


Abstract
Ribosome hibernation is a universal translation stress response found in bacteria as well as plant plastids. The term was coined almost two decades ago and despite recent insights including detailed cryo-EM structures, the physiological role and underlying molecular mechanism of ribosome hibernation has remained unclear. Here, we demonstrate that Escherichia coli hibernation factors RMF, HPF and RaiA (HFs) concurrently confer ribosome hibernation. In response to carbon starvation and resulting growth arrest, we observe that HFs protect ribosomes at the initial stage of starvation. Consistently, a deletion mutant lacking all three factors (ΔHF) is severely inhibited in regrowth from starvation. ΔHF cells increasingly accumulate 70S ribosomes harbouring fragmented rRNA, while rRNA in wild-type 100S dimers is intact. RNA fragmentation is observed to specifically occur at HF-associated sites in 16S rRNA of assembled 70S ribosomes. Surprisingly, degradation of the 16S rRNA 3′-end is decreased in cells lacking conserved endoribonuclease YbeY and exoribonuclease RNase R suggesting that HFs directly block these ribonucleases from accessing target sites in the ribosome.
INTRODUCTION
Translation is one of the most important cellular processes and consumes a considerable fraction of available resources (1,2). During translation in bacteria, free 30S and 50S ribosomal subunits are assembled into translating 70S ribosomes, which are mostly present in polysomic chains of ribosomes. Canonical translation has been studied extensively and is well understood (3). Recently, a more complex picture has emerged where translating ribosomes represent only a part of a heterogeneous ribosome population (4,5). When protein synthesis is reduced during stress conditions, ribosome capacity is generally adjusted by inhibition of de novo ribosome synthesis (6), degradation of excess ribosomes (7,8) and rapid modulation or inhibition of existing ribosomes by a variety of factors (4).
One of the most distinctive and striking examples of ribosome modulation is the dimerization of 70S ribosomes into inactive 100S complexes (5). Variations of this phenomenon, termed ribosome hibernation, are almost ubiquitously distributed in prokaryotes as well as in plant plastids (5,9). In most bacteria, including gram-positives, cyanobacteria and some gram-negatives, ribosome hibernation is induced by long hibernation promoting factor (lHPF), which binds 70S ribosomes and induces the formation of a 100S ribosome dimer (10–16). In some organisms, lHPFs stabilize ribosomes in a 70S assembled form as reported for mycobacteria and plant plastids (17–19). In most γ-proteobacteria, such as Escherichia coli, two distinct hibernation mechanisms exist: the formation of 100S dimers by the combined action of ribosome modulation factor (RMF) and hibernation promoting factor (HPF) (9,20,21), and the stabilization of 70S ribosomes by ribosome associated inhibitor A (RaiA) (22,23). Importantly, these two forms of hibernating ribosomes are thought to be simultaneously present in the cell and we will refer to these protein factors as hibernation factors (or HFs) (20). The formation of 100S dimers occurs in two steps: binding of RMF at the mRNA exit channel induces dimerization of 70S ribosomes to a 90S intermediate followed by joining of HPF to form a mature 100S complex (20,24). RaiA is a homolog of HPF, but contains a short C-terminal extension of 18 residues (20). Consistent with the highly similar sequence and structure of HPF and RaiA, both factors are thought to bind in the same position at the codon-anticodon interaction region of the ribosomal A- and P-site. The extended C-terminus present in RaiA is thought to protrude towards the mRNA channel and into the binding site of RMF, likely interfering with RMF binding and 100S formation (25,26).
HFs in E. coli are transcriptionally induced in a growth-rate dependent manner by major stress response pathways: rmf, hpf and raiA are all induced by starvation alarmone (p)ppGpp upon transition to stationary phase (27–32). In addition, rmf and raiA are also induced by cAMP in response to glucose starvation (33). Consistently, HFs are primarily present in stationary phase, while RaiA has also been detected at low levels during exponential growth (20,34,35). Interestingly, 100S dimers are exclusively formed in stationary phase or during stress and can rapidly dissociate when nutrients are replenished (27,34,36). This observation has led to the hibernation model in which 100S dimers serve as a translationally inactive, RNase-protected ribosome reservoir (21). The physiological significance of ribosome hibernation appears to differ from organism to organism (5). In E. coli, loss of hibernation factors has been implicated in reduced stationary phase survival and fitness, tolerance against heat, acid and osmotic stress (37–39), as well as increased susceptibility to aminoglycoside antibiotics gentamicin and netilmicin (40,41).
Recent high resolution structures of hibernation factors in complex with ribosomes have shed light on the structural basis of ribosome hibernation (9,18,25,42–44). However, the physiological role of ribosome hibernation on a mechanistic level is not clear. Here, we present a comprehensive analysis of the collective role of hibernation factors RMF, HPF and RaiA in E. coli. We show that ribosome hibernation confers a fitness advantage by ensuring efficient resuscitation from growth arrest in response to glucose starvation. Analysis of ribosome integrity, subunit distribution and rRNA fragment accumulation in a strain lacking HFs (ΔHF) shows increased release of free ribonucleotides during starvation, accompanied by a stark alteration of the ribosome profile. Intriguingly, hibernation-deficient cells exhibit increased accumulation of fragmented rRNA in free subunits and assembled 70S particles. Furthermore, mapping of 16S rRNA fragments of 70S particles in the absence of HFs revealed degradation taking place at the 30S-50S subunit interface and the 3′-terminus including the anti-Shine-Dalgarno sequence. Strikingly, the degraded rRNA domains all map to positions coinciding with binding sites of hibernation factors HPF/RaiA and RMF. Surprisingly, the degradation of the 16S rRNA 3′-end is dependent on the conserved ribonuclease YbeY and exonuclease RNase R, which have been reported to be involved in ribosome quality control and 16S rRNA maturation.
In conclusion, our data is consistent with HFs directly protecting ribosomes by preventing ribonuclease access to cleavage target sites within the ribosome. Furthermore, our study uncovers a new functional role of YbeY and RNase R in degradation of ribosomes disengaged from translation.
MATERIALS AND METHODS
Media and growth conditions
For growth on solid medium cells were routinely streaked out from frozen stocks on nutrient agar (NA; Oxoid) and incubated overnight (ON; ∼16 h) at 37°C. For cloning and recombination purposes, cells were cultured in lysogeny broth (LB; Oxoid) at 37°C and orbital shaking at 160 rpm in Erlenmeyer flasks or in 50 ml Falcon tubes or equivalent for overnight cultures. When required, solid and liquid culture medium was supplemented with ampicillin (30 μg/ml or 100 μg/ml for low-copy or high-copy plasmids, respectively), chloramphenicol (25 μg/ml) or kanamycin (25 μg/ml). As defined culture medium MOPS (3-morpholinopropane-1-sulfonic acid) minimal medium (MOPS MM) supplemented with 0.2% glucose, 1.32 mM K2HPO4 and 0.01 g/l of each nucleobase was used (45,46). Bacterial growth in liquid culture was determined by measuring optical density at 600 nm (OD600) in a DU730 spectrophotometer (Beckman Coulter).
Bacterial strains and plasmids
The strains used in this study are listed in Supplementary Table S1. Escherichia coli K-12 MG1655 Δrmf, Δhpf and ΔraiA were generated by P1 transduction (47) using E. coli K-12 derivative BW25113 Δrmf::kanR, Δhpf::kanR and ΔraiA::kanR as donor strains (48). The kanamycin resistance cassette (kanR) was removed by FLP-FRT recombination (49). Double and triple deletion mutants were generated by sequential P1 transduction of single and double mutants, respectively. Correct insertion and removal of the resistance cassette was confirmed by PCR with primers flanking the gene of interest and sequencing of the PCR product (for primer sequences see Supplementary Table S2 in supplementary).
Viability measurements
Viability in selected media was quantified as colony-forming units per ml of culture (CFU/ml) by spot-plating serial dilutions of cultures at indicated time points. To determine viability in defined media a single colony was inoculated into MOPS MM and incubated at 37°C and shaking at 160 rpm overnight. Overnight cultures were diluted 100-fold into 15 ml pre-warmed MOPS MM as pre-culture and grown to an OD600 of ∼0.3 before dilution into 30 ml pre-warmed medium to a final OD600 of 0.01. Cultures were incubated for up to 10 days at 37°C and shaking at 160 rpm. To determine viability in phosphate-buffered saline (PBS) an exponential phase culture prepared as described above was harvested by centrifugation at 4°C and 2700 x g, washed twice in cold PBS (Oxoid) and resuspended in pre-warmed PBS before continuing incubation for up to 10 days at 37°C and shaking at 160 rpm. At indicated time points OD600 of cultures was measured and serial dilutions of cells in PBS were spotted onto NA plates. Colonies were counted after overnight incubation at 37°C and after an additional 24 h of incubation at 37°C to include late-appearing colonies after prolonged starvation.
Regrowth assays
Cultures in defined media were prepared as described above: overnight cultures were diluted 100-fold in fresh medium, grown to exponential phase and back-diluted to an OD600 of 0.01 before incubation at 37°C and 160 rpm shaking for up to 5 days. At the indicated time points cultures were diluted 1:100 into 30 ml of pre-warmed MOPS MM and regrowth followed at 37°C and shaking at 160 rpm. Lag time was calculated as the intercept of the initial OD600 with the tangent of the growth curve at exponential growth (50).
In vivo rRNA degradation assay
To measure degradation of total RNA (and therefore approximate the degradation state of rRNA) we used a modified in vivo ribosome degradation assay (7,51,52). Overnight cultures in MOPS MM supplemented with 0.02 g/l uridine were diluted 100-fold in fresh medium, grown to exponential phase and back-diluted to an OD600 of 0.01 in MOPS MM supplemented with 0.02 g/l uridine and 1 μCi/ml [5-3H]-uridine (Perkin-Elmer). Incubation was continued at 37°C and 160 rpm shaking until cells reached growth arrest (∼8.5 h). The entire culture was rapidly cooled on ice, harvested by centrifugation at 4°C and 2700 x g, washed twice with cold 1× MOPS and re-suspended in an equal volume of filter-sterilized spent medium from a parallel culture grown in the same medium without [5-3H]-uridine. Incubation was continued at 37°C and 160 rpm for up to 5 days. Incorporation of labelled uridine in total RNA was determined by measuring radioactivity in the macromolecule fraction of cells. To precipitate all macromolecules 500 μl culture were added to 5 ml cold 5% TCA, left on ice overnight and filtered on pre-soaked glass fibre filters (Advantec) followed by three washes with 5 ml cold 5% TCA. Filters were dried for 30 min at 70°C in scintillation vials before adding 5 ml scintillation fluid (ProSafe FC+; Meridian) and counting in a HIDEX 300SL scintillation counter (Hidex). Free [5-3H]-nucleotide degradation products was measured by determining acid-soluble radioactivity. 500 μl culture were added to 250 μl cold 4 M formic acid, mixed and left on ice for 15 min before centrifugation for 15 min at 4°C and 15 000 × g. 200 μl of the supernatant was added to a scintillation vial and pH neutralized by addition of 260 μl 1M Tris base. 5 ml scintillation fluid was added and radioactivity was counted in the scintillation counter. The acid-soluble fraction was determined as the ratio of acid-soluble fraction to the combined counts of acid-soluble and TCA-precipitated fraction.
Sucrose density gradient centrifugation (SDGC)
SDGC experiments were performed according to a modified protocol from Beckert et al. (25). Cells were grown in MOPS MM as described above and 10–50 ml harvested at indicated time points by centrifugation at 5000 × g at 4°C for 15 min. The cell pellets were frozen in liquid nitrogen and stored at −80°C until further treatment. Cells were re-suspended in 500 μl ice-cold lysis buffer (25 mM HEPES, pH 7.5; 100 mM KOAc; 15 mM Mg(OAc)2; 1 mM DTT), added to an equal volume of 0.5 mm zirconia/silica beads (BioSpec) and lysed by vortexing in 5 cycles of 1 min vortexing and 1 min on ice. The lysate was cleared by centrifugation at 12 000 × g at 4°C for 5 min. The approximate yield was determined by measuring A260 with a NanoVue™ Plus Spectrophotometer (GE Healthcare). The lysate was diluted to 20 A260 units in 300 μl ice-cold lysis buffer and layered onto a 5–20% sucrose gradient (25 mM Hepes, pH 7.5; 100 mM KOAc; 15 mM Mg(OAc)2; 1mM DTT; 0.01% n-dodecyl-d-maltoside; 5–20% sucrose) prepared using a Gradient Master (BioComp). Ribosomal particles where separated by centrifugation at 37 000 rpm/209 627 g at 4°C for 2 h in a Thermo Sorvall WX90 ultracentrifuge with a TH-641 rotor. Obtained gradients were fractionated with a Gradient Fractionator (BioComp) and analysed with a Model EM-1 Econo UV monitor (Biorad) UV detection system at 254 nm.
Total RNA extraction from SDGC fractions
To extract total RNA from SDGC fractions, an equal volume of phenol (pH 4.3) was added to SDGC fractions and incubated on ice for 20 min with periodic vortexing. Samples were centrifuged at 12 000 × g at 4°C for 10 min to separate the aqueous phase from the phenolic phase. To the aqueous phase, 0.5 volumes of phenol (pH 4.3) and 0.5 volumes of chloroform were added, mixed by vortexing and centrifuged at 12 000 × g at RT for 5 min. RNA was precipitated from the aqueous phase by addition of sodium acetate to a final concentration of ∼0.2 M and 1.5 volumes of ice-cold 99% ethanol, followed by incubation at −80°C for at least 30 min or overnight. To collect the precipitate samples were centrifuged at 12 000 × g at 4°C for 20 min, the pellet washed twice in 70% ethanol and dried at RT for 5–10 min, before re-suspension in nuclease-free water and storage at −80°C.
Total RNA extraction from cell samples
Cells were grown in MOPS MM as described above and at indicated time points harvested into 1/5 volumes of ice-cold stop solution (5% water-saturated phenol in ethanol) and pelleted by centrifugation at 4°C and 2700 x g. Total RNA was routinely extracted using the hot phenol–chloroform method. In short, cells were re-suspended in ice-cold solution 1 (0.3 M sucrose; 0.01 M NaOAc, pH 4.5) and mixed with solution 2 (2% SDS; 0.01 M NaOAc, pH4.5) and 0.5 volumes phenol (pH 4.3) before incubation at 65°C for 3 min. Samples were frozen in liquid nitrogen and centrifuged at 12 000 × g at RT for 5 min. The aqueous phase was transferred to a new tube and the phenol treatment repeated. 0.5 volumes of phenol and 0.5 volumes of chloroform were added, the samples mixed and centrifuged at 12 000 × g at RT for 5 min. RNA in the aqueous phase was precipitated in 900 μl ice cold ethanol (96%) and 40 μl NaOAc (3M, pH4.8) at −80°C overnight. Precipitated RNA was pellet by centrifugation at 12 000 × g at 4°C for 30 min, the pellet washed twice in ethanol (70%) and the air-dried pellet re-suspended in 50 μl nuclease-free water.
Polyacrylamide gel electrophoresis and Northern blotting
RNA extracted from SDGC fractions or cell samples was separated using denaturing PAGE. RNA samples were mixed with an equal volume of formamide loading buffer (95% formamide, 0.025% bromophenol blue, 0.025 xylene cyanol, 5 mM EDTA, 0.025% SDS) and separated on 3% or 6% polyacrylamide gels with 8 M urea buffered in 1× TBE (100 mM Tris Base, 100 mM boric acid, 2 mM EDTA) at 300 or 450 V. When necessary, gels were stained for 30 min in 5 μg/ml ethidium bromide in 1× TBE. For Northern blotting, RNA was transferred to a Hybond-N+ blotting membrane (Amersham) in 1× TBE. Membranes were pre-hybridised in 6 ml hybridisation buffer (5 × SSPE, 5 × Denhardt's solution, 0.5% SDS, 0.55 mg/ml salmon sperm DNA) at 42°C for 1 h before addition of 30 pmol radiolabelled DNA oligonucleotide probe for overnight hybridization. Excess probe was removed by washing in 2 × SSC, 0.1% SDS and radiation detected by phosphor-imaging. Membranes were stripped for re-probing at 95°C in stripping buffer (0.1 × SSPE, 0.5% SDS) until no remaining radiation was detected.
Primer extension analysis of rRNA fragments
To determine the 5′-end of accumulating rRNA fragments primer extension analysis of RNA extracted from selected SDGC fractions was performed with primers adjacent to the approximate 5′ terminus as determined by Northern blotting. 4 pmol of oligonucleotide primer was radiolabelled with 6000 Ci/mmol (150 mCi/ml) 32P-γ-ATP (Perkin-Elmer) using polynucleotide kinase (PNK, Thermo). The kination reaction was carried out at 37°C for 30 min followed by inactivation for 15 min at 75°C. To remove excess unincorporated radiolabelled ATP, the reaction was centrifuged through an illustra™ MicroSpin™ G-25 Column (GE Healthcare) for 1 min at 750 x g. Sequencing reactions were performed using a gel-purified PCR product generated with primers 16S_5′ (17-36) and 16S probe f (978–997) as template: 0.1 pmol of template DNA was incubated with 0.4 pmol 32P-labelled primer and extension with 0.5 U DreamTaq DNA polymerase (Thermo Scientific) was performed in the presence of 5.625 μM of either ddGTP, ddATP, ddTTP or ddCTP (Roche). PCR was performed with the following program: initial denaturation (95.0°C, 2 min), 30 cycles of denaturation (95°C, 30 s), annealing (55°C, 30 s) and extension (72°C, 30 s), followed by final extension (72°C, 2 min). Reactions were stopped by addition of an equal volume of 2× formamide loading buffer (95% formamide, 0.025% bromophenol blue, 0.025 xylene cyanol, 5 mM EDTA, 0.025% SDS). For extension reactions 0.2 pmol radiolabelled primer was hybridized with 250 ng RNA by heating at 80°C for 5 min followed by rapid chilling on ice for 5 min. Reverse transcription was carried out with 2 U of SuperScript III Reverse Transcriptase (Invitrogen) in 1× First-Strand buffer, 10 mM DTT and 1 mM dNTPs at 54°C for 1 h. Reactions were stopped by addition of an equal volume of 2× formamide loading buffer. Sequencing and extension reactions were separated on 6% denaturing polyacrylamide gels. Obtained gels were fixed in 50% ethanol and 10% acetic acid, dried, and imaged using phosphor imaging.
Mapping of 3′-ends using miRcat-33 linker ligation followed by RT-PCR
3′-ends of detected rRNA fragments were estimated using a linker-based sequencing approach. In short, total RNA extracted from selected SDGC fractions was reverse transcribed into cDNA from an activated 3′-linker (miRcat-33, Integrated DNA Technologies), and cDNA amplified with selected primer pairs using PCR. Amplified DNA was then separated by agarose gel electrophoresis, bands of interest excised and DNA purified before sequencing. To ensure efficient ligation of the miRcat-33 activated primer 1 μg RNA was subjected to alkaline phosphatase treatment by incubation with 2 U FastAP (NEB) in 1× T4 RNA ligase buffer (NEB) supplemented with 40 U RNasin (Promega) in a total volume of 20 μl at 37°C for 10 min. Subsequently, 4 μl of EDTA (50mM) was added to chelate excess Mg2+, and FastAP deactivated by incubation at 75°C for 5 min.
miRCat 33 3′-linkers were ligated by incubation of the alkaline phosphatase-treated samples with 40 pmol miRCat-33 3′ RNA linker oligomers in 1× T4 RNA Ligase Buffer (NEB), 80 U RNasin, 20 U T4 RNA ligase (NEB) and 200 pmol MgCl2 in a total volume of 50 μl at 25°C for 120 min. Total RNA was then precipitated by addition of NaOAc to a concentration of 0.3 M and 2.5 volumes of ice-cold 99% ethanol, followed by incubation at −80°C for at least 30 min or overnight. Precipitated RNA was collected by centrifugation at 12 000 × g at 4°C for 20 min, washed twice in 70% ethanol and dried at RT for 5–10 min, before re-suspension in nuclease-free water and storage at −80°C. Reverse transcription was carried out with 1 mM 33-rev primer using 2 U of SuperScript III Reverse Transcriptase (Invitrogen) in 1× First-Strand buffer, 10 mM DTT, 1 mM dNTPs and 40 U RNasin (Promega) at 54°C for 1 h. Reverse transcriptase was inactivated by incubation at 65°C for 15 min and RNA was treated with 0.5 U RNase H (NEB) at 37°C for 60 min. cDNA was amplified using DreamTaq polymerase (Thermo Scientific) and 1 μM of miRCat-33-3′ reverse oligo and selected forward primers. Amplified cDNA was separated by 2% agarose gel electrophoresis and target bands in the expected size range excised, purified using QIAquick gel extraction kit (Qiagen), and sequenced using forward primers. Putative 3′-ends were defined as the last unambiguously detected nucleotide according to sequencing chromatograms.
RESULTS
Ribosome hibernation is necessary for efficient regrowth from starvation
In E. coli, hibernation factors have repeatedly been implicated in cell survival during stationary phase (28,29,53,54). However, other published results contradict these findings (55). We therefore investigated the physiological role of HFs in wild-type E. coli K-12 MG1655 (WT). For reproducibility we used chemically defined MOPS minimal medium containing 0.2% glucose (named MOPS MM). This medium has been optimized for enterobacteria and growth arrest in batch cultures is caused by glucose limitation (45). Using quantitative real-time PCR we confirmed that rmf, hpf and raiA are all up-regulated at the onset of growth arrest in response to glucose starvation (Supplementary Figure S1). Accordingly, we focused our investigation on a triple deletion strain (ΔHF) lacking rmf, hpf and raiA, to exclude previously proposed redundancy effects due to the reported overlapping ribosomal binding sites of the HFs (24–26). As HFs are induced in response to starvation and have been implicated in cell survival during long-term stationary phase we investigated the effect of ribosome hibernation on cell viability. When grown in MOPS MM, WT E. coli cultures reached growth arrest at an approximate OD600 of 2.2 or 8.5 h after inoculation (Supplementary Figure S1A). While the majority of cells remained viable for ∼1 week of starvation, the viability of ΔHF cells decreased ∼8-fold relative to WT after ten days of incubation in MOPS MM (Figure 1A). When cells were exposed to sudden nutrient starvation by transfer from exponential growth to phosphate-buffered saline (PBS), an abrupt 4-fold drop in viability was seen for both strains within 24 h. After four days of incubation, a significant viability defect was observed in ΔHF (approximately three-fold relative to WT), that increased to an approximately 14-fold difference after 10 days (Figure 1B). This suggests that the lack of HFs has a slight effect on viability during glucose starvation, and a more pronounced effect during the abrupt change to the nutrient-deprived PBS. However, gradual depletion of a nutrient is a more likely scenario for bacteria in their natural environment, therefore we opted to continue our investigation on glucose-starved cells. In either case, decreased relative viability of ΔHF was only observed after extended incubation times (at least 4 days), which does not correlate with the elevated expression of HFs during the growth state transition (7–8.5 h; Supplementary Figure S1).
Figure 1.
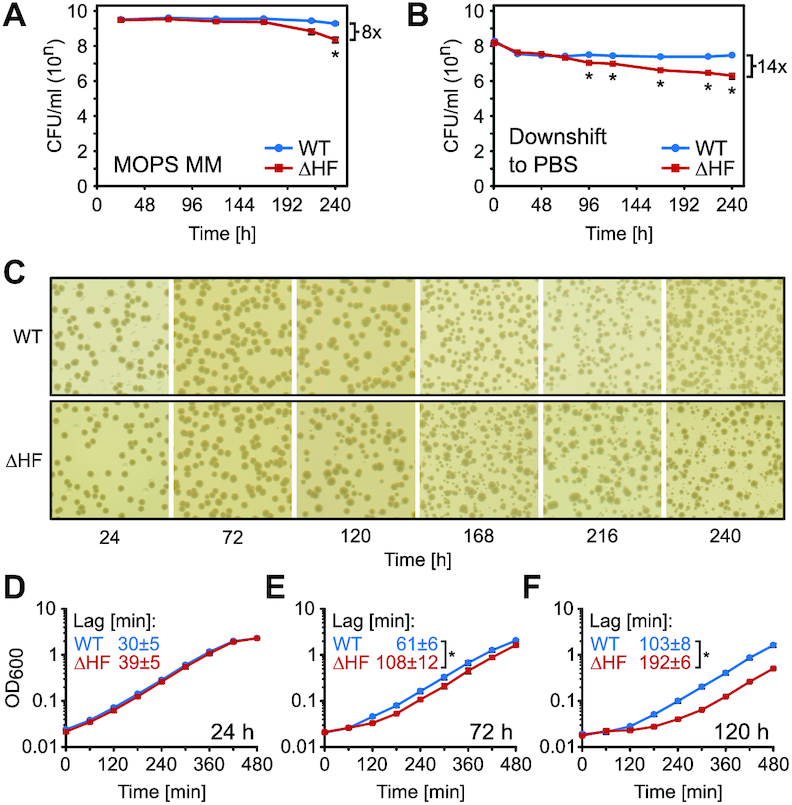
Hibernation factors only moderately affect long-term survival, but are essential for efficient regrowth. (A, B) Long-term viability of E. coli K-12 MG1655 (WT) and a hibernation factor-deficient mutant (ΔHF) in different starvation conditions. Cells were grown in batch culture in MOPS MM until growth arrest and incubation continued for 10 days (A). Exponentially growing cells were washed and transferred to phosphate-buffered saline (PBS) and incubation continued for ten days (B). At indicated time points the number of colony-forming units per ml of culture (CFU/ml) was determined by spot-plating of serial dilutions. Data points represent mean values of at least three independent biological replicates. Error bars represent the standard error of the mean (SEM). *P< 0.05 (two-tailed Student's t-test assuming unequal variances). The maximum fold difference in CFU/ml between WT and ΔHF at t = 120 h is indicated with a bracket. (C) Heterogeneity of colonies arising from cultures after prolonged starvation in (A). At the indicated time points appropriate dilutions of WT and ΔHF cultures in MOPS MM were plated on nutrient agar (NA) plates and incubated overnight. Images shown are representative of three independent biological replicates. (D–F) Regrowth of WT and ΔHF cells following nutrient replenishment after incubation in MOPS MM for 24, 72 and 120 h (D, E and F, respectively). Cells were collected at indicated time points, diluted into fresh MOPS MM, and regrowth was followed by optical density measurements at 600 nm (OD600). Data points represent mean values of at least four independent biological replicates. Error bars represent SEM. Lag times were calculated using the tangent method (50) on independent regrowth curves. Indicated values correspond to the mean lag times and the SEM of at least four independent biological replicates. *P< 0.05 (two-tailed Student's t-test assuming unequal variances).
We observed that colonies of the ΔHF mutant from glucose-depleted medium appeared to be heterogeneous in size when plated on nutrient rich plates after 120 h of starvation (Figure 1C). This heterogeneity could either be explained by accumulation of compensatory mutations in the ΔHF mutant or differential ability at the single-cell level to regrow upon nutrient replenishment. Re-inoculated heterogeneous colonies yielded comparable growth in solid and liquid medium (Supplementary Figure S2A), which suggested a regrowth defect. We tested this by measuring the lag-time preceding exponential growth when starved cells were diluted into fresh medium. After 24 h in MOPS MM, WT and ΔHF cells resumed exponential growth after 30 ± 5 and 39 ± 5 min, respectively (Figure 1D). However, after 72 h of incubation (Figure 1E), ΔHF cells exhibited significantly delayed regrowth (108 ± 12 min) when compared to WT (61 ± 6 min). This defect became increasingly severe with continued incubation, reaching 103 ± 8 and 192 ± 6 min, respectively, for WT and ΔHF cultures after 5 days (Figure 1F). Importantly, the regrowth defect of ΔHF cells could be almost completely rescued by expression of the HFs from a high-copy-number plasmid (Supplementary Figure S2B). When testing mutants deficient in only one form of hibernation, either 100S formation (ΔrmfΔhpf) or RaiA-mediated 70S stabilization (ΔraiA), no significant delay in regrowth could be detected (Supplementary Figure S2C), further corroborating a possible functional overlap between the two mechanisms. Taken together, these results show that hibernation factors jointly confer the ability to rapidly resume growth when glucose is replenished.
Hibernation factors protect ribosomes from degradation
It has long been speculated that ribosome hibernation serves as a reversible mechanism of inactivation or protection of ribosomes in response to adverse conditions (21,36). If the latter is the case, the regrowth defect could be caused by excessive degradation of ribosomes in the ΔHF mutant. To test this hypothesis, we performed an in vivo ribosome stability assay (Figure 2A, top). Total RNA was radiolabelled by growing cells in the presence of 3H-uridine until growth was arrested by nutrient depletion. At selected time points the fraction of acid-soluble radioactivity was determined as an approximation of total RNA degradation activity. As shown in Figure 2A (bottom), prolonged growth in MOPS MM and resulting glucose starvation led to an abrupt increase in acid soluble radioactivity counts in the first 16 h after growth arrest, reaching approximately 20% in wild-type cultures and almost 40% in ΔHF cultures, followed by a more gradual increase at similar rates in both strains (reaching approximately 50% and 70% after 5 days, respectively). Ribosomal RNA approximates the total RNA amounts in the cell (6), which suggests increased degradation of rRNA in the absence of HFs, especially during the initial phase following growth arrest.
Figure 2.
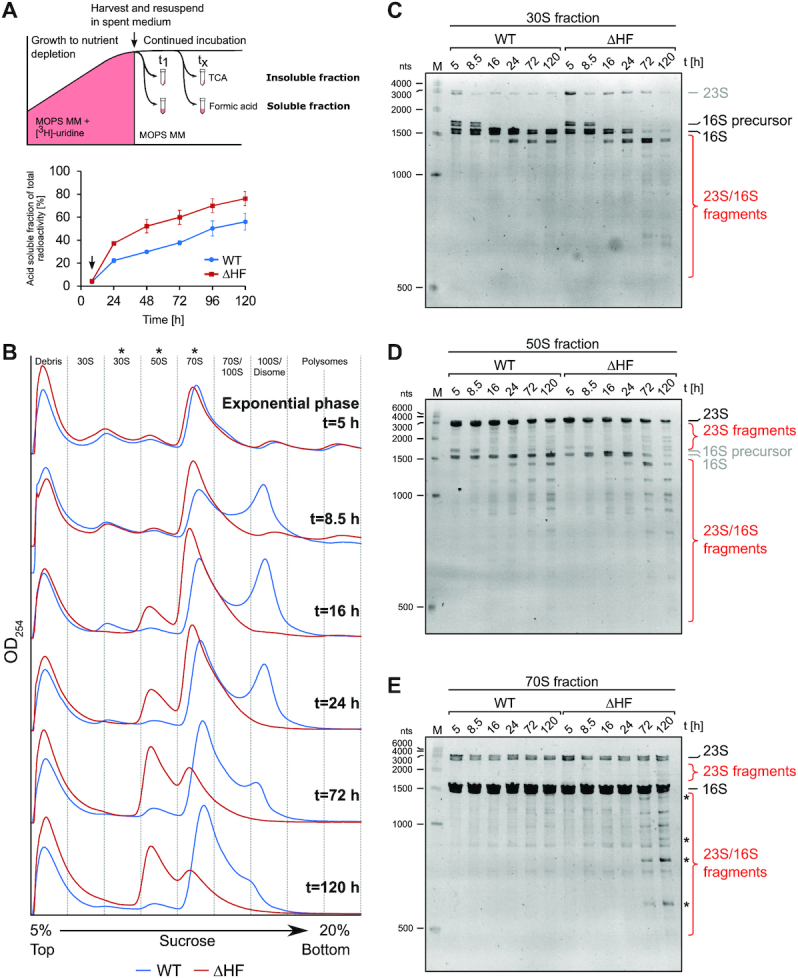
Ribosome hibernation protects ribosomal RNA from degradation and maintains ribosome particle homeostasis. (A) In vivo RNA stability during 5 days of starvation in MOPS MM. Top panel shows a schematic representation of the assay: WT and ΔHF cells were grown in MOPS MM supplemented with 1 μCi/ml 3H-uridine until growth arrest (indicated by arrow), washed and re-suspended in spent medium harvested from identical cultures grown in parallel but lacking radioactively labelled uridine. Incubation was continued for five days and at the indicated time points total degraded RNA was determined as the formic acid-soluble fraction of the sum of formic acid-soluble and trichloroacetic acid (TCA)-precipitated radioactivity. Bottom panel shows the acid-soluble fraction of radiolabeled RNA in WT and ΔHF cultures. Arrow indicates time of growth arrest, removal of 3H-uridine from the medium and re-suspension in spent, unlabelled medium. Data points represent mean values of three independent biological replicates. Error bars represent the standard error of the mean (SEM). (B) Time course of ribosome particle distribution from exponential phase to five days of incubation determined by sucrose density gradient centrifugation (SDGC). Cells were grown in MOPS MM batch culture and samples harvested at the indicated time points were lysed and equal amounts (A260 units) were layered onto 5–20% sucrose gradients for separation by ultracentrifugation. For ease of comparison, baselines of profiles corresponding to individual time points are stacked along the y-axis. Asterisks mark fractions selected for RNA purification in (C–E). The profiles shown are representatives of four technical replicates of two independent biological replicates. (C–E) Ribosomal RNA fragments within ribosome particles. Total RNA was purified from selected SDGC fractions corresponding to 30S (C), 50S (D) and 70S particles (E), and equal amounts of RNA was separated on denaturing urea-PAGE gels and stained with ethidium bromide. Asterisks mark fragments uniquely present in ΔHF fractions. M = size marker.
Ribosome hibernation is transitory and essential for maintaining ribosome particle homeostasis
The transitory increase in release of acid-soluble radioactivity counts in ΔHF cultures indicates that the protection of ribosomes by HFs may be transient (Figure 2A). To test this hypothesis, we determined the polysome profiles of WT and ΔHF cells over 5 days of glucose starvation by sucrose density gradient centrifugation (SDGC). Consistent with the low level of HF expression, the distribution of ribosomal particles in WT and ΔHF was close to identical during exponential growth (Figure 2B). Upon transition to growth arrest (t = 8.5 h), a distinct peak in the range of 100S appeared in WT cells, concomitant with decreased peak heights in the polysome fractions. The 100S peak reached a maximum at 16 h and, importantly, steadily declined to an almost undetectable level after 5 days of incubation, while the 70S peak increased. Free 50S subunits appeared to remain at a similar level throughout the incubation time, whereas free 30S subunits decreased. As expected, ΔHF cells did not exhibit formation of 100S dimers. Instead, an initial increase in the 70S fraction was observed, in accordance with the notion that 100S dimers contain surplus 70S ribosomes not engaged in translation. With continuing incubation, the 70S peak decreased to a lower level than in WT cells. 30S subunits were not detectable after 16 h of incubation, whereas particles in the 50S size fraction surprisingly accumulated, eventually forming the majority of particles present in the gradient. In summary, these observations suggest that 100S dimer formation is transitory and prevents an initial accumulation of 70S ribosomes that would otherwise dissociate into rapidly degraded 30S and accumulating 50S subunits as starvation continues.
ΔHF cells accumulate ribosomal particles containing fragmented rRNA species
The SDGC profiles did not only reveal a heavily altered ribosome particle distribution in the ΔHF mutant, but also subtle changes in the sedimentation of individual particles: the 100S peak in wild-type cells and 70S peak in ΔHF cells appear to be shifted towards the top of the gradient from 72 h onwards (Figure 2B). A shift from 100S towards a lower sedimentation coefficient may reflect the accumulation of 100S dissociation intermediates. The reduced sedimentation of ΔHF–70S particles is consistent with increased rRNA degradation in the ΔHF mutant (Figure 2A). We therefore investigated the integrity of rRNA in SDGC fractions corresponding to 30S, 50S or 70S particles over five days (Figure 2C–E). As expected from the similar exponential growth kinetics of both strains, wild-type and ΔHF rRNAs in exponential phase (5h) were comparable. In the 30S fraction, the majority of RNA present could be identified as 16S rRNA (Figure 2C), with an additional band above mature 16S in exponential and transition phase corresponding to 16S precursor-rRNA (Supplementary Figure S3A–C). With increasing incubation time, the level of intact 16S rRNA decreased and a smaller fragment appeared, consistent with a 16S degradation product. Interestingly, almost no intact 16S rRNA could be detected in ΔHF-30S from 72 h onwards, and instead additional, shorter RNA species appeared. A similar pattern was observed when examining the 50S fractions (Figure 2D). In both WT and ΔHF most rRNA was comprised of 23S rRNA, and several fragments likely derived from intact 23S rRNA as well as smaller fragments of either 23S or 16S rRNA (the latter most likely originating from fronting or tailing of the adjacent 30S and 70S gradient fractions) appeared over time. Intriguingly, RNA extracted from ΔHF–70S fractions contained several fragments that appeared to be unique to the ΔHF cells, alongside several fragments common to both strains (Figure 2E). Thus, lack of HFs leads to an overall increase in the accumulation of fragmented rRNA within subunits and assembled 70S ribosomes. Importantly, ΔHF cells exhibit an additional subpopulation of 70S containing unique rRNA fragments.
ΔHF-70S particles contain unique 16S rRNA fragments
To investigate the origin of the accumulating rRNA fragments, we used Northern blot analysis of RNA extracted from selected SDGC fractions (ranging from the debris fraction to the 100S fraction) obtained from cultures starved for five days. Probing for different parts of 23S rRNA revealed several 23S-derived degradation fragments in the ΔHF-50S and 70S fractions (Supplementary Figure S4A–D). These fragments were also present in WT-50S and 70S, and thus not solely a consequence of the lack of HFs. We therefore focused our investigation on fragments originating from 16S rRNA. When probing for different regions of 16S rRNA (Figure 3A), five distinct fragments (16S fragments I–V) could be detected in ΔHF-70S, three of which were uniquely present in the absence of HFs (Figure 3B; fragments I, III and V). ΔHF fragment I appeared to span most of the 16S, but could not be detected with probes h and i targeting the 16S 3′-terminus. Fragment I could be detected in WT-30S alongside intact 16S, whereas ΔHF-30S contained mostly fragment I and almost no intact 16S (Supplementary Figure S4F and G). Fragment III was only detected with probes a and b, and fragment V was only targeted by probes d to g. Intriguingly, when hybridizing with probe c (772–792) neither fragment III nor fragment V could be detected. This indicates a missing region between the two fragments in ΔHF-70S ribosomes. Fragment V also appeared to lack a large part of the 3′-end of the 16S rRNA, including the anti-Shine-Dalgarno (aSD) sequence (probes h and i). Importantly, an additional fragment of approximately 150 nts could be detected (fragment VI) with probe i (1522–1541; Figure 3C), likely corresponding to the missing 16S 3′-terminus of fragments I and V. Fragment VI was also highly enriched in the 30S fraction of both WT and ΔHF, alongside an additional, shorter fragment (VII), presumably originating from further degradation of fragment VI (Supplementary Figure S4H). Fragments II and IV, common to both strains, corresponded to approximately two thirds and one third of the 16S rRNA on the 5′- (probes a–e) and 3′-side (probe f-i), respectively (Figure 3A and B). The SDGC fraction containing WT-100S and the corresponding size-fraction in ΔHF (disomes) contained nearly exclusively intact 16S and 23S rRNA, regardless of which probe was used for detection (Figure 3D and E; Supplementary Figure S4E), suggesting that the rRNA cleavage events are unique to ribosomes in the 70S fraction.
Figure 3.
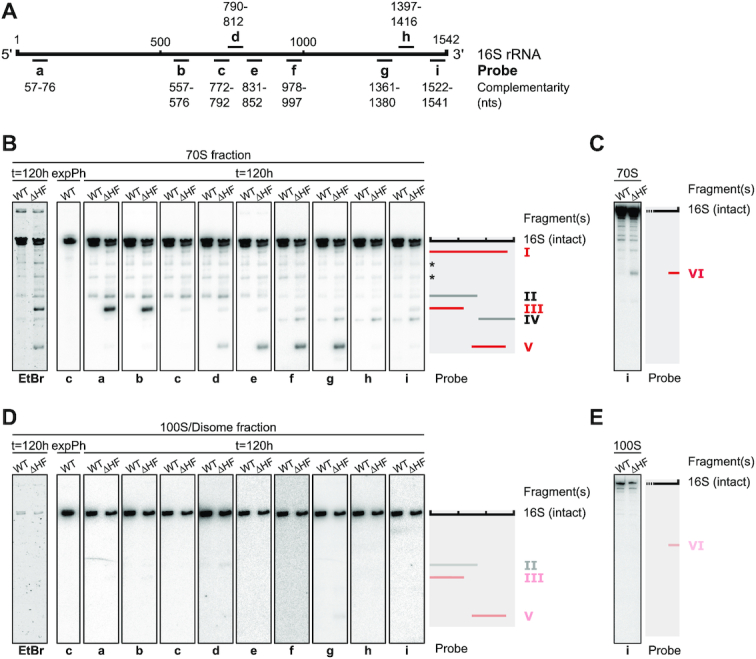
ΔHF cells accumulate 70S particles containing fragmented 16S rRNA. (A) Schematic of 16S rRNA and DNA oligonucleotide probes used for Northern blot analysis in (B-E). For the sequences of the indicated probes see Supplementary Table S2. (B) Northern blot analysis of 16S rRNA fragments found in the 70S fraction of five day old WT and ΔHF cultures. For orientation a cut-out of the ethidium bromide-stained gel (Supplementary Figure S4A) used for Northern blotting is shown (left-most panel). RNA purified from the 70S fraction of an exponentially growing WT culture served as a control for the detection of intact 16S rRNA. The schematic on the right shows fragments (I–V) mapped by urea–PAGE and Northern blot analysis. Gray bars indicate fragments common to both WT and ΔHF-70S particles (fragments II and IV), red bars indicate fragments only present in ΔHF-70S particles (fragments I, III and V). Asterisks mark additional unidentified 16S fragments or unspecific binding of labelled oligomers to fragments of 23S rRNA. (C) As described in (B). RNA was resolved on 6% urea–PAGE gels for detection of RNA in a lower size range. (D) Northern blot analysis of 16S rRNA found in the 100S/disome fraction of five day old WT and ΔHF cultures, as described in (B). Traces of fragments II, III and V are indicated by opaque bars in the schematic. (E) As described in (D). RNA was resolved on 6% urea–PAGE gels for detection of RNA in a lower size range.
Importantly, we could detect the 16S rRNA fragments found in SDGC fractions in total RNA extracted from 5 day old WT and ΔHF cultures, confirming that accumulation of the fragments is not due to degradation during sample preparation for SDGC experiments (Supplementary Figure S5). In addition, complementation of all three HFs in ΔHF from a high-copy plasmid resulted in an almost identical fragmentation pattern as observed in wild-type cells (Supplementary Figure S5).
In summary, both strains accumulate a subpopulation of 70S ribosomes that harbour fragmented 23S and 16S rRNA. The lack of HFs aggravates the accumulation of such ribosomes and, importantly, leads to accumulation of unique 70S particles that contain 16S rRNA with a missing or fragmented 3′-terminus including the aSD region, and/or miss a central region of the 16S rRNA spanning from at least nucleotide 772 to 792.
Hibernation factors protect 16S rRNA from degradation by occlusion of RNase cleavage and degradation sites
To determine the 5′- and 3′-ends of accumulating fragments we used primer extension analysis and an RT-PCR sequencing approach, respectively (Figure 4). Using probe e (831–852) as a primer we mapped the 5′-terminus of fragment V to nucleotides 798–799 (Figure 4A; Supplementary Figure S6A and B). Probe f (978–997) yielded multiple enriched bands common to WT and ΔHF stationary phase samples and additional bands exclusive to ΔHF samples (Figure 4B; Supplementary Figure S6A and C). These fragments possibly originate from an initial cleavage event followed by partial degradation. A band at nucleotide 842 was particularly enriched in both WT and ΔHF, and is therefore consistent with the 5′-terminus of fragment IV. Extension from primer d (790–812) did not yield any enriched bands when compared to the exponential phase control, indicating that the 5′-termini of 16S fragments are intact (Supplementary Figure S6A and D). Using RT-PCR and sequencing we determined the 3′-ends of fragments II and III to nucleotide 840 and 764, respectively (Figure 4C; Supplementary Figure S7A-D). Consistent with the results of the Northern blot analysis, RT-PCR analysis of fragments I and V yielded a single product with the 3′-terminus corresponding to nucleotide 1382 (Figure 4D; Supplementary Figure S7E and F). In summary, we mapped the five prevalent fragments found in ΔHF-70S particles to nucleotides 1–1382 (fragment I), 1–840 (fragment II), 1–764 (fragment III), 842–1542 (fragment IV) and 799–1382 (fragment V) (Figure 4E).
Figure 4.

Mapping of 16S rRNA fragments present in 70S ribosomes. (A, B) Primer extension analysis with RNA purified from 70S fractions of WT and HF cultures in exponential phase (expPh) and after 5 days of incubation (120 h), using primers e (panel A) and f (panel B). Red arrows indicate major 5′-ends that are exclusively present in ΔHF samples, black arrows indicate 5′-ends present in both WT and ΔHF samples. Light red and gray arrows mark additional 5′-ends in ΔHF and both strains, respectively. For full gels see Supplementary Figure S6. (C, D) RT-PCR analysis of RNA from samples described in (A, B). Shown are end sections of chromatograms obtained by sequencing with primer RT_a (C) and RT_b (D). Marked bases indicate the last base that could be assigned to 16S rRNA unambiguously. Red background indicates sequences corresponding to the 3′-linker used in the RT-PCR that are ambiguous due to a variable 3′-end of the fragment. For 3′-end sequencing workflow and extended results see Supplementary Figure S7. (E) Schematic summary of the mapped 16S rRNA fragments accumulating in 70S ribosomes after 5 days of starvation in MOPS MM. Red bars mark fragments exclusively or prevalently found in ΔHF-70S (fragments I, III, V and VI) or ΔHF-30S (fragment VII); gray bars mark fragments found in both WT- and ΔHF-70S (fragments II and IV). (F) Overlay of mapped degradation regions with the secondary structure of E. coli 16S rRNA (adapted from http://rna.ucsc.edu/rnacenter/images/figs/ecoli_16s.pdf). The 16S domains (5′ domain, 5′; Central domain, C; 3′ major domain, 3′M; 3′ minor domain, 3′m) are indicated. The gap between fragment I and III is coloured in orange; the missing 3′-terminus of fragments I and V is coloured in red. Inserts depict close-ups of selected regions. Light blue, magenta, green and brown background indicates residues that are interacting (according to Beckert et al. 2018) or in close proximity to RMF, HPF, ribosomal protein S1 or E-site tRNA in the 100S dimer, respectively. (G) 16S rRNA degradation sites in proximity to the binding site of RMF in the hibernating ribosome (PDB ID: 6H4N). The 3′-terminal region missing in fragments I and V (red) and h24 (orange) are in close proximity to RMF (dark blue) and ribosomal protein S1 (light green) at the mRNA exit channel and a tRNA in the E-site (brown). Inset shows close-up of the indicated region. Ribosomal proteins other than S1 have been omitted for the sake of clarity. (H) Top view of the 30S subunit of the hibernating ribosome with bound HPF (magenta) (PDB ID: 6H4N) interacting with h44, h45 and h24. Colour scheme as in (G).
Comparing the sequences of the 16S rRNA fragments with the known secondary structure of E. coli 16S rRNA and a cryo-EM structure of the hibernating ribosome (25) revealed that fragments I and V lack the entire 3′-minor domain (3′m) consisting of helix 44 (h44) and h45 and including the aSD sequence (Figure 4F–H). In addition, absence of nucleotides 1383–1396 destroys h28 which forms the bridge between the 3′-major domain (3′M) and the central and 3′-minor domains. The gap between fragments III and V spans most of h24 (Figure 4F–H). The respective 3′- and 5′-ends of fragments II (nucleotide 840) and IV (nucleotide 842) are located on h26 and the 3′ end of a possible additional fragment detected by RT-PCR and sequencing (Supplementary Figure S7C and D) maps to position 999 at the base of h33 (Supplementary Figure S8A). Both of these regions are located at the cytosolic face of the 30S subunit: h26 protrudes from the 16S platform, in close proximity to the 16S-3′-end and ribosomal protein S1 in its inactive 100S conformation, while h33 forms part of the 16S beak and remains exposed in the 100S complex (Supplementary Figure S8B and C). The additional 5′-ends observed by extension from primer f (Figure 4B) are located between the central and 3′-major domains and are mostly positioned in the neck and platform regions of the 16S rRNA (Supplementary Figure S8A and B).
YbeY and RNaseR are involved in the degradation of the 16S rRNA 3′-end
The observed fragmentation of 16S rRNA in ΔHF–70S raises the question which ribonuclease(s) are involved in the generation of the accumulating fragments. Fragments II and IV are likely generated by endonucleolytic cleavage at positions 840–842. Fragments III and V are likely generated by an initial endonuclease cleavage at position 799, followed by removal of helix 24 (765–798), either by additional endo- or exonuclease activity. Removal of the 16S 3′-end appears to occur by an endonuclease cleavage event resulting in fragment VI (Figure 3C).
The 3′-end of the 16S rRNA is a hotspot for degradation mechanisms during quality control of ribosome biogenesis and starvation-induced ribosome degradation (56–58). One of the ribonucleases implicated in processing and quality control of the 16S 3′-terminus is the single-strand specific endoribonuclease YbeY (56,57,59). Importantly, YbeY and the exonuclease RNase R jointly degrade assembled 70S containing misprocessed or otherwise compromised 30S subunits in a highly interdependent manner (56). We therefore speculated that YbeY and RNase R may be involved in the generation of 16S fragments and/or the degradation of ribosomes containing fragmented 16S rRNA. To test this we generated rnr or ybeY deletion mutants both in a WT and ΔHF background and analysed total RNA extracted from five day old cultures of WT and ΔHF strains as well as the mutants lacking ybeY or rnr. Both the Δrnr and in particular the ΔHFΔrnr mutant exhibited highly increased overall accumulation of 16S rRNA fragments roughly corresponding to fragments I-VII found in ΔHF (Figure 5). Intriguingly, both fragments I and V appeared to be slightly larger than in WT and ΔHF (thus designated I* and V*), and could be detected with a probe complementary to region 1378–1396 (probe g/h; Figure 5B; Supplementary Figure S9B), indicating that they are at least 20 nucleotides longer than their counterpart in WT and ΔHF, and still contain h28 of the 16S rRNA (Figure 4F).
Figure 5.
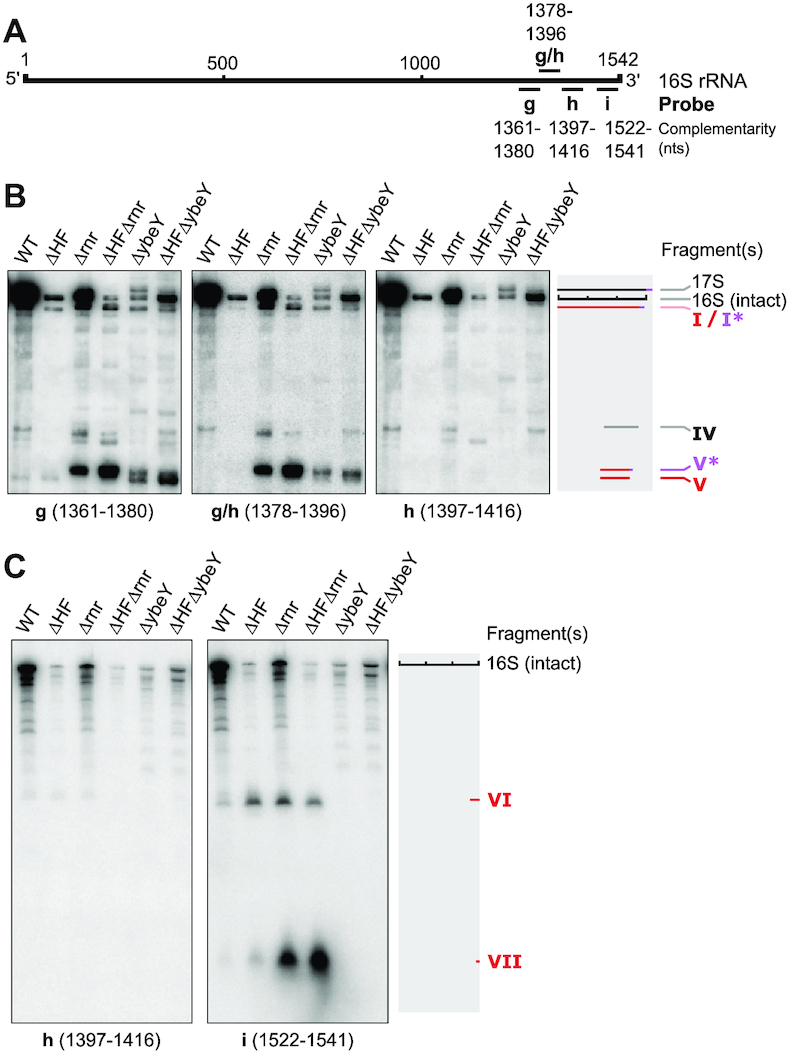
YbeY and RNase R are involved in generation and removal of accumulating fragments. (A) Schematic of 16S rRNA and DNA oligonucleotide probes used for Northern blot analysis in (B) and (C). For the sequences of the indicated probes see Supplementary Table S2. (B) Northern blot analysis of 16S rRNA fragments found in total RNA of hibernation factor mutants and RNase mutants after five days of starvation. The schematics on the right show detected fragments. Grey bars indicate fragments common to both WT and ΔHF, red bars indicate fragments only present in ΔHF. Pink bars indicate overhang in fragments I* and V* of Δrnr and ΔybeY mutants. For additional probes and magnified selected areas in the range of fragment I and fragment V see Supplementary Figure S9. (C) As described in (B). RNA was resolved on 6% urea–PAGE gels for detection of RNA in a lower size range. The schematic on the right shows fragments VI and VII mapped by urea–PAGE and Northern blot analysis. For additional probes see Supplementary Figure S9.
Deletion of ybeY led to the accumulation of a 17S rRNA fragment, likely due to the known 16S processing defect in strains lacking ybeY (56,57,59) (Figure 5B; Supplementary Figure S9B). Both ΔybeY and ΔHFΔybeY exhibited increased accumulation of fragments I–V. Fragments I and V were prevalently present as I* and V* but to some extent also as their short form (I/V). Strikingly, we could not detect fragments VI and VII, which approximately correspond to the 3′-terminus missing in fragments I and V (probe i; Figure 5C). In summary, deletion of rnr and ybeY strongly increased the accumulation of 16S rRNA fragments corresponding to fragments I–V observed in ΔHF–70S particles. Mutants lacking RNase R exclusively accumulate fragments I* and V*, which, in contrast to fragments I and V, contain the 16S region corresponding to h28. Most notably, deletion of ybeY leads to complete disappearance of both 3′-terminal fragments (VI and VII). In conclusion, YbeY is necessary for the generation of fragments corresponding to the 3′-end observed in the absence of HFs. Both YbeY and RNase R participate in the generation of fragments I and V, although degradation at the 3′-terminus can occur in the absence of either one of the RNases.
DISCUSSION
Here we have investigated the physiological role of hibernation factors. The triple hibernation factor mutant showed a significantly delayed regrowth upon nutrient replenishment after prolonged growth arrest (Figure 1C–F). Many bacteria, including E. coli, live in habitats with highly fluctuating nutrient availability. This feast-or-famine lifestyle requires rapid adaptability and a prolonged growth lag after nutrient replenishment constitutes a severe fitness defect in a competitive environment (10,60). A similar regrowth phenotype was observed in some gram-positive bacteria lacking the lHPF-mediated hibernation mechanism (10,14,42,61,62). We show here that in E. coli the two hibernation mechanisms, RMF–HPF-mediated 100S dimerization and RaiA–70S stabilization, appear to have overlapping functions, as only deletion of all three factors led to a significantly delayed regrowth phenotype (Figure 1C–F and Supplementary Figure S2C).
In several organisms, hibernation factors maintain viability during stationary phase and gram-positives S. aureus and M. smegmatis lacking their respective lhpf homolog exhibit decreased survival during prolonged starvation (18,63). In E. coli, reports have been inconclusive (28,54,55). We therefore investigated viability in a defined minimal medium leading to batch culture glucose starvation (MOPS MM) as well as abrupt growth arrest by transfer to PBS. Only moderate effects of deleting all three HFs could be observed, even after extended incubation times (Figure 1A and B). In addition, no direct correlation between the presence of hibernating ribosomes and cell viability was observed, given that 100S dimers in wild-type cells nearly disappeared after five days of starvation in MOPS MM, while the number of viable cells remained nearly constant for up to nine days (Figures 1A and 2B). Therefore, ribosome hibernation may not be needed to maintain viability even after extended periods of stationary phase.
The observed regrowth defect is likely caused by excessive breakdown of ribosomes: RNA degradation measured by the in vivo RNA stability assay showed an almost two-fold increased degradation in the ΔHF mutant over the WT during the first 16 h of growth arrest. Continued incubation led to more gradual degradation at similar rates in wild-type and ΔHF (Figure 2A). This is consistent with recently published results showing the degradation of rRNA in response to starvation for different nutrients, including glucose (7,8,51). Protection of ribosomes through hibernation could therefore be important during growth state transitions. This hypothesis is further supported by the transition-induced expression of hibernation factors (Supplementary Figure S1) and the high level of ribosome dimers during the first 16 h of incubation (Figure 2B).
We detected and mapped several distinct rRNA fragments (fragments I-VI) within assembled ΔHF–70S ribosomes (Figures 3 and 4). These results strongly suggests that ΔHF cells contain several subpopulations of 70S ribosomes, most importantly 70S containing intact 16S rRNA, 70S with 16S rRNA lacking the entire 3′-minor domain (fragment I, 1–1382) and 70S harbouring a split 16S consisting of fragment III (1–764) and fragment V (799–1382). In addition, a fraction of 70S containing fragment I or V appears to carry a remnant part of the missing 3′-minor domain (fragment VI).
16S rRNA contains multiple sites for inter-subunit bridges, which partly overlap with the missing regions in the fragmented 16S species. In particular h24, missing in the gap between fragment III and V (765–798) of ΔHF, contains conserved residues around loop 790, including bridge B2b, that are important for 30S assembly, ribosome function and 70S association (64–66). Similarly, the 3′-minor domain absent in fragments I and V comprises multiple residues forming inter-subunit bridges, including A1418 (bridge B5), A1483 (B3), G1486 (B3) and T1495 (B2a). Free 30 subunits originating from such 70S particles are highly unlikely to re-enter the translation cycle due to the lack of h24, h44 and h45 as well as the aSD sequence, which are essential for the formation of initiation complexes and translating 70S ribosomes (3).
Importantly, complementation of all three HFs from plasmid almost entirely rescued both the observed regrowth defect (Supplementary Figure S2B) and the fragmentation of 16S rRNA in ΔHF (Supplementary Figure S5). This further supports our conclusion that regrowth of ΔHF cells after starvation is delayed due to an overall decrease of intact 16S rRNA and the accumulation of translation-incompetent ribosomal particles containing fragmented 16S rRNA.
Intriguingly, degradation sites mapped uniquely to ΔHF–16S directly overlap with the interaction sites between hibernation factors RMF and HPF and 16S rRNA determined by cryo-EM (25). RMF directly interacts with the aSD sequence at nucleotides 1535–1537, and is enclosed by helices 28, 37, and 40. HPF binds at the A- and P-site at the interface between 30S and 50S subunit, contacting several 16S rRNA helices including h24 and 44 (Figure 4F–H). In addition, ribosomal protein S1 has been shown to adopt a distinct conformation in hibernating ribosomes, folding into the mRNA exit channel and interacting with nucleotides 1538–1540 (25). A tRNA present in the E-site may confer further protection (25). RaiA shares a binding site with HPF and is therefore likely to have a similar effect (23,26). The short C-terminal extension of RaiA reaches into the binding site of RMF (20,24–26). Thus, RaiA-bound 70S ribosomes may also be partially protected at the 3′-terminus of the 16S rRNA.
The observed fragmentation of rRNA indicates that excess ribosomes not protected by hibernation factors or ongoing translation are targeted by RNases. Recently it has been shown that the conserved endoribonuclease YbeY and RNase R jointly degrade assembled 70S ribosomes containing a compromised 30S subunit (56). When testing rRNA integrity in mutants lacking RNase R or YbeY we observed highly increased accumulation of 16S rRNA fragments both in a WT and ΔHF background, supporting a role of YbeY and RNase R in the degradation of 70S ribosomes containing fragmented 16S rRNA (Figure 5B and C; Supplementary Figure S9B and C). Importantly, the ybeY deletion mutants did not accumulate fragments VI and VII (Figure 5C). This indicates that YbeY generates these fragments, thereby producing 70S ribosomes lacking the 3′-end of the 16S rRNA. Consistently, YbeY interacts with ribosomal protein S11 and 16S rRNA h40 at the mRNA exit channel (67,68), which are occluded by RMF and S1 in the hibernating ribosome (25). Δrnr mutants exclusively accumulate extended versions of fragments I and V, that we designated I* and V*, respectively. Thus, RNase R is likely involved in the removal of this region (h28) after initial cleavage by YbeY, which is consistent with the intrinsic helicase activity of RNase R (69,70). ΔybeY mutants exhibit mainly fragments I* and V*, although fragments I and V are present to some extent, indicating reduced RNase R activity on h28 when YbeY is absent. This is in agreement with the proposed interdependence of YbeY and RNase R during quality control (56). Based on these observations we propose a novel degradation mechanism in which YbeY and RNase R take part in the initial fragmentation of the 16S 3′-terminus as well as in the subsequent degradation of the resulting compromised ribosomes (Figure 6). Hibernation factors prevent excessive degradation by this mechanism during starvation by preventing access of YbeY to crucial interaction and degradation regions on the ribosome. RMF, and S1 in its hibernation conformation occlude the 16S 3′-terminus, thereby prohibiting access of YbeY and RNase R.
Figure 6.
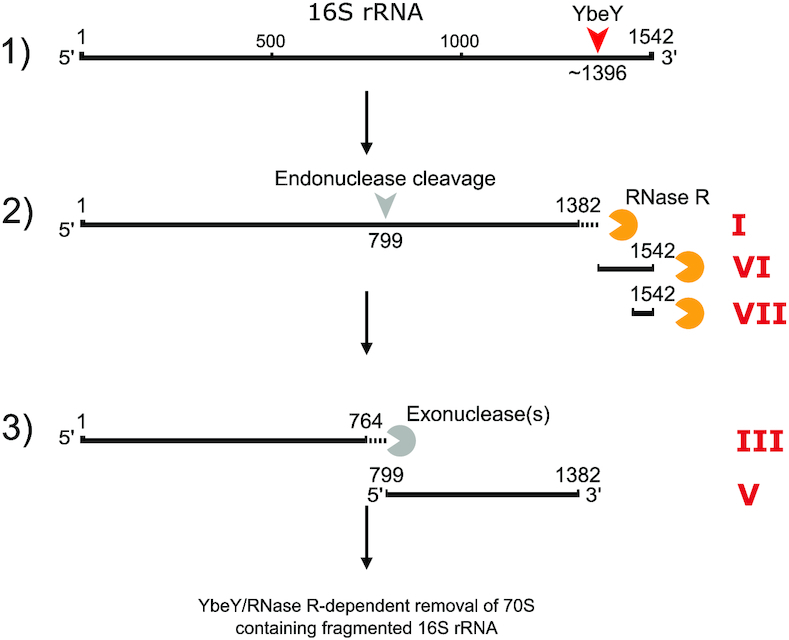
Model for degradation of unprotected 70S ribosomes during starvation. Degradation of 16S rRNA by YbeY and RNase R. 1) YbeY likely cleaves at or near position 1396 of the 16S rRNA in assembled 70S ribosomes, resulting in fragment I* (1 to approximately 1396) as well as fragment VI (and possibly VII). 2) RNase R trims fragment I* resulting in fragment I (1–1382), and degrades fragments VI and VII. An additional endonuclease cleavage takes place at position 799, generating fragments III* (1–799) and V. 3) One or several unidentified exonuclease(s) trim fragment III* resulting in fragment III (1–764). 70S ribosomes containing 30S subunits with fragmented 16S rRNA are likely removed by the concerted action of YbeY and RNase R.
It is important to note that ΔybeY mutants contained fragments similar to fragment I and V of ΔHF–70S, indicating that 3′-end removal by YbeY is not exclusive, and that in the absence of YbeY one or several exonucleases processively remove the 16S 3′-end. Intriguingly, a ybeY mutant exhibits increased accumulation of a fragment in the range of ΔHF–70S fragment I during stress (56) that is not present in double mutants of ybeY in combination with one of several exonucleases (56,59). Based on these results, different models of rRNA processing and quality control at the 3′-end of the 16S rRNA have been proposed (56,58,59). Our results are in agreement with the reported functional overlap between ribonucleases during rRNA processing and degradation (58,59,71).
It remains to be determined which ribonucleases are involved in the removal of h24 in ΔHF–70S and in the generation of the additional minor cleavage events (Supplementary Figure S8). We could not detect a fragment corresponding to the missing h24 region (probe c; Supplementary Figure S9C), indicating that endonuclease cleavage followed by exonuclease activity is responsible for the removal of h24, thus generating fragments III and V (Figure 6). The putative cleavage site between fragment II and IV (nucleotide 842) and an additional cleavage site at nucleotide 999 (Figure 4C and D; Supplementary Figure S7C and D) are not directly occluded by the second 30S subunit in the 100S dimer. However, they are in proximity to the 30S–30S interface, and may therefore still be protected to some extent by dimerization (Supplementary Figure S8). It is tempting to speculate that ribosomes are increasingly subjected to ribonuclease activity at exposed regions during prolonged starvation, that contribute to the destabilization and subsequent degradation of excess ribosomes.
It seems intuitive that when nutrients become scarce, bacteria not only downregulate the synthesis of macromolecules but also degrade excess components for recycling of building blocks. Fast growing cells can contain up to 70 000 ribosomes (6), and translation in stationary phase is reduced to ∼10% (72), thereby potentially freeing a large reservoir of nutrients in the form of excess ribosomes. A reduction of actively translating ribosomes during nutrient limitation can also contribute to the maintenance of elongation rates by reducing competition for ternary complexes (73). However, excessive degradation in response to nutrient stress would cause a prolonged growth lag when the stress is relieved.
To conclude, we propose ribosome hibernation as a regulatory mechanism that maintains a balance between the necessary recycling of ribosomal components and the upkeep of sufficient translational capacity. Hibernation factors RMF, HPF and RaiA appear to prevent excessive degradation by protecting 16S rRNA from both endo- and exonucleolytic attack in a novel pathway involving conserved ribonucleases YbeY and RNase R. This contributes to ribosome particle homeostasis and thereby efficient resumption of exponential growth after nutrient replenishment.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Sine L. Svenningsen for stimulating discussions and critical reading of the manuscript.
Author contributions: T.P., K.S.W. and M.A.S. designed experiments. T.P. performed the experiments. T.P., K.S.W. and M.A.S interpreted the data. T.P. wrote the initial manuscript. T.P., K.S.W. and M.A.S. edited the manuscript.
Contributor Information
Thomas Prossliner, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark.
Kenn Gerdes, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark.
Michael Askvad Sørensen, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark.
Kristoffer Skovbo Winther, Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Danish National Research Foundation [DNRF120]; Novo Nordisk Foundation. Funding for open access charge: Danish National Research Foundation [DNRF120]; Novo Nordisk Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Russell J.B., Cook G.M.. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol. Rev. 1995; 59:48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Szaflarski W., Nierhaus K.H.. Question 7: optimized energy consumption for protein synthesis. Orig. Life Evol. Biospheres. 2007; 37:423–428. [DOI] [PubMed] [Google Scholar]
- 3. Schmeing T.M., Ramakrishnan V.. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009; 461:1234–1242. [DOI] [PubMed] [Google Scholar]
- 4. Starosta A.L., Lassak J., Jung K., Wilson D.N.. The bacterial translation stress response. FEMS Microbiol. Rev. 2014; 38:1172–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prossliner T., Skovbo Winther K., Sørensen M.A., Gerdes K.. Ribosome hibernation. Annu. Rev. Genet. 2018; 52:321–348. [DOI] [PubMed] [Google Scholar]
- 6. Bremer H., Dennis P.P.. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus. 2008; 3:doi:10.1128/ecosal.5.2.3. [DOI] [PubMed] [Google Scholar]
- 7. Zundel M.A., Basturea G.N., Deutscher M.P.. Initiation of ribosome degradation during starvation in Escherichia coli. RNA. 2009; 15:977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fessler M., Gummesson B., Charbon G., Svenningsen S.L., Sørensen M.A.. Short-term kinetics of rRNA degradation in Escherichia coli upon starvation for carbon, amino acid, or phosphate. Mol. Microbiol. 2020; 113:951–963. [DOI] [PubMed] [Google Scholar]
- 9. Franken L.E., Oostergetel G.T., Pijning T., Puri P., Arkhipova V., Boekema E.J., Poolman B., Guskov A.. A general mechanism of ribosome dimerization revealed by single-particle cryo-electron microscopy. Nat. Commun. 2017; 8:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akanuma G., Kazo Y., Tagami K., Hiraoka H., Yano K., Suzuki S., Hanai R., Nanamiya H., Kato-Yamada Y., Kawamura F.. Ribosome dimerization is essential for the efficient regrowth of Bacillus subtilis. Microbiology. 2016; 162:448–458. [DOI] [PubMed] [Google Scholar]
- 11. Flygaard R.K., Boegholm N., Yusupov M., Jenner L.B.. Cryo-EM structure of the hibernating Thermus thermophilus 100S ribosome reveals a protein-mediated dimerization mechanism. Nat. Commun. 2018; 9:4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hood R.D., Higgins S.A., Flamholz A., Nichols R.J., Savage D.F.. The stringent response regulates adaptation to darkness in the cyanobacterium Synechococcus elongatus. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E4867–E4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kline B.C., McKay S.L., Tang W.W., Portnoy D.A.. The listeria monocytogenes hibernation-promoting factor is required for the formation of 100S ribosomes, optimal fitness, and pathogenesis. J. Bacteriol. 2015; 197:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puri P., Eckhardt T.H., Franken L.E., Fusetti F., Stuart M.C.A., Boekema E.J., Kuipers O.P., Kok J., Poolman B. Lactococcus lactis YfiA is necessary and sufficient for ribosome dimerization. Mol. Microbiol. 2014; 91:394–407. [DOI] [PubMed] [Google Scholar]
- 15. Ueta M., Wada C., Wada A.. Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells. 2010; 15:43–58. [DOI] [PubMed] [Google Scholar]
- 16. Ueta M., Wada C., Daifuku T., Sako Y., Bessho Y., Kitamura A., Ohniwa R.L., Morikawa K., Yoshida H., Kato T.et al.. Conservation of two distinct types of 100S ribosome in bacteria. Genes Cells. 2013; 18:554–574. [DOI] [PubMed] [Google Scholar]
- 17. Bieri P., Leibundgut M., Saurer M., Boehringer D., Ban N.. The complete structure of the chloroplast 70S ribosome in complex with translation factor pY. EMBO J. 2017; 36:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y., Sharma M.R., Koripella R.K., Yang Y., Kaushal P.S., Lin Q., Wade J.T., Gray T.A., Derbyshire K.M., Agrawal R.K.et al.. Zinc depletion induces ribosome hibernation in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:8191–8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sharma M.R., Dönhöfer A., Barat C., Marquez V., Datta P.P., Fucini P., Wilson D.N., Agrawal R.K.. PSRP1 is not a ribosomal protein, but a ribosome-binding factor that is recycled by the Ribosome-recycling Factor (RRF) and Elongation Factor G (EF-G). J. Biol. Chem. 2010; 285:4006–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maki Y., Yoshida H., Wada A.. Two proteins, YfiA and YhbH, associated with resting ribosomes in stationary phase Escherichia coli. Genes Cells. 2000; 5:965–974. [DOI] [PubMed] [Google Scholar]
- 21. Wada A., Igarashi K., Yoshimura S., Aimoto S., Ishihama Akira. Ribosome modulation factor: stationary growth phase-specific inhibitor of ribosome functions. Biochem. Biophys. Res. Commun. 1995; 214:410–417. [DOI] [PubMed] [Google Scholar]
- 22. Agafonov D.E., Kolb V.A., Nazimov I.V., Spirin A.S.. A protein residing at the subunit interface of the bacterial ribosome. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:12345–12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vila-Sanjurjo A., Schuwirth B.-S., Hau C.W., Cate J.H.D.. Structural basis for the control of translation initiation during stress. Nat. Struct. Mol. Biol. 2004; 11:1054–1059. [DOI] [PubMed] [Google Scholar]
- 24. Ueta M., Yoshida H., Wada C., Baba T., Mori H., Wada A.. Ribosome binding proteins YhbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli. Genes Cells. 2005; 10:1103–1112. [DOI] [PubMed] [Google Scholar]
- 25. Beckert B., Turk M., Czech A., Berninghausen O., Beckmann R., Ignatova Z., Plitzko J.M., Wilson D.N.. Structure of a hibernating 100S ribosome reveals an inactive conformation of the ribosomal protein S1. Nat. Microbiol. 2018; 3:1115–1121. [DOI] [PubMed] [Google Scholar]
- 26. Polikanov Y.S., Blaha G.M., Steitz T.A.. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012; 336:915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aiso T., Yoshida H., Wada A., Ohki R.. Modulation of mRNA stability participates in stationary-phase-specific expression of ribosome modulation factor. J. Bacteriol. 2005; 187:1951–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamagishi M., Matsushima H., Wada A., Sakagami M., Fujita N., Ishihama A.. Regulation of the Escherichia coli rmf gene encoding the ribosome modulation factor: growth phase-and growth rate-dependent control. EMBO J. 1993; 12:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Izutsu K., Wada A., Wada C.. Expression of ribosome modulation factor (RMF) in Escherichia coli requires ppGpp. Genes Cells. 2001; 6:665–676. [DOI] [PubMed] [Google Scholar]
- 30. Shimada T., Makinoshima H., Ogawa Y., Miki T., Maeda M., Ishihama A.. Classification and strength measurement of stationary-phase promoters by use of a newly developed promoter cloning vector. J. Bacteriol. 2004; 186:7112–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Durfee T., Hansen A.-M., Zhi H., Blattner F.R., Jin D.J.. Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol. 2008; 190:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Traxler M.F., Summers S.M., Nguyen H.-T., Zacharia V.M., Hightower G.A., Smith J.T., Conway T.. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol. Microbiol. 2008; 68:1128–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shimada T., Yoshida H., Ishihama A.. Involvement of cyclic AMP receptor protein in regulation of the rmf gene encoding the ribosome modulation factor in Escherichia coli. J. Bacteriol. 2013; 195:2212–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wada A. Growth phase coupled modulation of Escherichia coli ribosomes. Genes Cells. 1998; 3:203–208. [DOI] [PubMed] [Google Scholar]
- 35. Wada A., Yamazaki Y., Fujita N., Ishihama A.. Structure and probable genetic location of a ‘ribosome modulation factor’ associated with 100S ribosomes in stationary-phase Escherichia coli cells. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:2657–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCarthy B.J. Variations in bacterial ribosomes. Biochim. Biophys. Acta. 1960; 39:563–564. [Google Scholar]
- 37. El-Sharoud W.M., Niven G.W.. The activity of ribosome modulation factor during growth of Escherichia coli under acidic conditions. Arch. Microbiol. 2005; 184:18–24. [DOI] [PubMed] [Google Scholar]
- 38. Garay-Arroyo A., Colmenero-Flores J.M., Garciarrubio A., Covarrubias A.A.. Highly hydrophilic proteins in prokaryotes and eukaryotes are common during conditions of water deficit. J. Biol. Chem. 2000; 275:5668–5674. [DOI] [PubMed] [Google Scholar]
- 39. Niven G.W. Ribosome modulation factor protects Escherichia coli during heat stress, but this may not be dependent on ribosome dimerisation. Arch. Microbiol. 2004; 182:60–66. [DOI] [PubMed] [Google Scholar]
- 40. McKay S.L., Portnoy D.A.. Ribosome hibernation facilitates tolerance of stationary-phase bacteria to aminoglycosides. Antimicrob. Agents Chemother. 2015; 59:6992–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tkachenko A.G., Kashevarova N.M., Tyuleneva E.A., Shumkov M.S.. Stationary-phase genes upregulated by polyamines are responsible for the formation of Escherichia coli persister cells tolerant to netilmicin. FEMS Microbiol. Lett. 2017; 364:fnx084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beckert B., Abdelshahid M., Schäfer H., Steinchen W., Arenz S., Berninghausen O., Beckmann R., Bange G., Turgay K., Wilson D.N.. Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J. 2017; 36:2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matzov D., Aibara S., Basu A., Zimmerman E., Bashan A., Yap M.-N.F., Amunts A., Yonath A.E.. The cryo-EM structure of hibernating 100S ribosome dimer from pathogenic Staphylococcus aureus. Nat. Commun. 2017; 8:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khusainov I., Vicens Q., Ayupov R., Usachev K., Myasnikov A., Simonetti A., Validov S., Kieffer B., Yusupova G., Yusupov M.et al.. Structures and dynamics of hibernating ribosomes from Staphylococcus aureus mediated by intermolecular interactions of HPF. EMBO J. 2017; 36:2073–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neidhardt F.C., Bloch P.L., Smith D.F.. Culture medium for enterobacteria. J. Bacteriol. 1974; 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winther K.S., Roghanian M., Gerdes K.. Activation of the stringent response by loading of RelA-tRNA complexes at the ribosomal A-Site. Mol. Cell. 2018; 70:95–105. [DOI] [PubMed] [Google Scholar]
- 47. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning: A Laboratory Manual 2nd ed. 1989; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 48. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cherepanov P.P., Wackernagel W.. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995; 158:9–14. [DOI] [PubMed] [Google Scholar]
- 50. Pirt S.J. Principles of Microbe and Cell Cultivation. 1975; NY: Wiley. [Google Scholar]
- 51. Basturea G.N., Zundel M.A., Deutscher M.P.. Degradation of ribosomal RNA during starvation: comparison to quality control during steady-state growth and a role for RNase PH. RNA. 2011; 17:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cohen L., Kaplan R.. Accumulation of nucleotides by starved Escherichia coli cells as a probe for the involvement of ribonucleases in ribonucleic acid degradation. J. Bacteriol. 1977; 129:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Apirakaramwong A., Kashiwagi K., Raj V.S., Sakata K., Kakinuma Y., Ishihama A., Igarashi K.. Involvement of ppGpp, ribosome modulation factor, and stationary phase-specific sigma factor ςS in the decrease in cell viability caused by spermidine. Biochem. Biophys. Res. Commun. 1999; 264:643–647. [DOI] [PubMed] [Google Scholar]
- 54. Shcherbakova K., Nakayama H., Shimamoto N.. Role of 100S ribosomes in bacterial decay period. Genes Cells. 2015; 20:789–801. [DOI] [PubMed] [Google Scholar]
- 55. Bubunenko M., Baker T., Court D.L.. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J. Bacteriol. 2007; 189:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacob A.I., Köhrer C., Davies B.W., RajBhandary U.L., Walker G.C.. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell. 2013; 49:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Davies B.W., Köhrer C., Jacob A.I., Simmons L.A., Zhu J., Aleman L.M., RajBhandary U.L., Walker G.C.. Role of Escherichia coli YbeY, a highly conserved protein, in rRNA processing. Mol. Microbiol. 2010; 78:506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sulthana S., Basturea G.N., Deutscher M.P.. Elucidation of pathways of ribosomal RNA degradation: an essential role for RNase E. RNA. 2016; 22:1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ghosal A., Babu V.M.P., Walker G.C.. Elevated levels of Era GTPase improve growth, 16S rRNA processing, and 70S ribosome assembly of escherichia coli lacking highly conserved multifunctional YbeY endoribonuclease. J. Bacteriol. 2018; 200:e00278-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kolter R., Siegele D.A., Tormo A.. The stationary phase of the bacterial life cycle. Annu. Rev. Microbiol. 1993; 47:855–874. [DOI] [PubMed] [Google Scholar]
- 61. Akiyama T., Williamson K.S., Schaefer R., Pratt S., Chang C.B., Franklin M.J.. Resuscitation of Pseudomonas aeruginosa from dormancy requires hibernation promoting factor (PA4463) for ribosome preservation. Proc. Natl. Acad. Sci. 2017; 114:3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Galmozzi C.V., Florencio F.J., Muro-Pastor M.I.. The cyanobacterial ribosomal-associated protein LrtA is involved in post-stress survival in synechocystis sp. PCC 6803. PLoS One. 2016; 11:e0159346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Basu A., Yap M.-N.F.. Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic Acids Res. 2016; 44:4881–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee K., Varma S., SantaLucia J., Cunningham P.R.. In vivo determination of RNA Structure-Function relationships: analysis of the 790 loop in ribosomal RNA. J. Mol. Biol. 1997; 269:732–743. [DOI] [PubMed] [Google Scholar]
- 65. Pulk A., Maiväli Ü., Remme J.. Identification of nucleotides in E. coli 16S rRNA essential for ribosome subunit association. RNA. 2006; 12:790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Z., Culver G.M.. Differential assembly of 16S rRNA domains during 30S subunit formation. RNA. 2010; 16:1990–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McAteer S.P., Sy B.M., Wong J.L., Tollervey D., Gally D.L., Tree J.J.. Ribosome maturation by the endoribonuclease YbeY stabilizes a type 3 secretion system transcript required for virulence of enterohemorrhagic Escherichia coli. J. Biol. Chem. 2018; 293:9006–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vercruysse M., Köhrer C., Shen Y., Proulx S., Ghosal A., Davies B.W., RajBhandary U.L., Walker G.C.. Identification of YbeY-protein interactions involved in 16S rRNA maturation and stress regulation in Escherichia coli. mBio. 2016; 7:e01785-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hossain S.T., Malhotra A., Deutscher M.P.. The helicase activity of ribonuclease R is essential for efficient nuclease activity. J. Biol. Chem. 2015; 290:15697–15706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Awano N., Rajagopal V., Arbing M., Patel S., Hunt J., Inouye M., Phadtare S.. Escherichia coli RNase R has dual activities, helicase and RNase. J. Bacteriol. 2010; 192:1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sulthana S., Deutscher M.P.. Multiple exoribonucleases catalyze maturation of the 3′ terminus of 16S ribosomal RNA (rRNA). J. Biol. Chem. 2013; 288:12574–12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gefen O., Fridman O., Ronin I., Balaban N.Q.. Direct observation of single stationary-phase bacteria reveals a surprisingly long period of constant protein production activity. Proc. Natl. Acad. Sci. 2014; 111:556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dai X., Zhu M., Warren M., Balakrishnan R., Patsalo V., Okano H., Williamson J.R., Fredrick K., Wang Y.-P., Hwa T.. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat. Microbiol. 2016; 2:nmicrobiol2016231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.