

Abstract
In Staphylococcus aureus, de novo methionine biosynthesis is regulated by a unique hierarchical pathway involving stringent-response controlled CodY repression in combination with a T-box riboswitch and RNA decay. The T-box riboswitch residing in the 5′ untranslated region (met leader RNA) of the S. aureus metICFE-mdh operon controls downstream gene transcription upon interaction with uncharged methionyl-tRNA. met leader and metICFE-mdh (m)RNAs undergo RNase-mediated degradation in a process whose molecular details are poorly understood. Here we determined the secondary structure of the met leader RNA and found the element to harbor, beyond other conserved T-box riboswitch structural features, a terminator helix which is target for RNase III endoribonucleolytic cleavage. As the terminator is a thermodynamically highly stable structure, it also forms posttranscriptionally in met leader/ metICFE-mdh read-through transcripts. Cleavage by RNase III releases the met leader from metICFE-mdh mRNA and initiates RNase J-mediated degradation of the mRNA from the 5′-end. Of note, metICFE-mdh mRNA stability varies over the length of the transcript with a longer lifespan towards the 3′-end. The obtained data suggest that coordinated RNA decay represents another checkpoint in a complex regulatory network that adjusts costly methionine biosynthesis to current metabolic requirements.
Graphical Abstract
Graphical Abstract.
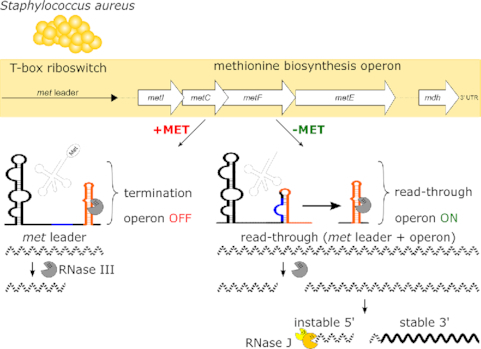
The MET-T-box riboswitch, controlling methionine biosynthesis in Staphylococcus aureus, is target of RNase III-mediated cleavage which in turn initiates met operon mRNA decay from the 5′-end involving RNase J.
INTRODUCTION
Methionine is the first N-terminal amino acid of nearly all newly synthesized proteins as well as the precursor for the vital methyl group donor S-adenosylmethionine (SAM), making the amino acid indispensable for all living organisms. Like many other bacteria, the versatile Gram-positive pathogen Staphylococcus aureus is capable of synthesizing methionine de novo when the amino acid becomes scarce. The methionine biosynthesis genes in staphylococci are organized in an operon whose size and arrangement is extremely rare among the Bacillales (1,2). Thus, except for metX, which is located elsewhere in the genome, the staphylococcal metICFE-mdh (met) operon encodes all enzymes required for de novo methionine biosynthesis, with the gene order matching the single synthesis steps (Figure 1A, B). Methionine is the amino acid with the highest synthesis costs regarding ATP consumption (3). Hence, the pathway is strictly controlled in various microorganisms to avoid methionine accumulation (1,2). Of note, staphylococci have limited metabolic capacities to reuse excess methionine as they lack methionine salvage and polyamine synthesis to recycle or redirect the amino acid into other pathways, further highlighting the need for effective control of de novo methionine biosynthesis in these organisms (4,5). We previously found that methionine biosynthesis in S. aureus indeed undergoes extremely tight regulation by a complex hierarchical network (6). Thus, the met operon is preceded by a 5′ untranslated region (met leader RNA) which represents a T-box riboswitch to control downstream met operon transcription. met leader RNA transcription itself is under control of the global transcription repressor CodY which in turn is sensitive to amino acid starvation and GTP levels in the cell via (p)ppGpp alarmone production through RelA/SpoT enzymes in a metabolic emergency circuit known as bacterial stringent response (7) (Figure 1A). The combination of stringent response-controlled CodY repressor activity with a T-box riboswitch to regulate gene expression is rare in bacteria, and staphylococcal methionine biosynthesis is further influenced by immediate RNase-mediated decay of met operon mRNA after transcription (6).
Figure 1.

(A) Schematic view of the organization of the S. aureus met operon including a CodY binding site and its 5′ UTR (met leader). (B) Methionine biosynthesis pathway in S. aureus, Me-THF, methylene-tetrahydrofolate. (C) Schematic of the binding interactions between tRNA and met leader RNA (T-box riboswitch). Left: system under methionine deprivation (‘−MET’), right: system under high intracellular methionine levels (‘+MET’). Base pairing of tRNA anticodon (‘CAU’) with specifier codon (‘AUG’) of met leader and of free 3′ tRNA end (‘ACCA’) with T-box (depicted in blue) sequence (‘UGGU’) is shown. Terminator sequence is highlighted in orange, gray ellipses symbolize RNA polymerase, ‘ON’: read-through into downstream genes, met mRNA transcription, ‘OFF’: premature transcription termination, no met mRNA transcription.
The crucial checkpoint in this hierarchical string of regulatory events, however, is the T-box riboswitch, which acts in a highly selective and strictly methionine-dependent manner to control met operon transcription. T-box riboswitches represent unique RNA-based bacterial transcription control platforms that interact with tRNAs as ligands to regulate downstream gene expression (see references (8,9) for recent reviews). In brief, T-box riboswitches are located in 5′ untranslated regions (UTRs) of genes where they (usually) control transcription by forming either a transcription terminator (system OFF: premature transcription termination) or an antiterminator (system ON: read-through into downstream genes). The ON/ OFF decision of a T-box riboswitch is being made upon binding of either a cognate uncharged or charged tRNA as ligand (Figure 1C). Thus, for example, when methionine is short in supply, methionyl-tRNAs become uncharged and the free 3′-CCA end of the tRNA basepairs with a nucleotide stretch within the 14-nucleotide T-box consensus sequence present in the terminator/antiterminator platform of the T-box riboswitch, resulting in antiterminator formation and ON switch of met operon transcription. Vice versa, if the 3′-end of the methionyl-tRNA is charged by methionine, no base pairing interaction with the T-box consensus sequence takes place and a terminator stem is formed, causing the RNA polymerase to fall off from its DNA template and to prematurely stop transcription without reading into downstream genes (system OFF) (Figure 1C). Thus, T-box riboswitches represent efficient systems to indirectly sense amino acid levels in the cell by using tRNA charging as a proxy. The tRNA 3′-CCA interaction with the T-box motif is conserved in all T-box riboswitches, while specificity for a distinct tRNA is usually conferred by interaction of the tRNA anticodon with a cognate codon present in the so-called specifier loop of the riboswitch (Figure 1C). However, other structural elements in the T-box riboswitch were shown to contribute to tRNA recognition and binding as well (10,11).
The T-box riboswitch preceding the S. aureus met operon, in the following referred to as met leader (RNA), has a number of interesting features. Thus, in comparison to most bacterial T-box riboswitches as well as to other known MET-T-box riboswitches (2,12), the met leader is larger in size (i.e. 440 versus ∼250 nt). The difference is mainly due to a long linker region of 226 nucleotides which spans between stem I at the 5′-end and the terminator/antiterminator platform at the 3′-end (6). The function of this exceptionally long linker is currently not known. Other striking features of the system were noticed with respect to met leader and met operon (m)RNA decay. Thus, upon activation of the S. aureus met operon T-box riboswitch, we previously found the metI mRNA to undergo rapid degradation immediately after transcription (6). Further, the met leader appeared as a distinct RNA species that was short-lived. Initial experiments suggested unexpected involvement of RNase III in met leader RNA decay, and of RNase J2 in met operon degradation (6). However, the molecular details and the biological significance of these findings remained elusive. Here, we determined the secondary structure of the met leader RNA in its OFF state and detected distinct structural elements that recruit RNase III to execute met leader RNA cleavage. We identified the exact cleavage site of RNase III within the terminator helix of the riboswitch and found the stability of liberated met operon mRNA to vary over the length of the transcript with the mRNA displaying a longer lifespan towards the 3′-end, which was also reflected by corresponding variations in protein amounts. We hypothesize that the uncommon, immediate physical separation of the met leader RNA by RNase III initiates targeted met operon mRNA decay by RNase J that represents an additional level in the hierarchical methionine biosynthesis control network to adapt enzyme expression to metabolic requirements.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The strains used in this study are listed in Table 1. Staphylococcus aureus strains were grown in chemically defined medium (CDM) with (CDM +MET) or without methionine (CDM –MET) described in Supplementary Material 1 when not otherwise stated. For strains carrying resistance genes on cloning vectors, antibiotics were used at the following concentrations for selection: 5 or 10 μg ml−1 erythromycin, 10 μg ml−1 chloramphenicol. Staphylococcus aureus conditional mutants of RNase J2 (rnjB::pMUTIN; ‘rnjBdepleted’) and RNase III (rnc::pMUTIN; ‘rncdepleted’), in which the respective RNase genes are under control of an IPTG-inducible promoter, were grown in absence of the inducing agent, rendering the strains functional RNase mutants.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| E. coli DC10B | E. coli DH10B Δdcm (cytosine methylase deficient) for direct cloning into S. aureus | (19) |
| S. aureus | ||
| RN4220 | Restriction-deficient derivative of S. aureus 8325-4, cloning host | (20) |
| Newman | Methicillin-sensitive isolate, NCTC 8178 | (21) |
| Newman 106 | RNase J2 mutant, rnjB::pMUTIN, Erm(5) | (6) |
| Newman 107 | RNase III mutant, rnc::pMUTIN, Erm(5) | (6) |
| Newman 217 | RNase Y mutant, rny::ermC, Erm(5) | (18) |
| PR01 (SA564RD ΔpyrFE) | hsdR type III mutant (restriction deficient), pyrimidine auxotroph | (22) |
| PR01 ΔRNaseJ1 | RNase J1 mutant, ΔrnjA, hsdR type III mutant, pyrimidine auxotroph | (22) |
| PR01 ΔRNaseJ2 | RNase J2 mutant, ΔrnjB, hsdR type III mutant, pyrimidine auxotroph | (22) |
| PR01 ΔRNaseJ1/J2 | RNase J1/J2 double mutant, ΔrnjA::ermC, ΔrnjB, hsdR type III mutant, pyrimidine auxotroph, Erm(10) | (22) |
| HG001 | Derivative of S. aureus 8325-4, rsbU restored RN1, agr positive | (23,24) |
| HG001 Δrnc | RNase III mutant, Δrnc region::cat86 | (25) |
| Newman ΔAntiTer&Ter met leader | ΔAntiTer&Ter met leader mutant | This work |
| Newman Ter_destab met leader | Ter_destab met leader mutant | This work |
| Newman Ter_mutated_1 met leader | Ter_mutated_1 met leader mutant | This work |
| Newman Ter_mutated_2 met leader | Ter_mutated_2 met leader mutant | This work |
| Newman Ter_mutated_3 met leader | Ter_mutated_3 met leader mutant | This work |
| Newman Ter_mutated_4 met leader | Ter_mutated_4 met leader mutant | This work |
| Plasmids | ||
| pBASE6 | ori pE194ts (temperature-sensitive); ori ColE1; G+/G- shuttle; CmR, AmpR; antisense secY under Pxyl/tet promoter | (14) |
| pBASE_met leader+1kb flanking (pFW001) | pBASE6 with wild type met leader sequence and 1 kb flanking regions | This work |
| pBASE_ΔAntiTer&Ter | pFW001 with ΔAntiTer&Ter met leader | This work |
| pBASE_Ter_destab | pFW001 with Ter_destab met leader | This work |
| pGEM-T-easy+Ter_mutated_1 | Ter_mutated_1 met leader | This work |
| pBASE6_Ter_mutated_1 | pFW001 with Ter_mutated_1 met leader | This work |
| pBASE_Ter_mutated_2 | pFW001 with Ter_mutated_2 met leader | This work |
| pBASE_Ter_mutated_3 | pFW001 with Ter_mutated_3 met leader | This work |
| pBASE_Ter_mutated_4 | pFW001 with Ter_mutated_4 met leader | This work |
| pEB01 | ori ColE1 (E. coli), ori pT181 (Gram+), bla (AmpR), cat (CmR), MCS to express gene of interest under native promoter, (pCN47 vector with ermC exchanged by cat) | (26) |
| pEB01-met leader-metI | met leader sequence from –35 signal to 5′ region of metI (215 nt) | This work |
| pJC1_tRNAi_deletion | pJC1 with met leader, yfp and cI repressor, deletion of tRNAi gene | This work |
Bacteria from overnight cultures (CDM +MET) were diluted to an initial optical density at 600 nm (OD600) of 0.05 in fresh medium and grown with shaking at 180 rpm at 37°C to mid-exponential growth phase. For methionine-deprived (−MET) conditions bacteria were then washed twice with 1× PBS, resuspended in CDM -MET and incubated for 2 h with shaking at 180 rpm at 37°C.
For rifampicin assays under methionine-deprived conditions, bacteria from overnight cultures were washed twice with 1× PBS, resuspended in 2× basic medium (25 mM Na2HPO4, 20 mM KH2PO4, 3.3 mM MgSO4, 18.5 mM NH4Cl, 17 mM NaCl) and diluted to an initial OD600 of 0.05 in CDM –MET. Bacteria were grown with shaking at 180 rpm at 37°C to mid-exponential phase.
For protein half-life and synthesis rate determinations bacteria from overnight cultures (CDM +MET) were washed twice with 1x PBS, resuspended in 2× basic medium and diluted in CDM –MET to an initial OD600 of 0.05. Bacteria were grown with shaking at 180 rpm at 37°C to mid-exponential growth phase. Then bacteria were washed once with CDM –MET without arginine and lysine and were resuspended in CDM –MET with 1 mM ‘heavy’ arginine (l-arginine HCl (13C,15N), Silantes GmbH, #201603902) and ‘heavy’ lysine (l-lysine HCl (13C, 15N), Silantes GmbH, #211603902) (instead of the normal ‘light’ (12C, 14N) arginine and lysine), t0 sample was taken and methionine or rifampicin was added to a final concentration of 10 mM and 100 μg ml−1, respectively. Bacteria were incubated with shaking at 180 rpm at 37°C. Samples were taken 15, 30, 60 and 240 min after methionine addition (half-life determination) and 15 and 30 min after rifampicin addition (synthesis rate determination), respectively. Samples were put on ice and split in two. One half was immediately pelleted, 1× washed with ice-cold TE-buffer (10 mM Tris (pH 8.0), 1 mM EDTA) supplemented with complete™, EDTA-free Protease Inhibitor Cocktail (Roche, #04693132001) according to manufacturer's instructions, pellet was snap-frozen in liquid nitrogen and stored at −80°C until further processing described under ‘Sample Preparation for Proteomics’. The other half of the sample was treated as described for ‘Preparation of total RNA’. In addition, the experiment was performed vice versa (switch from ‘heavy’ arginine and lysine to ‘light’ arginine and lysine containing CDM –MET). The number of biological replicates per time point was 4 (2× ‘light’ > ‘heavy’, 2× ‘heavy’ > ‘light’). For the protein quantification experiment, bacteria were resuspended in light medium after washing (no shifting), the t0 sample was taken and processed as described above.
Plasmid and strain construction
All plasmids and oligonucleotides used for this work are listed in Tables 1 and 2, respectively. Details on mutagenesis, cloning and oligonucleotides used for that purpose can be found in Supplementary Method 1 and Tables S1 and S2.
Table 2.
Oligonucleotides
| Oligonucleotides | |||
|---|---|---|---|
| Purpose | Template | Name | Sequence |
| cRACE | |||
| met leader | - | FW144 | CACTCCAAGGCCATTTTCAA |
| - | FW145 | GTGATAATTGTTCAGTAAGCAT | |
| metI | - | FW156 | CCTGTCGATTGTCCTAGTTT |
| - | FW157 | CACGTACTAAAAATCCTACA | |
| 5′-RACE | |||
| 5′ RACE RNA adapter | CUAGUACUCCGGUAUUGCGGUACCCUUGUACGCCUGUUUUAUA | ||
| 5′ RACE RNA adapter primer | GTATTGCGGTACCCTTGT | ||
| metI | - | FW200 | CGTGGCTGAATGTAAGACTATA |
| - | Sa_lgsm02-Lext-Rev | TCAGCACCTTCTGCTAGTGGT | |
| metF | - | FW149 | GCGATGTTGAAGTGTGTACGTTT |
| - | FW161 | CGGTGTGATAATGTATAAACCATT | |
| ivt for in-line probing | |||
| T7 met leader | pJC1_tRNAi_deletion | FW088 | TTAACTAATACGACTCACTATAGGGTCTTATAACAGTTTAATGAAACGTAAAC |
| Sa.R_met-sRNA | GAAAAAATAAAAAAAGCTTCCGTCCTTCG | ||
| T7 short met leader | pJC1_tRNAi_deletion | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| Sa.R_met-sRNA | GAAAAAATAAAAAAAGCTTCCGTCCTTCG | ||
| T7 short met leader Ter_destab | pBASE_Ter_destab | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| FW159 | GAAAAAATAAAAAAAGAGTCGTTGAATCGTCA | ||
| T7 short met leader Ter_mutated_1 | 2.2 kb overlap PCR met leader Ter_mutated_1 fragment | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| FW169 | GAAAAAATAAAAAAAGGATCGTTGCCTCGT | ||
| T7 short met leader Ter_mutated_2 | pBASE_Ter_mutated_2 | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| FW211 | GAAAAAATAAAAAAAGCTTCCGTGCTTCGT | ||
| T7 short met leader Ter_mutated_3 | pBASE_Ter_mutated_3 | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| FW212 | GAAAAAATAAAAAAAGCTTCCGTGCCTCGT | ||
| T7 short met leader Ter_mutated_4 | pBASE_Ter_mutated_4 | FW130 | TTAACTAATACGACTCACTATAGGGATTCTTTACGCACGATTTTTTGTT |
| FW213 | GAAAAAATAAAAAAAGCTTCCTGGCTTCGT | ||
| dsDNA probes | |||
| met leader | Newman | Sa_lgsm02_Fow | ATGTATTCTAAATGAGTCAGACAACC |
| Sa_lgsm02_Rev | CCGTCCTTCGTACCCGAATGA | ||
| metI | Newman | Sa_lgsm02-Lext_Fow | ACATCAAGTGGAATGTCAGCCA |
| Sa_0431_R | CTATTGGTGAAAGTGTTGCGCCA | ||
| metI 5′ | Newman | FW137 | |
| Sa_0431-R_RT | CGAATGATGCAATACCATGCTCA | ||
| metC | Newman | Sa_0430_F | GCTCGAACAAATCGAGGGTGCCA |
| Sa_0430_R | ACGAAAGCCAATAACGGCAC | ||
| metC 3′ | Newman | FW140 | GCCTCCTTTAATGCGTATTTGAT |
| FW141 | GCTGATGAGTCTAAAGCACAA | ||
| metF | Newman | Sa_0429_F | ACAACTCGTTCAATGTGGTGC |
| Sa_0429_R | TCTGCGAGTGTTACCGCATCTAC | ||
| metF 3′ | Newman | FW138 | CTCCTTGTGAGCAGTAATAGATT |
| FW139 | GGTATTAACACTGACGGTGAT | ||
| metE | Newman | Sa_metE_F | TGATGGTCGTAATGTATGGGCA |
| Sa_metE_R | CGTTTGTTCTTCCAATCTGCACG | ||
| mdh | Newman | FW051 | GCAACTTGGGTTGATTTAACGCAT |
| FW052 | GCCACGAGTTGGTAATTGATCTA | ||
| ssDNA probes | |||
| 16S rRNA | - | 16S_rDNA_R | TACGGCTACCTTGTTACGACTT |
| 5S rRNA | - | 5SrRNA | CAGTCCGACTACCATCGGCG |
Generation of markerless chromosomally encoded met leader mutants
Chromosomal integration of mutated met leader sequences was accomplished via allelic replacement with inducible counterselection adapted from Bae & Schneewind using the pBASE6 S. aureus/Escherichia coli shuttle vector (13,14).
Preparation of total RNA
Total RNA of bacteria was isolated as described previously (15). RNA was precipitated with 4.67× volume ethanol/3 M sodium acetate pH 6.5 (30:1 mix) at −20°C overnight or 1× volume isopropanol and 0.1× volume 3 M sodium acetate pH 6.5 for 10 min at room temperature. Pelleted RNA was washed with 70% ethanol and solved in RNase‐free ddH2O. The quantity was measured with the NanoDrop system (Thermo Scientific, ND‐2000). RNA was stored at −80°C.
Northern blot analysis
Unless otherwise specified, 10 μg of total RNA was size separated on 5% denaturing polyacrylamide gels with 7 M urea according to standard procedures. After gel electrophoresis, RNA was transferred onto a nylon membrane (GE Healthcare, Hybond‐XL, #RPN203S) using the wet electroblotting technique. RNA was then permanently cross‐linked to the membrane by UV light (2 × 120 mJ). For larger mRNA fragments, denaturing 1.2% agarose gels with 1.11% formaldehyde were run in 1x MOPS (3‐morpholinopropane‐1‐sulfonic acid) buffer. Blotting of RNA onto the nylon membrane after electrophoresis was done by classical capillary force technique with 10× SSC buffer (1.5 M NaCl, 150 mM tri-sodium citrate, pH 7.0) overnight or for 2.5 h using the rapid downward transfer method (TurboBlotter™ System, Whatman). RNA was permanently cross‐linked by UV light. Membranes were used for hybridization with sequence-specific, radioactively labeled probes. For dsDNA probes, PCR was performed with primer pairs listed in Table 2, and subsequently labeled using Klenow fragment (GE Healthcare, Amersham Megaprime DNA Labeling System, #RPN1606) and [α32P]‐dCTP (3000 Ci/mmol, 10 μCi/μl, Hartmann Analytic GmbH, Germany, #SRP-205). Oligonucleotides (ssDNA) listed in Table 2 were 5′‐end labeled using T4 polynucleotide kinase (PNK, Thermo Scientific #EK0031) and [γ32P]‐ATP (6000 Ci/mmol, 10 μCi/μl, Hartmann Analytic GmbH, Germany, #SRP-501). Hybridization of probes was done in Roti®‐Hybri‐Quick buffer (Carl Roth GmbH) overnight at 65°C with dsDNA and at 42°C with ssDNA probes. RNA sizes were estimated using the pUC mix marker 8 (Fermentas) as marker for small fragments (100–1000 nucleotides) and in vitro transcribed RNAs as ruler for large fragments (1–7.2 kb). Radioactive signals were detected by storage phosphor screens (BAS‐IP SR 2040 E) and the Typhoon™ FLA 7000 laser scanner (GE Healthcare).
Rifampicin assay for determination of transcript stability
Bacteria were grown to mid-exponential phase as described above. Then rifampicin to a final concentration of 100 μg ml−1 was added to the culture. Before (0) and after 0.5, 2, 4, 8, 16 and 32 min of rifampicin exposure, RNA was isolated and Northern blot analyses were performed as described in Supplementary Method 2. Quantification of bands was achieved using the Fiji software (16).
In-line probing
DNA templates that contain the T7 promoter sequence for in vitro transcription using the MEGAscript T7 Kit (Ambion, #AM1333) were generated by PCR. Oligos and DNA templates used to generate the individual T7 templates are listed in Table 2. Details about in vitro T7 transcription and in-line probing are given in Supplementary Method 3 and Table S3.
Rapid amplification of cDNA ends from circularized RNA (cRACE)
Rapid amplification of cDNA ends from circularized RNA (cRACE) was used to determine 5′- and 3′-ends of the met leader transcript as described previously (17). Briefly, DNA-free total RNA was treated with RNA 5′ pyrophosphohydrolase (RppH, NEB, #M0356S) to convert 5′ triphosphate RNA into 5′ monophosphate RNA, to allow for subsequent circularization of transcripts by ligation of 5′- and 3′-ends using T4 RNA ligase (NEB, #M0202). After phenol/chloroform extraction and ethanol precipitation, the circularized RNA was subjected to reverse transcription using a gene-specific primer oriented towards the 5′-end (FW144_cRACE_metleader_fw), followed by PCR amplification (FW144_cRACE_metleader_fw and FW145_cRACE_metleader_rv) (Table 2), cloning of the PCR products into pGEM®-T Easy Vector System I (Promega, #A1360) and transformation into E. coli DC10B. 5′-/ 3′-end fusions were analyzed by nucleotide sequencing and mapped to the met leader of S. aureus strain Newman as reference sequence using the CLC Main Workbench analysis tool (Version 6.6.1; www.clcbio.com).
5′ Rapid amplification of cDNA ends (5′ RACE)
5′ Rapid amplification of cDNA ends (5′ RACE) was performed as described previously (18), but omitting the 5′-triphosphate removal step (by RNA 5′ pyrophosphohydrolase treatment) to exclusively detect processed RNA molecules. Briefly, total RNA was isolated as described above. A specific RNA 5′ adapter (Table 2) was then ligated to the RNA. After phenol/chloroform extraction and ethanol precipitation, the RNA was subjected to reverse transcription using gene-specific primers (for metI: FW200, for metF: FW161) (Table 2), followed by PCR amplification (for metI: Sa_lgsm02-Lext-Rev and 5′ RACE RNA adapter primer, for metF: FW149 and 5′ RACE RNA adapter primer) (Table 2) and cloning of the PCR products into pCR-XL-2-TOPO vector (Thermo Fisher Scientific, #K8050) and transformation into E. coli DC10B. 5′- ends were analyzed by nucleotide sequencing and mapped to the met operon of S. aureus strain Newman as reference sequence using the CLC Main Workbench analysis tool (Version 6.6.1; www.clcbio.com).
Sample preparation for proteomics
Cell pellets were resuspended in TE-buffer (50 mM Tris–HCl pH 7.25, 10 mM EDTA) and disrupted with 0.1 mm glass beads and a homogenizer (4 cycles of 30 sec at 6.5 m/s). Between each cycle, samples were placed on ice for 5 min. Glass beads were removed by centrifugation and lysates were subsequently transferred to a new reaction tube. Protein concentration was determined with Roti-Nanoquant (Carl Roth GmbH) according to the manufacturer's protocol. For in solution digest 100 μg protein were reduced with 500 mM tris(2-carboxyethyl)phosphine for 45 min at 65°C and subsequently alkylated with 500 mM iodoacetamide in the dark for 15 min at room temperature before digestion was performed with trypsin (protein-to-enzyme-ratio 200:1) at 37°C for 14 h. Digestion was stopped by lowering the pH to 2 by adding trifluoroacetic acid before samples were desalted via C18 columns (Millipore ZipTips) according to the manufacturer's protocol.
LC–MS/MS measurements
Tryptic peptides were separated by liquid chromatography (LC) and measured online by ESI-mass spectrometry. LC–MS/MS analyses were performed with an EASY-nLC 1200 coupled to an Orbitrap Orbitrap Elite for the half-life and synthesis rate determination or an Orbitrap Velos Pro for the protein quantification (Thermo Fisher Scientific). Peptides were loaded on a self-made analytical column (3 μm particles, Dr Maisch GmbH, OD 360 μm, ID 100 μm, length 20 cm) and eluted by a binary nonlinear gradient of 5–53% acetonitrile in 0.1% acetic acid over 180 min with a flow rate of 300 nl min−1. For MS analysis a full scan in the Orbitrap (m/z 300–1700) with a resolution of 60 000 was followed by CID MS/MS experiments of the twenty most abundant precursor ions acquired in the linear ion trap.
Proteome data processing
Relative protein quantification was achieved using the MaxQuant software (version 1.6.1.0.) (27) and the Andromeda plug-in (28). The *.raw files were searched against a S. aureus strain Newman database (downloaded from Uniprot at 15 July 2018, 2584 entries). Additionally, MaxQuant's generic contamination list was included during the search. Database search was performed with following parameters: digestion mode, trypsin/P with up to two missed cleavages; variable modifications, methionine oxidation and acetylation of protein N-termini, a maximal number of five modifications per peptide and activated ‘match-between-runs’ feature. The false discovery rates of peptide spectrum match and protein were set to 0.01. Only unique peptides were used for protein quantification. The identified proteins from MaxQuant output files were filtered for contaminants, only identified by site and reverse hits with the Perseus software (v. 1.6.1.3). Proteins were accepted if at least two unique peptides could be identified in at least two of the four biological replicates. The LFQ-values were log2-transformed, exported and used for statistical analysis using TM4 (29). Statistical significance required a P-value <0.01 in an ANOVA applying standard Bonferroni correction. For comparison of protein amounts within one sample, the intensities of all identified peptides were normalized by dividing the respective intensity by the median of all peptide intensities of the same condition. According to Silva et al. (30) the normalized intensities of the three most abundant peptides of a proteins were summed to obtain the final quantitative value.
RESULTS
met leader RNA secondary structure
met leader RNA overall organization
To get an insight into the structural constraints of S. aureus met operon T-box riboswitch function, we determined the secondary structure of the met leader RNA in its OFF- state (i.e. without tRNA ligand) by in-line structural probing (31) (Figure 2A). The approach was augmented by computational folding predictions using the mfold algorithm (32), with experimental data being entered into the program as constraints to eventually build the met leader RNA secondary structure model (Figure 2B). Structural probing revealed that the met leader RNA harbors typical elements necessary for T-box riboswitch function such as stem I, stem II, stem IIA/B pseudoknot and stem III as well as a terminator helix (Figure 2). In contrast to other T-box riboswitches, however, the met leader RNA was found to harbor three additional stem–loops (designated L I–III) that span in a region between stem IIA/B and stem III (Figure 2). Also, a number of minor aberrations within conserved riboswitch elements were detected.
Figure 2.

Secondary structure of met leader. (A) In-line probing PAA gels used to build 2D model. Nomenclature of structural motifs according to (9). Additional stems within linker region are numbered L I to L III. Position of guanosines (G) is given on the left of each gel. Structural motifs are specified on right side of each gel. C: control reaction; untreated RNA, OH: alkaline hydrolysis reaction; ladder, T1: RNase T1-treated RNA; G-specific ladder, IL: in-line reaction. (B) 2D structure model of met leader in its OFF state, predicted antiterminator 2D structure is shown above. Color of dots (black, gray, white) next to nucleotides indicates cleavage intensity (high, medium, low) detected in (A). Interacting nucleotides of specifier loop are shown in yellow, base pairing nucleotides of AG-bulge and apical loop of stem I in orange, specifier codon in green, potentially interacting nucleotides of stem IIA/B pseudoknot in light blue, T-box sequence in dark blue. Linker region between stem I and T-box sequence and insertion region of stem L I–III are indicated by horizontal bars. Nucleotides of terminator base pairing with T-box sequence in antiterminator conformation are marked by asterisks (*). G 205 highlighted in gray indicates 5′ end of short met leader RNA used for in-line probing.
Stem I structural elements
Stem I (nt 27–129) harbors at its base a putative kink-turn element that consists of a typical asymmetric loop flanked by two short helices (Figure 2B). The overall arrangement of the met leader kink-turn matches the structures conserved in many T- and S-box riboswitches (9). Other than those elements, however, the kink-turn loop of the met leader lacks a 5′-GA-3′ motif that normally flanks the loop at either side (33). Immediately distal from the kink-turn, a canonical specifier loop was identified (nt 42–47 and nt 110–117) which harbors the AUG specifier triplet that is supposed to interact with the anticodon of the cognate met-tRNA (nt 114–116, marked in green in Figure 2B). Immediately 3′ of the AUG specifier codon a conserved purine (G117) is present which, in other T-box riboswitches, was shown to stabilize the specifier codon/ anti-codon interaction (10). Also, the specifier loop of the met leader harbors two conserved sequence motifs (marked in yellow in Figure 2B) that are known to undergo weak non-canonical base pairing (9) which is reflected by partial protection of these sequence stretches from degradation in the in-line probing approach (Figure 2A, gels #1–3). Finally, the distal region of stem I carries an AG-bulge (nt 63–71) with a 5′-AGAGA-3′ motif (nt 67–71) and the terminal loop (nt 78–88) with a 5′-GCUGAGA-3′ motif (nt 82–88) (both marked in orange in Figure 2B). These two sequence stretches are highly conserved in T-box riboswitches and were previously shown to undergo base-pairing with each other to form a platform for stem I/tRNA elbow interaction (10,34). In agreement with this function, the regions were found to be protected from degradation during in-line structural probing (Figure 2A, gels # 1 & 2).
Stem II and stem IIA/B pseudoknot
Next to stem I, structural probing demonstrated presence of stem II and a stem IIA/B pseudoknot which are both conserved features in most T-box riboswitches (9). Of note, stem II (nt 133–145) of the met leader is unusually short and lacks an S-turn internal loop typical for other T-box riboswitches (Figure 2B) (9). Stem II is followed by a canonical stem IIA/B (nt 150–163) whose apical loop sequence (nt 156–158) basepairs with a downstream single-stranded stretch (nt 169–171) thereby forming a pseudoknot that is found in many T-box riboswitches (Figure 2A, gel # 3; Figure 2B, nucleotides marked in light blue).
Insertion between stem IIA/B and stem III
The most striking and characteristic feature of the met leader is the presence of 150 additional nucleotides (nt 174–323) inserted between the stem IIA/B pseudoknot and stem III (Figure 2B). Structural probing revealed this region to harbor three stem-loops, designated stem L I (nt 174–205), L II (nt 217–283) and L III (nt 293–323) (Figure 2A, gels # 4 & 5). Stem L I is a 10 bp-long helix, carrying an internal 3+2 asymmetric loop (nt 180–182 and 198–199) and a 7-nt apical loop (nt 187–193). Stem L II represents a 22 nt-long helix, with a proximal 4+3 (nt 224–227 and 274–276) and a distal 3+2 (nt 237–239 and 263–264) asymmetric internal loop as well as an A/C mismatch pair (nt 232 & 269). The apical loop of stem L II consists of 11 nt (nt 246–256). Finally, stem L III is a 10 bp-long helix with an 11-nt apical loop (nt 303–313) (Figure 2B). The L I–III elements are absent in all other T-box riboswitches analyzed so far, and presence of this region expands the size of the linker region between stem I and the T-box sequence to 226 nt.
Stem III, T-box and terminator stem
Stem III is a highly conserved T-box riboswitch structure of variable sequence and length (9). Stem III of the met leader RNA (nt 330–351) consists of a 9-bp stem and an apical loop of four nucleotides (nt 339–342) (Figure 2A, gel # 5). The structure precedes the T-box sequence (nt 357–370, dark blue in Figure 2B) which forms (in the OFF-state of the met leader RNA) a small bulge (nt 361–365) that is stabilized at its base by two consecutive G/C base pairs (Figure 2A, gel # 5; Figure 2B). Downstream of the T-box motif a terminator stem is present which is an essential structural feature of all riboswitches that control downstream genes via transcription termination (Figure 2A gel #5, Figure 2B). The T-box transcription terminator of the met leader RNA consists of a 23-bp stem (nt 377–427) with an U/C mismatch pair (nt 392 & 412) and a small 5-bp apical loop (nt 400–404). Of note, the size of the met leader terminator stem is longer than most other known T-box riboswitch terminators (12). Also, due to its length and sequence, the met leader terminator is a thermodynamically highly stable structure (free energy ΔG = –32.60 kcal/mol). Similar to other T-box systems, the met leader terminator harbors seven (conserved) nucleotides (nt 384–390, marked by asterisks in Figure 2B) that are capable to basepair with the T-box sequence to support antiterminator formation once the system is ON (Figure 2B, top). Thus, in presence of uncharged met-tRNA, the proximal 5′-part of the terminator stem and the T-box sequence become part of the antiterminator, thereby forming the characteristic T-box bulge (nt 361–367) which is prone to interact with the free 3′-CCA end of tRNA ligands (Figure 2B, top; Figure 1C).
met leader RNA cleavage
Endonucleolytic cleavage releases the met leader RNA from met operon mRNA
We previously reported that also under methionine-rich conditions basic transcription of the met leader takes place as a stand-alone transcript of 440 nucleotides in size resulting from Rho-independent transcription termination (Figure 3A, first lane). On the contrary, no transcription of the met operon occurs (Figure 3A, second lane and (6)). Upon methionine depletion, met leader transcription is usually further induced with transcriptional read-through into the downstream met operon (6). In these read-through transcripts, we actually expected the met leader RNA to be attached to the met operon mRNA, which would give rise to a long transcript of ∼7.6 kb in size. However, we were never able to detect stable transcripts of this size (6). Instead, Northern blot hybridization of total RNA with a met leader-specific probe using 1.2% agarose gels shows a band whose size (∼400 nt) corresponds to that of the met leader RNA alone (Figure 3A, third lane). Re-hybridization of the same blot with a probe from the 3′-end of the met operon mRNA (i.e.metE) detected two signals. The faint 7.2 kb band in Figure 3A (fourth lane) would match the size of the met operon mRNA without the met leader, suggesting release of the latter by endonucleolytic cleavage, while the strong 4 kb 3′ met operon signal is unexpectedly small (for reasons see paragraphs below) (Figure 3A). Rifampicin stability assays (using 5% PAA gels for higher resolution) identified two met leader-specific RNA species, a faint 440-nt band and a more abundant 390-nt fragment, which are both short-lived and become rapidly degraded (Figure 3B). To substantiate the hypothesis of met leader RNA cleavage and release, we performed 5′-RACE with a metI-specific primer and native total RNA to determine the 5′-end of met operon mRNA (see Materials and Methods for details). Successful PCR amplification of met operon-specific fragments by this approach confirmed that, upon methionine depletion, transcriptional read-through occurs from the met leader into the met operon (data not shown). Nucleotide sequencing of the amplicons revealed that the 5′-ends of 50% (n = 12) of met operon transcripts are located at position 418 in the 3′-portion of the met leader (Figure 3C and Supplementary Table S4). We conclude from this experiment that the read-through transcripts must have undergone endonucleolytic cleavage, as the 5′-RACE data were obtained with (RNA 5′ pyrophosphohydrolase, RppH) untreated total RNA, indicating presence of 5′ monophosphate ends which are typically generated upon RNA cleavage events in native RNA molecules. Cleavage of the transcript separates the met leader RNA from the met operon mRNA, thereby leaving the last 23 nt of the 3′- met leader end attached to the met operon mRNA (Figure 3C).
Figure 3.

met leader is physically separated from met operon mRNA and rapidly degraded. (A) Total RNA isolated from S. aureus Newman grown in CDM with (‘+MET’) or without methionine (‘−MET’) was run on an agarose gel. Northern blot probed with a met leader-specific probe (lanes one and three) and re-probed with a metE-specific probe (lanes two and four). Positions of met leader RNA, met mRNA and 3′ region met mRNA are indicated on the left, approximate transcript sizes are given on the right. (B) Total RNA isolated from S. aureus Newman grown in CDM with (‘+MET’) and without methionine (‘−MET’) over a time course after rifampicin addition (0–32 min) was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control, transcript sizes are indicated on the right. (C) Schematic summary of met mRNA 5′ end determination by 5′ RACE. The scheme shows the very 3′ portion of the met leader with its terminator stem (marked in orange) and the beginning of met operon mRNA which is colored in black. Arrows mark the position of the met leader cleavage site identified within the terminator stem. Bold orange, black framed letters highlight the last 23 nucleotides of the met leader 3′-end that remain attached to met operon mRNA.
RNase III is involved in met leader RNA decay
As shown in Figure 3B, we found the met leader RNA species to be short-lived. In Gram-positive bacteria, main players of RNA decay are RNases III (a double-strand (ds)-specific endoribonuclease), J1/J2 (a bifunctional single-strand (ss)-specific endo- and 5′-3′ exoribonuclease) and Y (a ss-specific endoribonuclease), the latter known to be involved in riboswitch RNA turnover in Bacillus subtilis and S. aureus (35–37). To study the influence of staphylococcal RNase(s) on met leader stability, we subjected total RNA isolated from S. aureus conditional mutants of RNase J2 (rnjB::pMUTIN; ‘rnjBdepleted’), RNase III (rnc::pMUTIN; ‘rncdepleted’) and a deletion mutant of RNase Y (rny::ermC; Δrny) (all grown in CDM without methionine) to Northern blot hybridization using 5% PAA gels. Figure 4A confirms the detection of two met leader-specific fragments (∼440 and ∼390 nt) by this approach, with similar band patterns being displayed by the wildtype and the Δrny as well as rnjBdepleted mutants, indicating that neither RNase Y nor RNase J2 are involved in met leader RNA degradation. In the rncdepleted mutant background, however, enrichment of the larger 440 nt fragment was detectable (Figure 4A). Rifampicin stability assays and half-life determination further revealed that stability of the 440 nt transcript increased in the rncdepleted strain, resulting in longer half-life of the met leader RNA in comparison to the wildtype, while met leader RNA degradation remained unaffected in the Δrny and rnjBdepleted mutants (Figure 4B, C). A faint 390 nt band arising from cleavage of the stand-alone met leader transcript was still detectable in rncdepleted (Figure 4A). This is explicable by the nature of the rncdepleted strain as conditional mutant that still displays minimal residual RNase III activity. Indeed, when using a RNase III deletion mutant (kindly provided by Isabelle Caldelari), the cleaved form of the met leader is no longer traceable (Supplementary Figure S1). Together, the combined data suggest that RNase III, but not RNases Y or J2, is an important driver in met leader RNA decay.
Figure 4.

Influence of S. aureus RNases on met leader stability. (A) Total RNA isolated from S. aureus Newman and isogenic RNase J2 (rnjBdepleted), RNase III (rncdepleted) and RNase Y (Δrny) mutants grown in CDM without methionine (‘−MET’) was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control. Approximate transcript lengths are indicated on the right. (B) Quantification of met leader transcript levels over time calculated from rifampicin stability assays shown in (C). (C) Total RNA isolated from RNase mutant strains grown in CDM without methionine (‘−MET’) over a time course after rifampicin addition (0–8 and 0–32 min, respectively) was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control. (D) Graphical representation of cRACE data obtained for met leader transcript 3′ ends in the wild type ‘wt’ and the rncdepleted strain grown in CDM without methionine. Percentage of respective 3′ end positions of all analyzed transcripts are displayed in the respective column.
RNase III has a cleavage site within the met leader
To analyze the nature and exact size of the ∼390 and ∼440 nt met leader-specific RNA fragments, we determined their 5′- and 3′-ends. For this purpose, we performed cRACE with RNA isolated from S. aureus wildtype and the rncdepleted mutant grown under methionine-deprived conditions. In both strains, the 5′-end of the met leader mapped to identical positions (i.e. nt 404,416 of the S. aureus strain Newman genome; nt 1 in Supplementary Figure S2), demonstrating that wildtype and rncdepleted mutant initiate transcription of the riboswitch at the same site. Determination of 3′-ends by this approach, however, elucidated major differences between wildtype and the rncdepleted strain (Figure 4D). Thus, in the wildtype, 68% of transcripts ended at position 388 of the met leader which matches the size of the lower band detected in Figure 4A, while 5% of transcripts ended more downstream in a region between nt 425 and 440 (Figure 4D), corresponding to the upper band in Figure 4A. In contrast, in the rncdepleted strain (which still displays minor RNase III activity) only 30% of transcripts mapped to position 388, whereas 26% ended in the nt 425–440 region of the met leader (Figure 4D). Based on these results, we consider the lower (∼390 nt) met leader-specific RNA fragment to represent an RNase III-derived cleavage product of the met leader. The faint upper (∼440 nt) fragment, however, reflects non-cleaved met leader stand-alone RNA species which are likely to result from residual transcription termination events that continue to occur even upon activation of the system under methionine-deprived conditions. When combining these findings with the 5′-RACE data obtained for met operon mRNA 5′-end detection (see above), we conclude that cleavage occurs at positions 388 ^ 389 and 417 ^ 418 of the met leader RNA, generating a 2-nucleotide 3′ overhang that is typical for RNase III action (Figure 3C). According to the structural probing data, the identified cleavage site is located within the terminator stem of the riboswitch which is in good agreement with the function of RNase III as ds-specific endoribonuclease (Figure 2B).
RNase III targets the terminator stem structure of the met leader RNA
To further challenge the hypothesis of terminator cleavage by RNase III, we generated a series of markerless chromosomally encoded S. aureus mutants carrying alterations within the terminator/ antiterminator platform and studied the effects of these mutations on met leader RNA cleavage. In the first mutant, named Ter_mutated_1, we retained the terminator stem structure and the base-pairing strength, but altered the terminator nucleotide sequence (Figure 5A). The structure of the Ter_mutated_1 met leader RNA was assessed by in-line probing, confirming presence of a terminator stem similar to that of the wildtype (Figure 5B). Northern analysis of total RNA, isolated upon growth with and without methionine, demonstrates that met leader RNA cleavage is strongly impaired in mutant Ter_mutated_1, resulting in enrichment of non-cleaved 440 nt met leader RNA (Figure 5C). Next, we created mutants (i) Ter_destab in which introduction of mismatches prevented terminator stem formation (Figure 5A and B) as well as (ii) mutant ΔAntiTer&Ter in which the entire antiterminator and terminator sequence stretch (nt 352 to 439) was deleted. Although the met leader is transcribed (reflected by presence of multiple met leader RNA-specific bands), precise met leader RNA cleavage is completely abolished in both mutants (Figure 5C). Of note, no enrichment of non-cleaved stand-alone met leader RNA is detectable in mutants Ter_destab and ΔAntiTer&Ter which both lack the terminator stem structure (Figure 5C). Finally, when analyzing the mutants for met operon transcription (using a metE-specific probe), mutant Ter_mutated_1 was unable to switch on downstream gene transcription under methionine-deprived conditions, which is consistent with an impaired T-box function in this mutant (Figure 5D). In mutants Ter_destab and ΔAntiTer&Ter, however, met operon transcription was found to be activated independent of methionine supply, reflecting deregulation of the transcription control mechanism due to the lack of an effective transcription termination signal. From the combined data we conclude that the terminator stem is a necessary structure for met leader RNA degradation which, however, also involves sequence specificity to initiate physical separation of the met leader from met operon mRNA.
Figure 5.

Alteration of the terminator stem structure and influence on met leader cleavage. (A) Predicted secondary structures of terminator regions in met leader RNAs of wild type and mutants ‘Ter_mutated_1’ and ‘Ter_destab’. Point mutations introduced are highlighted in red, gray boxes indicate regions interacting with RNase III dimers as described in (40), P: proximal, M: middle, D: distal box. Nucleotides engaged to form the T-box bulge in antiterminator conformation (see also Figure 2B) are marked by asterisks (*). RNase III is depicted as pac-man. Position of RNase III cleavage site in wild type met leader is indicated by a dashed line. (B) In-line probing gel sections of the terminator region of met leader wild type, ‘Ter_mutated_1’ and ‘Ter_destab’ RNA are shown. Position of guanosines (G) as in ‘Ter_destab’ is given on the left of each gel, annotations as in Figure 2A. (C) Total RNA isolated from S. aureus Newman and the met leader mutants ‘Ter_mutated_1’, ‘Ter_destab’ and ‘ΔAntiTer&Ter’ grown in CDM with (‘+MET’) or without methionine (‘−MET’) was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control. (D) Total RNA same as in (C) was run on an agarose gel. Northern blot was probed with metE-specific probe and subsequently re-hybridized with a 16S rRNA-specific probe as loading control. Black arrowheads mark 3′ met mRNA transcripts.
Sequence constraints of met leader RNA terminator cleavage
RNase III is known to target RNA helices of 20–25 bp in size, with the homodimeric enzyme usually cleaving within a GC-rich sequence motif thereby generating 3′-overhangs of two nucleotides on the processed dsRNA products (38). Previous in vitro studies demonstrated that the sequence of the cleavage site itself is not required for recognition by RNase III and its catalytic activity (39). Instead, nucleotides outside the cleavage site, designated as proximal, middle and distal protein-interacting boxes, were shown to be required for substrate recognition and cleavage (Figure 5A) (40). As shown in Figures 3–5, cleavage of the met leader occurs between nucleotides 388 ^ 389 and 417 ^ 418, respectively. To analyze putative sequence-specific constraints for RNase III action on the met leader in vivo, three met leader mutants (mutants Ter_mutated_2, 3 and 4) with point mutations within or adjacent to the cleavage site were generated (Figure 6A). Structural probing confirmed integrity of the terminator stem structure (Figure 6B) and Northern analyses demonstrate that met leader RNA cleavage is still detectable in all three mutants (Figure 6C). However, in mutant Ter_mutated_3 (which carries three nucleotide exchanges), enrichment of non-cleaved met leader stand-alone RNA species occurs, suggesting that RNase III-mediated cleavage is diminished, but not completely abolished in this mutant (Figure 6C). The data corroborate that the sequence of the cleavage site itself is of minor importance for endonucleolytic processing by RNase III in vivo. Nevertheless, certain critical sequence constraints adjacent to the cleavage site seem to exist which influence activity of the enzyme.
Figure 6.

Sequence constraints of met leader RNA terminator cleavage. (A) Sequence and predicted secondary structure of terminator regions in met leader RNAs of wild type and mutants ‘Ter_mutated_2’, ‘Ter_mutated_3’ and ‘Ter_mutated_4’. Point mutations introduced are highlighted in red, gray boxes indicate regions interacting with RNase III dimers as described in (40), P: proximal, M: middle, D: distal box. Nucleotides engaged to form the T-box bulge in antiterminator conformation (see also Figure 2B) are marked by asterisks (*). Position of RNase III cleavage site in wild type met leader is indicated by a dashed line. (B) In-line probing gel sections of the terminator region of met leader wild type, ‘Ter_mutated_2’, ‘Ter_mutated_3’ and ‘Ter_mutated_4’ RNA are shown. Position of guanosines (G) as in ‘Ter_destab’ is given on the left and right of the gel, respectively, annotations as in Figure 2A. (C) Total RNA isolated from S. aureus Newman and the met leader mutants ‘Ter_mutated_2’, ‘Ter_mutated_3’ and ‘Ter_mutated_4’ grown in CDM with (‘+MET’) or without methionine (‘−MET’) was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control.
Fate of met operon (metICFE-mdh) mRNA and protein accumulation
Stability of the metICFE-mdh mRNA varies over the length of the transcript
Highly structured 5′ UTRs (such as riboswitches) usually protect mRNAs from degradation. The data obtained so far demonstrate that the met operon mRNA exists as a transcript that lacks the riboswitch (after met leader RNA cleavage) and which may undergo RNA decay. To study the stability of the met operon mRNA we performed rifampicin stability assays over the entire length of the transcript by using specific probes that target the single genes of the operon (Figure 7A–C). Figure 7B revealed that hybridization with a metI-specific probe, located in the 5′ portion of the transcript, resulted rather in a smear than a clear band. Only at time point t0 (prior to rifampicin addition), a blurry metI-specific band of 1 kb in size was detectable which, however, was highly instable as it was found to be degraded already 0.5 min after transcription arrest by rifampicin. Hybridization with a metC-specific probe, located in the 5′ portion of the transcript as well, did not result in any detectable signal (neither smear nor band). In contrast, hybridization with metE- and mdh-specific probes from the 3′-end of the operon revealed a fragment of ∼4 kb in size (Figure 7B). Compared to metI, this metE/ mdh-specific fragment was more stable as it was still detectable four minutes after rifampicin-induced transcription arrest (Figure 7B). The results of these experiments demonstrate that the stability of the metICFE-mdh mRNA varies over the length of the transcript with the 5′-portion being more instable than the 3′ part of the operon.
Figure 7.

Stability of the met operon mRNA varies over length of the transcript. (A) Schematic view of the organization of the met operon including its 5′ UTR (met leader). Lines below gene arrows indicate relative positions of the respective probes used in (B). (B) Total RNA isolated from S. aureus Newman grown in CDM without methionine (‘−MET’) over a time course after rifampicin addition (0–32 min) was run on an agarose gel. Northern blot was probed for metI, metC, metE and mdh. Open arrowhead marks metI mRNA and black arrowheads mark 3′ met mRNA. Approximate transcript lengths are indicated on the right of the respective blot. Re-hybridization with a 16S rRNA-specific probe was used as loading control. (C) Schematic view of the met operon 3′ region (without mdh) and relative positions of probes used. Total RNA isolated from S. aureus Newman grown in CDM without methionine (‘−MET’) was run on an agarose gel. Northern blot was probed for 3′ region of metC (‘metC 3′’), 3′ region of metF (‘metF 3′’) and metE. Open arrowhead marks full-length met operon mRNA (without met leader) and black arrowhead marks 3′ met operon mRNA. Approximate transcript lengths are indicated on the left. Re-hybridization with a 16S rRNA-specific probe was used as loading control. (D) Summary of 5′-RACE data. Upper diagram: Percentage of 5′ ends detected within distinct regions of metF. Lower part: Mapping of the detected 5′ ends to the metF region. Scale of the metF gene is illustrated at the bottom. Gray boxes represent 100 nucleotides each and the black arrow marks the approximate position of the primer used for cDNA synthesis. Transcripts characterized by 5′-RACE are depicted as wavy lines, dashed regions symbolize the range of detected 5′ ends. The percentage of each transcript group detected is given on the right.
The stable 3′-part of the met operon mRNA covers partly metF as well as the complete metE and mdh genes
Next, we further characterized the length and nature of the stable 4 kb metE/ mdh-specific fragment. Northern blot hybridization with various probes revealed that the stable transcript comprises the 3′ portion of metF as well as the metE and mdh genes (Figure 7C and B). Size calculation and Northern blot hybridization with a 5′-metF probe (data not shown) suggested that metF is only partly covered by the stable 4 kb transcript. Indeed, 5′-RACE experiments mapped the majority of the 5′-ends of the transcript to the 3′ portion of metF (Figure 7D). As shown in Figure 7D, the 5′ ends span a wide range within metF which speaks against the presence of a dedicated endoribonucleolytic cleavage site in this region. Finally, by performing cRACE experiments, we found the 3′ end of the stable 4 kb transcript to map 81 nucleotides downstream of the mdh stop codon, demonstrating presence of a 3′-UTR in met operon mRNA (data not shown). Together, the stable part of the met operon mRNA spans a region between the 3′-metF, metE and mdh genes as well as an 81 nt-long 3′-UTR.
RNase J is involved in metICFE-mdh mRNA decay
Next, we were interested in identifying the RNase(s) involved in the exonucleolytic degradation of the cleaved met operon mRNA from the 5′ end. The sole enzyme with known 5′ to 3′ exonuclease activity in S. aureus is RNase J (J1/J2). Although the J1/J2 enzyme complex is not essential in S. aureus, generation of J1 and J1/J2 double mutants by classical temperature-sensitive allele replacement vectors was notoriously difficult due to growth impairment of J1 mutants at 42°C (22). Eventually, employing a novel temperature-independent replacement vector allowed mutation of the RNase J-encoding genes in a dedicated pyrimidine auxotrophic S. aureus background (i.e. strain S. aureus PR01) (22). For assessing the influence of RNase J on met operon mRNA decay, we employed RNase J1 (ΔrnjA), J2 (ΔrnjB) and J1/J2 (ΔrnjA/B) mutants obtained in S. aureus PR01 (all kindly provided by Peter Redder). Initial experiments, however, revealed that S. aureus PR01 (for unknown reasons) is unable to activate its own chromosomal met operon upon methionine deprivation, making it impossible to study met leader/ met operon decay directly in the PR01 mutant strains (data not shown). We therefore cloned the met leader (under the control of its native promoter) along with the adjacent 5′-region of metI onto a vector which was transformed into S. aureus PR01 wildtype as well as into the three isogenic RNase J mutants (Figure 8A). Figure 8B demonstrates that methionine deprivation indeed induces met leader/ metI transcription from the vector (though only at low level). In the S. aureus PR01 wildtype background plasmid-derived metI-specific mRNA is not detectable which is probably due to its rapid degradation (Figure 8B). In contrast, the metI-specific transcript is enriched in the RNase J1 (ΔrnjA), J2 (ΔrnjB) and J1/J2 (ΔrnjA/B) mutants (Figure 8B). These data strongly suggest that the RNase J1/J2 complex is involved in the degradation of met operon mRNA by executing 5′ to 3′ exonucleolytic cleavage of the RNA.
Figure 8.
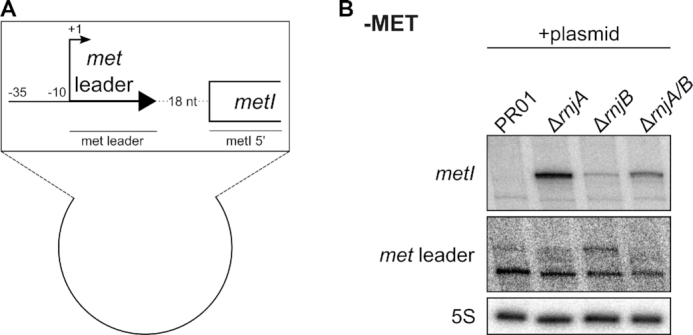
RNase J is involved in met operon mRNA degradation. (A) Scheme of the plasmid transformed into S. aureus SA564RD ΔpyrFE (‘PR01’) and its isogenic RNase J1 (‘ΔrnjA’), RNase J2 (‘ΔrnjB’) and RNase J1/J2 double (‘ΔrnjA/B’) mutants. ‘–35’ and ‘–10’ indicates the promoter region. Arrow with +1 marks the transcription start site of met leader. The met leader sequence is depicted as thick, black arrow. First 215 nt of metI are shown as open rectangle. Lines below genes indicate relative positions of probes used in (B). (B) Total RNA was isolated from S. aureus SA564RD ΔpyrFE (‘PR01’) and its isogenic RNase J1 (‘ΔrnjA’), RNase J2 (‘ΔrnjB’) and RNase J1/J2 double (‘ΔrnjA/B’) mutants. Bacteria were grown in MH medium to OD 0.5. Then cultures were washed twice with PBS, bacteria were shifted into CDM without methionine (‘−MET’) supplemented with pyrimidine for 30 min and samples were taken. 5 μg of total RNA was run on a PAA gel. Northern blot was hybridized with a met leader-specific probe, re-probed with a metI 5′-specific probe and subsequently re-hybridized with a 5S rRNA-specific probe as loading control.
Cleavage by RNase III facilitates met operon mRNA degradation
Next, we asked the question whether or not RNase III-mediated release of the met leader might have an influence on degradation of the met operon mRNA from the 5′-end. As detection of full-length met operon transcripts proved to be notoriously difficult (Figures 3 and 7), we focused on the stability of the very 5′-end of the met operon mRNA and transformed the met leader/ 5′ metI plasmid (described above) into S. aureus Newman and its isogenic RNase III (rnc::pMUTIN; rncdepleted) mutant. Northern hybridization using a 5′ metI-specific probe (Figure 9A) resulted in the detection of a larger (i.e. 970 nt) fragment in the rncdepleted mutant in comparison to the 550-nt band present in the wildtype (Figure 9B). The fragment size in the rncdepleted strain is in agreement with a non-cleaved plasmid-derived met leader/ 5′ metI transcript, confirming lack of met leader RNA release in a rncdepleted background (Figure 9B). Importantly, the non-cleaved met leader/5′ metI transcript becomes stabilized in the rncdepleted strain. Thus, rifampicin stability assays revealed that, in the RNase III-proficient wildtype, processed 5′ metI RNA is degraded after two minutes, while in the rncdepleted background the non-cleaved met leader/5′ metI transcript is still detectable after eight minutes (Figure 9C). These data led us to conclude that the met leader protects met operon mRNA from 5′ to 3′ exonucleolytic degradation. Cutting off the met leader through RNase III, however, abolishes this effect and favors degradation of the met operon mRNA.
Figure 9.

RNase III cleavage of terminator stem facilitates 5′ met operon mRNA exonucleolytic degradation. (A) Scheme of transcripts detected in (B); met leader sequence depicted as thick, black arrow, first 215 nt of metI shown as open rectangle, plasmid-derived transcription terminator shown as hairpin. Line below metI indicates relative position of the probe used in (B) and (C). RNase III cleavage site is shown as pac-man. Approximate transcript sizes are given below the scheme. (B) Total RNA was isolated from S. aureus Newman, its isogenic RNase III (rncdepleted) mutant and from both strains transformed with the plasmid (‘+p’) detailed in Figure 8A grown in CDM without methionine (‘−MET’). RNA was run on an agarose gel. Northern blot was probed with a metI 5′-specific probe. Open arrowhead marks ∼550 nt transcript and black arrowhead marks ∼970 nt transcript. Approximate transcript lengths are indicated on the right of the blot. Re-hybridization with a 16S rRNA-specific probe was used as loading control. (C) Total RNA was isolated from S. aureus Newman and its isogenic RNase III (rncdepleted) mutant strain transformed with the plasmid (‘+p’) grown in CDM without methionine (‘−MET’) over a time course after rifampicin addition (0–32 min). RNA was run on an agarose gel. Hybridization and labelling as described for (B).
Detection of met operon-encoded proteins
Next, we asked the question of whether mRNA stability might influence protein amounts or synthesis rates. Thus, we initially determined intracellular proportions of met operon-encoded MetI, MetC, MetF, MetE and Mdh enzymes by liquid chromatography (LC)/mass spectrometry (MS) (Figure 10A). Relative protein quantification by LC–MS/MS analysis, performed in mid-exponential growth phase, identified MetE as the most abundant protein of the met operon-encoded enzymes (69%), followed by Mdh (11%) and MetC (10%). The least abundant proteins were MetF (4%) and MetI (6%) (Figure 10A). As detectable cellular protein levels are the result of available transcript amounts, translation rate and protein degradation, we studied these processes as well. First, we analyzed protein stabilities by measuring the half-lives of the Met enzymes. For this purpose, we performed a pulse experiment by combining stable isotope labeling of amino acids (SILAC) with gel-free LC–MS/MS analysis (41). The SILAC approach allows to differentiate between ‘old’ and ‘newly’ synthesized proteins according to their respective isotope labels which are acquired upon a shift to media containing either stable isotope labeled (heavy) or unlabeled (light) amino acids (Figure 10B). In brief, following growth in CDM (without methionine, but containing light arginine and lysine) to mid-exponential phase, bacteria were washed with PBS to remove any light amino acids and shifted to CDM containing heavy arginine and lysine (label switch) (Figure 10B). Samples were taken at different time points after the label switch (t0′, t15′, t30′, t60′ and t240′) and analyzed by LC–MS/MS (Figure 10B, C). The label switch set a defined end-point of protein synthesis with light amino acids. Accordingly, proteins synthesized after the label switch exclusively contain heavy arginine and lysine residues. Hence, the label switch allows monitoring the stability of light proteins accumulated prior to the label switch thereby distinguishing them from newly synthesized heavy proteins. As this approach only considers proteins made prior to the switch, changes in transcription and translation that might occur after the switch, will not influence the results of the half-life determinations. The experiment revealed that, in contrast to its low abundance, MetI displayed a striking long half-life of 7.2 h (Figure 10C). Also Mdh, which exhibits a half-life of 8.2 h, is long-lived, while stability of MetE is lower (half-life 4.8 h). Finally, the lowest half-life in this experiment was displayed by MetC (i.e. 2.2 h) (Figure 10C). To investigate a putative direct influence of met operon mRNA stability on protein synthesis, we next determined the synthesis rates of the Met enzymes. Thus, we again performed a SILAC experiment, but now adding rifampicin to block further met operon transcription after the label switch (Figure 10B). In this experimental set up, Met enzymes will be exclusively translated from transcripts made prior to transcription arrest, with mRNA stability likely to influence protein synthesis rates. As described above, accumulation of these newly synthesized proteins can be readily monitored by measuring the incorporation of heavy arginine and lysine residues after the label switch (Figure 10B). Samples were taken before and after rifampicin addition at different time points after the label switch and analyzed by LC–MS/MS (t0′ and t30′) and Northern hybridization (t0′, t15′ and t30′) (Figure 10B&D). The latter confirmed immediate met operon transcription arrest after the addition of rifampicin as only traces of the stable metE-specific met operon mRNA were detectable after 15 min (Figure 10D, right). From the LC–MS/MS data we calculated the doubling times of the enzymes as a measure for the protein synthesis rates (Figure 10D, left). Among all Met enzymes, MetE and Mdh displayed the shortest doubling time (i.e.∼30 min), reflecting a high protein synthesis rate. Higher doubling times (and accordingly lower protein synthesis rates) were recorded for MetI and MetC (∼70 min) as well as for MetF (150 min) which displayed the lowest synthesis rate (Figure 10D, left). As these data are commensurate to met operon transcript stabilities, we conclude that varying mRNA degradation affects translation and the resulting protein amounts, with the latter being further influenced by variations in protein stability and possibly other (so far) unknown factors.
Figure 10.

Detection of met operon-encoded proteins by proteomics. (A) Relative cellular protein amounts of met operon-encoded enzymes as detected by LC–MS/MS analysis. (B) Experimental design for protein stability (C) and protein synthesis rate (D) determinations. Asterisks (*) indicate the heavy amino acids lysine and arginine added upon label switch (see text for details). (C) Half-lives of met operon enzymes as calculated from LC-MS/MS data of unlabeled proteins. t0′, t15′, t30′, t60′ and t240′ samples were used for half-life determination. (D) Synthesis rates of met operon enzymes after transcription arrest by addition of rifampicin (RIF) as calculated from LC–MS/MS data of heavy proteins. t0′ and t30′ samples were used for determination of doubling time. Corresponding total RNA was run on an agarose gel. Northern blot was probed with metE-specific probe. Arrow marks addition of rifampicin. 16S rRNA detected in Midorigreen-stained gel is shown as loading control. (h), hours; (min) minutes; n.d., not determined.
DISCUSSION
met leader RNA secondary structure
Secondary structure determination revealed that the met leader RNA indeed harbors all structural features known from classical T-box riboswitches. By its size, however, the element stands out from the majority of other T-box riboswitches, including those with methionyl-tRNA specificity (2,42). Interestingly, the met leader RNA T-box riboswitch was previously shown to prefer interaction with initiator-methionyl-tRNA over elongator-methionyl-tRNA to activate the system (6). As both methionyl-tRNA species possess a CAU anticodon to be recognized by the specifier AUG codon, discrimination is likely to involve additional structural features both on the tRNA and met leader RNA side. tRNA structures usually recognized by T-box-riboswitches comprise, in addition to the anticodon and the 3′ CCA sequence, the T- and D-arms which form the tRNA elbow in canonical L-shaped tRNAs (10). Sequence-specific differences between initiator- and elongator methionyl-tRNAs mainly exist in the D- and T-arms as well as in the acceptor stems of these tRNA species. Co-crystallization and NMR studies with glyQS and tyrS T-box riboswitches demonstrate that the tRNA elbow and anticodon stem closely interact with stem I of the T-box riboswitch (10,43–46). In this dynamic interaction, the kink-turn and hinge regions confer flexibility to stem I to enable bending over the tRNA (10,11,47). Except for the missing GA motif within the kink-turn, stem I of the met leader is not particularly different from other T-box riboswitches, suggesting similar structural interaction with methionyl-tRNAs, but making it unlikely that stem I considerably contributes to initiator/ elongator tRNA discrimination. The role of the adjacent stem II and stem IIA/B pseudoknot in T-box riboswitch function is still poorly understood, the more so as the structures may even lack in some systems (i.e. in glyQS) (9). Recent findings suggest that stem II bridges and latches the otherwise weak AU-rich specifier codon–tRNA anticodon interactions, making it dispensable for the GC-rich glyQS riboswitch codons (48). This lateral stabilization of the codon–anticodon interaction turned out to be essential in T-box riboswitches of the ultrashort stem I type, such as the ileS T-box riboswitch of Mycobacterium smegmatis, compensating for the absence of the stem I platform usually interacting with the tRNA elbow (49–51). It is therefore tempting to speculate that the short stem II of the met leader might be involved in the tRNA binding process as well. At least for the glyQS system, the linker region between stem I and stem III was suggested to play a role in exact tRNA positioning by acting as a ruler to monitor the tRNA length and overall shape (52,53). Interestingly, the additional stem-loops within the linker region seem to be specific for the staphylococcal MET-T-box riboswitch, and it is well conceivable that they may play a role in methionyl-tRNA interaction and discrimination. Species-specific structural features contributing to differential tRNA selectivity have been previously described for the S. aureus glyS T-box riboswitch, suggesting certain evolutionary flexibility in T-box systems which might allow for adaptation to distinct metabolic profiles of bacteria (54). Finally, through the insertion of stems LI to LIII into the linker region, the met leader becomes unusually long. In concert with the specific structural features of the linker, it is possible that the extended transcription time to synthesize the long met leader might further facilitate correct recognition and positioning of the tRNA. Recent single-molecule fluorescence resonance energy transfer (smFRET) studies dissected tRNA/ T-box riboswitch interaction as a highly dynamic process that proceeds in two steps, with the anticodon being recognized first, followed by 3′-CCA binding through the T-box bulge (11). The latter step is accompanied by a conformational change of the distal part of the T-box riboswitch to contact the tRNA (11). Postulating that tRNA binding is an induced-fit process that takes place while the T-box riboswitch RNA is transcribed (55), extended transcription time of the met leader may allow for exact shape recognition and binding of the cognate tRNA. However, in absence of three-dimensional stuctural data of the met leader RNA (or any other MET-T-box riboswitch) this is mere speculation at the moment. In general, T-box riboswitches are increasingly recognized as promising targets for anti-infective drugs to interfere with bacterial growth (56–59). In that sense and in the light of the ongoing antibiotic resistance crisis, detailed structural analyses of bacterial T-box riboswitches is certainly a worthwhile task.
RNase III mediates met leader RNA cleavage
Apart from the long linker region, the most striking structural feature of the met leader RNA was the identification of a terminator stem that represents a target for cleavage by RNase III (Figures 2B, 4A). This is rather unusual as bacterial riboswitch turnover is commonly associated with the action of RNase P and RNase Y (36,37,60). The met leader terminator stem comprises a helix of 23 bp which is above the average length of terminator stems usually present in other T-box riboswitches (including MET-T-box systems) (2,42). RNase III requires double-stranded RNA stretches of 20–25 bp for cleavage, and it is therefore reasonable to suggest that the length of the stem in combination with presence of a GC-rich region renders the terminator an RNase III target (38,39). Lack of met leader RNA cleavage in mutants that are devoid of the terminator stem structure (Ter_destab, ΔAntiTer&Ter) or possessing a terminator stem with an altered sequence (Ter_mutated_1) (Figure 5C) strongly support this hypothesis. In agreement with our previous studies, met leader RNA was overabundantly present also under methionine-rich conditions in which the system is actually OFF. Indeed, in presence of methionine no met operon expression took place while constitutive basic transcription of the met leader occurred regularly under all growth conditions. This is attributed to the somewhat ‘leaky’ CodY-mediated repression of the system which most likely supports immediate activation of met operon transcription when needed. As the prematurely terminated met leader RNAs (‘stand-alone transcripts’) carry the terminator stem, RNase III-driven cleavage and decay of the met leader under methionine-rich conditions comes as no surprise (Figures 3 and 5). However, met leader RNA cleavage was also detected under methionine-deprived conditions when met operon mRNA was transcribed upon antiterminator formation and read-through into downstream met operon genes (Figures 3–6). As antiterminator and terminator are mutually exclusive structures, read-through transcripts should actually lack the terminator stem as RNase III target. However, our combined experiments clearly speak in favor of terminator stem presence and RNase III-mediated cleavage of the read-through transcripts. Terminator stem presence in read-through transcripts might be explained by the high thermodynamic stability of the terminator stem (free energy ΔG = –32.60 kcal/mol) which is associated with its length and GC-richness (Figure 2B). Formation of the antiterminator is likely to be transient as it is the thermodynamically much weaker structure which is only required to allow the RNA polymerase to pass the distal expression platform of the T-box riboswitch to accomplish downstream gene transcription (8). At the same time, immediate terminator stem formation in read-through transcripts will release uncharged tRNAs from the T-box riboswitch which will then be available for amino acid charging, once sufficient amounts of the cognate amino acid is synthesized (8). In this respect, terminator stem formation in met leader/ met operon read-through transcripts makes sense.
RNase J mediates met operon mRNA decay
Cleavage of a (T-box) riboswitch by RNase III was a rather unexpected finding of this study. Commonly, riboswitch RNAs either remain attached to the mRNA or were shown to be cleaved by RNase P or RNase Y (36,37,60). In B. subtilis thrS, T-box riboswitch cleavage was found to occur immediately upstream of the terminator stem, leaving the structure as a protective element that stabilizes the mRNA and prevents degradation (61). In contrast, in the S. aureus met leader/met operon system we found the terminator stem to recruit RNase III to cleave off the met leader from the met operon mRNA, thereby considerably destabilizing the transcript (Figures 4 and 9). The data further show that met operon mRNA degradation occurs from the 5′-end (Figure 7), with the mRNA decay being accomplished by RNase J (Figure 8), the sole enzyme known to date in Gram-positive bacteria with 5′ to 3′ exoribonucleolytic activity (35). The RNase III cleavage removes the protective stem structure from the met mRNA and creates a 5′ monophosphate end that is prone to exonucleolytic degradation. Unexpectedly, mRNA degradation was not equally efficient over the length of the metICFE-mdh mRNA. Thus, the 5′ part of the transcript (encoding metI, metC and the 5′ part of metF) was more rapidly degraded than the 3′ end (covering the metF 3′, metE and mdh genes) (Figure 7). The molecular background of these differences in metICFE-mdh mRNA stability is currently not known. The scenario, however, is reminiscent of previous findings in E. coli and other bacteria showing that polycistronic mRNA can undergo differential RNA decay, leaving some genes within the operon more stable than others, with direct consequences for protein expression levels (62–65). In this respect, RNA decay was found to be regulated by mRNA secondary structures (62), and a recent study in S. aureus also identified distinct structural elements within a staphylococcal mRNA which influenced activity of RNase Y (66). It is well conceivable that similar functional elements might exist in the metICFE-mdh mRNA. However, preliminary bioinformatic analysis of the transcript did not reveal sufficient indications for that so far (data not shown). Another possibility to protect mRNAs from degradation is their decoration with ribosomes during translation which blocks access of RNases to cleavage sites within the RNA molecule (62,67). In this context, codon bias (i.e. the uneven use of synonymous codons in the genome) was shown to play a critical role for translation and mRNA turnover (68,69). Thus, rare codons are usually translated slower which causes pausing of the ribosomes, leading to stabilization of the mRNA by protecting the transcripts from RNases (68). Indeed, when performing a codon usage analysis (by employing the graphical codon usage analyzer 2.0 (70)) we identified a stretch of rare codons located within in the 3′ part of the metF gene (see Supplementary Figure S3 for details). 5′-RACE analysis showed that this region covers the majority of 5′-ends of the stable 3′-end transcript of the metICFE-mdh mRNA (Figure 7D). Thus, rare codon usage and protection by ribosomes might be involved in stabilization of the transcript towards the 3′-end. But, clearly more experimental work is needed to substantiate this hypothesis.
Met enzyme amounts are influenced by varying transcript and protein stabilities
Cellular protein levels are determined by a number of factors such as transcript availability and translation rate as well as by the stability of the synthesized protein. When quantifying intracellular levels of the single Met enzymes, we found considerable differences regarding relative protein amounts. Thus, enzymes engaged in the early steps of methionine biosynthesis such as transsulfuration (executed by MetI and MetC) are less abundant than MetE which performs the final and decisive methylation step to convert homocysteine into methionine, a reaction which is also involved in essential SAM recycling (Figure 10A). MetE is by far the most abundant protein encoded by the operon (i.e. 69 %). The enzyme represents a cobalamin-independent methionine synthase which, in E. coli, was described to be 100-fold less efficient than the cobalamin-dependent methionine synthase MetH (71). Under methionine-deprived conditions, MetE can account for up to 3% of total soluble protein in E. coli to compensate for the low efficiency of the enzyme (2,71,72). It is reasonable to suggest that staphylococci may accumulate high intracellular MetE levels for the same reason, for instance through stabilizing its mRNA. Indeed, when determining the protein synthesis rates after transcription arrest we found enzymes encoded towards the stable 3′-end of the met operon mRNA (i.e. MetE, Mdh) to have higher synthesis rates than those located at the instable 5′ end (i.e. MetI, MetC, MetF) (Figure 10D). However, transcript stability and protein synthesis rates are not the only factors influencing cellular protein amounts. In this respect, protein degradation is another important checkpoint and when determining the half-lives of the Met enzymes we found significant differences regarding individual Met enzyme stabilities (Figure 10C). Thus, for example, MetI has a long half-life (i.e. 7.2 h) and it is tempting to speculate that this might compensate for localization of the metI gene in the extremely short-lived 5′-portion of the transcript and the low protein synthesis rate (Figure 10D). Interestingly, not all of the enzymes encoded by the met operon seem to follow such a clear relationship between gene position, synthesis rate, protein stability and the detectable amount of protein. For example, Mdh (a protein of so far unknown function) which is encoded together with MetE in the stable 3′ transcript part, displays a similar protein synthesis rate as MetE. Despite of this and a very long Mdh half-life, the protein is by far less abundant than MetE (i.e. 11% versus 69%) (Figure 10A, C, D). The combined data suggest that transcript and protein stability are important, but certainly not the sole factors that adjust enzyme amounts to current metabolic requirements. Clearly, more future work is needed to fully understand staphylococcal methionine biosynthesis, for instance by addressing other regulatory levels such as translation initiation, enzyme activities and allosteric feedback control.
CONCLUSIONS
The strictly methionine-dependent and complex regulation of de novo methionine biosynthesis, involving a hierarchical string of regulatory cues, is probably due to the fact that staphylococci lack important methionine recycling pathways. Staphylococci therefore avoid overproduction of this metabolically costly amino acid. Coordinated mRNA decay seems to significantly contribute to the control of the pathway, thereby adding another layer of complexity to this sophisticated regulatory network. From an evolutionary point of view it is remarkable that staphylococci are the only genus among the Bacillales family to employ a T-box riboswitch to control the pathway. Even more astonishing, however, is the fact that this distinct MET-T-box riboswitch has unique structural features that distinguish it both from other methionyl-tRNA-specific systems as well as from T-box riboswitches in general. Notably, the unique terminator stem structure which renders this MET-T-box riboswitch an RNase III target, seems to be the key structural feature that sparks subsequent RNA decay events. Revealing the further details of this process, particularly regarding the mechanisms that stabilize the met operon mRNA towards the 3′-end, are interesting future experimental tasks.
DATA AVAILABILITY
Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (73). Data received from protein quantification and half-life determination experiments can be obtained with the dataset identifier PXD020619. Data of the proteome analysis for protein synthesis rate determination can be found with the dataset identifier PXD022677.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Peter Redder for providing the rnj mutants of S. aureus SA564RD, Christiane Wolz for providing the rnc, rny and rnjB mutants of S. aureus Newman and Isabelle Caldelari for providing the rnc mutant of S. aureus HG001. We also thank Tan Hock Siew for providing initial training with in-line probing. We thank Jürgen Bartel, Christine Wünsche and Thomas Sura for sample preparation prior to MS-analysis and MS-measurements.
Contributor Information
Freya D R Wencker, Institute of Molecular Infection Biology, University of Würzburg, Würzburg 97080, Germany.
Gabriella Marincola, Institute of Molecular Infection Biology, University of Würzburg, Würzburg 97080, Germany.
Sonja M K Schoenfelder, Institute of Molecular Infection Biology, University of Würzburg, Würzburg 97080, Germany.
Sandra Maaß, Institute of Microbiology, University of Greifswald, Greifswald 17489, Germany.
Dörte Becher, Institute of Microbiology, University of Greifswald, Greifswald 17489, Germany.
Wilma Ziebuhr, Institute of Molecular Infection Biology, University of Würzburg, Würzburg 97080, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Council (DFG) through SPP1617 [ZI 665/2]; Transregional Collaborative Research Centre 34 [INST 292/67, B04]; ShARE [ZI665/3-1]; German Federal Ministry of Education and Research (BMBF) [01KI1727E, 16GW0297]. Funding for open access charge: German Research Council (DFG).
Conflict of interest statement. None declared.
REFERENCES
- 1. Wencker F., Ziebuhr W.. Chapter 13: methionine synthesis in microbes. The Handbook of Microbial Metabolism of Amino Acids. 2017; Oxfordshire: CABI; 179–197. [Google Scholar]
- 2. Rodionov D.A., Vitreschak A.G., Mironov A.A., Gelfand M.S.. Comparative genomics of the methionine metabolism in Gram-positive bacteria: a variety of regulatory systems. Nucl. Acids Res. 2004; 32:3340–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaleta C., Schäuble S., Rinas U., Schuster S.. Metabolic costs of amino acid and protein production in Escherichia coli. Biotechnol. J. 2013; 8:1105–1114. [DOI] [PubMed] [Google Scholar]
- 4. Sekowska A., Ashida H., Danchin A.. Revisiting the methionine salvage pathway and its paralogues. Microb. Biotechnol. 2019; 12:77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Joshi G.S., Spontak J.S., Klapper D.G., Richardson A.R.. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol. Microbiol. 2011; 82:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schoenfelder S.M.K., Marincola G., Geiger T., Goerke C., Wolz C., Ziebuhr W. Methionine biosynthesis in Staphylococcus aureus is tightly controlled by a hierarchical network involving an initiator tRNA-Specific T-box riboswitch. PLoS Pathog. 2013; 9:e1003606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geiger T., Wolz C.. Intersection of the stringent response and the CodY regulon in low GC Gram-positive bacteria. Int. J. Med. Microbiol. 2014; 304:150–155. [DOI] [PubMed] [Google Scholar]
- 8. Suddala K.C., Zhang J.. An evolving tale of two interacting RNAs—themes and variations of the T-box riboswitch mechanism. IUBMB Life. 2019; 71:1167–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kreuzer K.D., Henkin T.M.. The T-box riboswitch: tRNA as an effector to modulate gene regulation. Microbiol. Spectrum. 2018; 6:RWR-0028-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J., Ferré-D’Amaré A.R.. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013; 500:363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J., Chetnani B., Cormack E.D., Alonso D., Liu W., Mondragón A., Fei J.. Specific structural elements of the T-box riboswitch drive the two-step binding of the tRNA ligand. eLife. 2018; 7:e39518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vitreschak A.G., Mironov A.A., Lyubetsky V.A., Gelfand M.S.. Comparative genomic analysis of T-box regulatory systems in bacteria. RNA. 2008; 14:717–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bae T., Schneewind O.. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006; 55:58–63. [DOI] [PubMed] [Google Scholar]
- 14. Geiger T., Francois P., Liebeke M., Fraunholz M., Goerke C., Krismer B., Schrenzel J., Lalk M., Wolz C.. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012; 8:e1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lerch M.F., Schoenfelder S.M.K., Marincola G., Wencker F.D.R., Eckart M., Förstner K.U., Sharma C.M., Thormann K.M., Kucklick M., Engelmann S.et al.. A non-coding RNA from the intercellular adhesion (ica) locus of Staphylococcus epidermidis controls polysaccharide intercellular adhesion (PIA)-mediated biofilm formation. Mol. Microbiol. 2019; 111:1571–1591. [DOI] [PubMed] [Google Scholar]
- 16. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B.et al.. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brenneis M., Hering O., Lange C., Soppa J.. Experimental characterization of cis-acting elements important for translation and transcription in halophilic archaea. PLos Genet. 2007; 3:e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marincola G., Schäfer T., Behler J., Bernhardt J., Ohlsen K., Goerke C., Wolz C.. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol. Microbiol. 2012; 85:817–832. [DOI] [PubMed] [Google Scholar]
- 19. Monk I.R., Shah I.M., Xu M., Tan M.-W., Foster T.J.. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio. 2012; 3:e00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair D., Memmi G., Hernandez D., Bard J., Beaume M., Gill S., Francois P., Cheung A.L.. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011; 193:2332–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baba T., Bae T., Schneewind O., Takeuchi F., Hiramatsu K.. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008; 190:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redder P., Linder P.. New range of vectors with a stringent 5-fluoroorotic acid-based counterselection system for generating mutants by allelic replacement in Staphylococcus aureus. Appl. Environ. Microbiol. 2012; 78:3846–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herbert S., Ziebandt A.-K., Ohlsen K., Schäfer T., Hecker M., Albrecht D., Novick R., Götz F.. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 2010; 78:2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caldelari I., Chane-Woon-Ming B., Noirot C., Moreau K., Romby P., Gaspin C., Marzi S.. Complete genome sequence and annotation of the Staphylococcus aureus strain HG001. Genome Announc. 2017; 5:e00783-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lalaouna D., Baude J., Wu Z., Tomasini A., Chicher J., Marzi S., Vandenesch F., Romby P., Caldelari I., Moreau K.. RsaC sRNA modulates the oxidative stress response of Staphylococcus aureus during manganese starvation. Nucleic Acids Res. 2019; 47:9871–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oun S., Redder P., Didier J.-P., François P., Corvaglia A.-R., Buttazzoni E., Giraud C., Girard M., Schrenzel J., Linder P.. The CshA DEAD-box RNA helicase is important for quorum sensing control in Staphylococcus aureus. RNA Biology. 2013; 10:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 28. Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M.. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011; 10:1794–1805. [DOI] [PubMed] [Google Scholar]
- 29. Saeed A. i., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M.et al.. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003; 34:374–378. [DOI] [PubMed] [Google Scholar]
- 30. Silva J.C., Gorenstein M.V., Li G.-Z., Vissers J.P.C., Geromanos S.J. Absolute quantification of proteins by LCMSE : a virtue of parallel ms acquisition. Mol. Cell. Proteomics. 2006; 5:144–156. [DOI] [PubMed] [Google Scholar]
- 31. Regulski E.E., Breaker R.R.. In-line probing analysis of riboswitches. Methods Mol. Biol. 2008; 419:53–67. [DOI] [PubMed] [Google Scholar]
- 32. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winkler W.C., Grundy F.J., Murphy B.A., Henkin T.M.. The GA motif: an RNA element common to bacterial antitermination systems, rRNA, and eukaryotic RNAs. RNA. 2001; 7:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grigg J.C., Ke A.. Sequence, structure, and stacking: specifics of tRNA anchoring to the T box riboswitch. RNA Biol. 2013; 10:1761–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Durand S., Condon C.. RNases and helicases in gram-positive bacteria. Microbiol. Spectrum. 2018; 6(2):RWR-0003-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shahbabian K., Jamalli A., Zig L., Putzer H.. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009; 28:3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khemici V., Prados J., Linder P., Redder P.. Decay-initiating endoribonucleolytic cleavage by RNase Y is kept under tight control via sequence preference and Sub-cellular localisation. PLos Genet. 2015; 11:e1005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lioliou E., Sharma C.M., Caldelari I., Helfer A.-C., Fechter P., Vandenesch F., Vogel J., Romby P.. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLos Genet. 2012; 8:e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Romilly C., Chevalier C., Marzi S., Masquida B., Geissmann T., Vandenesch F., Westhof E., Romby P.. Loop-loop interactions involved in antisense regulation are processed by the endoribonuclease III in Staphylococcus aureus. RNA Biology. 2012; 9:1461–1472. [DOI] [PubMed] [Google Scholar]
- 40. Gan J., Tropea J.E., Austin B.P., Court D.L., Waugh D.S., Ji X.. Structural insight into the mechanism of double-stranded RNA processing by Ribonuclease III. Cell. 2006; 124:355–366. [DOI] [PubMed] [Google Scholar]
- 41. Michalik S., Bernhardt J., Otto A., Moche M., Becher D., Meyer H., Lalk M., Schurmann C., Schlüter R., Kock H.et al.. Life and death of proteins: a case study of glucose-starved Staphylococcus aureus. Mol. Cell. Proteomics. 2012; 11:558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vitreschak A.G., Rodionov D.A., Mironov A.A., Gelfand M.S.. Riboswitches: the oldest mechanism for the regulation of gene expression?. Trends Genet. 2004; 20:44–50. [DOI] [PubMed] [Google Scholar]
- 43. Grigg J.C., Chen Y., Grundy F.J., Henkin T.M., Pollack L., Ke A.. T box RNA decodes both the information content and geometry of tRNA to affect gene expression. Proc. Natl Acad. Sci. U.S.A. 2013; 110:7240–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lehmann J., Jossinet F., Gautheret D. A universal RNA structural motif docking the elbow of tRNA in the ribosome, RNAse P and T-box leaders. Nucleic Acids Res. 2013; 41:5494–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J., Henkin T.M., Nikonowicz E.P.. NMR structure and dynamics of the specifier loop domain from the Bacillus subtilis tyrS T box leader RNA. Nucleic Acids Res. 2010; 38:3388–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Nikonowicz E.P.. Solution structure of the K-turn and specifier loop domains from the Bacillus subtilis tyrS T-box leader RNA. J. Mol. Biol. 2011; 408:99–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grigg J.C., Ke A.. Structural determinants for geometry and information decoding of tRNA by T box leader RNA. Structure. 2013; 21:2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang J. Unboxing the T-box riboswitches—a glimpse into multivalent and multimodal RNA–RNA interactions. WIREs RNA. 2020; 11:e1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sherwood A.V., Frandsen J.K., Grundy F.J., Henkin T.M.. New tRNA contacts facilitate ligand binding in a Mycobacterium smegmatis T box riboswitch. Proc. Natl Acad. Sci. U.S.A. 2018; 115:3894–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Battaglia R.A., Grigg J.C., Ke A.. Structural basis for tRNA decoding and aminoacylation sensing by T-box riboregulators. Nat. Struct. Mol. Biol. 2019; 26:1106–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Suddala K.C., Zhang J.. High-affinity recognition of specific tRNAs by an mRNA anticodon-binding groove. Nat. Struct. Mol. Biol. 2019; 26:1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yousef M.R., Grundy F.J., Henkin T.M.. tRNA requirements for glyQS antitermination: a new twist on tRNA. RNA. 2003; 9:1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang J., Ferré-DAmaré A.R.. Trying on tRNA for size: RNase P and the T-box riboswitch as molecular rulers. Biomolecules. 2016; 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Apostolidi M., Saad N.Y., Drainas D., Pournaras S., Becker H.D., Stathopoulos C.. A glyS T-box riboswitch with species-specific structural features responding to both proteinogenic and nonproteinogenic tRNAGly isoacceptors. RNA. 2015; 21:1790–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang J., Ferré-D’Amaré A.R.. Structure and mechanism of the T-box riboswitches. WIREs RNA. 2015; 6:419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stamatopoulou V., Apostolidi M., Li S., Lamprinou K., Papakyriakou A., Zhang J., Stathopoulos C.. Direct modulation of T-box riboswitch-controlled transcription by protein synthesis inhibitors. Nucleic Acids Res. 2017; 45:10242–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frohlich K.M., Weintraub S.F., Bell J.T., Todd G.C., Väre V.Y.P., Schneider R., Kloos Z.A., Tabe E.S., Cantara W.A., Stark C.J.et al.. Discovery of small-molecule antibiotics against a unique tRNA-Mediated regulation of transcription in gram-positive bacteria. Chem. Med. Chem. 2019; 14:758–769. [DOI] [PubMed] [Google Scholar]
- 58. Orac C.M., Zhou S., Means J.A., Boehm D., Bergmeier S.C., Hines J.V.. Synthesis and stereospecificity of 4,5-disubstituted oxazolidinone ligands binding to T-box riboswitch RNA. J. Med. Chem. 2011; 54:6786–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Deigan K.E., Ferré-D’Amaré A.R.. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc. Chem. Res. 2011; 44:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seif E., Altman S.. RNase P cleaves the adenine riboswitch and stabilizes pbuE mRNA in Bacillus subtilis. RNA. 2008; 14:1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Condon C., Putzer H., Grunberg-Manago M.. Processing of the leader mRNA plays a major role in the induction of thrS expression following threonine starvation in Bacillus subtilis. Proc. Natl Acad. Sci. U.S.A. 1996; 93:6992–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dar D., Sorek R.. Extensive reshaping of bacterial operons by programmed mRNA decay. PLos Genet. 2018; 14:e1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Newbury S.F., Smith N.H., Higgins C.F.. Differential mRNA stability controls relative gene expression within a polycistronic operon. Cell. 1987; 51:1131–1143. [DOI] [PubMed] [Google Scholar]
- 64. Belasco J.G., Beatty J.T., Adams C.W., Gabain A., Cohen S.N.. Differential expression of photosynthesis genes in R. capsulata results from segmental differences in stability within the polycistronic rxcA transcript. Cell. 1985; 40:171–181. [DOI] [PubMed] [Google Scholar]
- 65. Båga M., Göransson M., Normark S., Uhlin B.E.. Processed mRNA with differential stability in the regulation of E. coli pilin gene expression. Cell. 1988; 52:197–206. [DOI] [PubMed] [Google Scholar]
- 66. Marincola G., Wolz C.. Downstream element determines RNase Y cleavage of the saePQRS operon in Staphylococcus aureus. Nucleic Acids Res. 2017; 45:5980–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deana A., Belasco J.G.. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 2005; 19:2526–2533. [DOI] [PubMed] [Google Scholar]
- 68. Boël G., Letso R., Neely H., Price W.N., Wong K.-H., Su M., Luff J.D., Valecha M., Everett J.K., Acton T.B.et al.. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 2016; 529:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hanson G., Coller J.. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018; 19:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fuhrmann M., Hausherr A., Ferbitz L., Schödl T., Heitzer M., Hegemann P.. Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Mol. Biol. 2004; 55:869–881. [DOI] [PubMed] [Google Scholar]
- 71. Whitfield C.D., Steers E.J., Weissbach H.. Purification and properties of 5-Methyltetrahydropteroyltriglutamate-homocysteine transmethylase. J. Biol. Chem. 1970; 245:390–401. [PubMed] [Google Scholar]
- 72. Figge R.M. Wendisch V.F. Methionine biosynthesis in Escherichia coli and Corynebacterium glutamicum. Amino Acid Biosynthesis Pathways, Regulation and Metabolic Engineering, Microbiology Monographs. 2007; Berlin, Heidelberg: Springer Berlin Heidelberg; 163–193. [Google Scholar]
- 73. Vizcaíno J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T.et al.. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016; 44:D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (73). Data received from protein quantification and half-life determination experiments can be obtained with the dataset identifier PXD020619. Data of the proteome analysis for protein synthesis rate determination can be found with the dataset identifier PXD022677.