

Abstract
Background
Melanoma therapy has changed dramatically over the last decade with improvements in immunotherapy, yet many patients do not respond to current therapies. This novel vaccine strategy may prime a patient’s immune system against their tumor and work synergistically with immunotherapy against advanced-stage melanoma.
Methods
This was a prospective, randomized, double-blind, placebo-controlled, phase IIb trial of the tumor lysate, particle-loaded, dendritic cell (TLPLDC) vaccine administered to prevent recurrence in patients with resected stage III/IV melanoma. Patients were enrolled and randomized 2:1 to the TLPLDC vaccine or placebo (empty yeast cell wall particles and autologous dendritic cells). Both intention-to-treat (ITT) and per treatment (PT) analyses were predefined, with PT analysis including patients who remained disease-free through the primary vaccine/placebo series (6 months).
Results
A total of 144 patients were randomized (103 vaccine, 41 control). Therapy was well-tolerated with similar toxicity between treatment arms; one patient in each group experienced related serious adverse events. While disease-free survival (DFS) was not different between groups in ITT analysis, in PT analysis the vaccine group showed improved 24-month DFS (62.9% vs. 34.8%, p = 0.041).
Conclusions
This phase IIb trial of TLPLDC vaccine administered to patients with resected stage III/IV melanoma shows TLPLDC is well-tolerated and improves DFS in patients who complete the primary vaccine series. This suggests patients who do not recur early benefit from TLPLDC in preventing future recurrence from melanoma. A phase III trial of TLPLDC + checkpoint inhibitor versus checkpoint inhibitor alone in patients with advanced, surgically resected melanoma is under development.
Trial Registration
Supplementary information
The online version contains supplementary material available at (10.1245/s10434-021-09709-1).
The last decade has seen an increase in therapy options for the treatment of advanced melanoma. This was led by the emergence of checkpoint inhibitor (CPI) therapy,1,2 followed by the success of targeted therapies such as BRAF and MEK inhibitors.3 After showing promise in the once bleak field of metastatic melanoma, these therapies have recently been approved for adjuvant therapy in resectable disease.4–6 Given that recurrent or metastatic melanoma is rarely cured, this movement towards adjuvant therapy to prevent recurrence represents an important step forward.
While these therapies offer hope to patients who previously had few options, there is room for improvement. Most metastatic patients will eventually not respond to BRAF/MEK inhibitors when their tumors escape via novel mutations.7,8 Additionally, many patients do not respond to CPI therapy and few responses are durable.1 This failure of CPI therapy is likely, at least in part, due to a lack of endogenous antitumor immune response. Cancer vaccines may augment an immune response specific to a patient’s tumor when this has not happened innately. While cancer vaccine monotherapy has been mostly ineffective, one US FDA-approved dendritic cell (DC) vaccine is available for the treatment of prostate cancer.9 In melanoma, there has been no such success, even with DC vaccines,10 but few trials have studied such therapy in the adjuvant setting, where we expect vaccines to have a more meaningful effect.11 Additionally, while vaccine monotherapy may not be effective in those with intact metastatic disease, a vaccine-induced immune response may serve to improve responses to CPI therapy.12–14 Preclinical work has shown the promise of this combination strategy and early clinical trials are currently evaluating such combinations.15
Our group developed a novel vaccine for melanoma patients, the dendritoma vaccine, which utilizes a fusion of autologous tumor cells and DCs to activate a patient’s immune system against the unique collection of antigens from each individual’s tumor.16 While this strategy showed early success treating patients with metastatic melanoma, it is labor-intensive and expensive. Thus, we moved to a more scalable model—the tumor lysate, particle-loaded, dendritic cell (TLPLDC) vaccine. Briefly, this vaccine is created by ex vivo loading of autologous DC with yeast cell wall particles (YCWPs) containing a patient’s tumor lysate (TL), resulting in effective and efficient delivery of tumor antigens to the cytoplasm of the DC. An initial phase I trial demonstrated the safety and potential efficacy of this vaccine in patients with a variety of malignancies.17 We are currently conducting a phase IIb trial of the TLPLDC vaccine administered to prevent recurrence in patients with stage III/IV melanoma after successful resection of disease. In this study, we present the primary analysis for safety and efficacy.
Methods
Patient Characteristics
After approval by the Western Institutional Review Board, patients with completely resectable stage III/IV melanoma were identified prior to definitive surgery, then counseled and consented for tissue collection (consent #1). Consented patients underwent standard-of-care (SOC) resection as indicated. Any patients who were unable to be rendered disease-free surgically were considered screen failures. After surgery, patients received systemic therapy and/or radiation therapy at the discretion of their primary care team. If the patient received chemotherapy or interferon as part of their SOC therapies, then vaccination began after completion of these therapies. The protocol was amended partway through enrollment to allow concurrent CPIs after their approval for the adjuvant setting.
All enrolled patients were clinically disease-free after SOC therapies. Patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, were not involved in other clinical trials, and were capable of giving informed consent. Exclusion criteria included any evidence of residual disease after surgery and SOC therapies, insufficient tumor available to create the vaccine (<1 mg), active immunodeficiency disease, HIV, active hepatitis B or C infections, current corticosteroid or other immunosuppressant use.
After completion of SOC therapies, patients were recounseled and consented again for treatment (consent #2). Enrolled patients were randomized 2:1 to vaccine or control. Randomization tables were computer-generated with a site-balancing algorithm.
The protocol initially planned to enroll 120 patients, a sample size calculated with the assumption of a baseline recurrence rate of 60% at 2 years in a mixed group of stage III/IV resected melanoma, and treatment effect corresponding to a hazard ratio of 0.50. Size was calculated with 80% power to detect a statistical difference, with a two-sided alpha of 0.05. However, this protocol was amended to allow inclusion of additional patients randomized to the TLPLDC vaccine arm in a continuation trial in which patients received the same vaccine preparation method and dosing schedule, as described below, in a randomized, double-blind fashion. It was prespecified in the study protocol and statistical plan that these additional patients would be included in the primary analysis of this trial.
Vaccine Preparation and Administration
For DC isolation, patients received a single injection of granulocyte colony-stimulating factor (G-CSF) 300 µg subcutaneously 24–48 h prior to the collection of 50–70 mL of peripheral blood. Patients who could not tolerate G-CSF, or refused it, had 120 mL of blood drawn instead. Blood was sent to our central facility for DC isolation and vaccine preparation.
Vaccine preparation was completed as previously described.17 Briefly, for those randomized to the TLPLDC arm, TL was created through freeze/thaw cycling, then loaded into preprepared YCWPs along with stock CpG oligonucleotides and tetanus helper peptide (amino acids 948–968, sequence FNNFTVSFWLRVPKVSASHLE). Fifty milligrams of tumor was required for creation of sufficient TL, although 0.5 cm3 was preferred. The TL-loaded YCWP were introduced to the immature, monocytic-derived DC for phagocytosis, thus creating the TLPLDC vaccine. The vaccine was then frozen in single-dose vials, with each vial containing 1–1.5 × 106 TLPLDC and labeled with the patient’s unique study number. For patients randomized to the control group, unloaded YCWPs were introduced to a similarly prepared autologous DC, and the resultant placebo vaccine was appropriately frozen and labeled.
Based on patients’ randomization, autologous TLPLDC vaccine (vaccine group) or unloaded YCWPs + autologous DC (control group) were sent to each study site, with patients and site personnel blinded to allocation. Regardless of assigned group, the site received six single-dose vials. Patients received 3-monthly intradermal injections and then inoculations at 6, 12, and 18 months, all in the same lymph node draining area (preferably anterior thigh). The first three inoculations were considered the primary vaccine series (PVS) and the latter three were considered the booster series. Patients began vaccinations between 3 weeks and 3 months from resection after completion of SOC therapies; if started on CPIs, patients had to demonstrate tolerance of CPIs for 3 months prior to vaccination. Frozen tumor was maintained for all patients. Any patient in the control group who experienced a recurrence was offered active vaccine at the time of recurrence in a crossover fashion, in addition to repeat resection or other SOC therapies.
Toxicity
For both the primary inoculations and booster series, patients were monitored for 30 min after inoculation. Patients contacted research nurses as needed between visits, and were reassessed and questioned about any toxicity at their next visit. Local or systemic toxicities were collected and graded by each study site’s research personnel. The National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03 graded toxicity scale was utilized to assess local and systemic toxicity. Treatment-related adverse events (AEs) included toxicity classified as ‘definitely’, ‘probably’, or ‘possibly’ related by the study site primary investigator, while AEs classified as ‘unlikely’ or ‘unrelated’ were not considered treatment-related for this analysis. AEs were classified as serious if they met any of the following criteria: resulted in death, were life-threatening, required or prolonged inpatient hospitalization, were disabling, were a congenital anomaly/birth defect, or required medical or surgical intervention to prevent one of these outcomes.
Survival Analysis
Disease-free survival (DFS) and overall survival (OS) status were monitored per each patient’s treating provider as outlined by National Comprehensive Cancer Network guidelines. Suspected recurrences were confirmed with biopsy and pathologic assessment. Time to recurrence was based on the date of randomization to the time of confirmed recurrence. The primary endpoint of this trial was 2-year DFS. This primary analysis was prespecified at 24 months from the enrollment of the 120th patient. Per the statistical plan, survival analysis was performed on the intention-to-treat (ITT) and per treatment (PT) populations. The PT analysis included only patients who remained enrolled on the trial and disease-free at the fourth dose time point, 6 months from initiation of therapy. Secondary endpoints included 36-month DFS and OS, as well as safety measured using the CTCAE version 4.03.
Statistical Analysis
Demographic data were analyzed using Student’s t-test if quantitative and the Chi-square test if categorical. DFS was determined using Kaplan–Meier analysis. Comparison of toxicity between groups was performed using the Chi-square test or Fisher’s exact test. The proportion of subjects who recurred was compared using log-ranked analysis, with significance determined by the Mantel–Cox log-rank test. A p value <0.05 was considered significant. All statistics were calculated in SPSS version 22 (IBM Corp. Released 2013. IBM SPSS Statistics for Windows, Version 22.0. Armonk, NY, USA).
Results
Patients
From February 2015 to May 2019, 267 patients were screened and 144 patients were randomized, with 103 in the vaccine group and 41 in the control group (Fig. 1). The only significant clinical, pathologic, or treatment-related difference between the groups included a higher number of patients having primary disease in the TLPLDC versus placebo groups (70.9 vs. 53.7%, p = 0.049) compared with recurrent disease (29.1 vs. 46.3%) (Table 1). Although there was no difference in overall American Joint Committee on Cancer (AJCC) stage III versus stage IV disease between groups, there was a significant difference when compared by substage, namely a greater percentage of stage IIIc disease in the TLPLDC group and a greater percentage of stage IIIb disease in the placebo group (Table 2). There was an unexpectedly high rate of positive margins in both groups (25.2 vs. 31.7%, p = 0.756). This likely represented the initial biopsies, which were followed by definitive surgery to render patients disease-free surgically to meet the trial inclusion criteria, but final margin status was not captured in our data set. However, there was no difference between groups in margin status. As expected, most patients (94%) did not receive chemotherapy. Less than one-third received a CPI as these agents were not approved for the adjuvant setting until later in the enrollment period; the CPI agents used are shown in Table 3.
Fig. 1.

CONSORT diagram demonstrating patient flow through screening, randomization, treatment, and follow-up. ITT intention-to-treat, TLPLDC tumor lysate, particle-loaded, dendritic cell, PT per treatment
Table 1.
Demographic and pathologic data with comparison between TLPLDC versus placebo in the ITT and PT cohorts
| Category | ITT analysis | PT analysis | ||||
|---|---|---|---|---|---|---|
| TLPLDC | Placebo | p value | TLPLDC | Placebo | p value | |
| Age, years | ||||||
| Median | 63.6 | 59.4 | 0.076 | 64.9 | 60.9 | 0.036 |
| IQR | 55.6–73.0 | 49.5–67.8 | 58.2–74.9 | 50.3–70.2 | ||
| Race | ||||||
| Asian | 0 (0) | 1 (2.4) | 0.320 | 0 (0) | 1 (3.1) | 0.203 |
| Black | 0 (0) | 1 (2.4) | 0 (0) | 1 (3.1) | ||
| Hispanic | 3 (2.9) | 1 (2.4) | 0 (0) | 1 (3.1) | ||
| Native American | 1 (1.0) | 0 (0) | 1 (1.5) | 0 (0) | ||
| Unknown | 1 (1.0) | 0 (0) | 1 (1.5) | 0 (0) | ||
| White | 98 (95.1) | 38 (92.7) | 64 (97) | 29 (90.6) | ||
| Disease type | ||||||
| Primary | 73 (70.9) | 22 (53.7) | 0.049 | 45 (68.2) | 19 (59.4) | 0.390 |
| Recurrent | 30 (29.1) | 19 (46.3) | 21 (31.8) | 13 (40.6) | ||
| Ulceration | ||||||
| Present | 17 (16.5) | 12 (29.3) | 0.316 | 9 (13.6) | 10 (31.3) | 0.225 |
| Absent | 22 (21.4) | 7 (17.1) | 17 (25.8) | 6 (18.8) | ||
| Not available | 64 (62.1) | 22 (53.7) | 40 (60.6) | 16 (50.0) | ||
| TILs | ||||||
| Brisk | 3 (2.9) | 2 (4.9) | 0.462 | 2 (3.0) | 2 (6.3) | 0.319 |
| Mild | 2 (1.9) | 0 (0) | 2 (3.0) | 0 (0) | ||
| Non-brisk | 14 (13.6) | 10 (24.4) | 8 (12.1) | 7 (21.9) | ||
| Absent | 8 (7.8) | 1 (2.4) | 6 (9.1) | 0 (0) | ||
| Not available | 76 (73.8) | 28 (68.3) | 48 (72.7) | 23 (71.9) | ||
| In-transit disease | ||||||
| Present | 9 (8.7) | 1 (2.4) | 0.420 | 6 (9.1) | 1 (3.1) | 0.568 |
| Absent | 31 (30.1) | 10 (24.4) | 19 (28.8) | 7 (21.9) | ||
| Not available | 63 (61.2) | 30 (73.2) | 41 (62.1) | 24 (75.0) | ||
| Satellites | ||||||
| Present | 5 (4.8) | 1 (2.4) | 0.851 | 3 (4.5) | 1 (3.1) | 0.893 |
| Absent | 37 (35.9) | 13 (31.7) | 24 (36.4) | 10 (31.2) | ||
| Not available | 61 (59.2) | 27 (65.9) | 39 (59.1) | 21 (65.6) | ||
| Lymphatic invasion | ||||||
| Present | 15 (14.6) | 2 (4.9) | 0.442 | 8 (12.1) | 1 (3.1) | 0.502 |
| Absent | 33 (32.0) | 14 (34.2) | 20 (30.3) | 10 (31.2) | ||
| Not available | 55 (53.4) | 25 (60.9) | 38 (57.6) | 21 (65.6) | ||
| Primary location | ||||||
| Extremity | 39 (37.9) | 18 (43.9) | 0.779 | 25 (37.9) | 14 (43.8) | 0.805 |
| Trunk | 24 (23.3) | 7 (17.1) | 15 (22.7) | 6 (18.8) | ||
| Head and neck | 16 (15.5) | 6 (14.6) | 11 (16.7) | 6 (18.8) | ||
| Axillary | 12 (11.7) | 6 (14.6) | 9 (13.6) | 4 (12.5) | ||
| Inguinal | 5 (4.9) | 1 (2.4) | 3 (4.5) | 1 (3.1) | ||
| Intra-abdominal | 3 (2.9) | 1 (2.4) | 2 (3.0) | 0 (0) | ||
| Lung | 3 (2.9) | 1 (2.4) | 0 (0) | 1 (3.1) | ||
| Unknown | 1 (1.0) | 1 (2.4) | 1 (1.5) | 0 (0) | ||
| Margins | ||||||
| Positive | 26 (25.2) | 13 (31.7) | 0.756 | 15 (22.7) | 9 (28.1) | 0.413 |
| Negative | 46 (44.7) | 19 (46.3) | 27 (40.9) | 16 (50.0) | ||
| Not available | 31 (30.1) | 9 (22.0) | 24 (36.4) | 7 (21.9) | ||
| BRAF mutation | ||||||
| Yes | 48 (46.6) | 16 (39.0) | 0.538 | 30 (45.5) | 10 (31.3) | 0.385 |
| No | 38 (36.9) | 14 (34.1) | 24 (36.4) | 13 (40.6) | ||
| Not available | 17 (16.5) | 11 (26.8) | 12 (18.2) | 9 (28.1) | ||
Data are expressed as n (%) unless otherwise specified
TLPLDC tumor lysate, particle-loaded, dendritic cell, ITT intention-to-treat, PT per treatment, IQR interquartile range, TILs tumor-infiltrating lymphocytes
Table 2.
Staging data with comparison between TLPLDC versus placebo in the ITT and PT cohorts
| Category | ITT analysis | PT analysis | ||||
|---|---|---|---|---|---|---|
| TLPLDC | Placebo | p value | TLPLDC | Placebo | p value | |
| AJCC 7th edition overall stage | ||||||
| III | 80 (77.7) | 32 (78.0) | 0.961 | 52 (78.8) | 26 (81.3) | 0.777 |
| IV | 23 (22.3) | 9 (22.0) | 14 (21.2) | 6 (18.8) | ||
| AJCC 7th edition stage | ||||||
| IIIa | 9 (8.7) | 5 (12.2) | 0.013 | 6 (9.1) | 2 (6.2) | 0.211 |
| IIIb | 24 (23.3) | 19 (46.3) | 19 (28.8) | 16 (50.0) | ||
| IIIc | 47 (45.6) | 8 (19.5) | 27 (40.9) | 8 (25.0) | ||
| IV | 23 (22.3) | 9 (22.0) | 14 (21.2) | 6 (18.8) | ||
| T stage | ||||||
| T0 | 2 (1.9) | 0 (0) | 0.346 | 1 (1.5) | 0 (0) | 0.636 |
| Tis | 1 (1.0) | 0 (0) | 0 (0) | 0 (0) | ||
| T1 | 7 (6.8) | 7 (17.1) | 4 (6.1) | 4 (12.5) | ||
| T2 | 21 (20.4) | 4 (9.8) | 16 (24.2) | 3 (9.4) | ||
| T3 | 22 (21.4) | 8 (19.5) | 14 (21.2) | 7 (21.9) | ||
| T4 | 24 (23.3) | 7 (17.1) | 15 (22.7) | 7 (21.9) | ||
| TX | 11 (10.7) | 7 (17.1) | 7 (10.6) | 4 (12.5) | ||
| Unavailable | 15 (14.6) | 8 (19.5) | 9 (13.6) | 7 (21.9) | ||
| N stage | ||||||
| N1 | 18 (17.5) | 14 (34.1) | 0.168 | 13 (19.7) | 9 (28.1) | 0.595 |
| N2 | 26 (25.2) | 9 (22.0) | 17 (25.8) | 8 (25.0) | ||
| N3 | 32 (31.1) | 7 (17.1) | 22 (33.3) | 6 (18.8) | ||
| Unavailable | 27 (26.2) | 11 (26.8) | 14 (21.2) | 9 (28.1) | ||
| M stage | ||||||
| M0 | 75 (72.8) | 27 (65.9) | 0.868 | 48 (72.7) | 22 (68.8) | 0.714 |
| M1a | 5 (4.9) | 3 (7.3) | 3 (4.5) | 2 (6.3) | ||
| M1b | 4 (3.9) | 1 (2.4) | 3 (4.5) | 0 (0) | ||
| M1c | 5 (4.9) | 3 (7.3) | 3 (4.5) | 2 (6.3) | ||
| Unavailable | 14 (13.6) | 7 (17.1) | 9 (13.6) | 6 (18.8) | ||
Data are expressed as n (%)
TLPLDC tumor lysate, particle-loaded, dendritic cell, ITT intention-to-treat, PT per treatment, AJCC American Joint Committee on Cancer
Table 3.
Prior treatment data with comparison between TLPLDC versus placebo in the ITT and PT cohorts
| Category | ITT analysis | PT analysis | ||||
|---|---|---|---|---|---|---|
| TLPLDC | Placebo | p value | TLPLDC | Placebo | p value | |
| Wide local excision | ||||||
| Yes | 88 (85.4) | 29 (70.7) | 0.031 | 55 (83.3) | 24 (75.0) | 0.328 |
| No | 15 (14.6) | 12 (29.3) | 11 (16.7) | 8 (25.0) | ||
| Lymph node surgery | ||||||
| SLNB | 15 (14.6) | 8 (19.5) | 0.171 | 9 (13.6) | 8 (25.0) | 0.121 |
| LND | 30 (29.1) | 18 (43.9) | 19 (28.8) | 13 (40.6) | ||
| SLNB and LND | 29 (28.2) | 6 (14.6) | 21 (31.8) | 4 (12.5) | ||
| None | 29 (28.2) | 9 (22.0) | 17 (25.8) | 7 (21.9) | ||
| Immunotherapy | ||||||
| Yes | 41 (39.8) | 15 (36.6) | 0.721 | 30 (45.5) | 11 (34.4) | 0.297 |
| No | 62 (60.2) | 26 (63.4) | 36 (54.5) | 21 (65.6) | ||
| CPI | ||||||
| Yes | 34 (33.0) | 9 (22.0) | 0.229 | 24 (36.4) | 7 (21.9) | 0.148 |
| No | 69 (67.0) | 32 (78.0) | 42 (63.6) | 25 (78.1) | ||
| CPI agents | ||||||
| Ipilimumab only | 13 (12.6) | 6 (14.6) | 0.662 | 9 (13.6) | 5 (15.6) | 0.480 |
| Nivolumab only | 9 (8.7) | 1 (2.4) | 7 (10.6) | 1 (3.1) | ||
| Pembrolizumab only | 6 (5.8) | 1 (2.4) | 4 (6.1) | 1 (3.1) | ||
| Ipilimumab + pembrolizumab | 3 (2.9) | 0 (0) | 2 (3.0) | 0 (0) | ||
| Ipilimumab + nivolumab | 2 (1.9) | 1 (2.4) | 2 (3.0) | 0 (0) | ||
| Ipilimumab + PD1 (unspecified) | 1 (1.0) | 0 (0) | 0 (0) | 0 (0) | ||
| Other immunotherapy | ||||||
| Interleukin-2 | 4 (3.9) | 2 (4.9) | 0.090 | 1 (1.5) | 2 (6.2) | 0.266 |
| Interferon-α | 7 (6.8) | 3 (7.3) | 4 (6.1) | 1 (3.1) | ||
| T-VEC | 4 (3.9) | 2 (4.8) | 3 (4.5) | 2 (6.2) | ||
| BCG | 0 (0) | 3 (7.1) | 0 (0) | 2 (6.2) | ||
| Chemotherapy | ||||||
| Yes | 6 (5.8) | 1 (2.4) | 0.394 | 4 (6.1) | 0 (0) | 0.155 |
| No | 97 (94.1) | 40 (97.6) | 62 (93.9) | 32 (100) | ||
| Chemotherapy agents | ||||||
| Dacarbazine, cisplatin, vinblastine | 2 (1.9) | 0 (0) | 0.424 | 1 (1.5) | 0 (0) | 0.568 |
| Paclitaxel, carboplatin | 1 (1.0) | 0 (0) | 0 (0) | 0 (0) | ||
| Imatinib, dasatinib | 0 (0) | 1 (2.4) | 0 (0) | 0 (0) | ||
| Melphalan (isolated limb perfusion) | 3 (2.9) | 0 (0) | 3 (4.5) | 0 (0) | ||
| BRAF inhibitor | ||||||
| Yes | 8 (7.8) | 3 (7.3) | 0.927 | 6 (9.1) | 2 (6.2) | 0.630 |
| No | 95 (92.2) | 38 (93.7) | 60 (90.1) | 30 (93.8) | ||
| Radiation therapy | ||||||
| Yes | 26 (25.2) | 11 (26.8) | 0.844 | 16 (24.2) | 8 (25.0) | 0.935 |
| No | 77 (75.8) | 30 (73.2) | 50 (75.8) | 24 (75.0) | ||
Data are expressed as n (%)
TLPLDC tumor lysate, particle-loaded, dendritic cell, ITT intention-to-treat, PT per treatment, SLNB sentinel lymph node biopsy, LND lymph node dissection, CPI checkpoint inhibitor, PD1 programmed death 1, T-VEC talimogene laherparepvec, BCG bacillus Calmette–Guérin
The PT analysis included 66 patients in the vaccine group and 32 patients in the control group (Fig. 1) who remained enrolled on the trial and disease-free at the fourth dose time point, 6 months from initiation of therapy. A total of 28 patients in the vaccine group and 10 patients in the control group completed the full series of six vaccines. In PT analysis, the vaccine group was older (median age 64.8 vs. 60.9, p = 0.036), but there were no other significant differences between groups in terms of pathologic, staging, or treatment-related factors.
Vaccine Preparation
A full vaccination series was produced for all patients. For DC isolation, 71 patients received a dose of Neupogen® and had 50–70 mL of blood drawn, while 73 patients did not receive Neupogen® and instead had 120 mL of blood drawn. The median DC yield was higher in those patients receiving Neupogen® and smaller blood volume collection (9 × 106 cells vs. 7.2 × 106 cells, p = 0.021). A total of 12 vaccine production runs failed in 11 patients (one patient had two production runs fail and required a third blood draw). Of these 11 patients requiring redraw, five received Neupogen® and had 50–70 mL of blood drawn, while six patients had 120 mL of blood drawn. The reasons for production failure included six with inadequate cell production, four that failed to meet sterility requirements, and two with failed cell viability.
The median time to vaccine delivery was 18 days, with 3 days for production, 14 days for sterility/viability testing, and 1 day for shipping. This time was longer when the first production run failed.
Toxicity
AEs are summarized in Table 4. Study-wide, 97 (67.4%) patients experienced an AE, with no difference between groups (p = 0.880). Most (93.2%) AEs were grade 1 or 2. Maximum local and systemic toxicity was not significantly different between treatment groups (p = 0.35). A total of 20 patients (13.8%) experienced a grade 3 or higher AE, with no significant difference between groups (11.7% vaccine vs. 19.5% control, p = 0.218).
Table 4.
Adverse event data comparing TLPLDC and placeboa
| Adverse events | TLPLDC | Placebo | p value |
|---|---|---|---|
| Any AE | 69 (67.0) | 28 (68.3) | 0.880 |
| Serious AEb | 10 (9.7) | 6 (14.6) | 0.396 |
| Severe AE (grade 3 or higher)c | 12 (11.7) | 8 (19.5) | 0.218 |
| Related AEd | 37 (35.9) | 13 (31.7) | 0.632 |
| Related serious AE | 1 (1.0) | 1 (2.4) | 0.497 |
| Local adverse events | |||
| Injection site erythema | 17 (16.5) | 4 (9.8) | 0.300 |
| Skin induration | 11 (10.7) | 4 (9.8) | 0.870 |
| Systemic adverse events | |||
| Fatigue | 14 (13.6) | 5 (12.2) | 0.823 |
| Headache | 10 (9.7) | 3 (7.3) | 0.651 |
| Diarrhea | 10 (9.7) | 2 (4.9) | 0.344 |
| Cough | 7 (6.8) | 4 (9.8) | 0.546 |
| Fever | 5 (4.9) | 4 (9.8) | 0.273 |
| Hypertension | 5 (4.9) | 3 (7.3) | 0.560 |
| Nausea | 5 (4.9) | 3 (7.3) | 0.560 |
Data are expressed as n (%)
TLPLDC tumor lysate, particle-loaded, dendritic cell, AE adverse event, CTCAE Common Terminology Criteria for Adverse Events
aSpecific adverse events are categorized by Preferred Term
bDefined as resulting in death, life-threatening, requiring or prolonging inpatient hospitalization, disabling, a congenital anomaly/birth defect, or requiring medical or surgical intervention to prevent one of these outcomes
cDefined as grade 3 or higher according to the CTCAE version 4.03
dAEs classified as possibly, probably, and definitely related are included in related AEs
With regard to related AEs, 34.7% of patients experienced treatment-related AEs, with no difference in the rate between arms (35.9% vaccine vs. 31.7% control, p = 0.632). Of these related AEs, almost all (98.1%) were grade 1 or 2. No patients experienced a serious AE deemed likely related. Two experienced serious AEs deemed possibly related to treatment—one patient in the TLPLDC group (three severe AEs, i.e. cough, weight loss, and anemia) and one patient in the placebo group (unspecified systemic infection).
The most common local toxicities were injection site erythema (14.6%) and skin induration (10.4%). The most common systemic toxicities were fatigue (13.2%), headache (9.0%), diarrhea (8.3%), cough (7.6%), and fever (6.3%). There were no differences in the rates of these AEs between groups.
Recurrence Rates
In the ITT analysis, at a median follow-up of 19.1 months, 57/103 patients (55.3%) in the TLPLDC group had recurred compared with 28/41 (68.3%) in the placebo group (p = 0.154). In PT analysis, at a median follow-up of 19.5 months, 20/66 patients (30.3%) in the TLPLDC group had recurred compared with 19/32 (59.4%) in the placebo group (p = 0.006).
Survival Analysis
Disease-Free Survival
In the ITT analysis, at a median follow-up of 19.1 months, the estimated 24-month DFS did not show a significant difference in the ITT analysis (38.5% vs. 27.0%, p = 0.974), and there was no difference in the estimated 36-month DFS (34.2% vs. 21.6%, p = 0.889) (Fig. 2a).
Fig. 2.

Kaplan–Meier curves demonstrating 24- and 36-month disease-free survival comparing TLPLDC versus placebo in the (a) ITT analysis and (b) PT analysis. A summary of the survival table is included beneath each Kaplan–Meier curve. TLPLDC tumor lysate, particle-loaded, dendritic cell, ITT intention-to-treat, PT per treatment
In the PT analysis, at a median follow-up of 19.5 months, the vaccine group had significantly increased estimated 24-month DFS (62.9% vs. 34.8%; p = 0.041). Moreover, the vaccine group showed a significantly increased estimated 36-month DFS, with the vaccine group showing over twice the DFS of the placebo group (55.9% vs. 27.9%, p = 0.021) [Fig. 2b].
Overall Survival
In the ITT population, the 24-month OS was 86.4% in the TLPLDC arm and 75.1% in the control arm (p = 0.15) [Fig. 3]. The estimated 36-month OS for the ITT population was 67.8% in the TLPLDC arm and 49.3% in the control arm (p = 0.159).
Fig. 3.
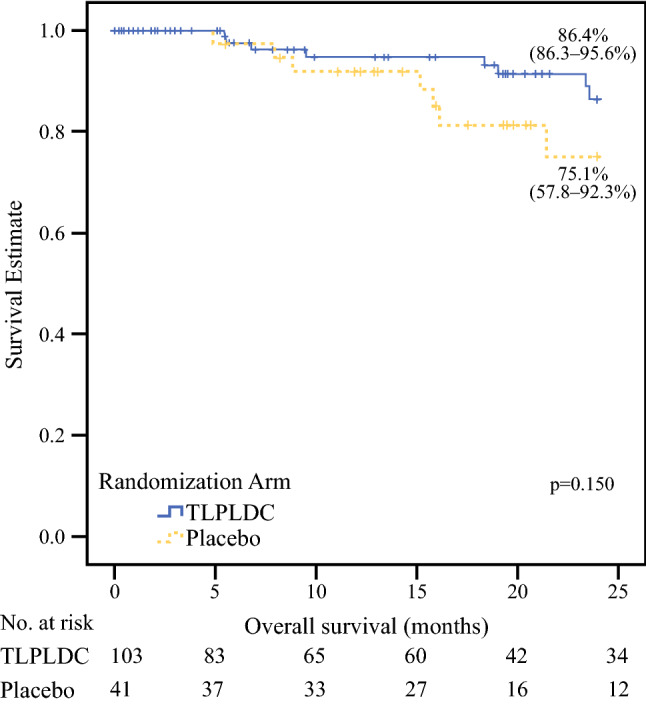
Kaplan–Meier curve demonstrating 24-month overall survival comparing TLPLDC versus placebo in the ITT analysis. A summary of the survival table is included beneath the Kaplan–Meier curve. TLPLDC tumor lysate, particle-loaded, dendritic cell, ITT intention-to-treat
Discussion
We report the primary analysis of the phase IIb trial of the TLPLDC vaccine administered to prevent recurrence in patients with stage III/IV melanoma after resection. Personalized vaccines were successfully prepared for all patients, and the vaccine was well-tolerated. While ITT analysis did not show a clear benefit from the vaccine, the predetermined PT analysis, limited to patients who did not recur during the PVS, showed a benefit from the vaccine, demonstrating a significantly increased 24-month DFS and doubling of the estimated 36-month DFS.
The recent emergence of CPIs has reinvigorated the field of immunotherapy. In addition to non-specific immunostimulatory CPIs, neoantigen-directed active specific immunotherapy, including adoptive cell therapies, is showing clinical benefit.9,18–20 Unfortunately, even these successful specific therapies have not been widely adopted, in part because their production is time- and labor-intensive, and prohibitively expensive.20 The TLPLDC vaccine was designed to stimulate a similarly specific immune response, but in a more efficient and scalable manner. Importantly, the TLPLDC vaccine incorporates all relevant antigens and neoantigens derived from a patient’s tumor in their personalized vaccine. This trial confirms that this vaccine can be successfully produced, with no failures in vaccine production for 144 consecutive patients and an over 90% success rate with the first-pass production run. Additionally, median time to delivery of the vaccines was only 18 days. This personalization and scalability could lead to broader application of immunotherapy for patients with any solid tumor, whether through vaccine monotherapy or in combination with a CPI.
A second factor limiting broader application of immunotherapies is toxicity. The first successful CPI, ipilimumab, has unquestionably changed the landscape of oncology, but this therapy came with a significant level of toxicity.21 The next generation of CPIs, to include pembrolizumab and nivolumab, have less, but still considerable, toxicity,6,22 and toxicity rates are even higher when CPIs are combined.23 While these high rates of toxicity are considered acceptable in the setting of metastatic melanoma, where options are limited and prognosis is poor, the acceptable threshold of toxicity is necessarily lower in the adjuvant setting. As progress continues in the treatment of non-metastatic melanoma, additional therapies need to add efficacy without adding to the already considerable toxicities of existing therapies. In line with this need for lower toxicity, the current study confirms that the TLPLDC vaccine is appropriately well-tolerated.
While safety is clearly important in the adjuvant setting, the ultimate goal is to improve outcomes. Although there was an 11% absolute difference in both 24-month DFS and OS favoring the TLPLDC vaccine in ITT analysis, this difference was not statistically significant. However, there was an encouraging trend in OS (p = 0.15), which is particularly meaningful in the context of growing evidence that evaluation of OS may show the benefit of a cancer vaccine even if DFS does not. This phenomenon is best demonstrated in the results of two phase III trials of the vaccine sipuleucel-T in metastatic castrate-resistant prostate cancer. In both trials, the vaccine led to an improvement in OS without a difference seen in progression-free survival.9,24 This result is explained by immunologic pressure created by vaccination, which may change the growth kinetics of the tumor that is targeted, changing the course of the disease even without preventing recurrence.25 Additionally, any real improvement in OS is likely blunted by the design of our trial as patients who recurred in the control arm were allowed to crossover and receive the TLPLDC vaccination in addition to repeat resection and other SOC therapies.
In addition to the promising signal in this ITT analysis, the PT analysis demonstrates a benefit from vaccine therapy, with a doubling of median DFS in the vaccine group. Given the mechanism of the vaccine’s efficacy, this result is not surprising. Patients who recurred early, during the PVS and shortly thereafter, are less likely to benefit from a vaccine, for multiple reasons. It is becoming increasingly clear that active immunotherapy takes time to show full effects, as was demonstrated in the recent adjuvant trial of pembrolizumab versus placebo in the adjuvant setting, where recurrence-free survival curves did not separate until approximately 6 months into the trial.5 Additionally, for vaccine therapy, if a patient recurs during the vaccine series, they did not receive the full course of therapy and therefore will not likely have a characteristic benefit. Thus, the PT analysis, excluding these patients with early recurrence, is a more meaningful representation of the full effects of vaccine therapy.
Beyond the time needed for therapy to work, the biology of patients’ disease must also be considered. Patients with very early recurrences are likely not ideal candidates for vaccine monotherapy as this more aggressive tumor biology generally does not respond to cancer vaccines.11 If the host immune system cannot keep the occult microscopic disease in check for even a short period, it is unlikely a vaccine will be capable of augmenting the immune system enough to overcome that tumor burden and aggressive biology. Patients with markers of aggressive disease may however respond well to CPI therapy. Subgroup analysis of a recent trial of ipilimumab in patients with stage III melanoma showed patients who benefited the most from this therapy were those with ulceration, stage IIIC disease, and four or more nodes.4 This efficacy of CPIs in more aggressive disease, where vaccines often fail, may indicate a role for the combination of a cancer vaccine and CPI, which could extend the reach of both agents without increased toxicity. This theoretical synergy is being studied in a number of ongoing clinical trials.15
During the time that this trial has been conducted, the landscape of melanoma treatment, particularly in the adjuvant setting, has shifted. The approval of both BRAF/MEK inhibitors and CPIs for the treatment of stage III melanoma has brought effective therapy to these patients, who have, until recently, had very few options. Vaccine monotherapy may still be a viable adjuvant option for early-stage disease, but will likely not be a relevant option for patients with more advanced stages of disease. However, the combination of a TLPLDC vaccine and a CPI should theoretically have a synergistic benefit for these patients. We are currently planning a phase III trial evaluating the efficacy of the vaccine in combination with CPIs in the adjuvant setting in clinically disease-free, high-risk melanoma patients, as this current study was especially limited by the introduction of CPIs in the midst of the trial.
There are several limitations to our study. First, while PT analysis was prespecified, this trial was powered based on ITT analysis. This raises the possibility of type II error in the underpowered PT analysis, which may explain the relatively modest p value coupled with a large improvement in DFS (55.9% vs. 27.9%, p = 0.021). Second, additional data on markers of immune response would augment our clinical response data, but no such markers are available. Assessing tumor response or immune infiltration is not possible as all patients were disease-free at the time of enrollment. Similarly, peripheral immunologic assays are not practical given the personalized nature of the vaccine, as we do not know each patient’s unique tumor antigens/neoantigens, and no one assay would be reliable across many patients. Next, this trial included a mixture of stage III and IV patients, which allowed us to enroll a broader range of patients and could potentially make this therapy applicable to more patients, but could make the results more difficult to interpret for a specific subgroup. Along these lines, we amended the protocol partway through the trial to allow concurrent CPI therapy. This was a necessary modification because the treatment of melanoma changed during the conduct of this trial. However, given the randomized nature of the trial, any effect from this should be seen in both the control and treatment arms. Finally, the primary endpoint of this trial was DFS, which may not be as meaningful as OS, as addressed above. We did however report OS as a secondary endpoint, mitigating the effects of this limitation.
Conclusions
The primary analysis of this phase IIb trial of the TLPLDC vaccine to prevent recurrence in resected stage III/IV melanoma shows that the vaccine is well-tolerated and improves DFS in patients who receive the PVS of 6 months of therapy. These results warrant further testing of this vaccine in a phase III trial.
Supplementary information
Acknowledgment
This work was funded by Elios Therapeutics.
Funding
This work was funded by Elios Therapeutics, a wholly-owned subsidiary of Perseus Holdings, USA.
Disclosures
This clinical study was sponsored by Elios Therapeutics, a wholly-owned subsidiary of Perseus Holdings, USA. which provided funding to the institution of Dr. Robert Andtbacka; Dr. James Jakub is a Advisory Board Member of a Melanoma Surgical Advisory Board, Norvartis Oncology; Dr. Thomas Wagner is an employee of Orbis Health Solutions; Dr. George Peoples is a part-time employeed of Orbis Health Solutions, has consulted for a number of companies broadly in the cancer vaccine space (none working in melanoma or with autologous DC-based vaccines), and has multiple patents for different versions of cancer vaccines, mostly peptide-based vaccines; Dr. Wagner is the inventor on several patents on the technology of the study. Timothy J. Vreeland, Guy T. Clifton, Diane F. Hale, Robert C. Chick, Annelies T. Hickerson, Jessica L. Cindass, Alexandra M. Adams, Phillip M. Kemp Bohan, Adam C. Berger, Jeffrey J. Sussman, Alicia M. Terando, and Mark B. Faries have no conflicts of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Callahan MK, Kluger H, Postow MA, et al. Nivolumab plus ipilimumab in patients with advanced melanoma: updated survival, response, and safety data in a phase I dose-escalation study. J Clin Oncol. 2018;36(4):391–398. doi: 10.1200/JCO.2017.72.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long GV, Eroglu Z, Infante J, et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J Clin Oncol. 2018;36(7):667–673. doi: 10.1200/JCO.2017.74.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845–1855. doi: 10.1056/NEJMoa1611299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggermont AMM, Blank CU, Mandala M, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378(19):1789–1801. doi: 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- 6.Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377(19):1824–1835. doi: 10.1056/NEJMoa1709030. [DOI] [PubMed] [Google Scholar]
- 7.Wahid M, Jawed A, Mandal RK, et al. Recent developments and obstacles in the treatment of melanoma with BRAF and MEK inhibitors. Crit Rev Oncol Hematol. 2018;125:84–88. doi: 10.1016/j.critrevonc.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 9.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 10.Bol K, Bloemendal M, van Willigen W, et al. MIND-DC: A randomized phase III trial to assess the efficacy of adjuvant dendritic cell vaccination in comparison to placebo in stage IIIB and IIIC melanoma patients. Ann Oncol. 2020;31:S732. doi: 10.1016/j.annonc.2020.08.1202. [DOI] [Google Scholar]
- 11.Hale DF, Vreeland TJ, Peoples GE. Arming the immune system through vaccination to prevent cancer recurrence. Am Soc Clin Oncol Educ Book. 2016;35:e159–167. doi: 10.1200/EDBK_158946. [DOI] [PubMed] [Google Scholar]
- 12.Schadendorf D, Ugurel S, Schuler-Thurner B, et al. Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol. 2006;17(4):563–570. doi: 10.1093/annonc/mdj138. [DOI] [PubMed] [Google Scholar]
- 13.Ozao-Choy J, Lee DJ, Faries MB. Melanoma vaccines. Surg Clin N Am. 2014;94(5):1017–1030. doi: 10.1016/j.suc.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Willigen WW, Bloemendal M, Gerritsen WR, Schreibelt G, De Vries IJM, Bol KF. Dendritic cell cancer therapy: vaccinating the right patient at the right time. Front Immunol. 2018;9:2265. doi: 10.3389/fimmu.2018.02265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vreeland TJ, Clifton GT, Herbert GS, et al. Gaining ground on a cure through synergy: combining checkpoint inhibitors with cancer vaccines. Expert Rev Clin Immunol. 2016;12(12):1347–1357. doi: 10.1080/1744666X.2016.1202114. [DOI] [PubMed] [Google Scholar]
- 16.Greene JM, Schneble EJ, Jackson DO, et al. A phase I/IIa clinical trial in stage IV melanoma of an autologous tumor-dendritic cell fusion (dendritoma) vaccine with low dose interleukin-2. Cancer Immunol Immunother. 2016;65(4):383–392. doi: 10.1007/s00262-016-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbert GS, Vreeland TJ, Clifton GT, et al. Initial phase I/IIa trial results of an autologous tumor lysate, particle-loaded, dendritic cell (TLPLDC) vaccine in patients with solid tumors. Vaccine. 2018;36(23):3247–3253. doi: 10.1016/j.vaccine.2018.04.078. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 20.Klingemann HG. Cellular therapy of cancer with natural killer cells-where do we stand? Cytotherapy. 2013;15(10):1185–1194. doi: 10.1016/j.jcyt.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Ascierto PA, Del Vecchio M, Robert C, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017;18(5):611–622. doi: 10.1016/S1470-2045(17)30231-0. [DOI] [PubMed] [Google Scholar]
- 22.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 23.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24(19):3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 25.Stein WD, Gulley JL, Schlom J, et al. Tumor regression and growth rates determined in Five Intramural NCI Prostate Cancer Trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res. 2011;17(4):907–917. doi: 10.1158/1078-0432.CCR-10-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.