


Abstract
Methylglyoxal (MG) is a byproduct of glycolysis that functions in diverse mammalian developmental processes and diseases and in plant responses to various stresses, including salt stress. However, it is unknown whether MG-regulated gene expression is associated with an epigenetic modification. Here we report that MG methylglyoxalates H3 including H3K4 and increases chromatin accessibility, consistent with the result that H3 methylglyoxalation positively correlates with gene expression. Salt stress also increases H3 methylglyoxalation at salt stress responsive genes correlated to their higher expression. Following exposure to salt stress, salt stress responsive genes were expressed at higher levels in the Arabidopsis glyI2 mutant than in wild-type plants, but at lower levels in 35S::GLYI2 35S::GLYII4 plants, consistent with the higher and lower MG accumulation and H3 methylglyoxalation of target genes in glyI2 and 35S::GLYI2 35S::GLYII4, respectively. Further, ABI3 and MYC2, regulators of salt stress responsive genes, affect the distribution of H3 methylglyoxalation at salt stress responsive genes. Thus, MG functions as a histone-modifying group associated with gene expression that links glucose metabolism and epigenetic regulation.
INTRODUCTION
Salt stress is a major environmental factor limiting plant growth and agricultural production; over 800 million hectares of land worldwide and ∼20% of irrigated land are affected by soil salinity (1). When a plant is subjected to salt stress, the Salt Overly Sensitive (SOS) system extrudes Na+, while abscisic acid (ABA)-dependent and -independent pathways improve plant stress tolerance by promoting the expression of salt stress responsive genes (2). Recent reports indicate that salt stress interrupts biological metabolic processes such as glycolysis, thereby changing the distribution of metabolites (1,3). For example, salt stress increases the accumulation of methylglyoxal (MG), a byproduct of the highly conserved glycolysis pathway in animals and plants; however, how MG acts is unknown (4).
MG is ubiquitous in cells and can be degraded through the glyoxalase I/II (GLY I/II)-mediated pathway. While GLY I catalyzes the formation of S-D-lactoylglutathione using MG and glutathione (GSH) as substrates, GLY II splits S-d-lactoylglutathione into d-lactate and GSH, thus degrading MG as d-lactate (5). In Arabidopsis, eleven GLY I and five GLY II genes have been identified and express in different development stages, tissues, and stress conditions according to bioinformatics analysis (6). While the overexpression of GLYI2, GLYI3, GLYI6 or GLYII4 confers plant salt stress tolerance, and mutation of GLYI2 results in reduced salt stress tolerance (7–9). In addition, GLYI4 mutation enhances plant resistance against necrotrophic pathogen (10). MG functions in diverse developmental processes and diseases in mammals (11,12). MG may play its roles through its modification of target proteins because many proteins such as catalase, glyceraldehyde-3-phosphate dehydrogenase, histones, heat shock proteins, superoxide dismutase, myoglobin, and Nav1.8 can be methylglyoxalated as shown in previous reports although these studies have not related the methylglyoxalation of these proteins except Nav1.8 to any biological process and disease (13,14). MG also functions with unknown mechanisms in plant growth and environmental responses (15). A GeneChip microarray analysis suggested that MG elicits changes in gene expression in plants, although the underlying mechanism is unclear (16).
Histones are easily modified because of their high proportion of both lysine and arginine residues. Histone post-translational modifications affect gene expression by altering chromatin structure and recruiting a variety of specific binding proteins, and thus play a crucial role in various physiological processes, including cell differentiation, tissue development, cancer induction, and environmental responses (17–19). Histone methylation and acetylation studied since the 1960s in both mammals and plants, participate in diverse physiological processes by regulating gene expression. For example, histone methylation silences stem/memory genes involved in mammalian immunity and FLC functioning in plant flowering, respectively (20,21). Histone acetylation acts in mammalian spermatogenesis and plant innate immunity by modulating gene expression (22,23).
Metaboloepigenetics has recently emerged to characterize the significance of interactions between epigenetics and metabolism (24–27). Many metabolites, including acetyl-CoA, S-adenosylmethionine, nicotinamide adenine dinucleotide and α-ketoglutarate, act as cofactors for chromatin-modifying enzymes, such as methyltransferases and deacetylases, and thereby determine epigenetic modifications and regulate gene expression (28–31). However, whether and how a particular metabolite can serve as a histone-modifying group to affect gene expression is largely unknown.
In this study, we report that MG, which accumulates in salt-stressed plants, modifies histone H3 associated with the expression of salt stress responsive genes. Following exposure to salt stress, the expression of salt stress responsive genes is higher in Arabidopsis glyI2 mutant, which accumulates more MG than the wild type and has increased H3 methylglyoxalation, but lower in 35S::GLYI2 35S::GLYII4 plants, which have reduced MG accumulation and H3 methylglyoxalation. In addition, we reveal that ABI3 and MYC2 change the distribution of H3 methylglyoxalation at salt stress responsive genes. Thus, we show that MG, a glycolysis metabolite, acts as a novel epigenetic modifier to affect gene expression in plants subjected to abiotic stress.
MATERIALS AND METHODS
Plant materials and growth conditions
Arabidopsis (Arabidopsis thaliana) ecotypes Columbia (Col), Landsberg erecta (Ler) and Wassilewskija (Ws) were used in this study, including mutant lines abi3–4 (CS6130, Ler) (32), abi4–1 (CS8104, Col) (33), abi5–1 (CS8105, Ws) (34), ein3–1 (CS8052, Col) (35), glyI2 (Salk_131547, Col) (8), jin1–9 (Salk_017005, Col) (36), and wrky33–1 (Salk_006603, Col) (37). The areb1 mutant (Salk_138853, Col) was obtained from the Arabidopsis Biological Resource Center and confirmed by PCR. Arabidopsis seeds were surface sterilized with 5% (w/v) bleach for 5 min, washed three times with sterile water, placed at 4°C for 3 days and then placed on agar medium containing 1/2 strength MS medium (supplemented with 0.8% (w/v) agar and 1% (w/v) sucrose, pH 5.8) at 23°C and 100 μmol m–2 s–1 illumination under 16 h light/ 8 h dark conditions.
To generate 35S::GLYI2 35S::GLYII4 transgenic plants, the coding sequences of GLYI2 (AT1G08110) and GLYII4 (AT2G43430) were amplified by PCR from cDNA of wild type (Col) Arabidopsis and cloned into pBARN and pCAMBIA1300S, respectively. The two constructs were successively transformed into Arabidopsis (Col) using Agrobacterium tumefaciens strain pGV3101 and the floral dip method. Primer sequences are listed in Supplementary Table S5.
Quantification of MG levels by HPLC
Endogenous MG levels were quantified by HPLC as previously described (38). Fresh Arabidopsis seedlings (0.5 g) were ground in liquid nitrogen, and the powder was homogenized in 1 ml PBS with 0.5 M HClO4 and 5% (w/v) insoluble polyvinylpyrrolidone. The homogenate was centrifuged at 12 000 g for 10 min, and the supernatant was incubated with 10 mM 1,2-diaminobenzene for 12 h at room temperature. The reaction product 2-methylquinoxaline (2-MQ) was purified using a C18 cartridge (Waters Sep-Pak) and detected at 315 nm using an Agilent 1100 HPLC system equipped with a C18 reversed-phase column (Hypersil ODS2, 5 μm, 4.6 mm × 250 mm, Elite-E2813968). 5-Methylquinoxaline (5-MQ) was used as an internal standard.
RNA extraction and RT-qPCR
As previously described (39), total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions and treated with RQ1 RNase-free DNase I (Promega) to remove contaminating DNA. Reverse transcription was carried out using ReverTra Ace (TOYOBO). qPCR was performed in 96-well plates on a Bio-Rad CFX96 apparatus with SYBR Green I dye (Invitrogen) under the following conditions: 3 min incubation at 95°C for complete denaturation, followed by 40 cycles of 95°C for 15 s and 60°C for 45 s. Protein Phosphatase 2A Subunit A3 (AT1G13320) and Eukaryotic Translation Initiation Factor 4A1 (AT3G13920) were chosen as the best reference genes based on analysis with geNorm software. All experiments included three independent biological replicates and three technical repetitions. Primer sequences are listed in Supplementary Table S5.
RNA-seq analysis
Five-day-old seedlings grown on medium containing 1/2 strength MS, 0.8% (w/v) agar and 1% (w/v) sucrose were transferred to agar medium containing 100 mM NaCl or 200 μM MG for 12 h. An RNA-seq assay was performed on a BGISEQ-500 by the Beijing Genomic Institution (http://www.genomics.org.cn, BGI, Shenzhen, China) with the A. thaliana genome from Tair10 (http://www.arabidopsis.org/) as reference. Data were normalized for variation in sequencing depth and gene length using the FPKM method, and differential expression analysis was performed using the DESeq2 R package (40). FDR < 0.05 and |log2 (fold change)| ≥ 1 defined the threshold for significant differential expression. MapMan software was used for hierarchical cluster analysis of gene expression patterns. RNA-seq experiments included three independent biological replicates.
Preparation of monoclonal antibodies
Monoclonal antibodies against MG-modified proteins were prepared according to a previously described protocol (41). Briefly, female mice were immunized with MG-treated keyhole limpet hemocyanin (Sigma) and the hybridomas were screened using an enzyme-linked immunosorbent assay (ELISA) with MG-treated BSA and native BSA as antigens. To obtain monoclonal antibodies against MG-modified histone H3, female mice were immunized with an MG-treated 40 amino acid peptide of the N-terminal of histone H3 (N’-ARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHR) and hybridomas were screened using ELISA with MG-treated BSA, native BSA, MG-treated peptide and native histone H3 N-terminal peptide as antigens.
Identification of MG-modified proteins
To identify endogenous MG-modified proteins, 5 g Arabidopsis seedlings treated or not with 200 μM MG was ground in liquid nitrogen and the powder was homogenized in two volumes of extraction buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 10% (v/v) glycerol, 0.1% (v/v) Nonidet P-40, 1 mM PMSF and 1× complete protease inhibitor cocktail, Sigma). The homogenate was filtered through four layers of cheesecloth and centrifuged at 12 000 g for 10 min. Homogeneous protein G-agarose suspension (20 μl) was added to the supernatant and incubated for 4 h at 4°C on a rocking platform. After centrifuging at 12 000 g for 20 s to remove pelleted agarose beads, 20 μl 1 mg/ml specific antibody was added to the supernatant and incubated for 2 h at 4°C on a rocking platform. Homogeneous protein G-agarose suspension (100 μl) was then added and incubated for 4 h at 4°C on a rocking platform. After centrifuging at 2000 g for 60 s, agarose–antibody–antigen complexes were collected and washed twice with washing buffer 1 (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, 1× complete protease inhibitor cocktail, Sigma), twice with washing buffer 2 (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 0.1% (v/v) Nonidet P-40, 0.05% (w/v) sodium deoxycholate) and once with washing buffer 3 (50 mM Tris–HCl, pH 7.5, 0.1% (v/v) Nonidet P-40, 0.05% (w/v) sodium deoxycholate). The agarose pellet was resuspended in 50 μl gel-loading buffer and heated to 100°C for 5 min. Proteins were identified by LC–MS/MS analysis from three independent biological replicates.
Preparation of histones from Arabidopsis
Arabidopsis seedlings were ground in liquid nitrogen, and the powder was homogenized in a solution containing 0.4 M sucrose, 10 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 5 mM β-mercaptoethanol and 5 mM sodium butyrate. The homogenate was filtered through four layers of cheesecloth and two layers of Miracloth (Millipore). The filtrate was centrifuged at 12 000 g for 10 min, and the pellet was suspended in 0.25 M sucrose, 10 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 1% (v/v) Triton X-100, 5 mM β-mercaptoethanol and 5 mM sodium butyrate. This centrifugation–suspension step was repeated, with the pellet finally homogenized in a medium containing 1.7 M sucrose, 10 mM Tris–HCl, pH 8.0, 2 mM MgCl2, 0.15% (v/v) Triton X-100, 5 mM β-mercaptoethanol and 5 mM sodium butyrate. The homogenized chromatin was layered over an equal volume of this buffer and centrifuged at 27 000 g for 60 min. The nuclei pellet was resuspended in 0.2 M H2SO4 overnight at 4°C. After centrifuging at 12 000 g for 10 min, histones in the supernatant were precipitated by the addition of 20% (w/v) trichloroacetic acid (TCA) and incubated at −20°C for 4 h. The precipitated histones were centrifuged at 12 000 g for 10 min, and the pellet was washed once with acidified acetone (0.1%, v/v, HCl) and twice with ice-cold acetone. After drying at room temperature, the pellet was dissolved in PBS (pH 7.4).
MNase chromatin-accessibility assays
MNase chromatin-accessibility assays were performed as described (42). Briefly, 2 g Arabidopsis seedlings treated or not with 200 μM MG was ground in liquid nitrogen, and the nuclei were obtained as described above. Nuclei were resuspended in buffer A (20 mM Tris–HCl, pH 7.5, 1.5 mM MgCl2, 340 mM sucrose, 10% (v/v) glycerol) with 5 mM CaCl2, 100 μg/ml BSA and digested with 20 gel units MNase (New England Biolabs) for 0, 2, 5, 10, 15 min. Reactions were terminated by adding 50 mM Tris–HCl, pH 8.0, 1% (w/v) SDS, 40 mM EDTA and 200 mM NaCl. Reactions were digested with 100 μg/ml RNase A and 200 μg/ml Proteinase K at 50°C for 1 h before DNA was extracted with phenol/chloroform.
Peptide synthesis
The peptides poly-l-lysine, poly-l-arginine, H3–40, H3–40-R2A, H3–40-K4A, P1, P2, P3, P4, H3–40-K4me1, H3–40-K4me2, H3–40-K4me3 and H3–40-K4ac were synthesized by the GenScript Corporation (Nanjing, China). Peptide sequences are listed in Supplementary Table S5.
Western blotting with competition assay
Histone proteins were extracted from 5-day-old wild-type seedlings treated or not with 200 μM MG for 12 h, then, 500 ng of histone protein extracts were resolved in SDS-PAGE. H3–40 peptide used for competition assay was incubated with or without 200 μM MG at 37°C for 6 h. To remove the excess MG, H3–40 peptide was precipitated by ice-cold acetone and incubated at −20°C for 4 h, the precipitated H3–40 peptide was centrifuged at 12 000 g for 10 min, and the pellet was washed twice with ice-cold acetone. After drying at room temperature, the pellet was dissolved to 100 μg/ml in PBS (pH 7.4). The competition assay was carried out by using the antibodies (2 μg), which were incubated with indicated peptides (4 μg) for 2 h.
Chromatin immunoprecipitation (ChIP) analysis
Nuclei were obtained as described above, and appropriately sized chromatin fragments were prepared by MNase (New England BioLabs) digestion as previously described (43). ChIP was performed with anti-MMP, anti-H3K4MG, anti-H3K4me1 (Abcam ab8895), anti-H3K4me2 (Abcam ab7766) or anti-H3K4me3 (Abcam ab8580) antibodies according to the protocol described (44).
ChIP DNA samples were sequenced using Illumina HiSeq2000 by the Beijing Genomic Institution (http://www.genomics.org.cn, BGI, Shenzhen, China) with the A. thaliana genome from Tair10 (http://www.arabidopsis.org/) as reference. ChIP-seq tag-enriched regions were identified by SICER with a window size of 200 bp and gap size of 600 bp. A sub-module of the SICER program was further used to predict genomic regions showing significant changes in chromatin accessibility using ChIP-seq data under stringent parameters (FDR < 0.01; fold change > 1). Peak annotation was performed using the R package ChIPseeker (45). ChIP-seq enrichment across TSS, gene body, and TES regions was calculated using deepTools (46). ChIP-seq data of histone marks were downloaded from Gene Expression Omnibus (GEO) (GSE112443: H3, H3K4me2, H3K4me3; GSE128434: H3K4me1).
For ChIP-qPCR, three batches of seedlings grown under identical conditions were mixed for one chromatin sample used for qPCR analysis, and qPCR analysis was repeated three times with ACTIN7 (AT5G09810) and PP2AA3 (AT1G13320) as the negative control. The above experiments were repeated three times as three biological replicates. Primer sequences are listed in Supplementary Table S5.
RESULTS
MG regulates salt stress responsive gene expression
Wild-type Arabidopsis plants have less MG and are less sensitive to salt stress than is glyI2 mutant, in which GLYI2 is mutated by T-DNA insertion and verified by complementary experiments (8). We showed that, similar to salt stress, exogenous MG treatment promoted the accumulation of endogenous MG (Figure 1A). Furthermore, MG accumulated to higher levels in the glyI2 mutant than in wild-type plants subjected to either salt stress or MG application (Figure 1A), suggesting that the GLY is required for salt-induced MG accumulation. Since the degradation of MG is a two-step enzyme-catalyzed reaction comprising GLY I and GLY II, and GLYI2 and GLYII4 are involved in plant salt stress response, we generated the 35S::GLYI2 35S::GLYII4 lines overexpressing both GLYI2 and GLYII4 (Supplementary Figure S1). Indeed, the higher MG accumulation seen in wild-type and glyI2 plants was repressed in 35S::GLYI2 35S::GLYII4 transgenic plants when treated with either salt stress or exogenous MG (Figure 1A).
Figure 1.

MG regulates salt stress responsive gene expression. (A) MG content in 5-day-old wild-type, glyI2, and 35S::GLYI2 35S::GLYII4 seedlings treated or not with 100 mM NaCl or 200 μM MG for 12 h. Data are means ± SD of three independent biological replicates. Different letters indicate significant differences between the annotated columns (P < 0.05 by Tukey's test). FW, fresh weight. (B) Venn diagrams representing the numbers of transcripts upregulated or downregulated by more than 2-fold, with genes shared between treatments overlapping. RNA-seq analysis of 5-day-old wild-type (WT) seedlings treated or not with 100 mM NaCl or 200 μM MG for 12 h. WT-NaCl/WT (NaCl-treated WT relative to WT), WT-MG/WT (MG-treated WT relative to WT). Significant level for the elements in common determined by Fisher's exact test: *P < 0.05; ***P < 0.001. (C and D) Hierarchical clustering of transcripts up- or down-regulated 2-fold or more in wild-type plants under NaCl (C) and MG (D) treatment, and fold changes of these transcripts in glyI2 and 35S::GLYI2 35S::GLYII4 plants. NaCl/Mock (NaCl-treated plants relative to untreated plants), MG/Mock (MG-treated plants relative to untreated plants). (E and F) Expression levels of salt stress responsive genes assayed by RNA-seq (E) and RT-qPCR (F) in 5-day-old wild-type, glyI2 and 35S::GLYI2 35S::GLYII4 seedlings treated or not with 100 mM NaCl for 12 h. FC (fold change of gene expression in NaCl-treated plants relative to untreated plants). Data are means ± SD of three independent biological replicates. Asterisks indicate significant differences in comparison with the wild type (Student's t-test): *P < 0.05; **P < 0.01; ***P < 0.001.
MG may participate in the plant salt stress response by regulating gene expression. We thus analyzed genome-wide transcription using RNA sequencing (RNA-seq). While 715 genes were upregulated and 473 genes were downregulated by >2-fold in salt-treated wild-type seedlings (Supplementary Table S1A and B), MG treatment resulted in upregulation of 994 genes and downregulation of 801 genes (Supplementary Table S1C and D). A total of 217 upregulated and 188 downregulated transcripts were common between NaCl and MG treatments (Figure 1B and Supplementary Table S1E and F) and 30 genes (15 upregulated by NaCl treatment but downregulated by MG treatment, 15 downregulated by NaCl treatment but upregulated by MG treatment) were reversely regulated by NaCl and MG treatments (Figure 1B and Supplementary Table S1G and H). A wide range of functional categories were simultaneously affected by salt stress and MG application, including stress, hormone metabolism, development, and signaling (Supplementary Figure S2), implying that MG may function by modulating gene expression in stressed plants.
This notion was further reinforced by our RNA-seq analysis of the glyI2 mutant and 35S::GLYI2 35S::GLYII4 lines. When wild-type plants were challenged with salt stress, 1183 genes were differentially expressed compared with the untreated control, and these were clustered into six groups by hierarchical clustering based on gene expression profiles (Supplementary Figure S3 and Supplementary Table S2A). When subjected to salt stress, 35S::GLYI2 35S::GLYII4 transgenic plants showed lower fold expression changes of genes in all six clusters than wild-type seedlings, correlated with less MG accumulation in 35S::GLYI2 35S::GLYII4 plants compared with wild-type plants (Figure 1C). Consistently, the glyI2 mutant with higher MG accumulation had higher or similar fold changes in expression of cluster I, IV and II genes (Figure 1C). However, the fold changes in expression of cluster III, V, and VI genes were lower in glyI2 than in the wild type (Figure 1C). This was probably due to excessive MG accumulation in glyI2, which may affect the expression of genes in these clusters. Thus, we subjected MG-treated wild-type, glyI2, and 35S::GLYI2 35S::GLYII4 plants to RNA-seq analysis. Gene expression patterns were similar in plants treated with MG or salt stress (Figure 1D, Supplementary Figure S4 and Supplementary Table S2B).
Finally, we assayed the expression of salt stress responsive genes, including LEA14, ATAF1, UGT74E2, COR15A, MYB2, NCED3, CAX3, ABCG6, KIN1 and RD29B, by reverse-transcription quantitative PCR (RT-qPCR), since changes in the expression of these genes by either overexpression or mutation results in increased or reduced plant salt stress tolerance, respectively. Similar to our RNA-seq data, expression of all these genes was induced in wild-type and glyI2 plants, but this induction was suppressed in 35S::GLYI2 35S::GLYII4 plants challenged with salt stress (Figure 1E and F), confirming the reliability of our RNA-seq data. These data suggest that MG mediates salt stress responsive gene expression.
MG modifies histone H3
MG participates in disease responses in mammals by modifying its target proteins to regulate their activity (13). To explore whether MG also functions in the plant salt stress response by modifying proteins, we generated two monoclonal antibodies (Ab2F8 and Ab8B6) against MG-modified keyhole limpet hemocyanin. These two antibodies recognized MG-modified but not native BSA and glyceraldehyde-3-phosphate dehydrogenase C subunit 1 (GAPC1) (Figure 2A and B). Furthermore, while both antibodies cannot react with the peptides composed of 20 l-arginine or l-lysine residues, Ab2F8 recognized only MG-modified lysine and Ab8B6 only MG-modified arginine, respectively (Figure 2C). Thus, Ab2F8 was renamed anti-MGK (specific for proteins having MG-modified lysine residue(s)) and Ab8B6 as anti-MGR (specific for proteins having MG-modified arginine). We mixed these two antibodies at equal antibody titers as anti-MMP (MG-modified proteins) for subsequent experiments.
Figure 2.

MG modifies histone H3. (A to C) Immunodetection of MG-modified BSA (A), GAPC1 (B), and indicated amounts of poly-l-lysine and poly-l-arginine (C); 10 μg/ml protein or peptide was incubated with or without 200 μM MG at 37°C for 6 h. (D and E) Immunodetection of MG-modified histone H3 using anti-MMP; the histone H3 was purified from E. coli and incubated with or without 200 μM MG at 37°C for 6 h (D), histones were extracted from 5-day-old wild-type seedlings treated or not with 200 μM MG for 12 h, western blotting was carried out with competition of a native or MG-modified H3–40 peptide (E), asterisk indicates the bands of histone H3. (F) Illustration of H3 methylglyoxalation sites in vivo. Gray frames represent sequences detected by LC–MS/MS analysis. All methylglyoxalation sites are shown in red and numbered.
Using anti-MMP and liquid chromatography–mass spectrometry (LC–MS)/MS analysis, we identified 71 potential MG-modified proteins (Supplementary Table S3) involved in glycolysis, the tricarboxylic acid cycle, fat metabolism, photosynthesis, and biotic and abiotic stress responses. In subsequent analyses, we focused on histone H3, one of these modified proteins, because histone modification such as methylation and acetylation is known to regulate gene expression.
To further verify MG modification of histone H3, we assessed methylglyoxalation of plant histone H3 heterologously expressed and purified from Escherichia coli. Anti-MMP reacted with MG-treated but not untreated histone H3 (Figure 2D). Further, endogenous plant histone H3 was MG-modified and this methylglyoxalation was promoted if the seedlings were treated with MG (Figure 2E). In addition, the signal could be efficiently competed away by a MG-modified histone H3 peptide (H3–40), but not an unmodified peptide (Figure 2E), further proving that anti-MMP do not cross react with unmodified histone H3. Combined with the above observation that MG treatment increases MG accumulation (Figure 1A), these data reveal that increased MG accumulation promotes H3 methylglyoxalation. Finally, we assayed methylglyoxalation sites of anti-MMP-enriched plant histone H3 using LC–MS/MS analysis. Fifteen H3 methylglyoxalation sites including K4 were identified (Figure 2F and Supplementary Figure S5). Similar to histone methylation and acetylation, histone methylglyoxalation occurred not only in the N-terminal tail domain, but also in the core domain of histone H3 (Figure 2F).
H3 methylglyoxalation positively correlates with gene expression
The above data showed that MG can modify histone H3 and regulate gene expression, we thus performed micrococcal nuclease (MNase) chromatin-sensitivity assays to explore the effects of MG on chromatin accessibility, an essential component of gene regulation (47). Chromatin from wild-type seedlings treated with MG appeared less condensed, as evidenced by the generation of more mono-, di-, and trinucleosomes than untreated control (Figure 3A). Consistently, we treated the nuclei directly with MG, and found that MG-treated nuclei were more sensitive to MNase digestion (Figure 3B). These results indicated that MG treatment increased chromatin accessibility. To assess whether methylglyoxalation is involved in regulating gene expression, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) using anti-MMP with MG-treated wild-type seedlings (Figure 4). Our experiments identified 8074 peaks (7812 ‘decorated’ genes) using SICER software (48) (Supplementary Table S4A), with peaks mostly in promoters (positioned −1 kb to 0; transcription start sites [TSS] designated as 0), 3′ untranslated regions (UTRs), and exon regions (Figure 5A). Further analyses of methylglyoxalation deposition relative to TSS and transcription end sites (TES) revealed that methylglyoxalation was enriched at gene bodies and peaked at ∼200 bp downstream of TSS (Figure 5B). To investigate the correlation between methylglyoxalation and gene expression level, we divided the 17 215 expressed genes (fragments per kilobase of transcript per million mapped reads [FPKM] ≥ 1) in wild-type seedlings treated with MG into five equal-sized groups according to their expression level. Methylglyoxalation around TSS and TES was found to be positively correlated with gene expression level, with the most highly expressed genes showing the highest level of modification and the least expressed genes showing the lowest level of modification (Figure 5C), suggesting a positive role for methylglyoxalation in regulating gene expression.
Figure 3.
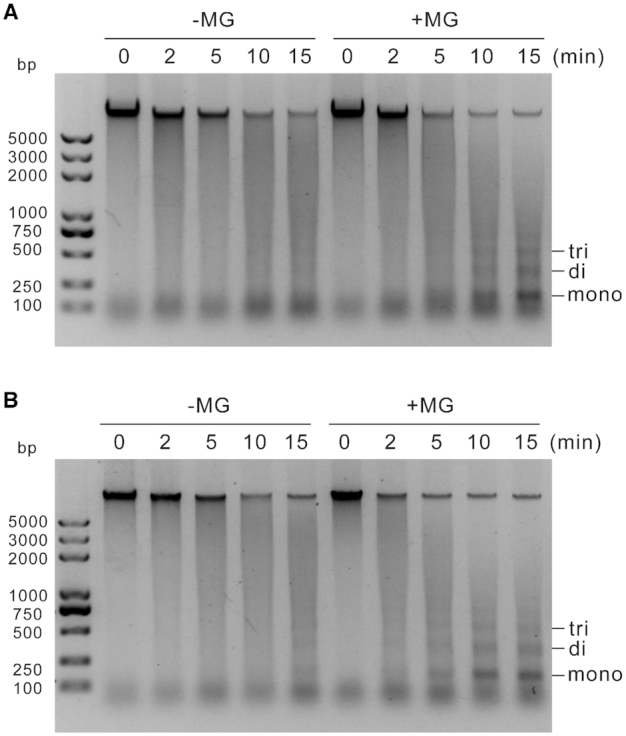
MG increases chromatin accessibility. (A) Chromatin-sensitivity assays were performed with nuclei from 5-day-old wild-type seedlings treated or not with 200 μM MG for 12 h. (B) Chromatin-sensitivity assays were performed with nuclei from 5-day-old wild-type seedlings. The nuclei were incubated with or without 200 μM MG at 37°C for 2 h. Digestions with MNase were performed for 0, 2, 5, 10 and 15 min. Mono-, di- and trinucleosomes are marked.
Figure 4.

ChIP-seq snapshots of MMP, H3K4MG, H3K4me1, H3K4me2, H3K4me3, H3 and input. (A) Genome browser traces on chromosome 1. (B and C) Genome browser traces on regions indicated with red boxes. The genome browser data was visualized with Integrative Genomics Viewer (IGV).
Figure 5.

H3 methylglyoxalation positively correlates with gene expression. (A) Peak annotation in 5-day-old wild-type seedlings treated with 200 μM MG for 12 h. (B) Average normalized ChIP-seq signals of anti-MMP, anti-H3K4MG, anti-H3, anti-H3K4me1, anti-H3K4me2 and anti-H3K4me3 in regions 2 kb upstream and downstream of genes. TSS and TES were aligned. RPKM is calculated as the number of reads mapping per kilobase of genomic region per million mapped reads. (C and H) Average normalized ChIP-seq signals of anti-MMP (C) and anti-H3K4MG (H) in regions 2 kb upstream and downstream of genes. All expressed genes (FPKM ≥1) in wild-type seedlings treated with MG are sorted into five equal-size groups according to their mRNA expression values (FPKM). Top20% stands for the highest expressed genes; 20–40%, 40–60%, and 60–80% are the medium expressed genes; 80–100% represents the lowest expressed genes. (D and E) Immunodetection of H3K4 methylglyoxalation in BSA, GAPC1, histone H3 peptide (H3–40) (D), and H3–40-R2A and H3–40-K4A (E), 10 μg/ml peptide was incubated with or without 200 μM MG at 37°C for 6 h. (F and G) Immunodetection of MG-modified histone H3 using anti-H3K4MG; the histone H3 was purified from E. coli and incubated with or without 200 μM MG at 37°C for 6 h (F), histones were extracted from 5-day-old wild-type seedlings treated or not with 200 μM MG for 12 h, Western blotting was carried out with competition of a native or MG-modified H3–40 peptide (G), asterisk indicates the bands of histone H3. (I) Venn diagram representing the number of target genes shared between anti-MMP and anti-H3K4MG. Significant level for the elements in common determined by Fisher's exact test: ***P < 0.001.
To further explore the role of site-specific methylglyoxalation of histone H3, we obtained a monoclonal antibody (Ab4A2) against MG-modified histone H3 peptide (H3–40). Immunoprecipitation analysis using native and MG-modified BSA, GAPC1 and histone H3 as antigens demonstrated that Ab4A2 recognized only MG-modified histone H3 (Figure 5D). To identify which MG-modified lysine or arginine residue was recognized by Ab4A2, we divided the 40 N-terminal amino acids of histone H3 into four overlapping segments (P1–P4) (Supplementary Figure S6A). Ab4A2 recognized MG-modified P1 but not P2–P4 (Supplementary Figure S6B), revealing H3R2 or H3K4 as the possible recognition site of Ab4A2. Then we synthesized histone H3 peptides in which R2 and K4 were individually replaced with an alanine residue (H3–40-R2A and H3–40-K4A) and the antibody reacted with MG-modified H3–40-R2A but not H3–40-K4A (Figure 5E), indicating that Ab4A2 specifically recognizes the methylglyoxalated K4 of H3 (hence Ab4A2 was renamed anti-H3K4MG). Anti-H3K4MG reacted with methylglyoxalated H3 but not unmodified histone H3 expressed in E. coli (Figure 5F). When histones extracted from wild-type seedlings treated or not with MG were used, anti-H3K4MG detected more histone H3 from MG-treated plants than untreated control, but did not detect any histone H3 when the MG-modified H3–40 peptide is present, demonstrating that this antibody can recognize MG-modified plant histone H3 (Figure 5G).
Then, this anti-H3K4MG was used in our ChIP-seq analyses (Figure 4). Similar to ChIP-seq data using anti-MMP, the 5339 peaks identified using anti-H3K4MG (5239 target genes) were mostly in promoters, 3′ UTRs, and exon regions (Figure 5A and Supplementary Table S4B). H3K4 methylglyoxalation was enriched at gene bodies, peaking ∼200 bp downstream of TSS (Figure 5B), and was positively correlated with gene expression levels (Figure 5H). Moreover, 65% of the H3K4 methylglyoxalation target genes identified with anti-H3K4MG co-localized with methylglyoxalation target genes detected with anti-MMP (Figure 5I). The above results suggested that H3K4 methylglyoxalation was representative of H3 methylglyoxalation.
Since methylation and acetylation generally occur at H3K4, we also tested whether our anti-MMP and anti-H3K4MG antibodies promiscuously recognized methylated or acetylated H3K4. Thus we synthesized H3–40-K4me1, H3–40-K4me2, H3–40-K4me3, and H3–40-K4ac peptides. Our experiments showed that these two antibodies recognized MG-modified H3–40 but not native H3–40, H3–40-K4me1, H3–40-K4me2, H3–40-K4me3 and H3–40-K4ac peptides (Supplementary Figure S7A). As expected, anti-H3K4MG did not react with MG-treated H3–40-K4me1, H3–40-K4me2, H3–40-K4me3 and H3–40-K4ac since H3K4 of these peptides was modified by methylation or acetylation respectively (Supplementary Figure S7B). Additionally, anti-MMP recognized MG-treated H3–40, H3–40-K4me1, H3–40-K4me2, H3–40-K4me3 and H3–40-K4ac but not untreated H3–40 (Supplementary Figure S7B), consistent with the above result that K4 is not the only methylglyoxalation site on histone H3 (Figure 2F). Taken together, our data indicated that anti-MMP and anti-H3K4MG antibodies do not recognize methylated or acetylated H3K4. Methylation of H3K4 is a well-known component of epigenetic regulation, which drives us to compare our ChIP-seq data with the reported H3K4me1, H3K4me2 and H3K4me3 data. The results showed that the distribution of H3K4 methylglyoxaltion was closer to H3K4me1 than to H3K4me2 or H3K4me3 (Figure 4), but the deposition relative to TSS and TES of H3K4 methylglyoxaltion was more similar to H3K4me2 than to H3K4me1 or H3K4me3 (Figure 5B), revealing that the characteristics of H3K4 methylglyoxaltion are different from H3K4 methylation.
H3 methylglyoxalation correlates with salt stress responsive gene expression
To explore whether H3 and H3K4 methylglyoxalation is involved in plant salt stress response, we subjected isolated histones from salt-stressed wild-type, glyI2 and 35S::GLYI2 35S::GLYII4 plants to immunoblot analysis using anti-MMP and anti-H3K4MG. H3 and H3K4 methylglyoxalation was increased under salt stress in wild-type plants, and this increase was further promoted in the glyI2 mutant, which had higher MG level than the wild type, but reduced in 35S::GLYI2 35S::GLYII4 transgenic plants, which had lower MG level (Figure 6A), indicating that salt stress enhances H3 and H3K4 methylglyoxalation by increasing MG.
Figure 6.

H3 methylglyoxalation correlates with salt stress responsive gene expression. (A) Immunodetection of H3 and H3K4 methylglyoxalation in 5-day-old wild-type, glyI2 and 35S::GLYI2 35S::GLYII4 seedlings treated or not with 100 mM NaCl for 12 h. The intensity of each band was measured with an image-processing and analysis software package (Image J). Relative protein levels were normalized against those in wild-type seedlings, which were set to 1. Data are means ± SD of three independent biological replicates. Different letters indicate significant differences between the annotated columns (P < 0.05 by Tukey's test). (B) Schematic representation of regions that were tested using ChIP-qPCR. The black boxes correspond to exons and white boxes to 5’ or 3’ UTR. (C–F) Enrichment of ABCG6 (C), ATAF1 (D), NCED3 (E), and RD29B (F) DNA fragments following ChIP using anti-MMP, anti-H3K4MG, anti-H3K4me1, anti-H3K4me2, and anti-H3K4me3 in 5-day-old wild-type seedlings. ChIP values were normalized to their respective DNA inputs. Experiments were repeated three times with similar results. Data shown represented one biological replicate, and data for the other two biological replicates were shown in Supplementary Figure S9. Error bars indicate ±SE of three technical replicates. Asterisks indicate significant differences in comparison with the negative control (No Ab) (Student's t-test): *P < 0.05; **P < 0.01; ***, P < 0.001. (G–J) Enrichment of ABCG6-P3 (G), ATAF1-P2 (H), NCED3-P6 (I), and RD29B-P1 (J) DNA fragments following ChIP using anti-MMP and anti-H3K4MG in 5-day-old wild-type, glyI2, and 35S::GLYI2 35S::GLYII4 seedlings treated or not with 100 mM NaCl for 12 h. ChIP values were normalized to their respective DNA inputs. Experiments were repeated three times with similar results. Data shown represented one biological replicate, and data for the other two biological replicates were shown in Supplementary Figure S10. Error bars indicate ±SE of three technical replicates. Different letters indicate significant differences between the annotated columns (P < 0.05 by Tukey's test).
Then we carried out ChIP-qPCR to analyze the salt stress responsive genes such as ABCG6, ATAF1, NCED3 and RD29B, which were also assayed by our ChIP-seq with anti-MMP and anti-H3K4MG (Supplementary Figure S8 and Supplementary Table S4). Our results showed that H3 methylglyoxalation was distributed at two or three of the six selected sequence regions within each gene locus (Figure 6B-F and Supplementary Figure S9). Thus, we chose the regions with high enrichment (ABCG6-P3, ATAF1-P2, NCED3-P6 and RD29B-P1) for ChIP-qPCR in wild-type, glyI2, and 35S::GLYI2 35S::GLYII4 plants under salt stress. We found that salt stress promoted H3 methylglyoxalation at these salt stress responsive genes compared with untreated controls (Figure 6G–J and Supplementary Figure S10). Further, consistent with expression levels of salt stress responsive genes in the glyI2 and 35S::GLYI2 35S::GLYII4 lines (Figure 1E and F), enrichment of H3 methylglyoxalation at these genes was higher in glyI2 but lower in 35S::GLYI2 35S::GLYII4 plants than in wild-type seedlings when challenged with salt stress (Figure 6G-J and Supplementary Figure S10). Taken together, our results reveal the positive correlation between H3 methylglyoxalation and MG-mediated expression of salt stress responsive genes.
To investigate whether total H3K4 methylation levels are affected in glyI2 mutant or 35S::GLYI2 35S::GLYII4 lines. We analyzed the changes of H3K4 methylation, and found that both glyI2 and 35S::GLYI2 35S::GLYII4 plants had similar H3K4me1, H3K4me2 and H3K4me3 levels as wild-type plants under stressed or unstressed conditions, respectively (Supplementary Figure S11), suggesting that MG does not affect total H3K4 methylation levels under salt stress. Then, the distributions of H3K4me1, H3K4me2 and H3K4me3 at salt stress responsive genes were detected. The data revealed that the most distribution regions of H3 methylglyoxalation and methylation were different (Figure 6C–F and Supplementary Figure S9), suggesting that H3 methylglyoxalation and methylation may mainly occur at different nucleosomes. We also noticed that few distributions of histone methylglyoxalation and methylation (ATAF1-P2, NCED3-P6, and RD29B-P1) were overlapped. To these overlapped regions, we carried out ChIP-qPCR of H3K4 methylation in wild-type, glyI2, and 35S::GLYI2 35S::GLYII4 plants under salt stress. We found that the enrichment of H3K4 methylation at most assayed regions in glyI2 and 35S::GLYI2 35S::GLYII4 plants was similar to that in wild-type plants (Supplementary Figure S12). Taken together, these results suggest that MG-regulated expression of salt stress responsive genes is highly correlated to histone methylglyoxalation but not or less related to histone methylation.
Transcription factors ABI3 and MYC2 affect the specific distribution of H3 methylglyoxalation at salt stress responsive genes
Like nitric oxide-mediated protein S-nitrosylation both in vitro and in vivo (49), MG can also methylglyoxalate histone H3 (Figure 2D–F). But, while recent reports have identified several enzymes required for protein S-nitrosylation by nitric oxide in vivo (50,51), whether some factors are involved in H3 methylglyoxalation in vivo remains to be verified. Sequence-specific, DNA-binding transcription factors (TFs) can interact with and direct epigenetic factors to specific genomic target loci for histone modification in both mammals and plants (52–54). We therefore examined H3 methylglyoxalation at salt stress responsive genes in seedlings harboring mutations of genes encoding TFs involved in plant salt stress responses, including ABI3, ABI4, ABI5, AREB1, EIN3, MYC2 and WRKY33. ChIP-qPCR analysis revealed that while abi4–1, abi5–1, areb1, ein3–1 and wrky33–1 mutants had similar levels of H3 methylglyoxalation at all tested genes (ABCG6, ATAF1, NCED3 and RD29B) (Supplementary Figure S13), H3 methylglyoxalation at ATAF1, NCED3 and RD29B was repressed under salt stress in jin1–9 (harboring a MYC2 mutation) and abi3–4 compared with wild-type plants (Figure 7A–D and Supplementary Figure S14), suggesting that ABI3 and MYC2 affect the specific distribution of H3 methylglyoxalation at salt stress responsive genes.
Figure 7.

Transcription factors ABI3 and MYC2 affect the specific distribution of H3 methylglyoxalation at salt stress responsive genes. (A–D) Enrichment of ATAF1-P2 (A), NCED3-P6 (B), RD29B-P1 (C) and ABCG6-P3 (D) DNA fragments following ChIP using anti-MMP and anti-H3K4MG in 5-day-old Col, Ler, abi3–4, and jin1–9 seedlings treated or not with 100 mM NaCl for 12 h. ChIP values were normalized to their respective DNA inputs. Experiments were repeated three times with similar results. Data shown represented one biological replicate, and data for the other two biological replicates were shown in Supplementary Figure S14. Error bars indicate ±SE of three technical replicates. Asterisks indicate significant differences in comparison with the wild type (Student's t-test): *P < 0.05; **P < 0.01; ***P < 0.001. (E) MG content in 5-day-old Col, Ler, abi3–4, and jin1–9 seedlings treated or not with 100 mM NaCl for 12 h. Data are means ± SD of three independent biological replicates. Different letters indicate significant differences between the annotated columns (P < 0.05 by Tukey's test).
These results, together with our observations that both jin1–9 and abi3–4 mutants had similar or higher MG accumulation but reduced expression of salt stress responsive genes compared with wild-type plants when subjected to salt stress (Figure 7E and Supplementary Figure S15), further support the notion that H3 methylglyoxalation correlates with MG-mediated expression of salt stress responsive genes in stressed plants.
DISCUSSION
Soil salinity seriously hinders plant growth and development. Many genes that improve survival under high salinity are upregulated in plants grown in saline soil. Overexpression of salt stress responsive genes such as ATAF1 and UGT74E2 confers tolerance to salt stress (55,56), and mutation of salt stress responsive genes, including ABCG6 and CAX3, reduces plant salt stress tolerance (57,58). Salt stress increases the accumulation of MG (4), and exogenous MG application modulates gene expression (16). In this study, we showed that salt stress upregulates the expression of the salt stress responsive genes via a mechanism that relies on MG accumulation. Under salt stress conditions, the expression of these genes was repressed in 35S::GLYI2 35S::GLYII4 overexpression lines, which accumulate less MG than the wild type, but elevated in the glyI2 mutant, which accumulates more MG than the wild type.
MG participates in diverse biological processes and diseases in mammals. For example, clinical studies show that MG is increased 5- to 6-fold and 3- to 4-fold in blood samples of patients with type 1 and type 2 diabetes mellitus, respectively (59), and overexpression of glyoxalase I improves diabetes-induced impairment of vasorelaxation and prevents damages of endothelial dysfunction in diabetic rats (60). MG may function by modifying its target proteins; for instance, methylglyoxalation of voltage-gated sodium channel Nav1.8 functions in diabetic hyperalgesia (i.e., exaggerated responses to painful stimuli) (13). MG also functions in various plant development processes and stress responses, but MG-modified proteins have not yet been identified in plants. In this study, we identified 71 potentially methylglyoxalated plant proteins involved in glycolysis, the tricarboxylic acid cycle, fat metabolism, photosynthesis, respiration, and biotic and abiotic stress responses. Thus, MG may act as a key node of sugar metabolism and other biological processes. One of these proteins is histone H3, which can be methylglyoxalated in vivo. Salt stress enhanced H3 methylglyoxalation at the salt stress responsive genes and the expression. Furthermore, the expression of these genes was elevated in glyI2 mutant plants with increased MG accumulation, but repressed in 35S::GLYI2 35S::GLYII4 plants having reduced MG accumulation, compared with salt-challenged wild-type plants, suggesting that MG functions as a histone epigenetic modifier to affect the expression of salt stress responsive genes. This greatly enhances our understanding of metaboloepigenetics; in addition to acting as cofactors for chromatin-modifying enzymes (29,30), certain metabolites may also modify histones and thereby regulate gene expression. Moreover, as histones, MG, and glycolysis are conserved in plants and mammals, we predict that MG may regulate gene expression through histone modification in mammals and contribute to the progression of various diseases.
Sequence-specific DNA-binding factors trigger specific histone modifications by recruiting epigenetic factors in both mammals and plants. For example, the Arabidopsis transcription factor VAL1 targets Polycomb repressive complex 2 to a region of FLC for trimethylation of H3K27, which represses FLC transcription (54). Thus, transcription factors involved in plant salt stress responses may also modulate H3 methylglyoxalation at salt stress responsive genes in stressed plants. Indeed, our results show that ABI3 and MYC2 affect the distribution of methylglyoxalated H3 at salt stress responsive genes, possibly by recruiting currently unidentified MG modifier(s) through their direct or indirect interaction.
DATA AVAILABILITY
All the sequencing data have been deposited in the Gene Expression Omnibus (GEO) with the accession code GSE124328.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Arabidopsis Biological Resource Center for providing seeds.
Authors’ contributions: Z.-W.F. and Y.-T.L. conceived and designed the experiments; Z.-W.F. performed most of the experiments; J.-H.L. measured the MG content; Y.-R.F. and X.Y. participated in the ChIP-qPCR experiments; Z.-W.F. and Y.-T.L. wrote the paper.
Contributor Information
Zheng-Wei Fu, State Key Laboratory of Hybrid Rice, College of Life Sciences, Wuhan University, Wuhan 430072, China.
Jian-Hui Li, State Key Laboratory of Hybrid Rice, College of Life Sciences, Wuhan University, Wuhan 430072, China.
Yu-Rui Feng, State Key Laboratory of Hybrid Rice, College of Life Sciences, Wuhan University, Wuhan 430072, China.
Xiao Yuan, State Key Laboratory of Hybrid Rice, College of Life Sciences, Wuhan University, Wuhan 430072, China.
Ying-Tang Lu, State Key Laboratory of Hybrid Rice, College of Life Sciences, Wuhan University, Wuhan 430072, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31830007]. Funding for open access charge: National Natural Science Foundation of China [31830007].
Conflict of interest statement. None declared.
REFERENCES
- 1. Che-Othman M.H., Millar A.H., Taylor N.L.. Connecting salt stress signalling pathways with salinity-induced changes in mitochondrial metabolic processes in C3 plants. Plant Cell Environ. 2017; 40:2875–2905. [DOI] [PubMed] [Google Scholar]
- 2. Zhu J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002; 53:247–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Silveira J.A.G., Carvalho F.E.L.. Proteomics, photosynthesis and salt resistance in crops: An integrative view. J. Proteomics. 2016; 143:24–35. [DOI] [PubMed] [Google Scholar]
- 4. Mustafiz A., Ghosh A., Tripathi A.K., Kaur C., Ganguly A.K., Bhavesh N.S., Tripathi J.K., Pareek A., Sopory S.K., Singla-Pareek S.L.. A unique Ni2+ -dependent and methylglyoxal-inducible rice glyoxalase I possesses a single active site and functions in abiotic stress response. Plant J. Cell Mol. Biol. 2014; 78:951–963. [DOI] [PubMed] [Google Scholar]
- 5. Kaur C., Singla-Pareek S.L., Sopory S.K.. Glyoxalase and methylglyoxal as biomarkers for plant stress tolerance. Crit. Rev. Plant Sci. 2014; 33:429–456. [Google Scholar]
- 6. Mustafiz A., Singh A.K., Pareek A., Sopory S.K., Singla-Pareek S.L.. Genome-wide analysis of rice and Arabidopsis identifies two glyoxalase genes that are highly expressed in abiotic stresses. Funct. Integr. Genomics. 2011; 11:293–305. [DOI] [PubMed] [Google Scholar]
- 7. Batth R., Jain M., Kumar A., Nagar P., Kumari S., Mustafiz A.. Zn2+ dependent glyoxalase I plays the major role in methylglyoxal detoxification and salinity stress tolerance in plants. PLoS One. 2020; 15:e0233493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaur C., Tripathi A.K., Nutan K.K., Sharma S., Ghosh A., Tripathi J.K., Pareek A., Singla-Pareek S.L., Sopory S.K.. A nuclear-localized rice glyoxalase I enzyme, OsGLYI-8, functions in the detoxification of methylglyoxal in the nucleus. Plant J. Cell Mol. Biol. 2017; 89:565–576. [DOI] [PubMed] [Google Scholar]
- 9. Devanathan S., Erban A., Perez-Torres R. Jr, Kopka J., Makaroff C.A.. Arabidopsis thaliana glyoxalase 2-1 is required during abiotic stress but is not essential under normal plant growth. PLoS One. 2014; 9:e95971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Proietti S., Caarls L., Coolen S., Van Pelt J.A., Van Wees S.C.M., Pieterse C.M.J. Genome-wide association study reveals novel players in defense hormone crosstalk in Arabidopsis. Plant Cell Environ. 2018; 41:2342–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moraru A., Wiederstein J., Pfaff D., Fleming T., Miller A.K., Nawroth P., Teleman A.A.. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metab. 2018; 27:926–934. [DOI] [PubMed] [Google Scholar]
- 12. Hanssen N.M.J., Scheijen J., Jorsal A., Parving H.H., Tarnow L., Rossing P., Stehouwer C.D.A., Schalkwijk C.G.. Higher plasma methylglyoxal levels are associated with incident cardiovascular disease in individuals with type 1 Diabetes: A 12-Year Follow-up study. Diabetes. 2017; 66:2278–2283. [DOI] [PubMed] [Google Scholar]
- 13. Bierhaus A., Fleming T., Stoyanov S., Leffler A., Babes A., Neacsu C., Sauer S.K., Eberhardt M., Schnolzer M., Lasitschka F.et al.. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012; 18:926–933. [DOI] [PubMed] [Google Scholar]
- 14. Galligan J.J., Wepy J.A., Streeter M.D., Kingsley P.J., Mitchener M.M., Wauchope O.R., Beavers W.N., Rose K.L., Wang T., Spiegel D.A.et al.. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. PNAS. 2018; 115:9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hoque T.S., Hossain M.A., Mostofa M.G., Burritt D.J., Fujita M., Tran L.S.. Methylglyoxal: an emerging signaling molecule in plant abiotic stress responses and tolerance. Front. Plant Sci. 2016; 7:1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaur C., Kushwaha H.R., Mustafiz A., Pareek A., Sopory S.K., Singla-Pareek S.L.. Analysis of global gene expression profile of rice in response to methylglyoxal indicates its possible role as a stress signal molecule. Front. Plant Sci. 2015; 6:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheung P., Vallania F., Warsinske H.C., Donato M., Schaffert S., Chang S.E., Dvorak M., Dekker C.L., Davis M.M., Utz P.J.et al.. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell. 2018; 173:1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siklenka K., Erkek S., Godmann M., Lambrot R., McGraw S., Lafleur C., Cohen T., Xia J., Suderman M., Hallett M.et al.. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science. 2015; 350:aab2006. [DOI] [PubMed] [Google Scholar]
- 19. Crevillen P., Yang H., Cui X., Greeff C., Trick M., Qiu Q., Cao X., Dean C.. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature. 2014; 515:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pace L., Goudot C., Zueva E., Gueguen P., Burgdorf N., Waterfall J.J., Quivy J.P., Almouzni G., Amigorena S.. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science. 2018; 359:177–186. [DOI] [PubMed] [Google Scholar]
- 21. Yang H., Berry S., Olsson T.S.G., Hartley M., Howard M., Dean C.. Distinct phases of Polycomb silencing to hold epigenetic memory of cold in Arabidopsis. Science. 2017; 357:1142–1145. [DOI] [PubMed] [Google Scholar]
- 22. Qian M.X., Pang Y., Liu C.H., Haratake K., Du B.Y., Ji D.Y., Wang G.F., Zhu Q.Q., Song W., Yu Y.et al.. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell. 2013; 153:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding B., Bellizzi Mdel R., Ning Y., Meyers B.C., Wang G.L.. HDT701, a histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell. 2012; 24:3783–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z., Long H., Chang C., Zhao M., Lu Q.. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: a comprehensive overview. Cell. Mol. Life Sci.: CMLS. 2018; 75:3353–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Netea M.G., Joosten L.A., Latz E., Mills K.H., Natoli G., Stunnenberg H.G., O’Neill L.A., Xavier R.J.. Trained immunity: a program of innate immune memory in health and disease. Science. 2016; 352:aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohli R.M., Zhang Y.. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013; 502:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaelin W.G. Jr, McKnight S.L.. Influence of metabolism on epigenetics and disease. Cell. 2013; 153:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thienpont B., Steinbacher J., Zhao H., D’Anna F., Kuchnio A., Ploumakis A., Ghesquiere B., Van Dyck L., Boeckx B., Schoonjans L.et al.. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016; 537:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ryall J.G., Dell’Orso S., Derfoul A., Juan A., Zare H., Feng X., Clermont D., Koulnis M., Gutierrez-Cruz G., Fulco M.et al.. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015; 16:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shyh-Chang N., Locasale J.W., Lyssiotis C.A., Zheng Y., Teo R.Y., Ratanasirintrawoot S., Zhang J., Onder T., Unternaehrer J.J., Zhu H.et al.. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013; 339:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wellen K.E., Hatzivassiliou G., Sachdeva U.M., Bui T.V., Cross J.R., Thompson C.B.. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009; 324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giraudat J., Hauge B.M., Valon C., Smalle J., Parcy F., Goodman H.M.. Isolation of the Arabidopsis ABI3 gene by positional cloning. Plant Cell. 1992; 4:1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finkelstein R.R., Wang M.L., Lynch T.J., Rao S., Goodman H.M.. The Arabidopsis abscisic acid response locus ABI4 encodes an APETALA 2 domain protein. Plant Cell. 1998; 10:1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finkelstein R.R., Lynch T.J.. The Arabidopsis abscisic acid response gene ABI5 encodes a basic leucine zipper transcription factor. Plant Cell. 2000; 12:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo H., Ecker J.R.. Plant responses to ethylene gas are mediated by SCF(EBF1/EBF2)-dependent proteolysis of EIN3 transcription factor. Cell. 2003; 115:667–677. [DOI] [PubMed] [Google Scholar]
- 36. Anderson J.P., Badruzsaufari E., Schenk P.M., Manners J.M., Desmond O.J., Ehlert C., Maclean D.J., Ebert P.R., Kazan K.. Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell. 2004; 16:3460–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang Y., Deyholos M.K.. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 2009; 69:91–105. [DOI] [PubMed] [Google Scholar]
- 38. Dhar A., Desai K., Liu J., Wu L.. Methylglyoxal, protein binding and biological samples: are we getting the true measure. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009; 877:1093–1100. [DOI] [PubMed] [Google Scholar]
- 39. Fu Z.W., Wang Y.L., Lu Y.T., Yuan T.T.. Nitric oxide is involved in stomatal development by modulating the expression of stomatal regulator genes in Arabidopsis. Plant Sci. 2016; 252:282–289. [DOI] [PubMed] [Google Scholar]
- 40. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oya T., Hattori N., Mizuno Y., Miyata S., Maeda S., Osawa T., Uchida K.. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999; 274:18492–18502. [DOI] [PubMed] [Google Scholar]
- 42. Sakabe K., Wang Z., Hart G.W.. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. PNAS. 2010; 107:19915–19920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O’Neill L.P., Turner B.M.. Immunoprecipitation of native chromatin: NChIP. Methods. 2003; 31:76–82. [DOI] [PubMed] [Google Scholar]
- 44. Gendrel A.V., Lippman Z., Martienssen R., Colot V.. Profiling histone modification patterns in plants using genomic tiling microarrays. Nat. Methods. 2005; 2:213–218. [DOI] [PubMed] [Google Scholar]
- 45. Yu G., Wang L.G., He Q.Y.. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015; 31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 46. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ucar D., Márquez E.J., Chung C.H., Marches R., Rossi R.J., Uyar A., Wu T.C., George J., Stitzel M.L., Palucka A.K.et al.. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J. Exp. Med. 2017; 214:3123–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zang C., Schones D.E., Zeng C., Cui K., Zhao K., Peng W.. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009; 25:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee S.B., Kim C.K., Lee K.-H., Ahn J.-Y.. S-nitrosylation of B23/nucleophosmin by GAPDH protects cells from the SIAH1–GAPDH death cascade. J. Cell Biol. 2012; 199:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu C., Parrott A.M., Liu T., Beuve A., Li H.. Functional proteomics approaches for the identification of transnitrosylase and denitrosylase targets. Methods. 2013; 62:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morris G., Berk M., Klein H., Walder K., Galecki P., Maes M.. Nitrosative stress, hypernitrosylation, and autoimmune responses to nitrosylated proteins: new pathways in neuroprogressive disorders including depression and chronic fatigue syndrome. Mol. Neurobiol. 2017; 54:4271–4291. [DOI] [PubMed] [Google Scholar]
- 52. Deng X., Qiu Q., He K., Cao X.. The seekers: how epigenetic modifying enzymes find their hidden genomic targets in Arabidopsis. Curr. Opin. Plant Biol. 2018; 45:75–81. [DOI] [PubMed] [Google Scholar]
- 53. Li S., Lin Y.-C.J., Wang P., Zhang B., Li M., Chen S., Shi R., Tunlaya-Anukit S., Liu X., Wang Z.et al.. The AREB1 transcription factor influences histone acetylation to regulate drought responses and tolerance in Populus trichocarpa. Plant Cell. 2019; 31:663–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Questa J.I., Song J., Geraldo N., An H., Dean C.. Arabidopsis transcriptional repressor VAL1 triggers Polycomb silencing at FLC during vernalization. Science. 2016; 353:485–488. [DOI] [PubMed] [Google Scholar]
- 55. Liu Y., Sun J., Wu Y.. Arabidopsis ATAF1 enhances the tolerance to salt stress and ABA in transgenic rice. J. Plant Res. 2016; 129:955–962. [DOI] [PubMed] [Google Scholar]
- 56. Tognetti V.B., Van Aken O., Morreel K., Vandenbroucke K., van de Cotte B., De Clercq I., Chiwocha S., Fenske R., Prinsen E., Boerjan W.et al.. Perturbation of indole-3-butyric acid homeostasis by the UDP-glucosyltransferase UGT74E2 modulates Arabidopsis architecture and water stress tolerance. Plant Cell. 2010; 22:2660–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yadav V., Molina I., Ranathunge K., Castillo I.Q., Rothstein S.J., Reed J.W.. ABCG transporters are required for suberin and pollen wall extracellular barriers in Arabidopsis. Plant Cell. 2014; 26:3569–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao J., Barkla B.J., Marshall J., Pittman J.K., Hirschi K.D.. The Arabidopsis cax3 mutants display altered salt tolerance, pH sensitivity and reduced plasma membrane H+-ATPase activity. Planta. 2008; 227:659–669. [DOI] [PubMed] [Google Scholar]
- 59. Lu J., Randell E., Han Y., Adeli K., Krahn J., Meng Q.H.. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin. Biochem. 2011; 44:307–311. [DOI] [PubMed] [Google Scholar]
- 60. Geoffrion M., Du X., Irshad Z., Vanderhyden B.C., Courville K., Sui G., D’Agati V.D., Ott-Braschi S., Rabbani N., Thornalley P.J.et al.. Differential effects of glyoxalase 1 overexpression on diabetic atherosclerosis and renal dysfunction in streptozotocin-treated, apolipoprotein E-deficient mice. Physiol. Rep. 2014; 2:e12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the sequencing data have been deposited in the Gene Expression Omnibus (GEO) with the accession code GSE124328.