP inside the catalytic pocket in an orientation that may be initially sterically unreactive but a simple conformational rearrangement may lead to a more reactive bound form upon binding of tRNA.
MATERIALS AND METHODS
Protein purification and preparation of site-directed mutants
Proteins for kinetic assays and binding analysis were overproduced and purified as previously described (30). Site-directed mutants of E. coli TsaD were prepared using Quikchange mutagenesis (Agilent Technologies) with tsaD cloned into pET28a as the template (30) and pfuTurbo DNA polymerase. Oligonucleotides for mutagenic PCR are from Integrated DNA Technologies, Inc. and are listed in Supplementary Table S1. Plasmids were purified using the QIAprep spin miniprep kit (QIAgen) and both strands of the plasmid were sequenced across the entire gene to confirm the accuracy of the desired mutation (UW Biotechnology Center). For assays, proteins were quantified using the BioRad Bradford Protein Assay.
Preparation of tRNA transcripts
Unmodified tRNA was prepared by in vitro runoff transcription using overlapping oligonucleotides essentially as described (46,47). T4 DNA polymerase Klenow fragment (Promega) was used to extend overlapping oligonucleotides containing a 12-bp overlapping region and a fused T7 promoter sequence. For E. coli tRNALys, the oligos utilized were the following: Plus strand: 5′-TAATACGACTCACTATAGGGTCGTTAGCTCAGTTGGTAGAGCAGTTGACTTTTAATC and minus strand: 5′-TGGTGGGTCGTGCAGGAATTGAACCTGCGACCAATTGATTAAAAGTCA (T7 promoter underlined and overlapping bases in italics). The extension reaction used 1 mM each oligo and 0.4 mM dNTPs with 10 units/ml polymerase in the manufacturer supplied buffer at 37°C for 2 h. DNA was isolated by phenol then chloroform extraction and ethanol precipitation. The redissolved DNA was then used directly for in vitro transcription. Transcription reactions contained 1 μM template DNA in a reaction mixture consisting of 50 mM Tris, pH 7.8, 35 mM MgCl2, 1 mM spermidine, 5 mM of each NTP, 20 units/ml RNasin (Promega) and 1000 units of T7 RNA polymerase (Lucigen). After incubating at 37°C overnight, 10 units of RQ1 RNase-free DNase was added directly to the mixture and incubated at 37°C for 30 min. After addition of an equal volume of 7.5 M urea with 0.1% bromophenol blue, the RNA was purified by polyacrylamide gel electrophoresis using 8% slab gels and run at 20 W for 1.5 h. RNA bands were visualized by UV shadowing on a fluorescent TLC plate (Merck). The RNA was excised and eluted in 0.5 M NaCl, filtered and then precipitated with 2.5 vol ethanol at –20°C. After centrifugation at 14 000 × g at 4°C (Beckman Avanti-J 25.50 rotor), the RNA pellet was washed with cold 70% ethanol, air-dried briefly and resuspended in deionized water and stored at –80°C. Typical yields were 1.6 mg of tRNA transcript/ml of transcription reaction.
In vitro assay for t6A formation
An HPLC assay was used to quantify t6A by UV detection at 260 nm. Full reaction mixtures (50 μl) contained 50 mM MOPS pH 7.5, 20 mM NaHCO3, 25 mM KCl, 50 mM l-threonine, 1 mM ATP, 10 μM unmodified E. coli tRNALys, and 1 μM each of TsaC, TsaB, TsaD and TsaE. (For substrates with apparent Km values <10 μM, 0.5 μM enzyme concentrations were used.) For kinetic studies, one of the substrates was varied while all other substrates were kept at the concentrations stated above. Reactions were initiated with enzymes by adding TsaE, TsaB, TsaD and then TsaC as the last component followed by mixing by vortexing for 1 s. Reaction mixtures were incubated at 23°C for 5 min and quenched by adding 25 μl of the reaction mixture to a new tube containing 4 μl 0.25 M EDTA pH 8 followed by vortexing and placing on dry ice. Once a set of reactions was complete, tubes were thawed and placed in a microcentrifuge (Eppendorf) and spun at 14 000 rpm for 10 s. Small molecules were removed by addition of the quenched reaction mixtures to micro biospin columns (BioRad) containing 0.7 ml (settled volume) Sephadex G-50 (fine), which had been drained by centrifugation at 7000 rpm for 1 min using a microcentrifuge. After careful addition of the reaction mixture to the top of the gel, the column was eluted by centrifugation at 7000 rpm for 1 min. To the resulting RNA eluate was added 0.1 volume of 0.3 M NaOAc, 1 mM Zn(OAc)2, pH 6, and the tRNA was digested by addition of 1.5 units of nuclease P1 and 0.05 units of alkaline phosphatase for ≥2 h at 37°C. The digested RNA (∼45 μl) was analysed directly by HPLC using a Supelco LC-18S column (250 × 4.6 mm) similar to reported methods (48,49). The HPLC analysis used an isocratic buffer system consisting of 5 mM ammonium acetate pH 5.3, containing 4% (v/v) acetonitrile. Under these conditions the nuclease P1 reaction was the rate determining enzyme in the digestion. If insufficient nuclease or digestion time was used for this step, an additional peak is observed at a longer retention time (24 min versus 21 min for t6A). Material isolated from this peak was consistent with the dinucleotide 5′-t6ApA-3′ as determined by enzymatic analysis. For kinetic analysis of TC-AMP as the substrate, TsaC, l-threonine and bicarbonate were omitted, and the reaction was initiated by addition of TC-AMP.
Synthesis of TC-AMP mimic BK951 (1)
Figure 3 shows the overall synthetic scheme. Experimental details for the synthesis of analogs 2–5 can be found in the Supplementary Material.
Figure 3.

Synthesis of phosphonate inhibitor BK951. See Materials and Methods for experimental details.
Nα-(2-Chloroacetyl)-O-benzyl-l-threonine benzyl ester (7)
Chloroacetyl chloride (3.19 ml, 0.040 mol) was added drop wise to a solution of K2CO3 5.8 g (0.041 mol) and O-benzyl-l-threonine benzyl ester 6 (10.91 g, 0.036 mol) in DCM (73 ml) at 0°C. Following the addition, the ice bath was removed and the mixture was allowed to reach room temperature before heating to reflux. After stirring for 2.5 h at reflux, TLC verified that all of the starting material was consumed. The mixture was allowed to cool to room temperature and partitioned between DCM (300 ml) and H2O (300 ml). The organic layer was separated and set aside while the aqueous layer was extracted with DCM (3 × 200 ml). The combined organic layers were then washed with saturated NH4Cl solution (1 × 300 ml) and brine (1 × 300 ml), dried with MgSO4, and filtered. The solvent was removed under reduced pressure to give 7 as a white solid (12.84 g, 94%). 1H NMR (400 MHz, CDCl3) δ 7.07–7.44 (m, 11H), 5.15, 5.09 (ABq, JAB = 12.20 Hz, 2H), 4.67 (dd, J = 9.16, 2.32 Hz, 1H), 4.52, 4.30 (ABq, JAB = 12.00 Hz, 2H), 4.19 (qd, J = 6.19, 2.32 Hz, 1H), 4.11 (s, 2H), 1.22 (d, J = 6.19 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.0, 166.9, 138.0, 135.4, 128.9, 128.8, 128.7, 128.1, 127.9, 74.3, 71.1, 67.7, 57.4, 42.8, 16.7. MS (ESI): m/z 398 (M+Na)+. HRMS calculated for C20H22ClNO4 (M+Na)+: 398.112957; Found: 349.11309.
Nα-[2-(Dimethoxyphosphinyl)acetyl]-O-benzyl-l-threonine benzyl ester (8a)
A round bottom flask equipped with a stir bar and condenser was charged with 7 (1.88 g, 0.005 mol) and trimethyl phosphite (5.14 ml, 0.03 mol), and heated to reflux at 130°C. The reaction was allowed to stir at these conditions overnight. The next morning TLC was used to verify the absence of starting material. Excess trimethyl phosphite was distilled off and the crude product was purified by column chromatography on silica gel, using 3:1 EtOAc:Hexanes to 100% EtOAc gradient to yield 8a as a yellow oil (1.94 g, 86%). 1H NMR (400 MHz, CDCl3) δ 7.22–7.35 (m, 8H), 7.16 - 7.22 (m, 2H), 7.04 (d, J = 8.30 Hz, 1H), 5.14, 5.09 (ABq, JAB = 12.20 Hz, 2H), 4.69 (dd, J = 8.30, 2.20 Hz, 1H), 4.51, 4.29 (ABq, JAB = 11.96 Hz, 2H), 4.17 (qd, J = 6.30, 2.20 Hz, 1H), 3.71–3.85 (m, 6H), 2.89–3.04 (m, 2H), 1.25 (d, J = 6.30 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 170.3, 164.6, 138.1, 135.5, 128.8, 128.6, 128.5, 127.9, 127.9, 74.5, 71.0, 67.4, 57.4, 16.5. 31P NMR (162 MHz, CDCl3) δ 23.90. MS (ESI): m/z 472 (M+Na)+. HRMS calculated for C22H28NO7P (M+Na)+: 472.14956; Found: 472.14959.
Nα-[2-(Methoxyhydroxyphosphinyl)acetyl]-O-benzyl-l-threonine benzyl ester (8b)
A round bottom flask equipped with a stir bar was charged with 8a (1.94 g, 0.00432 mol) and CH3CN (85 ml). After the addition of LiBr (0.750 g, 0.00864 mol), the mixture was brought to 75°C and stirred for 3 h. Upon completion, the solvent was removed under reduced pressure and the crude oil was diluted with H2O (200 ml) and 1M HCl solution (50 ml). This aqueous layer was extracted with DCM (4 × 75 ml). The organic fractions were combined, washed with brine (1 × 100 ml), dried with MgSO4, filtered, and the solvent was removed under reduced pressure to give 8b as a hard, pale white film (1.5 g, 80%). 1H NMR (400 MHz, CDCl3) δ 11.05 (br. s., 1H), 6.96–7.53 (m, 11H), 4.95–5.22 (m, 2H), 4.74 (d, J = 8.89 Hz, 1H), 4.45, 4.24 (ABq, JAB = 11.75 Hz, 2H), 4.11 (m, 1H), 3.65 (m, 3H), 2.85–3.10 (m, 2H), 1.01–1.36 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 170.2, 166.5, 138.0, 135.5, 128.6, 128.5, 128.5, 128.4, 127.8, 74.5, 70.8, 67.3, 57.2, 16.2. 31P NMR (162 MHz, CDCl3) δ 21.48. MS (ESI): m/z 434 (M-H)−. HRMS calculated for C21H26NO7P (M-H)−: 434.137412; Found: 434.13863.
5′-O-[(Methoxy)[(2-oxo-2(O-benzyl-l-threoninebenzylester))ethyl)phosphinyl]-6-chloro-9-(2′,3′-O-isopropylidene-d-ribofuranosyl)purine (9)
To 8b (0.233 g, 0.54 mmol), 9 (0.175 g, 0.54 mmol) and PPh3 (0.238 g, 0.00108 mol) under argon was added distilled THF (1.3 ml). Upon dissolution, DEAD (40% in toluene, 0.424 ml, 0.00108 mol) was added drop wise. Stirring was continued for 20 h at room temperature at which point nearly all starting material was consumed according to TLC. The solvent was removed under reduced pressure and the crude product was taken directly to purification by column chromatography on silica gel using a 2:1 EtOAc:Hexanes to 100% EtOAc gradient. Compound 10 was isolated as a mix of diastereomers (2:1 ratio) as a white foam (0.315 g, 78%). 1H NMR (400 MHz, CDCl3) δ 8.78 (m, 3H), 8.41 (s), 8.28 (s), 7.22–7.35 (m), 7.13–7.20 (m), 6.99–7.05 (m), 6.86 - 6.93 (m), 6.19 (d, J = 2.67 Hz, 2H), 6.17 (d, J = 2.94 Hz, 1H), 5.31–5.38 (m), 5.04–5.17 (m), 4.62–4.70 (m), 4.57 (m), 4.48 (m), 4.30–4.43 (m), 4.21–4.30 (m), 4.10–4.20 (m), 3.77 (m), 3.72 (m), 3.68 (m), 2.90 - 2.99 (m), 2.82–2.90 (m), 1.59–1.67 (m), 1.39 (s), 1.16–1.26 (m). 13C NMR (126 MHz, CDCl3) δ 169.9, 164.2, 164.2, 152.1, 152.0, 151.1, 151.0, 151.0, 144.5, 144.3, 137.6, 137.6, 135.1, 132.1, 132.1, 132.0, 132.0, 131.9, 131.8, 128.5, 128.4, 128.3, 128.2, 127.7, 127.6, 127.5, 114.9, 114.7, 90.8, 85.2, 85.2, 85.1, 85.0, 84.2, 84.0, 81.0, 74.4, 70.9, 70.8, 67.2, 66.0, 66.0, 65.7, 65.7, 57.2, 53.2, 53.2, 35.4, 35.3, 34.4, 34.2, 27.3, 27.2, 25.5, 16.3. 31P NMR (162 MHz, CDCl3) δ 23.97, 23.60. MS (ESI): m/z 766 (M+Na)+. HRMS calculated for C34H39ClN5O10P (M+Na)+: 766.201527; Found: 766.20110.
5′-O-[(Hydroxy)[(2-oxo-2(O-benzyl-l-threoninebenzylester))ethyl)phosphinyl]-6-azido-9-(2′,3′-O-isopropylidene-d-ribofuranosyl)purine sodium salt (11)
A round bottom flask was charged with a diastereomeric mix of 10 (0.4 g, 0.54 mmol) and stir bar. After addition of DMF (5.83 ml) and NaN3 (0.087 g, 1.35 mmol), the reaction was allowed to stir overnight at 60°C. TLC the following morning confirmed the absence of starting material. Following the removal of solvent under reduced pressure, the crude oil product was purified directly by column chromatography on silica gel using 10% H2O in CH3CN as a running solvent. 11 was isolated as a single diastereomer, resembling an off white solid (0.3 g, 73%). It should be noted that a small fraction (>10%) of the sample appears to exist as the N1, C6 tetrazole tautomer in CD3OD. 1H NMR (400 MHz, CD3OD) δ 9.81 (s, 1H), 8.81 (s, 1H), 7.10–7.28 (m, 10H), 6.40 (d, J = 2.64 Hz, 1H), 5.39 (br. s., 1H), 5.18 (d, J = 4.70 Hz, 1H), 5.04 (s, 2H), 4.66 (m, 1H) 4.55 (br. s., 1H), 4.47, 4,25 (ABq, JAB = 11.75 Hz, 2H), 4.06–4.18 (m, 3H), 2.68–2.93 (m, 2H), 1.56–1.65 (m, 3H), 1.38 (s, 3H), 1.17 (m, 3H). 13C NMR (126 MHz, CD3OD) δ 147.2, 145.0, 143.7, 139.9, 137.4, 130.0, 129.9, 129.8, 129.8, 129.4, 129.1, 115.7, 93.8, 88.4, 88.3, 87.1, 83.9, 76.2, 72.5, 68.7, 66.8, 28.2, 26.3, 17.2. 31P NMR (202 MHz, CD3OD) δ 14.17. MS (ESI): m/z 735 (M)−. HRMS calculated for C33H36N8O10P (M)−: 735.22975; Found: 735.22854.
5′-O-[(Hydroxy)[(2-oxo-2(O-benzyl-l-threoninebenzylester))ethyl)phosphinyl]-adenosine sodium salt (1, BK951)
A round bottom flask equipped with a stir bar was charged with 11 (0.085 g, 0.112 mmol) and PdCl2 (0.02 g, 0.112 mmol), then subsequently flushed with nitrogen. MeOH (2.25 ml) and H2O (2.25 ml) were added via glass syringe. Following dissolution, the round bottom flask was equipped with a hydrogen balloon and the reaction was allowed to stir for 26 h. At this point, crude NMR revealed that a majority of the reaction mixture was the desired product. The minor product present was the debenzylated 2′, 3′-O-isopropylideneadenosine analogue. TFA (0.2 ml) was added and the reaction was stirred for an additional hour in order to convert the remaining isopropylidene analogue to the desired product. The solvent was then removed under reduced pressure and the crude reaction mixture was taken up in MeOH and filtered through celite. The filtered product was then further purified by HPLC using a Vydac C18 prep column (250 × 30 mm) and the following gradient: A = 10 mM ammonium bicarbonate; B = 10 mM ammonium bicarbonate, 20% (v/v) MeOH. 0–12 min: 100% A. 12–40 min: 0→100% A→B to give (0.02 g, 36%) of BK951 (1) as a transparent foam. 1H NMR (500 MHz, D2O) δ 8.48 (s, 1H), 8.22 (s, 1H), 6.13 (d, J = 5.74 Hz, 1H), 4.77 (dd, J = 5.74, 4.31 Hz, 1H), 4.52 (m, 1H), 4.41 (br. s., 1H), 4.12–4.30 (m, 4H), 2.80–3.02 (m, 2H), 1.16 (d, J = 6.25 Hz, 3H). 13C NMR (126 MHz, D2O) δ 176.3, 168.7, 155.0, 152.3, 148.5, 139.3, 118.1, 86.4, 83.5, 73.8, 70.0, 67.8, 63.6, 60.3, 36.3, 35.3, 18.9. 31P NMR (162 MHz, D2O) δ 16.21. MS (ESI): m/z 513 (M+Na)+. HRMS calculated for C16H23N6O10P (M+Na)+: 513.110548; Found: 513.10904.
Measurement of IC50 values for inhibitors
Reaction mixtures and the procedure for measuring IC50 values for the overall reaction were the same as described above, except the concentration of l-threonine was reduced to 1 mM and the inhibitors were preincubated with enzymes in the reaction mixture for 1 min. Reactions were initiated by the addition of ATP. For measuring IC50 values for the isolated second step of the reaction, the reaction mixtures contained 10 μM TC-AMP, 1 mM ATP, 0.5 μM of EcTsaBDE and variable amounts of BK951 in a total volume of 50 μl. Reactions were initiated by addition of TC-AMP and incubated at 23°C for 5 min and worked up and analysed as described for the full reaction. Measurement of IC50 values for TsaC were performed with reaction mixtures that were the same as for the overall assays except TsaBDE and tRNA were omitted. TsaC was incubated with all components except for ATP for 2 min at 23°C before initiating the reaction by the addition of ATP. After incubating for 1 min at 23°C, the mixture was injected directly onto a Supelco LC-18T HPLC column (4.6 × 250 mm). Isocratic elution was used with 50 mM potassium phosphate pH 6 and 1% acetonitrile and the amount of TC-AMP was measured by UV detection at 260 nm.
Enzymatic preparation of TC-AMP and SC-AMP
Reaction mixtures (80 μl) contained 50 mM MOPS pH 7.5, 10 mM MgCl2, 25 mM KCl, 2 mM ATP, 50 mM l-threonine, 40 mM NaHCO3 and 42 μg (37 μM) of EcTsaC. After incubating at 23°C for 10 min, the reaction mixture was injected onto a Supelco LC-18T column (250 mm × 4.6 mm) and eluted with an isocratic buffer of 50 mM potassium phosphate pH 6.0. Typical yields were 25 nmol at a concentration of 20–30 μM for TC-AMP. Solutions could be carefully concentrated by rotary evaporation (water bath < 30°C) and used directly or aliquoted and stored at –80°C. (Approximately 10% degradation was typically observed for each freeze/thaw cycle.) The serine analog of TC-AMP (SC-AMP) was also prepared by substituting l-serine for l-threonine and purifying by an analogous method. Typical yields for SC-AMP were 10 nmol per reaction. The identity of SC-AMP was confirmed by accurate mass determination (HRMS Calcd for MH+ 479.09222; Found 479.09214) and purity was assessed by HPLC (>95%, Supplementary Figure S5).
Data analysis for determining kinetically derived parameters
For kinetic analysis, concentrations for all fixed substrates were the same as described above for the full reaction mixture. Reactions were run in duplicate and values for Km and kcat were derived from fitting to the standard Michaelis–Menten model (Equation 1). For substrates with Km values near the enzyme concentrations (≤5-fold), kinetic values were derived from Morrison's equation (50) for tight binding substrates (Equation 2). For kinetics that show substrate inhibition, the data were fit to a simple model for complete substrate inhibition in which the complex containing two bound substrates is inactive (Equation 3). Error values reported are the standard error for the fit. All data analysis was performed using Prism 8 (GraphPad) software. Values for kcat in Tables 1 and 2 were obtained by dividing V0 (Vmax) values obtained from curve fitting by the known enzyme concentration.
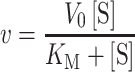 |
(1) |
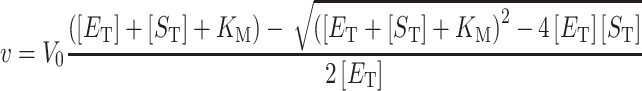 |
(2) |
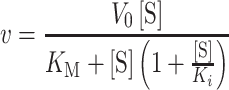 |
(3) |
Table 1.
Summary of kinetically-derived parameters for t6A overall and partial reactions
| Ligand | Overall reaction | TsaBDE reaction | TsaC reaction | |||
|---|---|---|---|---|---|---|
| K m (μM) | k cat (min−1) | K m (μM) | k cat (min−1) | K m (mM) | k cat (min−1) | |
| L-Thr | 590 ± 90 | 0.42 ± 0.02 | — | — | 1.77 ± 0.26 | 9.0 ± 0.4 |
| ATP | 60 ± 6 (Kis = 2200)a | 0.40 ± 0.4 | 14 ± 3 (Kis = 1800)a | 0.12 ± 0.1 | 0.13 ± 0.01 | 7.9 ± 0.2 |
| tRNALysUUU | 0.5 ± 0.2 (Kd = 87 ± 11) | 0.50 ± 0.03 | ndb | nd | ||
| TC-AMP | 1.5 ± 0.4 | 0.31 ± 0.02 | ||||
aCalculated for observed substrate inhibition.
bNot determined.
Table 2.
Summary of kinetically-derived inhibition and thermodynamically-derived binding constants of TCAMP and its analogs to t6A proteins
| IC50 (μM) | |||
|---|---|---|---|
| Ligand | Overall reactiona | TsaBDE reactionb | TsaC reactionc |
| BK951 | 1.3 ± 0.5 | 7.5 ± 1.1 (Ki = 2.5)d (Kd = 3.4 ± 0.6)e |
>180 |
| 2 | 51.2 ± 1.4 | 123 ± 1.2 (Ki = 41)d | 31 ± 1.1 |
| 3 | 45.5 ± 1.1 | 99 ± 1.1 (Ki = 33)d | 97 ± 1.4 |
| 4 | >1000 | ndf | nd |
| 5 | >1000 | Nd | nd |
| SC-AMP | 9.3 ± 1.2 | Nd | nd |
| 5′-ADP | 160 ± 20 | Nd | nd |
| 5′-AMP | 113 ± 10 | Nd | nd |
aATP = 1 mM, l-Thr = 0.6 mM.
bATP = 1mM.
cATP = 1.2 mM, l-Thr = 1.8 mM.
dCalculated using Cheng–Prusoff equation (59).
eObtained using MST with fluorescently labelled EcTsaBD.
fNot determined.
Microscale thermophoresis (MST) binding studies
MST measurements were performed using a Monolith NT115.1 Pico instrument (Nanotemper Technologies). EcTsaD was fluorescently labelled with the Monolith Red-NHS second generation dye (cat. no. MO-L011) using the manufacturer's protocol. Briefly, purified His6-TsaD (20 μM) in buffer (50 mM sodium phosphate pH 7.5, 300 mM NaCl, 5% glycerol) was mixed with an equal volume (100 μl) of dye reagent (60 μM) in same buffer containing 5% DMSO for 25 min at room temperature in the dark. Excess reagent was removed using gel exclusion chromatography with buffer described above and column provided in the kit to give the cyan-colored protein. Fluorescently labelled TsaD was analysed for t6A activity using the full reaction HPLC assay as described above and found to be 95% active relative to the unlabelled enzyme. Aliquots of the labelled protein were frozen in dry ice/acetone and stored at –80°C. Binding measurements were conducted in t6A Buffer (equal to full assay buffer described above) containing PBS with 0.05% Tween 80. Full t6A activity was observed in this buffer. A stock concentration of 1 μM EcTsaD: 2 μM EcTsaB was prepared in binding buffer and dilutions were carried out to give aliquots of added to separate micro (PCR) striptubes containing 10 nM of the labelled TsaBD complex. After an initial addition of an equal volume (10 μl) of 1 mM of the ligand BK951 was made to the first tube, serial dilutions were prepared to span the range of 0.1–500 μM BK951 and 5 nM EcTsaBD complex. A glass capillary was then filled with the binding reaction mixture from each tube and placed in the MST tray. The capillaries were analysed for thermophoretic mobility. The instrument software analyses for fluorescence homogeneity in the samples and artefacts such as ligand induced autobleaching, as well as protein aggregation, which could lead to erroneous results. When suitable conditions are found, binding data is collected and analysed by the software package included with the instrument. Binding measurements were performed in triplicate.
TCAMP stability studies in complex with EcTsaBD and EcTsaC
To study the effects of TsaBD on TC-AMP stability, HPLC-purified TC-AMP in 50 mM potassium phosphate was added to a solution of TsaBD in 50 mM HEPES pH 7.5, 0.3 M NaCl to give 100 μl of 10 μM TC-AMP with or without 40 μM TsaBD. The mixtures were incubated at 37°C and 10 μl timed aliquots were withdrawn and analysed by HPLC for AMP and TC-AMP by measuring peak areas using absorbance at 260 nm. Plots of ln [TC-AMP]/[TC-AMP + AMP] vs time were analysed by linear regression (GraphPad Prism 8) to get a series of rate constants for first order TC-AMP decomposition to AMP. To measure the effect of TsaC on TC-AMP stability, EcTsaC was overproduced in BL21(DE3) cells from IPTG induction of the gene cloned in pET15b. The protein was purified using Ni-NTA chromatography and buffer exchanged with a 10 ml Sephadex G-50 column as previously described (30). This procedure typically gives 1 ml of 100–150 μM EcTsaC, which contains 70–80% bound TC-AMP. The experiments described in this work used 130 μM TsaC with 100 μM (77%) bound TC-AMP as determined by HPLC and Bradford protein assay. The stability of this complex at different concentrations at 37°C in the same reaction buffer was analysed using the same HPLC protocol as described for TC-AMP:TsaBD mixtures to get a series of rate constants for each concentration of the complex.
Fluorescence anisotropy binding measurements
The binding of the E. coli TsaBD complex to 3′Fluorescein-labelled tRNALysUUU was monitored with a TECAN INFINTE F200PRO microplate reader by using a Nunc F96 microwell plate (Thermo Scientific). 40 nM of 3′Fluorescein-labelled tRNA probe was incubated in each well (final volume of 200 μl) with increasing amounts of TsaBD (final concentrations between 0 and 3000 nM) in 20 mM HEPES pH 7.5, 4 mM NaCl and 2 mM DTT supplemented or not with BK951 (final concentration 50 μM). The plate was incubated at 22°C in the dark for 30 min and then fluorescence anisotropy was recorded at 535 nm (λexc = 495 nm). Titration curves have been obtained by fitting experimental values to the following hyperbolic saturation function: A = Bmax.[TsaBD]/(KD + [TsaBD]) using SigmaPlot (Systat) software. Measurements have been performed in triplicate and mean values have been used for fitting.
Cloning, expression, purification and crystallization of TsaD and TsaB from Escherichia coli
Genes coding EcTsaD and EcTsaB have been cloned in pET9a expression plasmids and proteins have been produced and purified as previously described by Zhang et al. (37). TsaBD complex was concentrated to 10.1 mg.ml−1 for crystallization trials. Crystals of heterodimer TsaBD from Escherichia coli were obtained by hanging-drop vapor diffusion after at 18°C by mixing 1 μl of the protein solution pre incubated with 2.5 mM BK951 and 5 mM MgCl2 with 1 μl of 0.25 mM lithium sulfate 0.11 M HEPES pH 7.5 0.15 mM sodium acetate and 25% PEG 4K. The crystallization drop was equilibrated against a 500 μl reservoir solution containing 0.25 mM lithium sulfate, 0.11 M HEPES pH 7.5, 0.15 mM sodium acetate and 25% PEG 4K. Crystals were cryo-protected by quick-soaking in crystallization buffer supplemented with 25% ethylene glycol, 3 mM BK951 and 6 mM MgCl2 prior to flash freezing in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data collection was carried out on beamline Proxima2 at the SOLEIL Synchrotron (Saint-Aubin, France) at 100 K. Data were processed, integrated and scaled with the XDS program package. TsaBD belonged to space group P1 with unit cell parameters of a = 63.23 Å, b = 69.25 Å, c = 86.54 Å; α = 109.38°, β = 92.116°, γ = 116.94°. The structure was solved by molecular replacement using the PHASER module implemented in the CCP4 software package (search model was EcTsaBD, PDB ID: 4YDU). Two copies of the TsaBD dimer were found into the asymmetric unit of the crystal. The 2Fo – Fc electronic density indicated the presence of the BK951 inhibitor bound to a zinc ion. The initial structure was refined using the BUSTER program and completed by interactive and manual model building using COOT. Data collection and refinement statistics are gathered in Table 3. The refined structure was deposited at the Protein Data Bank (code 6Z81). All figures of structures were generated with Pymol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Table 3.
Data collection and refinement statistics
| EcTsaBD + BK951 + Zn | |
|---|---|
| PDB code | 6Z81 |
| Number of copies in asymmetric unit | 2 |
| Space group cell parameters | P1: a = 63.23 Å, b = 69.25 Å, c = 86.54 Å, α=109.38°, β = 92°, γ = 116.94° |
| Resolution (Å) | 42.47–2.31 (2.33–2.31) |
| No. of total reflections | 181 321 (15 816) |
| No. of unique reflections | 51 130 (1023) |
| Multiplicity | 3.5 (3.4) |
| R pim | 0.77 (0.525) |
| Completeness (%) | 96.1 (88.76) |
| I/σ(I) | 7.75 (1.71) |
| CC1/2 | 0.992 (0.75) |
| R cryst (%) | 18.84 (26.95) |
| R free (%) | 24.31 (32.70) |
| RMSD bond lengths (Å) | 0.014 |
| RMDS bond angles (°) | 1.81 |
| Average B (Å2) Protein Ligand Solvent | 38.47 38.38 50.27 31.60 |
| Clashscorea MolProbity scorea | 3.63 1.85 |
| Ramachandran plot (%) Favoured Allowed Outliers | 96.64 3.36 0.00 |
Values for the highest resolution shell are in parentheses.CC1/2 = percentage of correlation between intensities from random half‐dataset.
aCalculated with MolProbity.
Molecular docking of adenine into the active site of TsaD
To find out how A37 of tRNA substrate could be oriented into the active site, molecular docking was performed using AutoDockTools 4.2. Adenine was retrieved from the PDB and converted using the Babel format molecule converter (http://openbabel.org/). We removed TsaB from the present crystal structure (keeping TsaD bound to BK951) and added Kollman charges. The Lamarckian Genetic Algorithm (LGA) implemented in Autodock 4.2 was used for docking experiments. The docking parameters were as follows: 50 docking trials, population size 300 and 2 500 000 energy evaluations. We used a grid consisting of twenty 0.5 Å spacings in each dimension centered onto the active site.
RESULTS
Rationale and preparation of stable mimics of TC-AMP
Because TC-AMP is unstable, we sought to prepare chemically stable analogs that are close structural mimics in order to obtain inhibitors that could provide structural information on the bound conformation of TC-AMP itself when bound to TsaBD. The structures of the mimics are shown in Figure 2. We began with the isosteric phosphonate BK951 (1). Simple replacement of the ester oxygen of the carbamate in TC-AMP gives a molecule that is stable to all of the observed TC-AMP degradation pathways that result in the formation of AMP. These include cyclization, direct hydrolysis or (potential) elimination pathways (30). For the synthesis (Figure 3), we envisioned a convergent strategy, wherein l-threonine and inosine would provide the sources of stereochemistry in the target molecule. The retrosynthesis employs a key disconnection between one of the phosphonate ester oxygens and the ribose 5′-carbon, which may be linked via Mitsonobu coupling (51). 6-Chloropurine riboside was used instead of an adenosine derivative to minimize the tendency of adenosine to cyclize at N3 during activation of the 5′-hydroxyl group (52). Protecting groups included an isopropylidine for the 2′,3′-cis-diol of the riboside, while l-threonine was protected using benzyl groups at both the β-hydroxyl and carboxylic acid groups. The 6-chloro could be converted to a 6-azido functionality using azide, and the benzyl groups could then be removed during azide reduction to regenerate the adenosine moiety in a single step. As shown in Figure 3, the route begins with the starting dibenzyl protected l-threonine derivative 6, which was reacted with chloroacetyl chloride to give 94% of the chloride 7, which itself was converted to the dimethylphosphonate by Arbuzov reaction with trimethylphosphite in 86% yield. Dealkylation with LiBr in acetonitrile gave the phosphonate monoester, which was coupled with the isopropylidine protected 6-chloroinosine 10 in 78% yield. Reaction of 10 with sodium azide in DMF with heating gave the 6-azido purine derivative with concomitant demethylation to the phosphonic acid monoester. Global deprotection and reduction of the azide to the amine at N6 was achieved by hydrogenation using PdCl2 in MeOH, followed by acidification with TFA to complete the removal of the isopropylidine group to give crude 1. Purification of 1 by reversed phase HPLC gave the pure material in 36% yield from 10 on 100 mg scale. This material was >99% pure (Supplementary Figure S5) and used for both the enzymatic inhibition assays and structural analysis with TsaBD.
Figure 2.

Chemical structures of TC-AMP and inhibitors prepared and analysed in this work.
A series of other bioisosteres of TC-AMP were prepared (see Supplementary Data and Figures S1-S4 for experimental details). These include sulfamate isosteres 2 and 3, in which either the phosphonate only or both phosphonate and the amide of the intermediate were replaced by sulfamoyl and sulfonylurea groups, respectively. Sulfamoyl groups are well described successful substitutions as phosphodiester isosteres for the preparation of adenylate mimics in the inhibition of aminoacyl-tRNA synthetases and many other adenylate intermediates (53,54). For both 2 and 3, the bridging nitrogen is expected to be deprotonated under physiological conditions. The alkylaminosulfonyl-sulfamate ester moiety of 3 is unprecedented and was found to be chemically stable. We chose this group due to the likelihood that the carbonyl carbon of the TC-AMP carbamate would form a tetrahedral sp3-hybridized geometry during attack of N6 of A37 of the tRNA in the carbamoyl transfer reaction catalysed by TsaBDE. Such geometry could be selectively stabilized by TsaBD binding. Compounds 4 and 5 employed malonates as the bridging group between the ribose and threonine and were designed to promote potential Zn binding to the active site of TsaD, via the enol tautomer of the beta-amido ester (4) or amide (5). Final products were purified by HPLC and characterized by NMR and High Resolution MS, which were consistent with the assigned structures (see Supplementary Material).
Kinetic analysis of E. coli t6A reaction using in vitro assays
Before the TC-AMP mimics were tested for inhibition of t6A formation, we carried out an initial kinetic investigation of the overall reaction, as well as the half reaction for TC-AMP conversion to t6A, using a previously described HPLC assay (30). The assay is based on isolation and enzymatic digestion of the modified tRNA product and HPLC quantitation of t6A directly. This assay shows linear reaction rates (Supplementary Figure S6) and allowed us to approximate initial rates for fitting data to the Michaelis-Menten (MM) model (see Table 1). Because of the relatively weak affinity of the TsaBDE ternary complex, protein concentrations of 0.5 μM or above were required for the assays. We are thus reporting apparent kinetic parameters, due to the likely heterogeneity in protein complex formation and its effects on the initial rates for some substrate and inhibitor concentrations. For L-threonine in the overall reaction, fitting the kinetic data to the classical MM model gave an apparent value for Km = 0.59 ± 0.09 mM (Figure 4A, Table 1). The kinetic data for ATP as the variable substrate show a good fit to MM kinetics up to a concentration of 1 mM with a value for Km = 60 ± 6 μM (Figure 4B). However, severe substrate inhibition was observed for higher ATP concentrations (Figure 4C). This inhibition shows a 50% reduction in the Vmax at 5 mM ATP (90% at 10 mM). The physiological relevance of this inhibition is not clear. Although average intracellular ATP concentrations in E. coli are In the millimolar range (55,56), free ATP is unknown. Single cell analysis in E. coli has also shown a wide distribution of intracellular concentrations from 0.1 to 2 mM (57). The substrate inhibition observed is consistent with a similar observation recently reported by Swinehart et al. (58). Kinetic data with tRNA as the variable substrate was somewhat limited by the apparent low values of Km for most tRNA transcripts. We were able to measure an apparent Km = 0.5 ± 0.2 μM for unmodified tRNALys (Figure 4D) by fitting data to the Morrison quadratic equation (50), which is used to account for substrate depletion in reactions in which the tRNA substrate is close to the enzyme concentration.
Figure 4.

Kinetics of E. coli t6A formation in the full reaction and isolated second step with purified TC-AMP as substrate. (A) Full reaction mixture with L-threonine as variable substrate fit to MM kinetics (Equation 1). (B) Full reaction mixture with ATP as variable substrate ≤1 mM. (C) Full reaction with ATP as variable substrate showing substrate inhibition at high concentrations (fit to Equation 3) (D) Full reaction with tRNALysUUU as the variable substrate fit to Morrison Equation (2). (E) Isolated second step of reaction with TC-AMP as variable substrate and 1 mM ATP fit to Morrison equation (Equation 2). (F) Second step of the reaction with TC-AMP (10 μM) as substrate and variable ATP displaying substrate inhibition as in (C). (G) Second step of the reaction with constant TCAMP (10 μM) and ATP as the variable substrate (≤1 mM) fit to MM Equation (1).
In addition to measuring t6A formation with the full reaction mixture, we could also isolate the second step of the modification reaction by using HPLC-purified TC-AMP as the substrate with TsaBDE in the presence of ATP (30). Using TsaBDE at a concentration of 0.5 μM, we used the Morrison equation to fit kinetic data for reaction rates with TC-AMP in the concentration range of 0.5–10 μM. Under these conditions, with ATP = 1 mM, we observe an apparent Km = 1.5 ± 0.4 μM for TC-AMP (Table 1, Figure 4E). Furthermore, with ATP as the variable substrate and [TC-AMP] = 10 μM, we see similar substrate inhibition as observed in the overall reaction (Figure 4F). This suggests that the substrate inhibition observed in the overall reaction is due to ATP interference with the TsaBDE-catalysed transfer of the carbamoyl group to tRNA. Although ATP binding was not observed by ITC with EcTsaBD, likely due to hydrolysis, isosteric ATP analogs have been observed to bind to the heterodimer with low micromolar Kd values (37). By fitting the data to a substrate inhibition model (Equation 3 above), we measured a Km = 14 μM and a Kis = 1.8 mM for ATP in the second step with TC-AMP as substrate (Table 1). Figure 4G shows more clearly the fit of the data for variable ATP concentrations ≤1 mM. Interestingly, this Km value is 40-fold lower than the Km value (0.64 mM) for TsaE-catalyzed ATPase activity by the EcTsaBDE complex (37). Thus, ATP is utilized more efficiently for t6A formation at low ATP concentrations, albeit with a turnover that is ∼80-fold lower than the ATPase activity. It is not yet clear what the implications of these data are for the mechanistic role of TsaE in t6A formation or its physiological relevance.
Inhibition of t6A formation by TCAMP analogs
In testing the TC-AMP mimics for inhibition of the t6A reaction, we first used the full reaction assay to measure IC50 values with concentrations of ATP and l-threonine at 1 mM each. These substrate concentrations were necessary to achieve an optimum rate of t6A formation in our in vitro assay and are close to the estimated physiological concentrations in E. coli (55). The ATP concentration was also chosen to avoid substrate inhibition. The IC50 values were obtained by varying the inhibitor concentration and measuring as close to the initial rates as possible, given limitations on the lower range of enzyme concentration that could be used. The IC50 values for the inhibitors are listed in Table 2. The most potent inhibitor was the phosphonate BK951, which had an IC50 value of 1.3 ± 0.5 μM (Figure 5A). The sulfamate derivative 2 (IC50 = 51 μM) showed 40-fold weaker inhibition under these conditions, as did the aminosulfonylsulfamate 3 (IC50 = 45.5 μM). We also prepared the serine analog of TC-AMP, SC-AMP, which is a poor substrate, and gives an IC50 = 9.3 μM (Supplementary Figure S7A–C). This shows the relative importance of the methyl group of the threonine moiety in binding. Both the malonate ester derivative 4 and the malonate amide 5 showed no observable inhibition at concentrations as high as 1 mM (data not shown). The ester analog 4 was also particularly unstable during storage at –20°C or in solution.
Figure 5.

Characterization of BK951 inhibition of t6A formation. (A) IC50 determination for BK951 with full reaction mixture. (B) IC50 determination for BK951 with 10 μM TC-AMP, 0.5 μM EcTsaBDE and 1 mM ATP. (C) Kd measurement of TsaB binding to fluorescently labelled TsaD using Microscale Thermophoresis (MST). (D) Kd measurement of BK951 binding to labelled TsaBD heterodimer using MST. (E) Kd measurement of TsaE to labelled TsaBD in the presence of 100 μM ATP. For the MST studies, fluorescently labelled TsaD = 10 nM, TsaB = 0.5 μM (10 × Kd) and TsaE = 20 μM (∼10 × Kd).
Since TC-AMP is a product of TsaC in the first step and a substrate for TsaBDE in the second step of t6A formation, we tested the analogs as inhibitors for each step of the reaction. For the phosphonate BK951, we found an IC50 = 7.5 ± 1.2 μM for the second reaction step with 10 μM TC-AMP and 1 mM ATP (Figure 5B). By assuming competitive inhibition with TC-AMP, the Ki for BK951 can be estimated using the Cheng–Prusoff equation (59) to give a calculated value of 2.5 μM. This Ki calculation assumes that ATP concentration does not affect inhibitor binding. We did observe that the ATP concentration does have a measurable effect on the IC50 value for BK951 in the overall reaction (Supplementary Figure S8A). At 0.1 mM ATP, the IC50 for BK951 is reduced to 0.4 μM, while increasing ATP to 5 mM raises the IC50 of BK951 to 5.6 μM. Thus, the Ki calculation is only an estimate due to ATP competition with TC-AMP in the active site. Similar measurements at 1mM ATP with analogs 2 and 3 gave IC50 values of 123 and 99 μM, respectively, showing that the phosphonate is a better inhibitor of the TsaBDE catalyzed second step in t6A formation.
To provide an additional, direct measurement of BK951 binding to EcTsaBD, we used a recently described technique, microscale thermophoresis (MST), which measures the difference of thermal motion of fluorescently labelled targets as a function of ligand binding (60). The technique is highly versatile, and can be used to measure protein-protein interactions, as well as small molecule and ion binding to proteins or nucleic acids (61). For these experiments, we used fluorescently labelled EcTsaD, prepared using a probe supplied by the manufacturer that covalently reacts with accessible amines on the protein surface. We found that labelled EcTsaD showed 95% of the activity of the nonlabelled protein (data not shown) in our t6A assay. In addition, measurement of the affinity of fluorescently tagged EcTsaD to EcTsaB by MST gave a value of 45 ± 6 nM (Figure 5C), which is in good agreement with the previously reported measurement of 120 nM by isothermal calorimetry (34). Measurement of the binding affinity of BK951 with the preformed TsaBD heterodimer (using 0.5 μM unlabelled TsaB) gave a Kd value of 3.4 ± 0.6 μM (Figure 5D). We also used MST to confirm the binding of EcTsaE to the EcTsaBD heterodimer (Kd = 2.2 μM, Figure 5E). An attempt to measure binding of BK951 to the EcTsaBDE ternary complex was unsuccessful and gave only scattered data points (data not shown). ATP (1 mM) was included in these experiments to promote ternary complex formation (37), although it was found to be hydrolyzed to ADP during the lifetime of the binding measurements, which was confirmed by HPLC. It is thus possible that the high ADP concentration could be inhibiting BK951 binding to EcTsaBDE. To answer this question, we measured the IC50 values for both ADP and AMP as inhibitors. As shown in Table 2 (and Supplementary Figure S8B,C), these nucleotides had IC50 values of 160 and 113 μM, respectively. Although ADP is likely inhibiting BK951 binding in the MST experiments, it is unlikely that either AMP or ADP are formed in high enough concentrations during the kinetic assays to cause the ATP substrate inhibition we observe.
We also looked at the effect of high concentrations of TsaE on the binding of Bk951 in the kinetic assay of the overall reaction. We find that high relative concentrations of TsaE (>2.5 μM with the other enzymes at 0.5 μM) strongly inhibit t6A formation (Supplementary Figure S9A). However, this has little effect on BK951 binding (IC50 = 2.4 ± 0.8 μM at 10 μM TsaE, Supplementary Figure S9B). On the other hand, 10 μM TsaE increases the Km of tRNALys over 20-fold to 12.0 μM (Supplementary Figure S9C). This latter observation is consistent with the lack of observed tRNA binding by the EcTsaBDE ternary complex (37) and with the rapid loss of tRNA binding upon increasing TsaE to give the TsaB2D2E2 complex of T. maritima. These results suggest that TsaE competes with tRNA binding to EcTsaBD but not for BK951 binding. Because of the differential effects of TsaE on the binding of tRNA and BK951, we also asked if the binding of BK951 affects tRNA binding to EcTsaBD. Preparation of a fluorescently labelled tRNALys allowed us to use fluorescence anisotropy to measure the binding of tRNA to the EcTsaBD heterodimer. By titrating a solution of labelled tRNA with increasing concentrations of EcTsaBD, either alone or in the presence of 50 μM BK951, we measured Kd values of 87 ± 11 and 112 ± 13 nM, respectively (Supplementary Figure S10). This shows that the presence of bound BK951 has no significant effect on the binding affinity of the tRNA substrate.
Finally, since TC-AMP has been observed as a bound product to TsaC during isolation (34, vide infra), we tested the most potent inhibitors of the overall reaction for the ability to inhibit EcTsaC as product analogs. Table 2 shows that BK951 surprisingly does not bind with significant affinity to this enzyme. This contrasts with the sulfamates 2 and 3, which bind with IC50 values of 31 and 97 μM, respectively (Supplementary Figure S7D, E). Thus, the inhibition of the overall reaction by phosphonate BK951 is due solely to its binding to TsaBD, whereas for the sulfamate derivatives, it is a combination of inhibition of both enzymatic steps.
Structure of BK951 bound to EcTsaBD
Knowledge of the interactions between the TC-AMP substrate and TsaD is essential for the understanding of the t6A reaction mechanism. We therefore determined the crystal structure of EcTsaBD in complex with our highest affinity inhibitor BK951. Crystallization of EcTsaBD in presence of 2 mM BK951 yielded crystals that diffracted to a resolution of 2.4 Å. The structure was solved by molecular replacement, revealing two identical copies of the EcTsaBD complex in the asymmetric unit. The structure of EcTsaBD is nearly identical to those of apo-form or nucleotide complexes of StTsaBD and EcTsaBD (36,37) (RMSD of about 0.39 Å for 337 superposed Cα positions). The presence of the inhibitor bound to EcTsaD was supported by an extended cloud of residual electron density near the metal sites of TsaD for the two copies in the asymmetric unit (Figure 6A). A complete BK951 molecule could be easily fitted into this density. At full occupancy, the refined B-factors of the BK951 atoms are on average comparable to those of the surrounding side chains. BK951 binds as expected in the active site groove between the N- and C-terminal domains of EcTsaD (Figure 6B). The inhibitor is bound by an extensive network of hydrogen bonding and van der Waals interactions between active site residues, the bound metal ion and the inhibitor (Figure 6C). The purine and ribose engage in identical interactions as those observed for the corresponding moieties of the ATPγS complex with StTsaD (36): the ribose oxygens are H-bonding with the Asp167 carboxylate and the N1 of adenine interacts with the Asn272 side chain (Figure 6D). Figure 6E superposes ATPγS (StTsaD) onto BK951, showing that (i) their α-PO4 moieties superpose very well and (ii) the carbamoyl carbon of BK951 superposes onto the β-P position. The active site Zn coordination sphere is composed of a phosphonate-oxygen and the carboxylate of BK951, His111, His115 and Asp300. The interactions of the threonylcarbamoyl moiety of BK951 are discussed below.
Figure 6.

Structural characteristics of the BK951–EcTsaBD complex. (A) Stereoview of the residual 2Fo-Fc electron density contoured at 1σ levels for the bound BK951 (green sticks) with metal ion (gray sphere) and surrounding side chains (red sticks). (B) Cartoon presentation of the TsaBD heterodimer bound to BK951; TsaB in blue, TsaD in red, BK951 in green sticks and Zinc as a grey sphere. (C) BK951 (green sticks) bound in the active site of TsaD, Zinc ion as grey sphere. H-bonds and ligand-metal interactions: black dotted lines. (D) LigPlot+ (69) representation of the inhibitor BK951 bound into the active site of EcTsaD (E) Superposition of ATPγS bound to StTsaD (orange sticks) onto BK951 bound to EcTsaD (green sticks). Only residues from EcTsaD are labelled.
Binding pocket of the l-threonine moiety bound to EcTsaBD
For the first time, the BK951-EcTsaBD structure allows a detailed look at the binding pocket for the threonine moiety. Asp300 binds to the Zn and is conformationally restricted by hydrogen bonding with Ser9, while also making a weak hydrogen bond with the carbamoyl NH of the phosphonate inhibitor. The threonine side chain specificity for the bound inhibitor appears to be the result of hydrogen bonding to the β-hydroxyl by Ser9 and a pocket formed by van der Waals interactions between the β-methyl group of the inhibitor with His111, Cys10, and a small loop containing residues Pro82, Gly83 and Leu84 (Figure 6C and D). Our kinetic studies show that (±)-3-hydroxynorvaline (hNV) is a good substrate with a Km = 0.5 mM to give the methyl homolog hn6A (Supplementary Figure S11), which has been observed in the tRNA of both bacteria (62) and archaea (63). In vitro substrate activity of hNV with SUA5 and KEOPS has been shown in the archaeal system by Perrochia, et al. (31) and also recently shown for E. coli and T. maritima by Swinehart et al. (58). In the structure, the binding pocket for the threonine side chain has an open face into the ‘tunnel’ formed in TsaD such that extended alkyl chains may be accommodated if they can project into the tunnel but not if they are restricted in the direction of Cys10. The insertion of a methyl group for one of the β-methyl hydrogens is likely tolerated by projecting the methyl into this open space.
The threonine carboxylate is both coordinated to the active site Zn atom and also bound by hydrogen bonds from Ser136 and His139. One of the possible strategies to catalyze the reaction would be an activation of the carbamoyl group by the bound metal ion. In the present configuration, the carbamoyl is ideally positioned for nucleophilic attack by the N6 of A37. One can imagine upon tRNA binding a simple rotation about the Cα-amine C–N bond could position the carbonyl oxygen of the TC-AMP carbamoyl group to replace the carboxylate inside the coordination sphere of the Zn. This would allow Lewis acid activation of the carbonyl and facilitate acylation of the N6 of A37 of the tRNA by TC-AMP. The plausibility of such a conformational rotation is suggested by the comparison of the structure of BK951/EcTsaBD and ATPγS/StTsaBD (Figure 6E). The α and β-phosphates of ATPγS superpose onto the αP and carbamoyl carbon of BK951, but the dihedral configuration of the bonds is different. One can easily imagine a rotation of the Cα–N bonds that leads to an orientation comparable to the P–O configurations of ATPγS to bring the carbamoyl into a catalytic competent orientation. Alternatively, the metal could impose a reactive conformation of the TC-AMP without actively participating in chemical catalysis of the reaction pathway. Indeed, as discussed further, catalytic assistance is offered by active site residues other than the metal ion. By initially binding TC-AMP in a stabilizing conformation the enzyme could protect it from decomposition until the tRNA is bound. Further kinetic studies would be useful to determine if the binding of TC-AMP precedes tRNA binding which would lend support to this hypothesis.
Functional analysis of residues in the TsaD active site
We used site-directed mutagenesis to explore the functional significance of EcTsaD amino acid side chains in the active site binding region of the threonine moiety of the inhibitor (Figure 7). As in the previously reported structure of EcTsaBD bound to ADP, the Zn atom is coordinated by three universally conserved residues His111, His113 and Asp300 (37). These residues were each mutated to alanine and found to give purified proteins that were inactive in the in vitro t6A assay. We also mutated His139, because of its conservation in bacterial TsaD and its position as a Zn ligand in TmTsaBDE (41). In the EcTsaBD-ADP structure, His139 is not liganded to the Zn, and in the BK951 bound structure reported here, it is also not chelated to the Zn but it is making a relatively short hydrogen bond to the non-liganded oxygen of the carboxylate of L-threonine (Figure 6C). Surprisingly, both the His139Ala and His139Asn mutants retained activity, with the alanine mutant indistinguishable from wild type. It is possible that a compensatory interaction in the mutants makes up for the loss of this hydrogen bond. In the T. maritima carboxy-AMP bound TsaB2D2E2 structure, the analogous His137 is a ligand for the active site Zn ion and found to be required for protein stability. These results likely reflect subtle differences in the organization of the active site in these quite different organisms and may have mechanistic implications.
Figure 7.
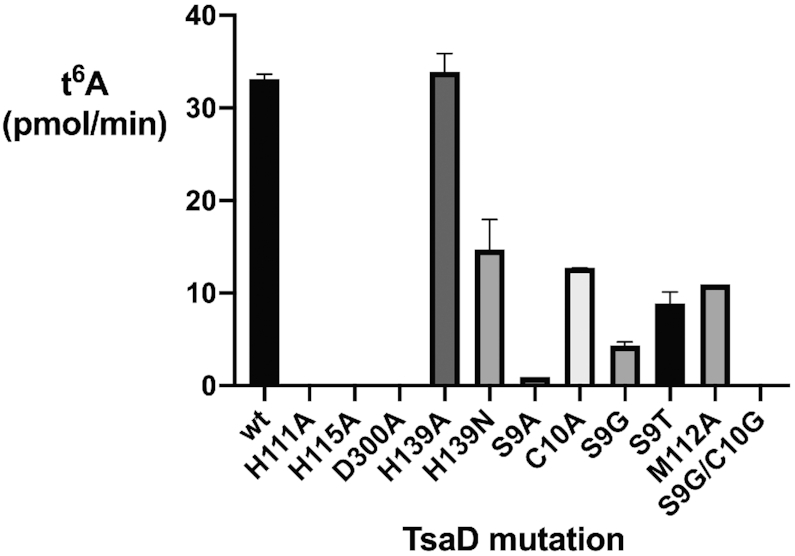
t6A activity of active site TsaD mutants using the full assay mixture (see Materials and Methods for details).
We also investigated possible specificity determinants for the threonine moiety of BK951. Interactions between EcTsaD and the threonyl moiety of BK951 are largely made of weak hydrogen bonding interactions (>3.5 Å) with Ser9, His111 and the amide nitrogen of Gly83, as well as van der Waals interactions with the β-carbon of Cys10 and atoms of the Pro82 side chain (Figure 6C and D). Ser9 also makes a hydrogen bonding interaction (3.3 Å) with the unliganded imidazole nitrogen of His111 and likely acts to restrict its conformation for interaction with the threonine moiety of the inhibitor. The Ser9Ala and Ser9Gly mutants have significantly reduced activity, while the Cys10Ala mutant is reduced only by about a third. The Ser9Gly, Cys10Gly double mutant is inactive, which indicates additive effects of removing van der Waals interactions with the threonine moiety. These mutants suggest that the specificity for the l-threonyl part of TC-AMP is enforced by a number of weak interactions that together lead to selection of threonine/hydroxynorvaline over other potential amino acids and provides a structural explanation for the recent report of TsaD as the main guardian of such specificity (58).
Modeling A37 of the tRNA substrate into the active site of EcTsaD
Assuming the BK951 adopts the position of the TC-AMP in the active site just before transfer of the threonylcarbamoyl group onto acceptor tRNA, we wondered how the incoming A37 of the latter could be positioned. We therefore modeled an adenine into the active site occupied by BK951 using the Autodock 2 software (64). The protein was kept rigid during the docking process and the distance between the N6 atom of the adenine and the carbamoyl carbon was restrained to 2.5 Å (Supplementary Figure S12). In this model the adenine ring is held between the loops encompassing residues 9–12, 40–44, 137–138 and 176–179. The adenine N9 atom points towards an empty space above the active site, leaving room for an attached tRNA molecule. Because tRNAs that are modified in their stem loops often undergo important conformational changes when bound in the active site of the corresponding enzymes, we did not attempt to model a complete tRNA into the active site of TsaD. Residues Cys10, Asp11, Val42 and Tyr177 are surrounding the adenine. Mutation of Cys10 into Ala causes a more that 50% drop in t6A activity. To provide full access of a flipped-out adenine connected to a complete tRNA substrate, the 40–44 loop probably would have to move from its present position in the TsaBD–BK951 complex. Interestingly, this loop was shown to be very flexible and completely disordered in the context of the TmTsaBDE complex (39,40).
Stabilization of TCAMP bound by EcTsaBD and EcTsaC
The inhibitor BK951 is bound to TsaBD in an extended conformation that likely precludes certain decomposition pathways. However, in this structure, the molecule appears to be readily exposed to solvent water. To understand more about the potential stabilizing effect of TsaBD on bound TC-AMP, we carried out a stability study. A mixture of 10 μM purified TC-AMP was incubated with or without 20 μM EcTsaBD at 37°C and timed aliquots were removed for HPLC analysis of TC-AMP conversion to AMP. As shown in Figure 8A, there is only a small protective effect (kobs = 6.0 ± 0.2 × 10−2 versus 7.3 ± 0.2 × 10−2 min−1) at a concentration of EcTsaBD where TC-AMP should be mostly bound. For comparison, the stabilising effect of 13 μM TsaC is nearly an order of magnitude higher (kobs = 4.3 ± 0.1 × 10−3 min−1). The rate constants correspond to a t1/2 = 9.4 min without enzyme, compared to t1/2 = 11.5 min in the presence of TsaBD and t1/2 = 160 min in the presence of TsaC. These data suggest that either TC-AMP is transferred to tRNA rapidly after binding to TsaBD, or additional binding events involving TsaC, or tRNA may help to protect bound TC-AMP from hydrolysis before it is transferred by shielding it from solvent water.
Figure 8.
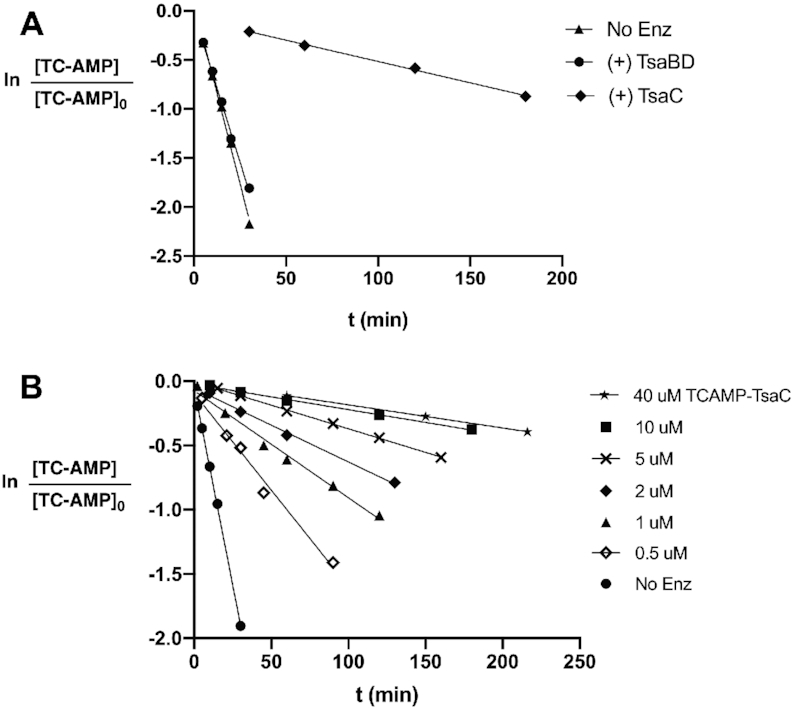
Stability of TC-AMP bound to either EcTsaBD or EcTsaC. (A) Comparison of rate of decomposition of 10 μM TC-AMP at 37°C in 50 mM HEPES pH 7.5, 300 mM NaCl with 20 μM TsaBD (filled circles); 13 μM TsaC (filled diamonds); or in the absence of enzyme (filled triangles). (B) Concentration dependence of the rate of decomposition of a 1.3:1 molar ratio of TsaC:TC-AMP at varying concentrations under the same conditions as (A).
For the TsaC experiments, we utilized the TC-AMP bound form of EcTsaC that can be isolated from the purification of highly overexpressed protein. We find that the amount of TC-AMP bound to TsaC can be quantified directly by HPLC and typically approaches 70% occupancy after affinity and size exclusion chromatography (Supplementary Figure S13A). Bound TC-AMP can also be converted to ATP quantitatively by the addition of 1.5 equivalents of pyrophosphate in the presence of Mg ion (Supplementary Figure S13B) and subsequently removed by size exclusion chromatography to provide the unliganded form of the enzyme (Supplementary Figure S13C). Further investigation of TsaC stabilization of TC-AMP in these complexes shows a significant reduction in protection as the concentration of the complex is lowered (Figure 8B). This concentration dependence is likely due to an increased fraction of unbound TC-AMP. A plot of the t1/2 values for TC-AMP stability versus TC-AMP concentration gives a crude estimated half-maximal value of ∼5 μM (data not shown), which may reflect a similar magnitude for the Kd of TC-AMP binding to EcTsaC. If this were true, it would suggest that unless the concentration of TsaC or TC-AMP in E. coli is greater than 5 μM, the majority of TsaC in the cell will be free of bound TC-AMP.
DISCUSSION
The phosphonate BK951 is a potent inhibitor of t6A biosynthesis in vitro
The phosphonate BK951 was the most potent analog in this study, with an IC50 value of 1.3 μM for the overall t6A reaction. We measured inhibitor IC50 values in the presence of L-threonine and ATP concentrations of 1 mM each. For E. coli, 1 mM ATP is in the lower part of the physiological range of up to 10 mM. The intracellular L-threonine concentration has been recently reported to be 1.3 mM (55). Substitution of the phosphonate with a sulfamoyl group in 2 results in a loss of inhibition in the kinetic assay by 40-fold, which is possibly due to the weaker binding of the sulfonyl oxygen with Zn compared to the fully ionized phosphonate. Further substitution of the carbamoyl group of TC-AMP with a tetrahedral sulfonyl gives an alkylaminosulfonyl-N-sulfamate 3, which is an intended transition state mimic of the attack by N6 of A37. The unique pharmacophore structure of this inhibitor has surprisingly not been previously described in the literature. Similar to a few known alkylaminosulfonamides, this structure likely forms an ‘inner salt’, with the bridging nitrogen deprotonated and the amine of the L-threonine protonated to give a zwitterion structure. Despite the novelty of its structure, compound 3 has virtually the same IC50 as sulfamate analog 2. Since the carbonyl oxygen of BK951 is making only one hydrogen bond in the EcTsaBD bound structure, there does not appear to be amino acid side chains in place to distinguish the sp3 orientation of the sulfonyl group in this area of the active site in this conformation of the protein. Finally, both the malonate ester and the malonate amide bridge between the adenosine ribose and threonine (4 and 5, Figure 2) failed to give molecules that showed any observable binding to EcTsaBD. Both molecules were designed to examine the hypothesis that the Zn could chelate the phosphonate and the carbamate carbonyl of TC-AMP, which in this structure is not observed. Hence, the lack of observed binding of the malonates supports the overall significance of the current structure.
If we assume the affinity of TC-AMP to EcTsaBD is similar in magnitude to the Km value of 1.5 μM, the binding data for the phosphonate analog suggests it is a good mimic that is taking advantage of most of the protein binding interactions. This conclusion is supported by the similarity of the conformation of atoms of the adenosyl 5′-phoshate/phosphonate group in the active site of TsaD of both BK951 and ATP. In the recently modelled binding of TC-AMP to TmTsaD based on the structure of bound carboxy-AMP, Luthra et al. proposed a similar binding of the threonine carboxylate moiety to the active site Zn (41). Their model differs from our structure in the lack of chelation between the phosphonate and the Zn. It is possible that there are species-specific differences in the orientation of some amino acids, as well as rearrangements of the substrates along the reaction coordinate. Thus, many of the various structures may each be relevant for a distinct part of the transformation.
The sulfamate analogs are better mimics of TC-AMP binding to TsaC
Although BK951 is more potent in inhibiting the second step of t6A synthesis, it surprisingly shows no inhibition of the first step of the reaction catalysed by TsaC. The sulfamates 2 and 3, on the other hand, show significant inhibition of both enzymatic steps in t6A formation. A reexamination of the crystal structure of TC-AMP bound to TsaC2 from S. tokodaii (34) shows a hydrogen bond between the backbone amide of conserved Ser144 to the adenylate ester oxygen of TC-AMP that is replaced by a methylene in BK951. There are also close van der Waals interactions likely between the backbone carbonyl and alpha carbons of Pro143 (usually a Thr in TsaC1). A possible steric clash between these carbon atoms and the methylene of the phosphonate analog, in addition to the loss of hydrogen bond would be sufficient to explain its lack of binding to TsaC. Conversely, the amide nitrogen of the sulfamate group in analogs 2 and 3 is acidic enough to be deprotonated, which makes it an excellent mimic of the adenylate ester oxygen as previously documented (53,54). Therefore, TsaC appears to use these interactions to closely select for an ester at this position and analogs that preserve the integrity of these interactions will be generally more potent as inhibitors.
The binding conformation of BK951 suggests a precatalytic complex
The binding interactions between BK951 and the active site of the TsaD protein suggests that at one point in the reaction coordinate, the Zn interacts directly with the phosphonate oxygen and carboxylate oxygens of TC-AMP. The carbamoyl oxygen does not form a ligand to the metal ion, but establishes two H-bonds with mainchain NH groups of Ser136GlyGly138, the β-hairpin connection between strands β6–β7. This is a similar binding mode as the carbamoyl adenylate bound to the homologous TobZ protein, which catalyses the carbamoylation of tobramycin (34). This position of the carbonyl suggests that the amide main chain NH groups of Ser136GlyGly138 could provide an oxyanion hole during the nucleophilic attack of the A37 6-amino group during the transfer reaction. As shown in Figure 9, this loop is extremely well conserved in the TsaD/Kae1/Qri7 family, suggesting its importance for catalytic activity. This loop is also present in the related carbamoylating enzymes TobZ and HypF, reinforcing the hypothesis of its role in the stabilization of the intermediate. This means that the main role of the metal is to provide a scaffold for productive binding of the substrate. In the TobZ active site, a histidine is positioned near the carbamoyl group that could act as a general base to subtract a proton from the incoming tobramycin OH nucleophile. This is not the case for TsaD, probably because the A37 NH2 nucleophile is deprotonated. Alternatively, the carbamoyl group of the TC-AMP could slightly rotate and engage in a Lewis acid interaction with the metal to increase its reactivity. The latter scenario seems less likely to us because it would render the carbon less accessible for an incoming nucleophile. Clearly, structural data in presence of the tRNA substrate are needed for a better understanding of the mechanism. Assuming that the binding mode of the BK951 carbonyl mimics that of the TC-AMP substrate, its access by the bulky tRNA substrate would likely be hampered by some of the surrounding active site loops and helices, especially the region contained between residues 32 and 45. Interestingly this region becomes completely disordered upon TsaE binding (40), suggesting that flexibility plays a major role in the catalytic cycle.
Figure 9.

Multiple sequence alignment of the TsaD/Kae1/OSGEP family members was realized using the blosum 45 with MAFFT (online version 7) (70). Structure-based alignment was performed using the ESPRIPT web-server (71). The SGG peptide conserved in all homologs is highlighted in pink.
The ability of EcTsaBD and EcTsaC to stabilize bound TC-AMP differ significantly
Previous work showed that TC-AMP is unstable with a half-life of 3.5 minutes at pH 7.5 in the presence of 2 mM Mg ion (30). Both the instability and the magnitude of the Km value for TC-AMP (1.5 μM) for the isolated TsaBDE-catalysed t6A reaction are similar in magnitude for the structurally analogous primary metabolite carbamoyl phosphate (CP). The Km values for CP for the enzymes aspartate and ornithine transcarbamoylases are ∼4–10 μM. Wang et al. (65) have shown that CP is stabilized by binding to these enzymes in the absence of the other substrates, and the effect is more pronounced for hyperthermophiles to the point of requiring substrate channelling (66). Such binding prevents the major mechanism of degradation of CP which involves intramolecular transfer of the carbamoyl NH proton to the phosphate and subsequent rapid formation of cyanate. In our previous analysis of TC-AMP decomposition, NMR data showed l-threonine and the cyclic oxazolidinone as the only two detectable final products produced in an equimolar ratio (30). The oxazolidinone forms via attack by the β-hydroxyl oxygen on the carbamoyl carbonyl carbon (or isocyanate intermediate) to release AMP and give the cyclic product. The structure reported here suggests that TC-AMP is bound by TsaBD in an extended conformation which would minimize the cyclization pathway. Although we did find that EcTsaBD stabilizes TC-AMP in solution, the effect is small. This is likely due to TC-AMP remaining accessible to solvent water when bound to TsaBD, which allows for direct hydrolysis. The fact that the half-life of TC-AMP bound to EcTsaBD is more similar to free TC-AMP in the absence of metal ion (16 min) than in its presence (3.5 min) is consistent with the conformation of bound BK951, in which the carbamoyl carbonyl is not chelated to the Zn. Retaining stability of TC-AMP may be another reason for avoiding such coordination. In other words, if the carbamoyl carbonyl were coordinated to the Zn, the enzyme could catalyze wasteful transfer of the carbamoyl group to water.
In contrast to EcTsaBD, we find an order of magnitude greater stabilizing effect on TC-AMP by binding to EcTsaC. Previous work has shown that overproduced EcTsaC retains bound TC-AMP after purification (30) and that crystallized TsaC2 from S. tokodaii (45) contained bound TC-AMP (34). In this structure, the threonylcarbamoyl group of TC-AMP is sequestered from solvent in a long, narrow binding pocket. The observed greater stabilization by TsaC may be used to prevent wasteful hydrolysis of TC-AMP in the cell (34). However, we find that as the concentration of EcTsaC is lowered into the low micromolar range, the stabilizing effect of the enzyme on TC-AMP is diminished significantly. It thus may be less of an imperative for mesophilic organisms to stabilize this metabolite which could be a relic of enforced stabilization in hyperthermophilic organisms, considering the ancient nature of this enzyme system (34). Harris, et al. (67) have reported that the C-terminus of EcTsaC contains sites that are highly dynamic in structure and this region may be involved in induced fit of ATP binding, but also in protein-protein interaction with TsaD. This flexibility of the C-terminus is also consistent with the crystal structure of unliganded EcTsaC (68). Thus, a major question still exists of whether TsaC presents bound TC-AMP to the TsaBD heterodimer (in mesophiles) or if the intermediate diffuses freely as suggested by in vitro activity experiments (27,30). It will be interesting to see if the channelling that is observed for CP enzymes from hyperthermophiles is also operative for TC-AMP in t6A formation in organisms such as T. maritima. For example, if strict channelling of TC-AMP occurs in the hyperthermophilic organisms, the kinetic inhibition of BK951 may be observed to be very weak due to restricted access to the active site. The structure reported in this paper suggests that TsaBD-bound TC-AMP, if also protected from solvent water in a higher order complex, would likely provide considerable stabilization to the intermediate.
In summary, we report here the synthesis, biochemical characterization and cocrystal structure of a potent inhibitor of bacterial t6A formation. The inhibitor appears to bind in the active site of the heterodimeric TsaBD complex in a manner similar to the natural substrate TC-AMP and could therefore be useful as a mechanistic probe for the reaction in a variety of biological contexts. The crystal structure should be useful for the rational design of more potent, cell permeable inhibitors of t6A modification that may find application as biochemical probes in a variety of organisms given the universal importance of this modification. Inhibitors optimized for the bacterial enzymes may find use as potentially novel antibiotics.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number 6Z81.
Supplementary Material
ACKNOWLEDGEMENTS
Sophia Missoury received a scholarship from the ‘Fondation de la Recherche Médicale’. We thank the staff of the Proxima1 and 2 beamlines at the SOLEIL synchrotron for assistance during the diffraction data collection. This work received support from the French Infrastructure for Structural Biology (FRISBI ANR-10-INSB-0501).
Notes
Present address: Brett J. Kopina, Pfizer Inc. 275 N. Field Dr., Lake Forest, IL 60035, USA.
Contributor Information
Brett J Kopina, Pharmaceutical Sciences Division, School of Pharmacy, University of Wisconsin, Madison, WI 53705, USA.
Sophia Missoury, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France.
Bruno Collinet, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France; Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), Sorbonne-Université, UMR7590 CNRS, MNHN, Paris, France.
Mark G Fulton, Pharmaceutical Sciences Division, School of Pharmacy, University of Wisconsin, Madison, WI 53705, USA.
Charles Cirio, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France.
Herman van Tilbeurgh, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France.
Charles T Lauhon, Pharmaceutical Sciences Division, School of Pharmacy, University of Wisconsin, Madison, WI 53705, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French Infrastructure for Integrated Structural Biology (FRISBI) [ANR-10-INSB-0501 to H.v.T.]; University of Wisconsin School of Pharmacy and Wisconsin Alumni Research Fund (to C.L.). Funding for open access charge: University of Wisconsin School of Pharmacy.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gustilo E.M., Vendeix F.A.P., Agris P.F.. tRNAs modifications bring order to gene expression. Curr. Opin. Microbiol. 2008; 11:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Björk G.R. Söll D., RajBhandary U.. Biosynthesis and function of modified nucleosides. tRNA: Structure, Biosynthesis, and Function. 1995; Washington, DC: American Society for Microbiology; 165–205. [Google Scholar]
- 3. El Yacoubi B., Bailly M., de Crecy-Lagard V.. Biosynthesis and function of post-transcriptional modifications of transfer RNAs. Ann. Rev. Gen. 2012; 46:69–95. [DOI] [PubMed] [Google Scholar]
- 4. Urbonavicius J., Qian Q., Durand J.M., Hagervall T.G., Bjork G.R.. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001; 20:4863–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schweizer M.P., Chheda G.B., Baczynskyj L., Hall R.H.. Aminoacyl nucleosides. VII. N(purin-6-ylcarbamoyl)threonine. A new component of transfer ribonucleic acid. Biochemistry. 1969; 8:3283–3289. [DOI] [PubMed] [Google Scholar]
- 6. Powers D.M., Peterkofsky A.. The presence of N-(purin-6-ylcarbamoyl)threonine in transfer ribonucleic acid species whose codons begin with adenine. J. Biol. Chem. 1972; 247:6394–6401. [PubMed] [Google Scholar]
- 7. Miller J.P., Hussain Z., Schweizer M.P.. The involvement of the anticodon adjacent modified nucleoside N-(9-(Beta-D-ribofuranosyl)-purin-6-ylcarbamoyl)-threonine in the biological function of E. coli tRNAIle. Nuc. Acids Res. 1976; 3:1185–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thiaville P.C., Iwata-Reuyl D., de Crécy-Lagard V.. Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t6A) a universal modification of tRNA. RNA Biol. 2015; 12:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murphy F.V., Ramakrishnan V., Malkiewicz A., Agris P.F.. The role of modifications in codon discrimination by tRNALysUUU. Nat. Struct. Mol. Biol. 2004; 11:1186–1191. [DOI] [PubMed] [Google Scholar]
- 10. Handford J.I., Ize B., Buchanan G., Butland G.P., Greenblatt J., Emili A., Palmer T.. Conserved network of proteins essential for bacterial viability. J. Bacteriol. 2009; 191:4732–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El Yacoubi B., Lyons B., Cruz Y., Reddy R., Nordin B., Agnelli F., Wil- liamson J.R., Schimmel P., Swairjo M.A., de Crécy-Lagard V.. The universal YrdC/Sua5 family is required for the formation of threonyl- carbamoyladenosine in tRNA. Nuc. Acids Res. 2009; 37:2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin C.A., Ellis S.R., True H.L.. The Sua5 protein is essential for normal translational regulation in yeast. Mol. Cell. Biol. 2010; 30:354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thiaville P.C., El Yacoubi B., Kohrer C., Thiaville J.J., Deutsch C., Iwata- Reuyl D., Bacusmo J.M., Armengaud J., Bessho Y., Wetzel C.et al.. Essentiality of threonylcarbamoyladenosine (t6A), a universal tRNA modification, in bacteria. Mol. Microbiol. 2015; 98:1199–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyauchi K., Kimura S., Suzuki T.. A cyclic form of N6-threonylcarbamoyladenosine as a widely distributed tRNA hypermodification. Nat. Chem. Biol. 2103; 9:105–113. [DOI] [PubMed] [Google Scholar]
- 15. Mateszewski M., Wojciechowski J., Miyauchi K., Gdaniec Z., Wolf W.M., Suzuki T., Sochacka E.. A hydantoin isoform of cyclic N6-threonylcarbamoyladenosine (ct6A) is present in tRNAs. Nuc. Acids Res. 2017; 45:2137–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J.-P., Iwata- Reuyl D., Murzin A.G., de Crécy-Lagard V.. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011; 30:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E.V., Karzai A.W., Sternglanz R.. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011; 30:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daugeron M.C., Lenstra T.L., Frizzarin M., El Yacoubi B., Liu X., Baudin-Baillieu A., Lijnzaad P., Decourty L., Saveanu C., Jacquier A.et al.. Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs. Nuc. Acids Res. 2011; 39:6148–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang W., Collinet B., Graille M., Daugeron M.C., Lazar N., Libri D., Durand D., van Tilbeurgh H.. Crystal Structures of the Gon7/Pcc1 and Bud32/Cgi121 complexes provide a model for the complete yeast KEOPS complex. Nuc. Acids Res. 2015; 43:3358–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kisseleva-Romanova E., Lopreiato R., Baudin-Baillieu A., Rousselle J.C., Ilan L., Hofmann K., Namane A., Mann C., Libri D.. Yeast homolog of a cancer-testis antigen defines a new transcription complex. EMBO J. 2006; 25:3576–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Downey M., Houlsworth R., Maringele L., Rollie A., Brehme M., Galicia S., Guillard S., Partington M., Zubko M.K., Krogan N.J.et al.. A genome-wide screen identifies the evolutionarily conserved KEOPS complex as a telomere regulator. Cell. 2006; 124:1155–1168. [DOI] [PubMed] [Google Scholar]
- 22. Meng F.L, Hu Y., Shen N., Tong X.J., Wang J., Ding J., Zhou J.Q.. SUA5p is a single-stranded telomeric DNA binding protein that facilitates telomere replication. EMBO J. 2009; 28:1466–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He M.-H., Liu J.-C., Lu Y.-S., Wu Z.-J., Liu Y.-Y., Wu Z., Peng J., Zhou J.-Q.. KEOPS complex promotes homologous recombination via DNA resection. Nuc. Acids Res. 2019; 47:5684–5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braun D.A., Rao J., Mollet G., Schapiro G., Daugeron M.C., Tan W., Gribouval O., Boyer O., Revy P., Jobst-Schwan T.et al.. Mutations in KEOPS complex genes cause nephrotic syndrome with primary microcephaly. Nat. Genet. 2017; 49:1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edvardson S., Prunetti L., Arraf A., Haas D., Bacusmo J.M., Hu J.F., Ta-Shma A., Dedon P.C., de Crecy-Lagard V., Elpeleg O.. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3(OGSEP) mutation manifest by neurodegeneration and renal tubulophathy. Eur. J. Hum. Genet. 2017; 25:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arrandel C., Missoury S., Snoek R., Patat J., Menara G., Collinet B., Liger D., Durand D., Gribouval O., Boyer O.et al.. Defects in t(6)A tRNA modification due to GON7 and YRDC mutations lead to Galloway-Mowatt syndrome. Nat. Comm. 2019; 10:3967–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wan L.C., Mao D.Y., Neculai D., Strecker J., Chiovitti D., Kurinov I., Poda G., Thevakumaran N., Yuan F., Szilard R.K.. Reconstitution and characterization of eukaryotic N6-threonylcarbamoylation of tRNA using a minimal enzyme system. Nuc. Acids Res. 2013; 41:6332–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin H., Miyauchi K., Harada T., Okita R., Takeshita E., Komaki H., Fujioka K., Yagasaki H., Goto Y., Yanaka K.et al.. CO2-Sensitive tRNA modification associated with human mitochondrial disease. Nat. Comm. 2018; 9:1875–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deutsch C., El Yacoubi B., de Crécy-Lagard V., Iwata-Reuyl D.. Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 2012; 287:13666–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lauhon C.T. Mechanism of threonylcarbamoyladenosine (t6A) biosynthesis in bacteria: isolation and characterization of the intermediate threonylcarbamoyl-AMP. Biochemistry. 2012; 51:8950–8961. [DOI] [PubMed] [Google Scholar]
- 31. Perrochia L., Crozat E., Hecker A., Zhang W., Bareille J., Collinet B., van Tilbeurgh H., Forterre P., Basta T.. In vitro biosynthesis of a universal t6A tRNA modification in Archea and Eukarya. Nucleic Acids Res. 2013; 41:1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kostuch A.P., Zheng W., Liger D., Daugeron M.-C., Letoquart J., Sierra-Galley I.L.D.L., Forterre P., Collinet B., van Tilbeurgh H., Basta T.. Structure-function analysis of SUA5 protein reveals novel functional motifs required for the biosynthesis of the universal t6A tRNA modification. RNA. 2018; 24:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wan L.C.K., Pillin M.C., Thevakumaran N., Sun Y., Chakrabartty A., Guarne A., Kurinov I., Durocher D., Sicheri F.. Structural and functional characterization of KEOPS dimerization by Pcc1 and its role in t6A biosynthesis. Nucleic Acids Res. 2016; 44:6971–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parthier C., Görlich S., Jaenecke F., Breithaupt C., Bräuer U., Fandrich U., Clausnitzer D., Wehmeier U.F., Böttcher C., Scheel D.et al.. The O- Carbamoyltransferase TobZ catalyzes an ancient enzymatic reaction. Angew. Chem. Int. Ed. Eng. 2012; 51:4046–4052. [DOI] [PubMed] [Google Scholar]
- 35. Thiaville P.C., El Yacoubi B., Perrochia L., Hecker A., Prigent M., Thiaville J.T., Forterre P., Namy O., Basta T., de Crécy-Lagard V.. Cross kingdom function of the core universally conserved threonylcarbamoyladenosine synthesis enzymes. Eukaryot. Cell. 2014; 13:1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nichols C.E., Lamb H.K., Thompson P., El Omari K., Lockyer M., Charles I., Hawkins A.R., Stammers D.K.. Crystal structure of the dimer of two essential Salmonella typhimurium and proteins, YgjD and YeaZ and calorimetric evidence for the formation of a ternary YgjD-YeaZ-YjeE complex. Protein Sci. 2013; 22:628–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang W., Collinet B., Perrochia L., Durand D., van Tilbeurgh H.. The ATP-mediated formation of the YgjD–YeaZ–YjeE complex is required for the biosynthesis of tRNA t6A in Escherichia coli. Nuc. Acids Res. 2015; 43:1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luthra A., Swinehart W., Bayooz S., Phan P., Stec B., Iwata-Reuyl D., Swairjo M.A.. Structure and mechanism of a bacterial t6A modification system. Nuc. Acids Res. 2018; 46:1395–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Missoury S., Plancqueel S., de la Sierra-Galley I.L., Zhang W., Liger D., Durand D., Dammak R., Collinet B., van Tilbeurgh H.. The structure of the TsaB/TsaD/TsaE complex reveals an unexpected mechanism for the bacterial t6A modification (erratum). Nucleic Acids Res. 2018; 47:9464–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Missoury S., Plancqueel S., de la Sierra-Galley I.L., Zhang W., Liger D., Durand D., Dammak R., Collinet B., van Tilbeurgh H.. The structure of the TsaB/TsaD/TsaE complex reveals an unexpected mechanism for the bacterial t6A modification. Nucleic Acids Res. 2019; 46:5850–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luthra A., Paranagama N., Swinehart W., Bayooz S., Phan P., Quach V., Schiffer J.M., Stec B., Iwata-Reuyl D., Swairjo M.A.. Conformational communication mediates the reset step in t6A biosynthesis. Nucleic Acids Res. 2019; 47:6551–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Teplyakov A., Obmolova G., Tordova M., Thanki N., Bonander N., Eisenstein E., Howard A.J., Gilliland G.L.. Crystal structure of the YjeE protein from Haemophilus influenza: a putative ATPase involved in cell wall synthesis. Proteins. 2002; 48:220–226. [DOI] [PubMed] [Google Scholar]
- 43. Allali-Hassani A., Campbell T.L., Ho A., Schertzer J.W., Brown E.D.. Probing the active site of YjeE, a vital Escherichia coli protein of unknown function. Biochem. J. 2004; 384:577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karst J.C., Foucher A.E., Campbell T.L., DiGuilmi A.M., Stroebel D., Mangat C.S., Brown E.D., Jault J.M.. The ATPase activity of an “essential” Bacillus subtilis enzyme, YdiB, is required for its cellular function and is modulated by oligomerization. Microbiology. 2009; 155:944–956. [DOI] [PubMed] [Google Scholar]
- 45. Agari Y., Sato S., Wakamatsu T., Bessho Y., Ebihara A., Yokoyama S., Karamitsu S., Shinkai A.. X-ray crystal structure of a hypothetical SUA5 protein from Sulfolobus tokodaii strain 7. Proteins. 2008; 70:1108–1111. [DOI] [PubMed] [Google Scholar]
- 46. Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C.. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987; 15:8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peterson E.T., Uhlenbeck O.C.. Determination of recognition nucleotides for Escherichia coli phenylatlanine tRNA synthetase. Biochemistry. 1992; 31:10380–10389. [DOI] [PubMed] [Google Scholar]
- 48. Gehrke C.W., Kuo K.C., McCune R.A., Gerhardt K.O.. Ribonucleoside analysis by reversed phase high performance liquid chromatography. J. Chromatogr. 1982; 230:297–308. [PubMed] [Google Scholar]
- 49. Pomerantz S.C., McCloskey J.A.. Analysis of RNA hydrolyzates by liquid-chromatography-mass spectrometry. Methods Enzymol. 1990; 193:796–824. [DOI] [PubMed] [Google Scholar]
- 50. Morrison J.F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight- binding inhibitors. Biochim. Biophys. Acta - Enzymol. 1969; 185:269–286. [DOI] [PubMed] [Google Scholar]
- 51. Kimura J., Yagi K., Suzuki H., Mitsunobu O.. Studies on nucleosides and nucleotides. VIII. Preparations and reactions of triphenylphosphorandiylnucleosides. Bull. Chem. Soc. Jpn. 1980; 53:3670. [Google Scholar]
- 52. Klein E., Mons S., Valleix A., Mioskowski C., Lebeau L.. Synthesis of enzymatically and chemically non-hyrolyzable analogs of dinucleoside triphosphates Ap3A and Gp3G. J. Org. Chem. 2002; 67:146–153. [DOI] [PubMed] [Google Scholar]
- 53. Duckworth B.P., Nelson K.M., Aldrich C.C.. Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top. Med. Chem. 2012; 12:766–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lux M.C., Standke L.C., Tan D.S.. Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors. J. Antibiot. 2019; 72:325–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park J.O., Rubin S.A., Xu Y.F., Amador-Noguez D., Fan J., Schlomi T., Rabinowitz J.D.. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat. Chem. Biol. 2016; 12:482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Buckstein M.H., He J., Rubin H.. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacterol. 2008; 190:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yaginuma H., Kawai S., Tabata K.V., Tomiyama K., Kakizuka A., Komatsuzaki T., Noji H., Imamura H.. Diveristy in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 2014; 4:6522–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swinehart W., Deutsch C., Sarachan K.L., Luthra A., Bacusmo J.M., de Crecy-Lagard V., Swairjo M.A., Agris P., Iwata-Reuyl D.. Specificity in the biosynthesis of the universal tRNA nucleoside N6-threonylcarbamoyl adenosine (t6A) – TsaD is the gatekeeper. RNA. 2020; 26:1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng Y., Prusoff W.H.. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973; 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- 60. Weinken C.J., Baaske P., Rothbauer U., Braun D., Duhr S.. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Comm. 2010; 1:100. [DOI] [PubMed] [Google Scholar]
- 61. Amari M., Ratih R., Alhazmi H.A., Deeb S.E.. Thermophoresis for characterizing biomolecular interaction. Methods. 2018; 146:107–119. [DOI] [PubMed] [Google Scholar]
- 62. Reddy D.M., Crain P.F., Edmonds C.G., Gupta R., Hashizume T., Stetter K.O., Widdel F., McCloskey J.A.. Structure determination of two new amino acid-containing derivatives of adenosine from tRNA of thermophilic bacteria and archaea. Nuc. Acids Res. 1992; 20:5607–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu N., Jora M., Solivio B., Thakur P., Acevedo-Rocha C.G., Randau L., de Crecy-Lagard V., Addepalli B., Limbach P.A.. tRNA modification profiles and codon-decoding strategies in Methanocaldococcus jannaschii. J. Bacteriol. 2019; 201:e00690-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Morris G.M., Goodsell G.S., Huey R., Olson A.J.. Distributed automated docking of fleixible ligands to proteins. Parallel applications of Autodock 2.4. J. Comput.-Aided Mol. Des. 1996; 10:293–304. [DOI] [PubMed] [Google Scholar]
- 65. Wang Q., Xia J., Guallar V., Krilov G., Kantrowitz E.R.. Mechanism of thermal decomposition of carbamoyl phosphate and its stabilization by aspartate and ornithine transcarbamoylases. Proc. Nat. Acad. Sci. U.S.A. 2008; 105:16918–16925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Purcarea C., Evans D.R., Herve G.. Channeling of carbamoyl phosphate to the pyrimidine and arginine biosynthetic pathways in the deep sea hyperthermophilic archeon Pyrococcus abysii. J. Biol. Chem. 1999; 274:6122–6129. [DOI] [PubMed] [Google Scholar]
- 67. Harris K.A., Bobay B.G., Sarachan K.L., Sims A.F., Bilbille Y., Deutsch C., Iwata-Reuyl D., Agris P.F.. NMR-based structural analysis of threonylcarbamoyl-AMP synthase and its substrate interactions. J. Biol. Chem. 2015; 290:20032–20043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Teplova M., Tereshko V., Sanishvili R., Joachimiak A., Bushueva T., Anderson W.F., Egli M.. The structure of the YrdC gene product from Escherichia coli reveals a new fold and suggests a role in RNA binding. Prot. Sci. 2000; 9:2557–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Laskowski R.A., Swindells M.B.. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J .Chem. Inf. Model. 2011; 51:2778–2786. [DOI] [PubMed] [Google Scholar]
- 70. Katoh M., Kuma M.. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Robert X., Gouet P.. Deciphering key features in protein structures with the new Endscript server. Nucleic. Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number 6Z81.