

Abstract
Infections are the second major cause of mortality in patients with kidney disease and accompanying uremia. Both vascular access and non-access related infections contribute equally to the infection-related deaths in patients with kidney disease. Dialysis is the most common cause of systemic infection by Candida albicans in these patients. C. albicans also reside in the gastro-intestinal tract as a commensal fungus. However, the contribution of gut-derived C. albicans in non-access related infections in kidney disease is unknown. Using a mouse model of kidney disease, we demonstrate that uremic animals showed increased gut barrier permeability, impaired mucosal defense and dysbiosis. The disturbance in gut homeostasis is sufficient to drive the translocation of microbiota and intestinal pathogen Citrobacter rodentium to extra-intestinal sites, but not C. albicans. Interestingly, majority of uremic animals showed fungal translocation only when the gut barrier integrity is disrupted. Our data demonstrate that uremia coupled with gut mucosal damage may aid in the translocation of C. albicans and cause systemic infection in kidney disease. Since most of the individuals with kidney disease suffer from some form of gut mucosal damage, these results have important implications in the risk-stratification and control of non-access related opportunistic fungal infections in these patients.
Introduction
Kidney disease is a major public health problem in the 21st century (1). Infection is the 2nd leading cause of mortality (20%) in patients with kidney disease (2, 3). Although vascular access-related infections are the primary cause of infection-related deaths in kidney disease, non-access related infectious complications while equally important are often overlooked (4). A study showed that infection-related hospitalizations are not due to vascular access in 77% of identified cases, rather the most common reason was an infection of unknown source (5). Even though advances in dialysis techniques have lowered the rate of access-related infections over the past decade, prevention of non-access related infections is still a major problem. In most cases, the source of non-access related infections in kidney disease remains unidentified, which is a major constraint in controlling infections in these patients.
Several predisposing factors make patients with kidney disease susceptible to infection. (6–8). These include dialysis access, presence of coexisting illnesses, vaccine hypo-responsiveness, immunosuppressive therapy and uremia. Uremia is characterized by the retention of ~900 metabolites in the blood in the absence of kidney function. A subset of these (∼100) have profound impact on various biological systems and are termed uremic toxins (9). Uremia is implicated in immune dysfunction and is considered as an independent risk factor for infections independent of vascular access (7, 10). While the role of uremia in access-related infections has been extensively studied (11, 12), nothing is known about the impact of uremia in non-access related infections.
The gastro-intestinal (GI) tract harbors trillions of microorganisms referred to as the gut microbiota, which play important role in digestion and host metabolism (13). The microbiota is also implicated in the development of metabolic, inflammatory and infectious diseases, if gut homeostasis is altered (14, 15). The intestinal epithelial cells maintain the symbiotic relationship between the microbiota and the host by separating them. The mucosal barrier function is regulated by continuous interaction between the microbiota and host immune cells. Such interactions are also critical for maintaining the balance between symbionts and pathobionts and prevent dysbiosis. Hence, altered gut homeostasis aids in the leakage of microorganisms into the circulation leading to systemic infections and inflammatory diseases.
Accumulating evidence indicates that crosstalk between host and microbiota is patho-physiologically relevant in kidney disease. Uremia impacts both the composition and metabolism of the gut microbiota (16). Additionally, 80% of all uremic patients show GI manifestations, where the gut epithelial lining is damaged (17, 18). On the other hand, many uremic toxins originated from microbial metabolism cause damage to renal and vascular cells (16, 19). The past decade has seen major effort in understanding the role of gut-derived uremic toxins in kidney and vascular disease. However, not much emphasis has been placed on investigating whether gut microorganisms can cause non-access related opportunistic infections in uremia.
Candida albicans causes a severe nosocomial systemic infection known as disseminated candidiasis (20). C. albicans accounts for 79% of all systemic fungal infections in patients with kidney disease (21). Invasive medical procedures are the major sources of disseminated candidiasis in these patients (21). C. albicans is also a commensal fungus in the GI tract of humans (22). In mouse models, the fungus generally resides in the GI tract and does not translocate unless the animals show neutropenia and compromised gut barrier integrity (23–25). However, it is unclear whether commensal C. albicans can cause systemic infection in uremia.
In this report, using a well validated mouse model of kidney disease and associated uremia, we demonstrate that disruption of gut mucosal layer in uremia may aid in the translocation of C. albicans and cause systemic infection. Collectively, these results highlight the role of commensal C. albicans in causing non-vascular access related opportunistic infection in a subset of kidney disease patients, where uremia is coupled with gut mucosal damage.
Materials and Methods
Mice
C57BL/6NTac (WT) male mice were purchased from Taconic Biosciences, Inc. (Germantown, NY). All mice were housed under specific pathogen-free conditions under the supervision of Division of laboratory animal resources (DLAR). All animal experiments were conducted following National Institute of Health (NIH) guidelines under protocols approved by the University of Pittsburgh IACUC (Protocol # 20087922).
Aristolochic acid I nephropathy (AAN)
To induce kidney dysfunction, mice were injected intraperitoneally with 10 mg/kg of Aristolochic acid I (AAI) (Sigma). Mice in chemical control group were injected intraperitoneally (ip) with 10 mg/kg of Aristolochic acid II (AAII) (Sigma) and control animals received intraperitoneal injection of equal volume of PBS. For the pharmacological inhibition study, mice were treated as follows: (1) A single ip injection of AAI at 10 mg/kg. (2) The probenecid + AAI group mice were ip injected with probenecid at 150 mg/kg bodyweight, 30 min before the administration of AAI. (3) The probenecid only group mice were ip injected with probenecid at 150 mg/kg.
DSS mediated gut epithelial injury
Mice received 2.5% Dextran sulphate sodium (DSS) (36,000–50,000 M.W) (MP Biomedicals) in their drinking water for 5 days. Control animals received autoclaved water for the entire period. Mice were monitored daily for body weight.
C. albicans intestinal colonization
Mice were orally gavaged with 2 × 108 CFU of C. albicans (strain SC5314). For establishment of C. albicans intestinal colonization, mice were supplemented with ampicillin (1 mg/ml) in drinking water two days prior to oral C. albicans gavage. Thereafter, mice were maintained on ampicillin supplemented drinking water throughout the experiment. C. albicans colonization in GI tract was enumerated by culturing fecal contents on YPD agar supplemented with 0.010 mg/ml vancomycin and 0.100 mg/ml gentamicin. Liver and spleen tissues were homogenized and plated on YPD agar for determination of systemic dissemination of C. albicans.
C. rodentium infection
Mice were infected by oral gavage with 2×109 colony-forming units of Citrobacter rodentium (strain DBS100). Mice were weighed daily and monitored for signs of illness or distress. Bacterial counts were determined in freshly collected fecal pellets or aseptically collected liver and spleens by homogenization in PBS, followed by plating of serial dilutions of the homogenate on MacConkey agar plates.
Unilateral ureteral obstruction (UUO)
A unilateral ureteral obstruction (UUO) kidney disease model was induced in 8 weeks old WT male mice by left ureteral ligation.
Measurement of serum BUN and Creatinine levels
Serum blood urea nitrogen (BUN) was measured using Blood Urea Nitrogen Enzymatic kit (Bioo Scientific Corp.) and Creatinine levels were measured with QuantiChrom Creatinine Assay kit (BioAssay Systems).
FITC Dextran Assay for Intestinal Permeability
Mice were off fed for four hours prior to FITC dextran gavage. FITC-dextran (4kDa, Sigma) was resuspended in PBS at a concentration of 100 mg/ml and orally gavaged to each mouse at a dose of 40 mg/100 g body weight. Four hours later, mice were euthanized, and blood was collected immediately by retro-orbital bleeding in tubes containing anti-coagulant. Plasma was isolated from blood samples. 100 μL of plasma was added to a black 96-well microplate in duplicate. Concentration of FITC in plasma was determined by spectrophoto fluorometry with an excitation of 485 nm (20 nm band width) and an emission wavelength of 528 nm (20 nm band width) using as standard serially diluted FITC-dextran. Plasma from mice gavaged with PBS was used to determine background.
Analysis of bacterial or C. albicans translocation
Mice were euthanized and aseptically collected liver and spleen samples were homogenized in PBS. Tissue homogenates were plated on Brain Heart Infusion (BHI) agar and YPD agar plates analysis of bacterial and fungal translocation, respectively.
Preparation of tissue cell suspensions and flow cytometric analysis
Spleen and MLN were harvested from mice and subjected to mechanical dissociation to prepare single cell suspensions, followed by RBC lysis by ACK lysing buffer (Thermo Fisher Scientific). SILP leukocytes were isolated. Briefly, peyer’s patches were carefully excised from 10 cm piece of terminal part of small intestine. Intestinal tissues were opened longitudinally and washed with HBSS to remove luminal contents. Tissues were incubated with 20 ml of HBSS containing 5mM EDTA for 20 min at 37°C in a shaking incubator to remove epithelial cells. Tissues were cut into small pieces and incubated in 10 mL of RPMI1640 containing 0.3 mg/mL collagenase D and 0.1 mg/mL DNase I for 20 min at 37°C in a shaking water bath. 10% FBS was added to stop activity of digesting enzymes and tissue suspension was passed through 70 microns cell strainer. Cell suspension was washed twice with RPMI containing 10% FBS and passed through 40 microns cell strainer for removal of tissue debris.
RNA extraction and Real time PCR
RNA was extracted from intestinal tissues using RNeasy kits (Qiagen, Valencia, CA). Complementary DNA was synthesized by SuperScript III First Strand Kits (Thermo Fisher Scientific, Waltham MA). Quantitative real-time PCR (qPCR) was performed with the PerfeCTa SYBR Green FastMix (Quanta BioSciences, Beverly, MA) and analyzed on an ABI 7300 real-time instrument. Primers were from obtained QuantiTect Primer Assays (Qiagen). The expression of each gene was normalized to that of GAPDH.
Gut microbiome sequencing and analysis
Fecal DNA were isolated using QiAMP stool DNA extraction kit. Microbial community analysis utilized PCR amplification of the of 16S rRNA followed by sequencing on an Illumina MiSeq. Sequences from the Illumina MiSeq were deconvolved and then processed through the Center for Medicine and the Microbiome (CMM) in house sequence quality control pipeline, which includes dust low complexity filtering, quality value (QV) trimming, and trimming of primers used for 16S rRNA gene amplification, and minimum read length filtering. Forward and reverse reads were merged into contigs then processed through the CMM’s Mothur based 16S clustering and sequence annotation pipeline. Sequence taxonomic classifications was performed with the Ribosomal Database Project (RDP). Naive Bayesian Classifier with the Silva reference database. Microbiota profiles were statistically quantified and analyzed using three distinct methods at the genus taxonomic level: Distance-based methods (intersample difference) utilized the Manhattan distance metric, distribution-based methods (e.g. diversity) utilized the Tail statistic and Shannon index, and abundance-based methods utilized the additive-log ratio (alr) transformation. Sample time points (baseline and 10 days post AAI injection) from the same subject were paired and linear models were fit with the paired differences as the response (paired analyses).
Histology
The terminal ileum and colon tissues was fixed with 10% buffered formaldehyde and embedded in paraffin. Slices of 4 μm thick were made, stained with hematoxylin and eosin (H&E), and then observed by pathologist blinded to this study design with light microscopy.
Immunofluroscence staining
Sections from frozen SI and colon were fixed in 4% paraformaldehyde and stained with ZO-1 primary antibody (Invitrogen). The secondary antibody used was goat anti-rabbit Cy3. Sections were subsequently stained with DAPI nuclear stain. Images acquired with EVOS FL Auto microscope (Life Technologies).
Statistical analysis
All data are expressed as Mean ± SD. Statistical analyses were performed using the ANOVA, Mann-Whitney or unpaired Student’s t test through Graphpad Prism 7 program. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. All experiments were performed a minimum of twice in independently performed replicates.
Results
Uremic mice show increased gut permeability
Using a mouse model of Aristolochic Acid I nephropathy (AAN) (26), we tested whether uremia negatively impacts gut homeostasis and cause disseminated candidiasis. Here, mice injected with Aristolochic Acid I (AAI) show kidney dysfunction and uremia (Fig 1A). Control animals received PBS. AAII was used as a non-nephrotoxic control. We first measured the impact of uremia on intestinal barrier function in mice with AAN. Uremic mice showed a ten-fold increase in the gut barrier permeability than controls (Fig 1B). The increase in barrier permeability was highest around 6–10 days post AAI injection, timepoint at which blood urea nitrogen (BUN) level peaked in the uremic group, as shown before (26) (Fig 1C and D). Consequently, uremic animals showed reduced expression of ZO-1 tight junction protein in the epithelial lining of small intestine (SI) and colon (Fig 1E). There was an increase in the expression of few tight junction protein genes (ZO1, Occludin and Claudin4), signifying the onset of repair responses in dysfunctional gut epithelium, as previously described (27) (Fig 1F). The loss of barrier function in uremia was not due to mucosal damage of SI and colon (Fig 1G).
Fig 1: Increased gut barrier permeability in AAI-injected mice.
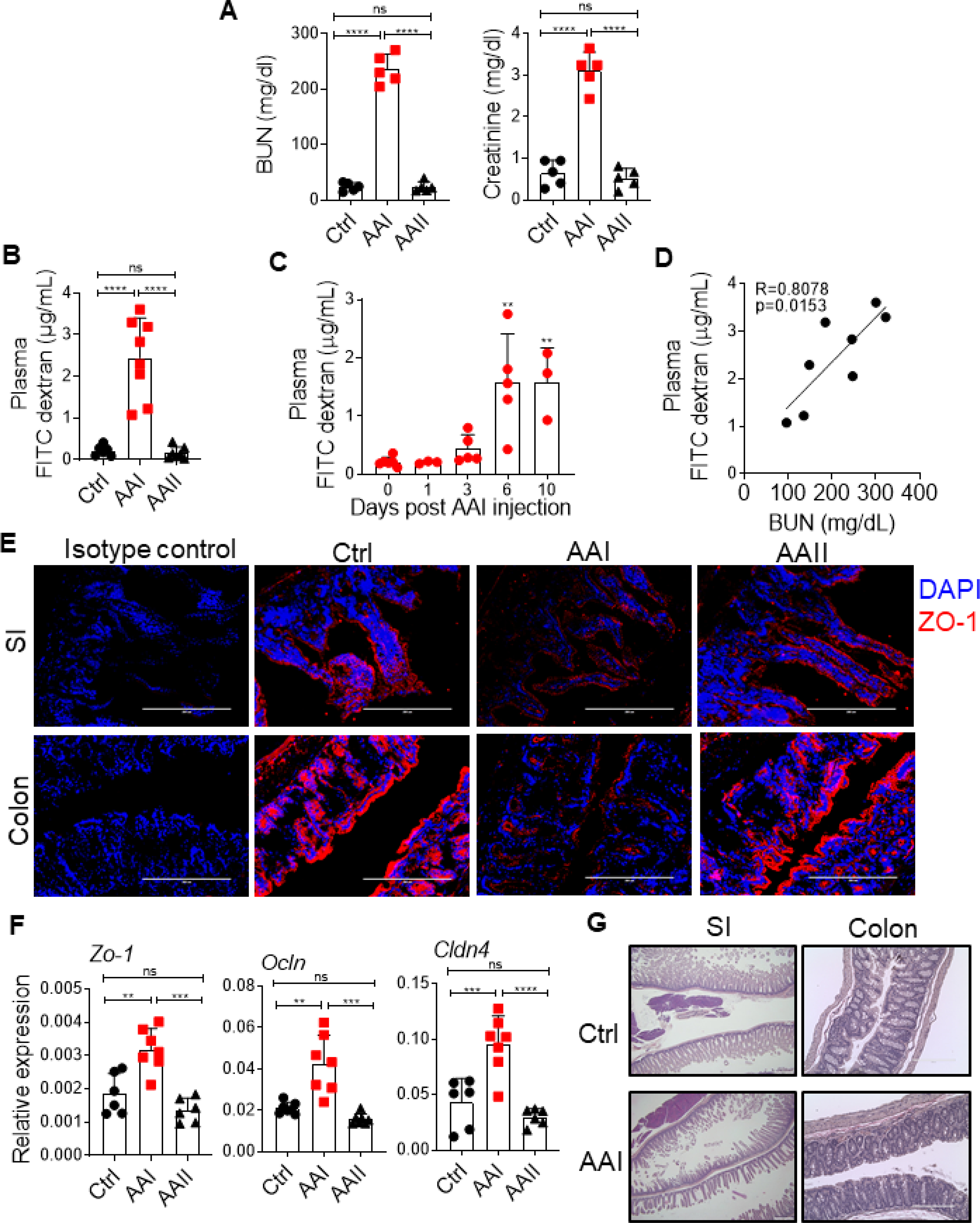
C57Bl/6 (WT) mice (n=6–8) were either injected with AAI, PBS (Ctrl) or AAII. (A) Serum BUN and creatinine levels (n=5) were measured at day 10 post AAI injection. At (B) day 10 (n=6–8), and (C) indicated time points post-AAI injection (n=3–6), mice were gavaged with FITC-dextran and assessed for FITC-dextran concentration in the plasma. (D) Correlation between gut barrier permeability and BUN level (n=8). (E) SI and colon sections were stained for ZO-1 expression. (F) Transcript expression of tight junction protein genes were evaluated by qPCR (n=6–7). (G) H&E staining of SI and colon of uremic and control mice. Images from 1 of 3 mice/group for (E) and (G). Magnification: 200X. Data pooled from atleast 2 independent experiments for A-D and F and expressed as mean ± S.D (A, B, C, and F). Statistical analyses by Pearson correlation (D) and One-way ANOVA (A-C, and F).
We next determined whether kidney damage is responsible for the alteration in gut barrier permeability independent of uremia. To test this issue, we adapted a unilateral ureteral obstruction (UUO) model of renal fibrosis, in which one of the ureters is ligated below the kidney while the contralateral ureter is left intact, allowing for normal function by the non-ligated kidney (28). Consequently, UUO causes unilateral kidney damage but not uremia (Fig 2A–C). Mice with UUO showed no changes in the gut barrier permeability in comparison to non-UUO control (Fig 2D), indicating that the increase in gut permeability is due to uremia and not kidney damage per se. Moreover, to determine whether increased permeability is due to a direct effect of AAI on the intestinal epithelial cells, AAI-injected mice were treated with probenecid, an organic anion transporter inhibitor that prevents kidney dysfunction and uremia without neutralizing AAI (29) (Fig 2E–G). Uremic mice treated with probenecid showed reduced FITC-dextran in the plasma, indicating that AAI does not exert any direct effect on the barrier permeability (Fig 2H). These data suggest that uremia drives increased barrier permeability in mice with kidney disease (12).
Fig 2: Uremia drives increased gut permeability.
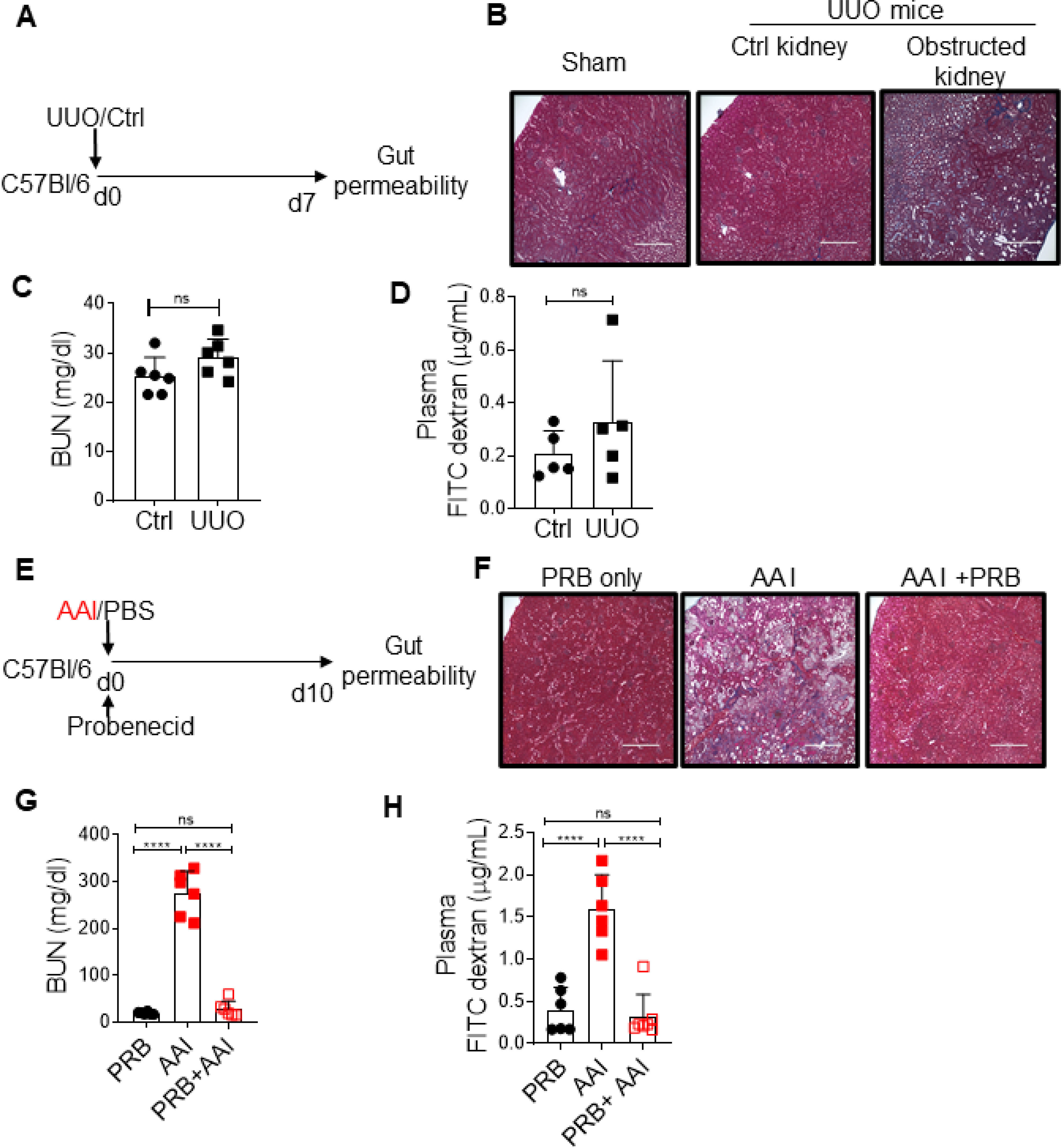
(A) Schematic diagram of the experimental design. WT mice (n=5–6) were subjected to UUO. At day 7 post-surgery, UUO and non-UUO control mice were gavaged with FITC-dextran and assessed for barrier permeability. (B) Kidney histopathology following Massone-trichome staining, (C) serum BUN level, and (D) plasma FITC-dextran concentration were measured at day 7 post-surgery. (E) Schematic diagram of the experimental design. Groups of uremic mice (n=6–7) were either treated with probenecid (AAI+PRB) or left untreated (AAI). Control mice received probenecid only (PRB only). Mice were evaluated for (F) kidney fibrosis, (G) serum BUN level, and (H) gut barrier permeability. Images from 1 of 3 mice/group for (B) and (F). Magnification: 200X. The data is pooled from atleast 2 independent experiments for C, D, G and H and expressed as mean ± S.D (C, D, G and H). Statistical analyses by Students T-test (C and D) and One-way ANOVA (G and H). ns: statistically not significant.
Uremia causes alterations in the mucosal defense and composition of gut microbiota
The innate and adaptive immune cells have distinct, yet complementary role in maintaining gut homeostasis and mucosal immune defense (30). In the small intestinal lamina propria (SILP), the percentages of macrophage and dendritic cell subsets were comparable between the uremic and control mice (Fig 3A and B). Strikingly, we observed a reduction in the percentage of neutrophils in the SILP of uremic mice than controls (Fig 3C). Uremic mice also demonstrated a reduction in the percentage of T-helper 17 (Th17) but not Th1 cells (Fig 3D). Markedly, the percentage of IgA+CD11b+ plasmablasts in the SILP of uremic mice was reduced than control animals (Fig 3E). There was a trend towards diminished IgA+CD11b- plasmablasts in uremia although the difference between the groups did not achieve statistical significance. However, the frequency of regulatory T cells (Treg) was similar between the groups (Fig 3F). We observed no difference in the frequencies of Th17, Th1, and Treg cells in the mesenteric lymph nodes (MLN) between uremic and non-uremic groups (Fig 3G and H). These results indicate that uremia negatively impacts the number of neutrophils, Th17 and IgA producing plasmablasts in the SILP.
Fig 3: Compromised gut mucosal immunity and dysbiosis in uremia.
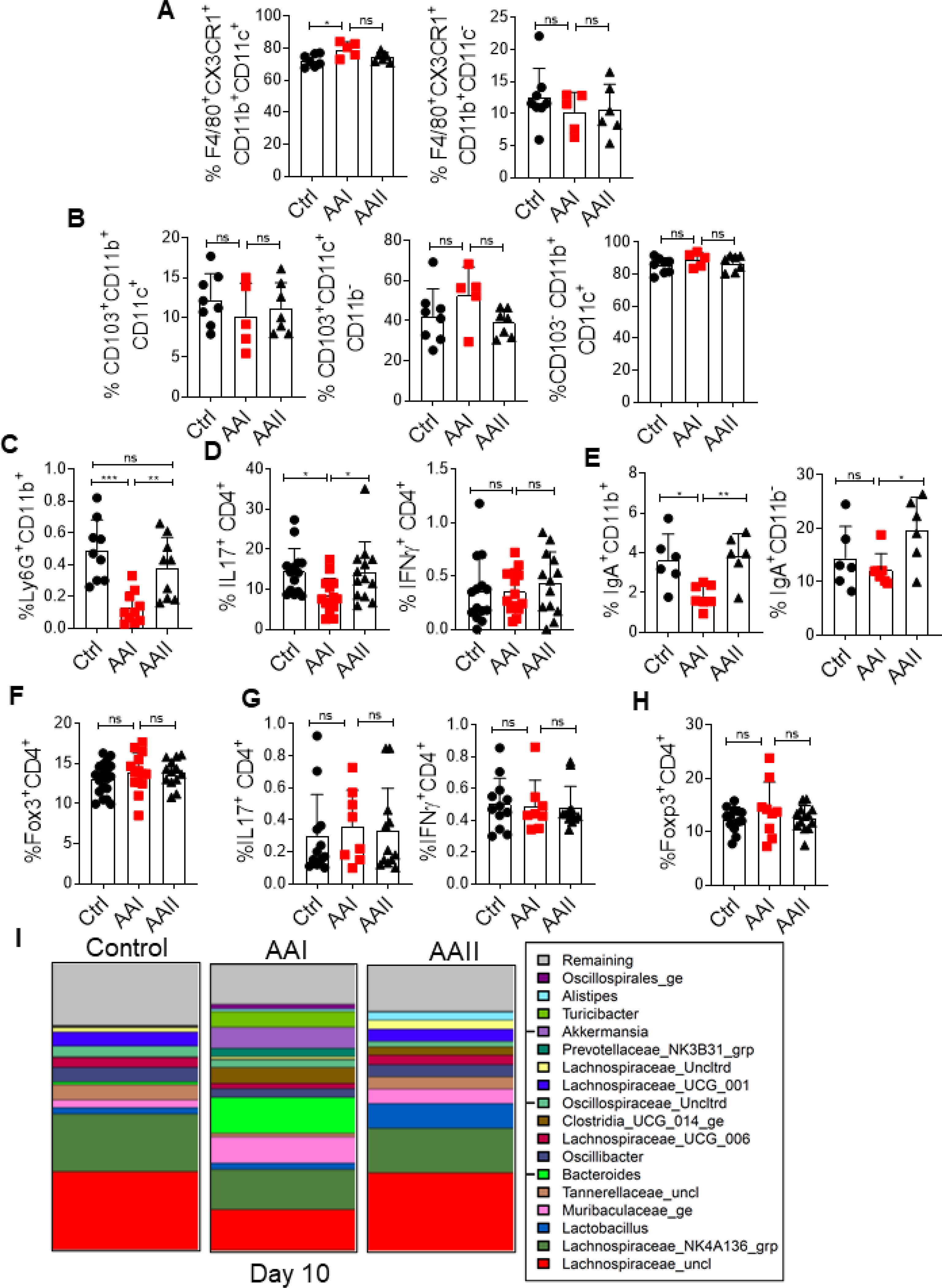
At day 10 post AAI injection, SILP (n=5–19) were evaluated for the frequency of (A) macrophages (liveCD45+CD11b+ F4/80+CX3CR1+CD11c+; liveCD45+CD11b+F4/80+CX3CR1+CD11c-), (B) dendritic cells (liveCD45+CD11b+CD103+CD11c+, liveCD45+CD11b-CD103+CD11c+; liveCD45+CD11b+ CD103-CD11c+) (C) neutrophils (liveCD45+CD11b+Ly6G+), (D) Th17 (liveCD45+CD4+IL-17+) and Th1 (liveCD45+CD4+IFNγ+), and (E) IgA producing plasmablasts (liveCD45+CD11b+IgA+; liveCD45+CD11b-IgA+) cells, (F) T regulatory cells (liveCD4+Foxp3+) by FACS at day 10 post AAI injection. (G) Percentages of Th17 and Th1 cells in the MLN (n=8–12) were determined by intra-cellular cytokine staining following in vitro stimulation with PMA/Ionomycin. (H) Frequency of T regulatory cells was determined in the MLN by FACS. (I) At day 10 post AAI injection, fecal pellets from uremic and control (n=5) mice were subjected to targeted 16S rRNA sequencing. Data pooled from at least 2 independent experiments for A-H and expressed as mean ± S.D (A-H). Statistical analyses by One-way ANOVA (A-H) and pairwise using Wilcoxon Rank Sum Test (I).
Next, we assessed the relative change in the diversity and composition of gut microbiota in uremic and control animals between days 0 (baseline) and 10 post AAI injection. There was no significant difference in the diversity of microbiota between uremic and control groups at day 10 post AAI injection (Fig 3I). Both control and AAII-injected animals demonstrated altered microbiota composition between days 0 and 10. Interestingly, uremic mice showed significantly greater change (p=0.0094) in the overall microbiota composition than control animals at day 10 post injection. While the control group showed a reduction in the abundance of Tannerellaceae and Lactobacillus, uremic animals exhibited an increase in the Tannerellaceae. Collectively, these results show a modest impact of uremia on the composition of gut microbiota in uremic mice.
Uremic mice exhibit translocation of gut microbiota and C. rodentium
We assessed whether uremia-driven disturbance in gut hemostasis can drive the translocation of gut microbiota. While we were unable to detect any bacterial colonies in the control or AAII mice, 30–50% of uremic animals showed translocation of microbiota in the liver and spleen at day 10 post AAI injection (Fig 4A). Moreover, probenecid-treated uremic mice did not show any bacterial colony in the liver and spleen (Fig 4B). The translocation of microbiota caused activation of T cells in the MLN and spleen of uremic mice, as shown before (Fig 4C and D) (12).
Fig 4: Uremic mice exhibit translocation of gut microbiota.
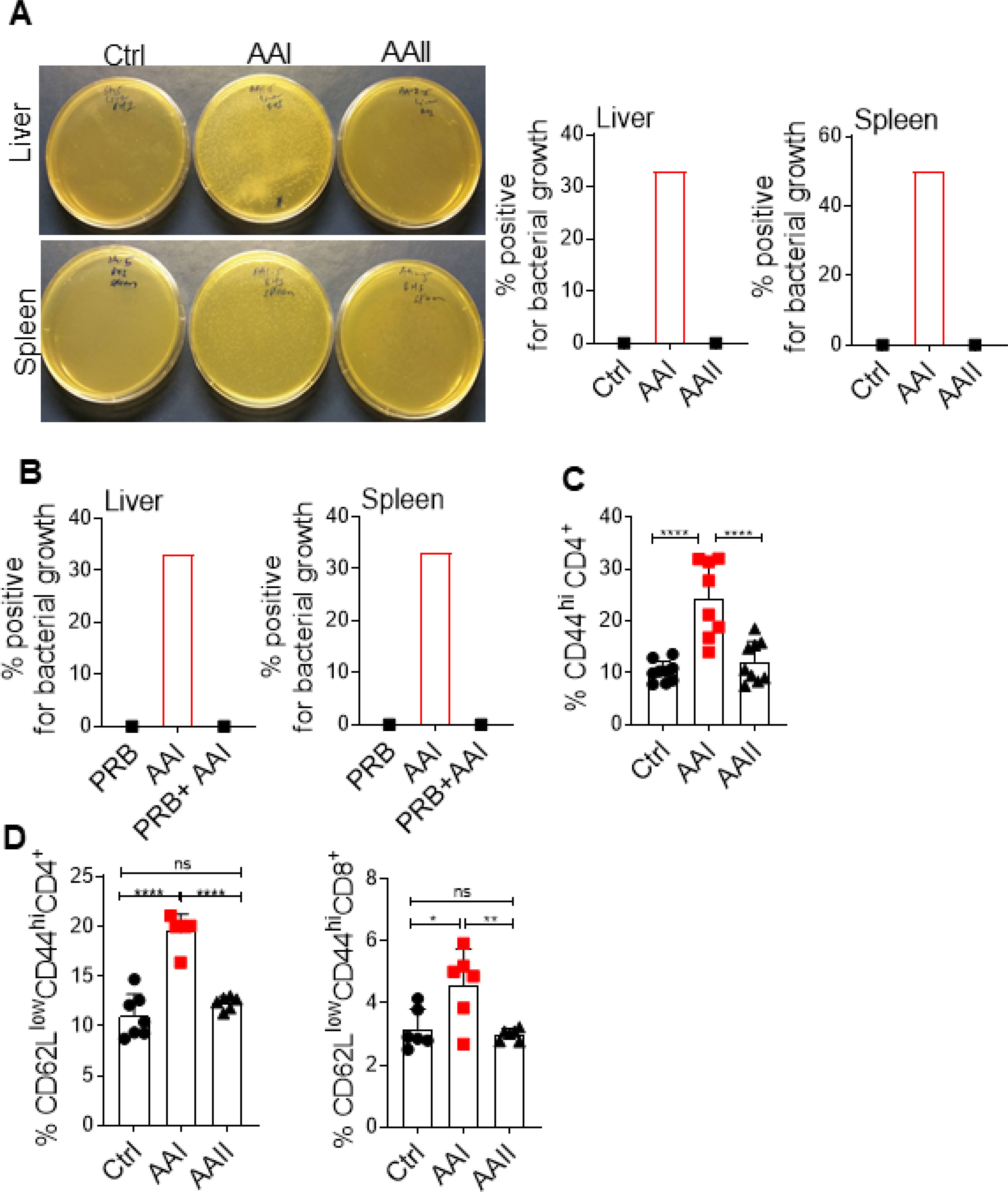
(A) Mice (n=6) were subjected to AAN and evaluated for the translocation of gut microbiota in the liver and spleen at day 10 post AAI injection. Images from 1 of 6 mice/group. (B) Uremic mice (n=6) were either treated with probenecid (AAI+PRB) or left untreated (AAI) and assessed for microbiota translocation in the liver and spleen. Uremic and control groups (n=6–9) were evaluated for the activation of T cells in the (C) MLN (liveCD4+CD44hi), and (D) spleen (liveCD4+CD62LloCD44hi and liveCD8+CD62LloCD44hi) by FACS. Pooled data from atleast 2 experiments for A-D and expressed as mean ± S.D (C and D). Statistical analyses by One-way ANOVA (C and D).
We next evaluated the ability of harmful bacteria C. rodentium to cause systemic infection in uremic animals (Fig 5A). C. rodentium is a murine intestinal pathogen that is closely related to human pathogens including enteropathogenic E. coli and enterohemorrhagic E. coli (31). Following oral infection, we observed an increased number of C. rodentium in the fecal pellet and cecal contents of uremic mice (Fig 5B). Few control mice exhibited very low colony counts of C. rodentium in the liver and spleen, as shown before (32) (Fig 5C). In contrast, 60–70% of the uremic animals showed higher C. rodentium burden in the liver and spleen. Our data suggest that gut microbiota and C. rodentium can translocate from the gut and cause systemic infection in uremia.
Fig 5: Uremic mice exhibit translocation of C. rodentium.
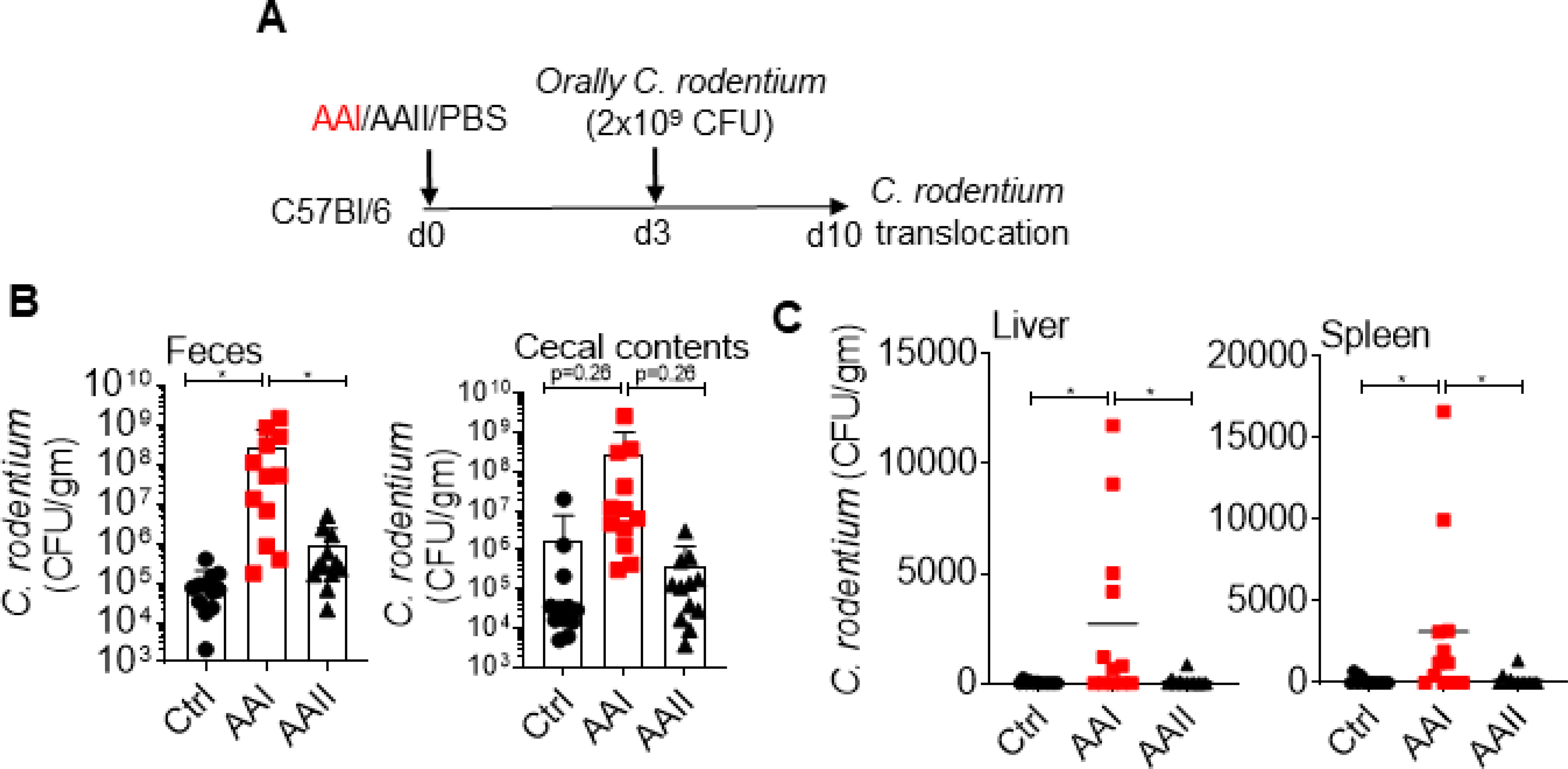
(A) Schematic diagram of the experimental design. AAI, control and AAII-injected mice (n=12) were gavaged with C. rodentium at day 3 post AAI injection. At day 7, C. rodentium burden in the (B) fecal pellet and cecal content, and (C) liver and spleen were measured. Pooled data from atleast 2 experiments for B and C and expressed as mean ± S.D (B and C). Statistical analyses by One-way ANOVA (B and C).
Uremia and mucosal damage are required for the translocation of C. albicans
The role of gut-derived C. albicans in non-access related infections in kidney disease is unknown. Based on our data (Fig 4 and 5), we hypothesize that C. albicans could translocate and cause disseminated candidiasis in uremia. C. albicans is not a commensal fungus in mice. Hence, colonization of the fungus requires alteration in gut homeostasis (33). Following oral gavage, uremic mice showed a modest increase in fungal colonization in the gut at day 7 p.i. (Fig 6A and B). We were unable to detect any fungal colony in the liver and spleen of uremic animals at this time point (Fig 6C). We also treated uremic and control mice with oral antibiotic to aid in robust colonization (34) (Fig 6D and E). However, uremic animals showed no fungal dissemination in the organs (Fig 6F). Thus, unlike C. rodentium, C. albicans cannot translocate from the gut in uremia.
Fig 6: Uremic mice show fungal translocation following DSS treatment.
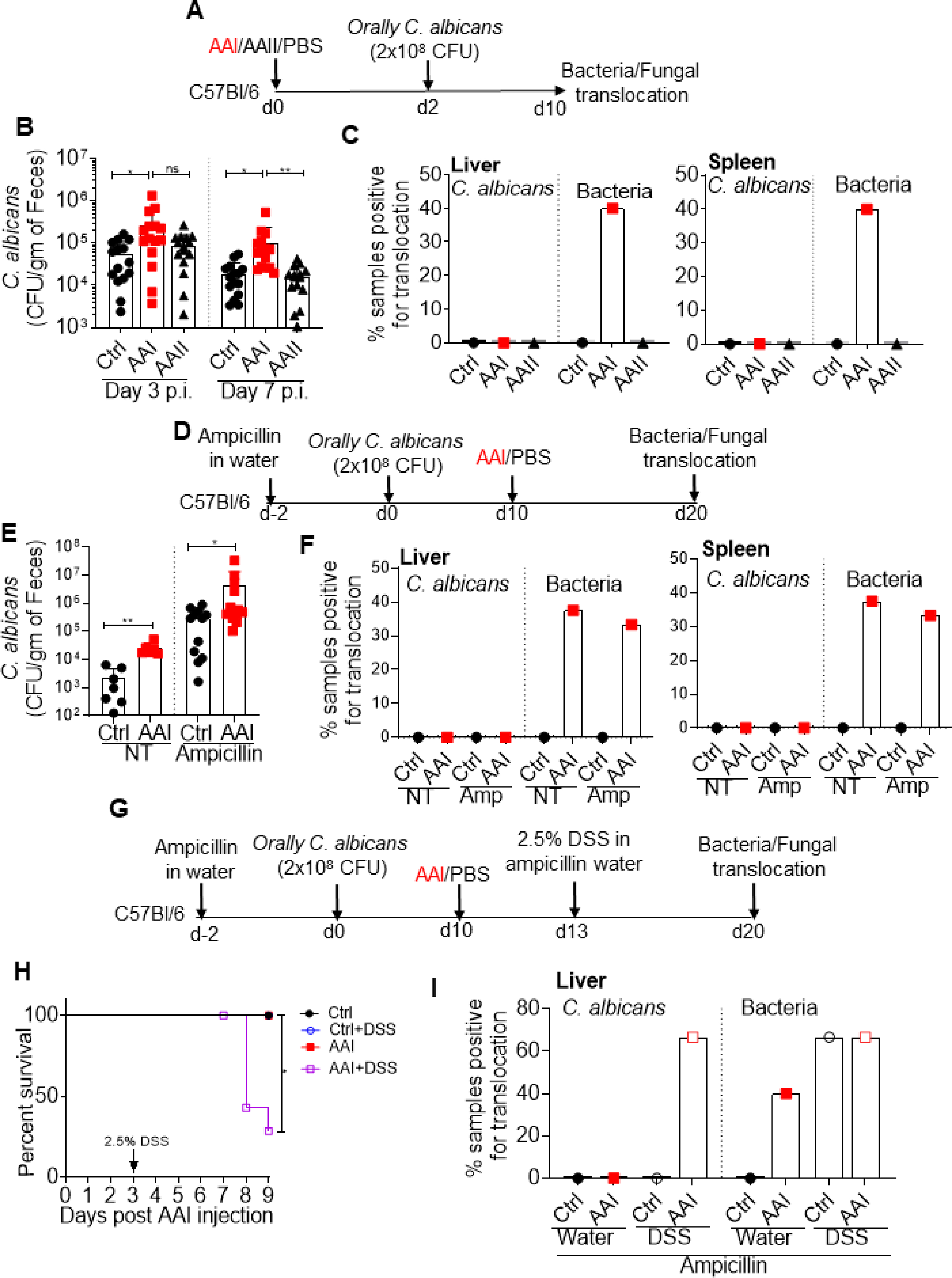
(A) Schematic representation of the experimental design. Mice (n=10–15) were gavaged with C. albicans at day 2 post AAI injection. At day 8, C. albicans and microbiota burden in the (B) fecal pellet, and (C) liver and spleen were evaluated. (D) Schematic diagram of the experimental plan. Mice (n=6–13) were fed with ampicillin in the drinking water throughout the experiment. At day 10 post oral fungal infection, mice were either injected with AAI or PBS. Fungal and bacterial CFU in the (E) fecal pellet, and (F) liver and spleen were determined. (G) Schematic representation of the experimental plan. Oral antibiotic treated animals were fed with 2.5% DSS in water 3 days after AAI injection. (H) Survival (n=4–8) was evaluated for 9 days post AAI injection. (I) Mice were evaluated for the translocation of C. albicans in the liver. Pooled data from atleast 2 experiments for B, C, E, F, H and I and expressed as mean ± S.D (B and E). Statistical analyses by One-way ANOVA (B), Mann-Whitney T test (E), Log rank test (H).
Most kidney disease patients suffer from some form of GI manifestations, where the gut mucosa is damaged (17). These include perforation of intestine, intestinal ulcer, uremic colitis, ischemic colitis, diverticular disease and intestinal hemorrhage. Hence, we tested whether C. albicans can cause systemic infection when the gut mucosa is disrupted in uremia. Accordingly, uremic and control mice were fed with antibiotic to favor colonization followed by oral gavage with 2.5% DSS to damage the epithelial lining (23) (Fig 6G). AAI-injected mice treated with DSS showed reduced survival (30%) in comparison to AAI only, control only and control + DSS animals (100%) (Fig 6H). Control animals with DSS showed no fungal dissemination in the liver, as shown before (23) (Fig 6I). There was no fungal translocation in uremic mice receiving no DSS. Interestingly, 65% of the uremic animals fed with DSS showed fungal translocation in the liver. These data indicate that C. albicans can translocate and cause systemic infection only when the mucosal barrier is damaged in majority of uremic animals. Thus, it is reasonable to speculate that uremic individuals with GI manifestations may have a higher risk of systemic infection by gut-derived C. albicans.
Discussion
Prevention of infection is one of the few avenues to reduce hospitalizations, control costs, and improve quality of life for patients with kidney disease. In the past decade, technological advances in the dialysis procedure have lowered the incidence of access-related infections. However, it has not been successful in preventing infection-related hospitalizations in these patients (2, 3). This is partly due to an alarming rise in the incidence of previously unappreciated non-access related infections. Currently, our knowledge about the risk and source of non-access related infections in patients with kidney disease is surprisingly rudimentary. Using a clinically relevant mouse model of kidney disease and associated uremia, we show that gut commensal C. albicans causes systemic infection when uremia is coupled with mucosal damage. These results identify gut-derived commensal C. albicans as a source of non-access related systemic infection in patients with kidney disease and showing GI damage.
Our data show that the loss of barrier function in uremic mice is due to an increase in the gut barrier permeability and mucosal damage. The level of uremia correlates with the loss of barrier function, indicating that uremia is directly implicated in the increased gut permeability. Although our study provided evidence of leaky gut in 100% of the uremic mice, only 30–50% of the animals consistently showed translocation of microbiota. These data imply that factors other than increase in gut permeability play a major role in bacterial translocation in uremia. Moreover, when microbiota and C. rodentium showed translocation, C. albicans failed to do so. The fungal yeast (2–10 μm) and hyphae (>10 μm) are bigger in size than bacteria (0.5–5 μm), which may act as an impediment for the fungal yeast and hyphal form to pass though the tight junctions of GI epithelial cells. Additionally, control or uremic mice do not show any fungal translocation without mucosal damage. These data argue against the fact that C. albicans can translocate by causing damage to GI epithelial cells, as proposed by others (35).
Dysbiosis observed in this study may be due to iatrogenic causes or uremia per se. Loss of kidney function leads to diffusion of urea in the GI tract. Subsequent hydrolysis of urea by urease expressed by some gut microbes, results in the formation of large quantities of ammonia, which could affect the growth of commensal bacteria (16, 19). We observed a modest change in the gut microbiota in our mouse model of AAN. This is in contrast to chronic kidney disease patients, where a markedly altered change in terms of quantity and quality of microbiota is evident (16, 19). There may be several reasons for this discrepancy. First, AAN is an acute kidney injury model, where the gut bacteria are exposed to uremic toxins for a relatively short period of time ie. 7–10 days. Second, mice and human microbiota differ considerably and their susceptibility to uremia may reflect the difference in alterations in gut bacterial content (36). Finally, uremic toxins differ between mice and human making it difficult to compare their impact on the gut microbiota (37).
We observed a decrease in the percentages of innate and adaptive immune cells in the SILP of uremic mice. Uremia induced neutrophil apoptosis may account for the reduced number of neutrophils in the uremic gut (38). This observation is in line with a previous report showing that immunosuppressive treatment-induced neutropenia is required for the fungus to translocate and cause systemic infection in non-uremic animals (23). Additionally, IgA producing plasmablasts deficiency in uremic mice can be simultaneously mediated by increased B cell apoptosis and reduced expression of BAFF-R, as demonstrated before (39). The role of IgA in C. albicans colonization and translocation is poorly understood. Interestingly, we did not see any change in Treg cells in the gut, a hallmark of CKD patients (40).
The GI symptoms are reported in up to 80% kidney disease patients (17). Some of these symptoms including intestinal necrosis, spontaneous colonic perforation, uremic colitis, gastric ulcer, GI hemorrhage, acute diverticulitis result in damage of mucosa and development of sepsis. Thus, it is reasonable to speculate that uremic individuals with GI manifestations may have a higher risk of systemic infection by commensal C. albicans. To date, no studies have looked at the prevalence of disseminated candidiasis in uremic patients with GI manifestations. Additionally, these findings have compelling implications in the risk stratification and clinical management of infection control and prevention in patients with kidney disease.
Acknowledgments
We would like to thank Drs. Sarah Gaffen, Tim Hand and Mandy McGeachy for suggestions.
Funding source: This work is supported by grants from National Institute of Health (AI142354, DK104680 and R21AI45242) to P.S.B.
References
- 1.Nugent RA, Fathima SF, Feigl AB, and Chyung D. 2011. The burden of chronic kidney disease on developing nations: a 21st century challenge in global health. Nephron. Clinical practice 118: c269–277. [DOI] [PubMed] [Google Scholar]
- 2.Sarnak MJ, and Jaber BL. 2000. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney international 58: 1758–1764. [DOI] [PubMed] [Google Scholar]
- 3.Dalrymple LS, Johansen KL, Chertow GM, Cheng SC, Grimes B, Gold EB, and Kaysen GA. 2010. Infection-related hospitalizations in older patients with ESRD. American journal of kidney diseases : the official journal of the National Kidney Foundation 56: 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins AJ, Foley RN, Herzog C, Chavers BM, Gilbertson D, Ishani A, Kasiske BL, Liu J, Mau LW, McBean M, Murray A, St Peter W, Guo H, Li Q, Li S, Li S, Peng Y, Qiu Y, Roberts T, Skeans M, Snyder J, Solid C, Wang C, Weinhandl E, Zaun D, Arko C, Chen SC, Dalleska F, Daniels F, Dunning S, Ebben J, Frazier E, Hanzlik C, Johnson R, Sheets D, Wang X, Forrest B, Constantini E, Everson S, Eggers PW, and Agodoa L. 2010. Excerpts from the US Renal Data System 2009 Annual Data Report. American journal of kidney diseases : the official journal of the National Kidney Foundation 55: S1–420, A426–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allon M, Depner TA, Radeva M, Bailey J, Beddhu S, Butterly D, Coyne DW, Gassman JJ, Kaufman AM, Kaysen GA, Lewis JA, Schwab SJ, and H. S. Group. 2003. Impact of dialysis dose and membrane on infection-related hospitalization and death: results of the HEMO Study. Journal of the American Society of Nephrology : JASN 14: 1863–1870. [DOI] [PubMed] [Google Scholar]
- 6.Pastan S, Soucie JM, and McClellan WM. 2002. Vascular access and increased risk of death among hemodialysis patients. Kidney international 62: 620–626. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri ND, Pahl MV, Crum A, and Norris K. 2012. Effect of uremia on structure and function of immune system. Journal of renal nutrition : the official journal of the Council on Renal Nutrition of the National Kidney Foundation 22: 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Syed-Ahmed M, and Narayanan M. 2019. Immune Dysfunction and Risk of Infection in Chronic Kidney Disease. Advances in chronic kidney disease 26: 8–15. [DOI] [PubMed] [Google Scholar]
- 9.Dobre M, Meyer TW, and Hostetter TH. 2013. Searching for uremic toxins. Clinical journal of the American Society of Nephrology : CJASN 8: 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen G, and Horl WH. 2012. Immune dysfunction in uremia—an update. Toxins 4: 962–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen K, Kesper MS, Marschner JA, Konrad L, Ryu M, Kumar Vr S, Kulkarni OP, Mulay SR, Romoli S, Demleitner J, Schiller P, Dietrich A, Muller S, Gross O, Ruscheweyh HJ, Huson DH, Stecher B, and Anders HJ. 2017. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. Journal of the American Society of Nephrology : JASN 28: 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders HJ, Andersen K, and Stecher B. 2013. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney international 83: 1010–1016. [DOI] [PubMed] [Google Scholar]
- 13.Amato KR, Martinez-Mota R, Righini N, Raguet-Schofield M, Corcione FP, Marini E, Humphrey G, Gogul G, Gaffney J, Lovelace E, Williams L, Luong A, Dominguez-Bello MG, Stumpf RM, White B, Nelson KE, Knight R, and Leigh SR. 2016. Phylogenetic and ecological factors impact the gut microbiota of two Neotropical primate species. Oecologia 180: 717–733. [DOI] [PubMed] [Google Scholar]
- 14.Rooks MG, and Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nature reviews. Immunology 16: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tilg H, Zmora N, Adolph TE, and Elinav E. 2020. The intestinal microbiota fuelling metabolic inflammation. Nature reviews. Immunology 20: 40–54. [DOI] [PubMed] [Google Scholar]
- 16.Meijers B, Evenepoel P, and Anders HJ. 2019. Intestinal microbiome and fitness in kidney disease. Nature reviews. Nephrology 15: 531–545. [DOI] [PubMed] [Google Scholar]
- 17.Ala-Kaila K, and Pasternack A. 1989. Gastrointestinal complications in chronic renal failure. Digestive diseases 7: 230–242. [DOI] [PubMed] [Google Scholar]
- 18.Carrera-Jimenez D, Miranda-Alatriste P, Atilano-Carsi X, Correa-Rotter R, and Espinosa-Cuevas A. 2018. Relationship between Nutritional Status and Gastrointestinal Symptoms in Geriatric Patients with End-Stage Renal Disease on Dialysis. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramezani A, and Raj DS. 2014. The gut microbiome, kidney disease, and targeted interventions. Journal of the American Society of Nephrology : JASN 25: 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lionakis MS, Iliev ID, and Hohl TM. 2017. Immunity against fungi. JCI insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandhi BV, Bahadur MM, Dodeja H, Aggrwal V, Thamba A, and Mali M. 2005. Systemic fungal infections in renal diseases. Journal of postgraduate medicine 51 Suppl 1: S30–36. [PubMed] [Google Scholar]
- 22.Limon JJ, Skalski JH, and Underhill DM. 2017. Commensal Fungi in Health and Disease. Cell host & microbe 22: 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, and Pier GB. 2008. Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS pathogens 4: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi-Sakamoto M, Tamai R, Isogai E, and Kiyoura Y. 2018. Gastrointestinal colonisation and systemic spread of Candida albicans in mice treated with antibiotics and prednisolone. Microbial pathogenesis 117: 191–199. [DOI] [PubMed] [Google Scholar]
- 25.Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, Simms-Waldrip TR, Xie Y, Hooper LV, and Koh AY. 2015. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nature medicine 21: 808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jawale CV, Ramani K, Li DD, Coleman BM, Oberoi RS, Kupul S, Lin L, Desai JV, Delgoffe GM, Lionakis MS, Bender FH, Prokopienko AJ, Nolin TD, Gaffen SL, and Biswas PS. 2020. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Science translational medicine 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kigerl KA, Hall JC, Wang L, Mo X, Yu Z, and Popovich PG. 2016. Gut dysbiosis impairs recovery after spinal cord injury. The Journal of experimental medicine 213: 2603–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramani K, Tan RJ, Zhou D, Coleman BM, Jawale CV, Liu Y, and Biswas PS. 2018. IL-17 Receptor Signaling Negatively Regulates the Development of Tubulointerstitial Fibrosis in the Kidney. Mediators of inflammation 2018: 5103672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baudoux TE, Pozdzik AA, Arlt VM, De Prez EG, Antoine MH, Quellard N, Goujon JM, and Nortier JL. 2012. Probenecid prevents acute tubular necrosis in a mouse model of aristolochic acid nephropathy. Kidney international 82: 1105–1113. [DOI] [PubMed] [Google Scholar]
- 30.Belkaid Y, and Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157: 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bouladoux N, Harrison OJ, and Belkaid Y. 2017. The Mouse Model of Infection with Citrobacter rodentium. Current protocols in immunology 119: 19 15 11–19 15 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallance BA, Deng W, Jacobson K, and Finlay BB. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infection and immunity 71: 3443–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh AY 2013. Murine models of Candida gastrointestinal colonization and dissemination. Eukaryotic cell 12: 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, Pham G, Burg AR, Ruff B, Gonzalez T, Khurana Hershey GK, Haslam DB, and Way SS. 2019. Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell host & microbe 25: 404–417 e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allert S, Forster TM, Svensson CM, Richardson JP, Pawlik T, Hebecker B, Rudolphi S, Juraschitz M, Schaller M, Blagojevic M, Morschhauser J, Figge MT, Jacobsen ID, Naglik JR, Kasper L, Mogavero S, and Hube B. 2018. Candida albicans-Induced Epithelial Damage Mediates Translocation through Intestinal Barriers. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hugenholtz F, and de Vos WM. 2018. Mouse models for human intestinal microbiota research: a critical evaluation. Cellular and molecular life sciences : CMLS 75: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, and Niwa T. 2013. Correlation between Serum Levels of Protein-Bound Uremic Toxins in Hemodialysis Patients Measured by LC/MS/MS. Mass spectrometry 2: S0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majewska E, Baj Z, Sulowska Z, Rysz J, and Luciak M. 2003. Effects of uraemia and haemodialysis on neutrophil apoptosis and expression of apoptosis-related proteins. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 18: 2582–2588. [DOI] [PubMed] [Google Scholar]
- 39.Pahl MV, Gollapudi S, Sepassi L, Gollapudi P, Elahimehr R, and Vaziri ND. 2010. Effect of end-stage renal disease on B-lymphocyte subpopulations, IL-7, BAFF and BAFF receptor expression. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 25: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meier P 2009. FOXP3+ regulatory T-cells in chronic kidney disease: molecular pathways and clinical implications. Advances in experimental medicine and biology 665: 163–170. [DOI] [PubMed] [Google Scholar]