

Abstract
Objectives.
Intravenous iloprost improves Raynaud’s phenomenon (RP) and promotes healing of digital ulcers (DU) in scleroderma (SSc). Despite a short half-life, its clinical efficacy lasts weeks. Endothelial adherens junctions, which are formed by VE-cadherin clustering between endothelial cells (ECs), regulate endothelial properties including barrier function, endothelial to mesenchymal transition (Endo-MT), and angiogenesis. We hypothesized that junctional disruption contributes to vascular dysfunction in SSc, and that the protective effect of iloprost is mediated by strengthening of those junctions.
Methods.
Dermal ECs from SSc patients and healthy controls were isolated. The effect of iloprost on ECs was examined using immunofluorescence, permeability assays, Matrigel tube formation, and quantitative PCR.
Results.
Adherens junctions in SSc were disrupted compared to normal ECs, as indicated by reduced levels of VE-cadherin and increased permeability in SSc ECs (p<0.05). Iloprost increased VE-cadherin clustering at junctions and restored junctional levels of VE-cadherin in SSc ECs (37.3 ± 4.3 fluorescent unit, mean ± SD) compared to normals (29.7 ± 3.4, p<0.05) after 2 hours of iloprost incubation. In addition, iloprost reduced permeability of monolayers, increased tubulogenesis, and blocked Endo-MT in both normal and SSc ECs (n≥3, p<0.05). The effects in normal ECs were inhibited by a function-blocking antibody that prevents junctional clustering of VE-cadherin.
Conclusions.
Our data suggests that the long-lasting effects of iloprost reflect its ability to stabilize adherens junctions, resulting in increased tubulogenesis and barrier function, and reduced Endo-MT. These results provide a mechanistic basis for the use of iloprost in treating SSc patients with RP and DU.
Introduction
Systemic sclerosis (SSc) is an autoimmune disease characterized by immune activation, widespread fibrosis, and a structural and functional vasculopathy (1). Vascular involvement includes Raynaud’s phenomenon (RP), digital ulcers (DU), scleroderma renal crisis and pulmonary arterial hypertension. RP is the most typical vascular manifestation that occurs earliest in SSc and generally precedes organ involvement. DU are present in approximately 50% of SSc patients and responsible for significant disability and poor quality of life. Iloprost is a synthetic analogue of prostacyclin (PGI2). Similar to PGI2, iloprost has vasodilatory and antiplatelet effects, but it is more stable than PGI2 with a longer half-life (20 to 30 minutes) and better solubility (2). Intravenous iloprost is marketed in Europe for multiple indications, including the treatment of patients with severe, disabling RP unresponsive to other therapies. In addition, 2017 EUSTAR recommendations assigned Grade A recommendation for treatment of severe SSc-related RP attacks and for treatment of DU (3). It is also widely used in the management of peripheral vascular complications unrelated to SSc-related RP. Pharmacologically, iloprost activates PGI2 receptors, which stimulate adenylate cyclase to produce cyclic adenosine monophosphate (cAMP). PGI2 receptors on smooth muscle cells and platelets inhibit smooth muscle constriction and platelet aggregation. PGI2 receptors are also expressed on endothelial cells (ECs), where they initiate numerous protective effects including augmentation of endothelial adherens junctions and reduced monolayer permeability (4, 5).
Vascular and endothelial pathology is present in SSc patients. These functional and structural defects include increased vascular permeability, reduced nitric oxide (NO) activity, elevated inflammation, EC apoptosis, impaired angiogenesis, endothelial to mesenchymal transition (Endo-MT), intravascular fibrosis, and microvascular rarefaction (6–13). ECs also have lower VE-cadherin expression (12). Adherens junctions, which are formed by clustering of VE-cadherin on neighboring ECs, regulate numerous endothelial properties, including cell morphology, signaling and phenotype. The vascular protective effects of adherens junctions include increased barrier function, amplifying NO signaling, inhibiting apoptosis, and reducing inflammation (14). In contrast, when junctions are disrupted, VE-cadherin and β-catenin are disengaged from the cell membrane and contribute to vascular dysfunction and Endo-MT. It is shown that VE-cadherin clustering at adherens junctions is increased by iloprost and PGI2 by activation of PGI2 receptor (4, 15).
Despite its short half-life, iloprost is beneficial for RP and healing of DU that can extend for weeks after cessation of treatment (16). To dissect the mechanisms involved, we hypothesized that vascular dysfunction in SSc reflects disruption of EC adherens junctions and that the vascular protective effect of iloprost is mediated by strengthening of these junctions in SSc ECs. In this study we report the beneficial effect of iloprost in SSc ECs. Iloprost was shown to enhance the impaired barrier dysfunction in these cells, promote angiogenesis, and inhibit Endo-MT, all of which were potentially dependent on increased VE-cadherin clustering at adherens junctions, as blockade of VE-cadherin in normal ECs blocked these effects.
Methods
Patients and Controls.
All patients recruited met the 2013 ACR/EULAR criteria for the classification of SSc (17). We obtained two 4 mm punch biopsies from the distal forearm of subjects for EC isolation. The healthy controls and patients were matched with age, ethnicity, and gender (Supplemental Table 1). Thirteen healthy controls were recruited (age 50.8 ± 3.8 years, mean ± SEM). All 10 patients had diffuse cutaneous SSc (age 54.9 ± 4.9 years, mean ± SEM), and the disease duration was 2.6 ± 0.4 years (mean ± SEM). Their skin scores ranged from 0 to 28 with a mean of 11.1 ± 2.9 (mean ± SEM). All patients had RP but none had active DU at the time of biopsy. These patients were being treated with immunosuppressive drugs, vasodilators, or proton pump inhibitors, among others at the time of biopsy (Supplemental Table 1). This study was approved by the University of Michigan Institutional Review Board.
Cell culture and treatment.
Dermal ECs were isolated from skin biopsies obtained from the forearms of the subjects. Although the skin scores ranged from 0 to 28 in SSc patients, the skin scores at the site of biopsies ranged between 0 and 2 (6 patients with a score of 0, 3 with a score of 1, and a with a score of 2). The cells from patients were randomized in each experiments, with at least 3 patient lines used in each assay. Skin digestion and cell purification were described previously (6, 18). The CD31 MicroBead Kit (Miltenyi Biotech) was used to purify ECs. Cells were maintained in EBM-2 media supplemented with growth factors (Lonza). For all experiments ECs between passages 3 and 6 were used. Before all experiments, cells were cultured in EGM media supplemented with bovine brain extract for at least 1 day. Cells were treated with 150 nM of iloprost (Cayman) for various time points. There are currently no published pharmacokinetic studies in SSc patients receiving iloprost by IV infusion. In healthy subjects, infusion of iloprost at the dose of 1 and 3 ng/kg/min achieved steady state concentrations of 0.13 and 0.37 nM (19). However, SSc patients showed increased systemic exposure to iloprost after oral doses, suggesting reduced clearance of the drug, with a 1.8 fold increase in Cmax after 8 days of iloprost administration (20). The concentration of iloprost in this study was chosen based on published in vitro studies (21–23).
Inhibition of VE-cadherin clustering at endothelial junctions was achieved by pre-treating cells with 25 μg/ml VE-cadherin function-blocking antibody (BV9, LSBio) for 30 min in culture. This approach prevents new VE‐cadherin trans‐interactions without affecting existing junctional or monolayer integrity (21). To induce Endo-MT in normal ECs, cells were treated with 10 ng/ml TGFβ and/or 150 nM iloprost for 3 days. For the groups incorporating BV9, the cells were pre-treated with 25 μg/ml of this antibody for 30 min before TGFβ and/or iloprost were added. BV9 was present the duration of the experiment.
Immunofluorescence staining.
ECs were cultured in gelatin-coated chambers and treated with iloprost for up to 2 hours. Anti-VE-cadherin antibodies (R&D Systems), anti-β-catenin antibodies (Abcam), and Texas Red™-X Phalloidin (Thermo Fisher) were used to visualize VE-cadherin, β-catenin, and F-actin. VE-cadherin and β-catenin were then probed with Alexa Fluoro secondary antibodies. The nuclei were stained using 4’,6-diamidino-2-phenylindole. Fluorescence was detected using a Nikon A1 confocal microscope. Visualization and analysis of images were done using the ND2 reader plugin in ImageJ.
Permeability assay.
Permeability was assessed by measuring horseradish peroxidase (HRP) movement through EC monolayers in a transwell system (Cell Biologics). Briefly, ECs were plated at 50,000 cells/ml in the transwells and allowed to grow to confluence, then cultured in EBM-2 media with 1% fetal bovine serum (FBS) in the upper and lower chambers. Treatments including iloprost (150 nM) and/or TGFβ (10 ng/ml) were added to the upper chambers along with HRP, and aliquots of the media in the lower chambers were collected at various time points. When analyzing the effect of the function-blocking antibody to VE-cadherin, cells were pre-treated with BV9 (25 μg/ml) for 30 min before addition of iloprost and/or TGFβ. BV9 was present throughout the experiment. The amount of HRP was quantified by addition of 3,3’,5,5’-Tetramethylbenzidine and stop solution and measured at 450nm in a plate reader.
Matrigel tube formation assay.
To examine whether iloprost affects EC angiogenesis, we pre-treated ECs with iloprost for 24 hours and performed Matrigel tube formation assays. Growth factor reduced Matrigel (BD Biosciences) was coated in 8-well Lab-Tek chambers before adding treated ECs, which were suspended in EBM-2 with 1% FBS in the presence of iloprost. Cells were cultured for 6 hours before they were fixed and stained. Pictures were taken using the EVOS XL Core Cell Imaging System. The Angiogenesis Analyzer function in ImageJ was used to quantify the tubes. In a separate experiment, we adopted a different approach where blocking antibodies to VE-cadherin (25 μg/ml) were added in the Matrigel coated chambers with the ECs for 30 min before adding iloprost. The ECs were then cultured in the presence of iloprost and/or BV9 for an additional 8–10 hours.
mRNA extraction and qRT-PCR.
Extraction of RNA was done using the Direct-zol™ RNA MiniPrep Kit and cDNA was prepared using the Verso cDNA synthesis kit. Primers were mixed with Power SYBR Green PCR master mix (Applied Biosystems). The ViiA™ 7 Real-Time PCR System was used to quantify cDNA.
Statistical analysis.
Results were expressed as mean ± S.D except stated otherwise. To determine the differences between the groups, Mann–Whitney U test, Kruskal–Wallis test, or two-way ANOVA were performed using GraphPad Prism version 6 (GraphPad Software, Inc). P-values of less than 0.05 were considered statistically significant.
Results
Disruption of adherens junctions in SSc ECs compared to normal ECs.
To examine the effect of iloprost on adherens junctions, we treated ECs with 150 nM of iloprost for various time periods and visualized VE-cadherin, β-catenin, and F-actin via immunofluorescence. Under control conditions, in the absence of iloprost (NT or time 0), adherens junctions were disrupted and F-actin filaments were disorganized in SSc compared to normal ECs (Figures 1A and 1B). Indeed, junctional levels of VE-cadherin were significantly lower in SSc ECs compared to normal ECs at baseline (0 min, *p<0.05, Figure 1C). In normal ECs, iloprost caused a transient increase in the clustering of VE-cadherin (purple) and β-catenin (green) at cell junctions, which peaked at 10 mins and returned to control levels by 60 mins (Figures 1A). Indeed, VE-cadherin was significantly elevated at 10 mins of iloprost stimulation compared to baseline (Figure 1C, #p<0.05 10 mins vs. 0 min). Staining of F-actin (red) showed that iloprost induced accumulation of peripheral F-actin at the adherens junctions (Figure 1A). In contrast, in SSc ECs, iloprost caused a delayed but more sustained increase in clustering of β-catenin and VE-cadherin at cell junctions (Figure 1B and 1C). The increase in VE-cadherin clustering was evident at 60 mins, and remained significantly elevated after 120 mins (Figure 1C, #p<0.05, 60 or 120 mins vs. NT). After iloprost, the clustering of VE-cadherin at cell junctions in SSc ECs was not only normalized, but was significantly higher than observed in normal ECs (Figure 1C, *p<0.05 at 120 mins, normal vs. SSc). Consistent with increased clustering of VE-cadherin and strengthening of adherens junctions, iloprost also promoted sustained increases in cortical F-actin in SSc ECs (Figure 1B).
Figure 1. Effect of iloprost on VE-cadherin localization and cell permeability.
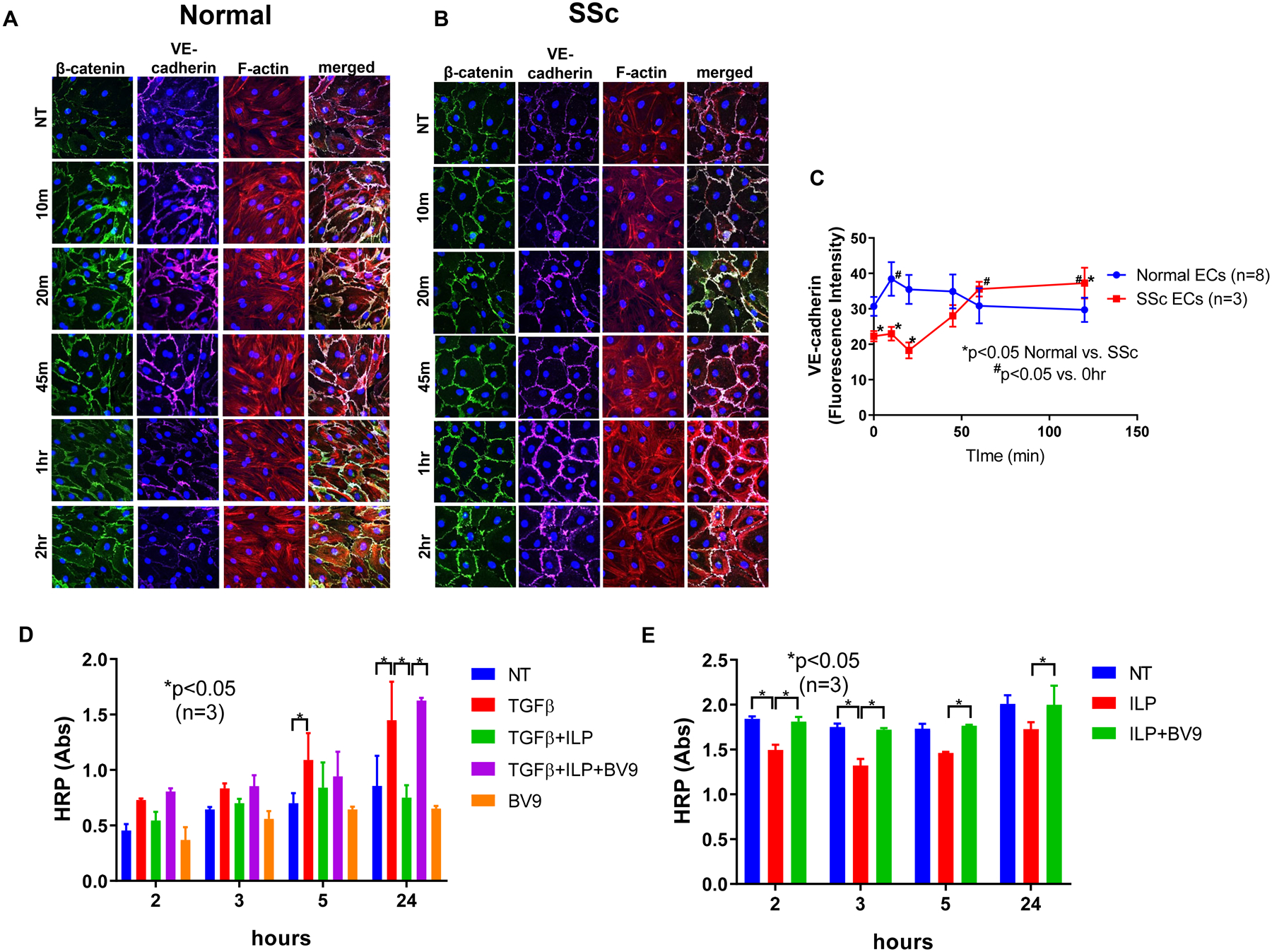
Immunofluorescence of VE-cadherin, β-catenin, and F-actin in ECs were visualized using a Nikon A1 confocal microscope and pictures were taken at 600x. Cell permeability was assessed by measuring HRP movement through EC monolayers using the Endothelial Transwell Permeability Assay Kit. Cells were treated with iloprost (150nM) and/or TGFβ (10ng/ml) at various time points. BV9 (25 μg/ml) was used to pre-treat the cells for 30 min before iloprost and TGFβ were added. (A) Iloprost increased junctional clustering of VE-cadherin and β-catenin as soon as 10 min of incubation in normal ECs; (B) Iloprost showed a delayed but more prolonged effect on VE-cadherin and β-catenin clustering in SSc ECs. Disorganized F-actin filaments in SSc ECs were also observed; (C) Quantification of fluorescent signal of VE-cadherin showed significant reduction of VE-cadherin in SSc ECs compared to normal ECs at baseline. After iloprost, VE-cadherin intensity was greater in SSc compared to normal ECs; (D) In normal ECs, TGFβ increased permeability as measured by HRP movement through EC monolayers. Iloprost inhibited permeability of EC monolayers while blockade of VE-cadherin by BV9 reversed it. (E) In SSc ECs, iloprost inhibited the increased permeability of these cells while the function blocking antibody to VE-cadherin (BV9) prevented the effects of iloprost. Experiments were done with at least 3 subject-derived lines. Results are expressed as mean +/− SD and p<0.05 was considered significant. NT: not treated; ILP: iloprost; Abs: absorbance
Effect of iloprost on endothelial monolayer permeability.
To examine the effect of iloprost on permeability of EC monolayers, we first treated normal ECs with TGFβ which is known to increase permeability in ECs (24). TGFβ increased HRP permeability significantly across EC monolayers at 5 and 24 hours (Figure 1D). Co-incubation of iloprost prevented the TGFβ-induced increase in permeability at 24 hours. To examine whether VE-cadherin is involved in the response to iloprost, we used BV9, a blocking antibody to VE-cadherin that prevents its clustering at EC junctions. Pretreatment with BV9 did not affect EC monolayer integrity (Figure 1D). However, the effect of iloprost to reduce the TGFβ-induced increase in permeability was prevented by BV9 at 24 hours, suggesting that the protective effect of iloprost on EC monolayer integrity was dependent on VE-cadherin clustering at adherens junctions.
Because microvascular abnormalities and vascular leakage are prominent hallmarks of SSc (25), we postulated that SSc ECs would show barrier dysfunction in vitro. Indeed, under control conditions in the absence of iloprost, HRP permeability was significantly higher in SSc ECs compared to normal ECs (Figures 1D and 1E, HRP Abs 1.843 ± 0.027 vs 0.454 ± 0.059 at 2 hour; 1.751 ± 0.038 vs. 0.644 ± 0.022 at 3 hour; 1.733 ± 0.054 vs. 0.700 ± 0.091 at 5 hour; 2.010 ± 0.096 vs. 0.855 ± 0.272 at 24 hour, NT groups in normal vs. SSc ECs, all p<0.05), confirming that SSc ECs have impaired endothelial barrier function. Iloprost significantly decreased the permeability of SSc ECs at 2 and 3 hours; an effect that was prevented by the function-blocking antibody to VE-cadherin (BV9, Figure 1E). Taken together, these results suggest that iloprost potentiates formation of adherens junctions, augmenting endothelial barrier function and reducing the barrier dysfunction of SSc ECs in a VE-cadherin-dependent manner.
Effect of iloprost on EC tubulogenesis.
In normal ECs, iloprost significantly increased tube formation (Figure 2A). This angiogenic effect was prevented by the function-blocking antibody BV9 (Figure 2B), suggesting that it was dependent on the clustering of VE-cadherin at endothelial junctions. We previously showed that angiogenesis is deficient in SSc ECs (6, 18). Pre-treatment of SSc ECs with iloprost enhanced their ability to form tubes on Matrigel (Figure 2C).
Figure 2. Effect of iloprost on EC angiogenesis.
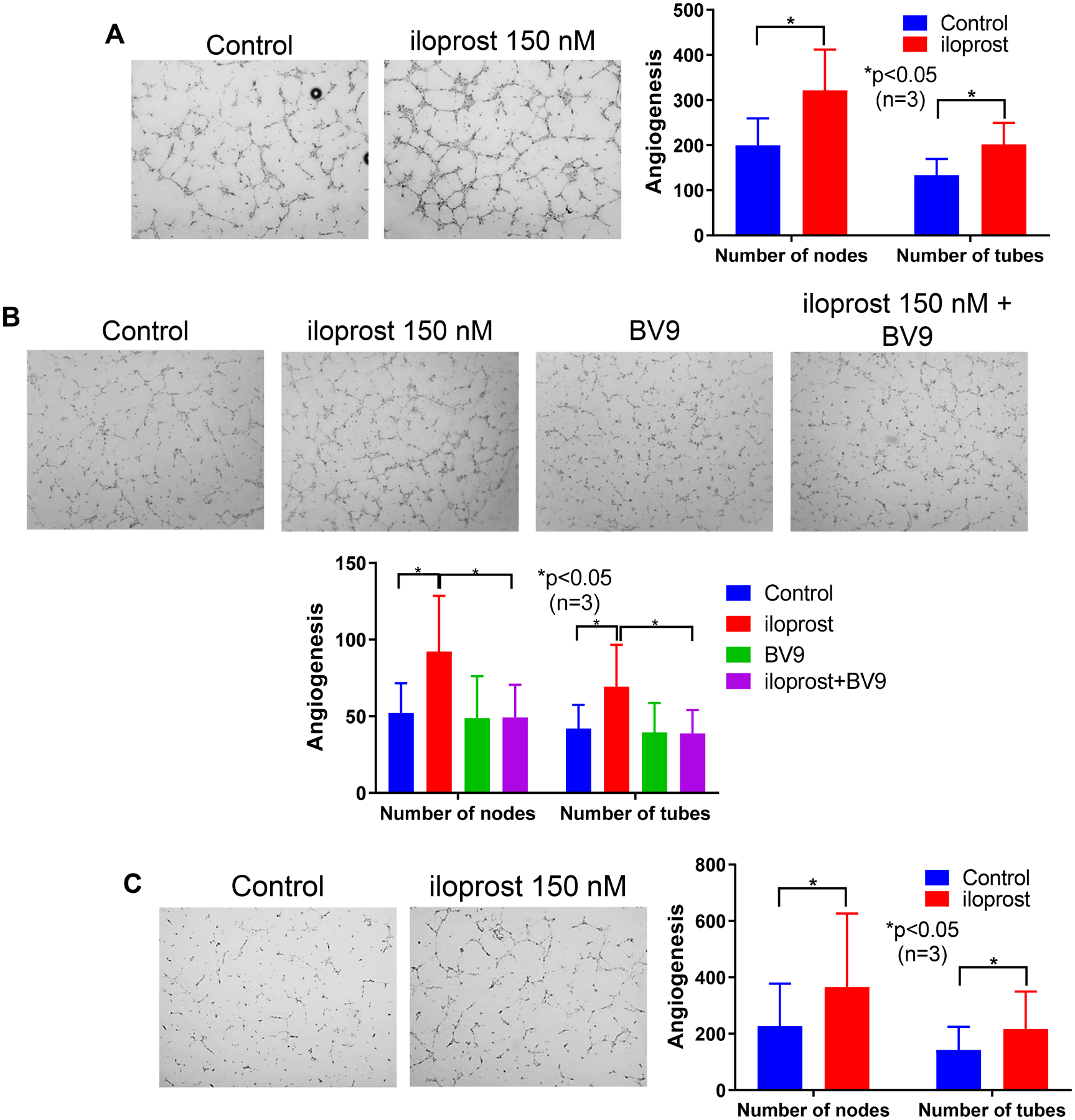
Angiogenesis of ECs was measured using an in vitro Matrigel tube formation assay. For A and C, ECs were pre-treated with iloprost (150nM) overnight before they were plated on Matrigel. After 6 hours, cells were fixed and stained. For B, BV9 (25 μg/ml) was used to pre-treat the ECs for 30 min before iloprost was added. The cells were cultured for 8 hours before they were fixed and stained. (A) and (B) Iloprost increased tubulogenesis in normal ECs and this was reduced by a function blocking antibody to VE-cadherin (BV9); (C) Iloprost induced tubulogenesis in SSc ECs. Experiments were done with 3 subject-derived lines. Results are expressed as mean +/− SD and p<0.05 was considered significant.
Effect of iloprost on Endo-MT.
Endo-MT appears to be a prominent feature of SSc, with SSc ECs having increased expression of Endo-MT markers and reduced expression of EC marker proteins (12). During Endo-MT, ECs lose cell-cell adhesion disrupting endothelial junction stability and increasing vascular permeability. We hypothesized that iloprost, by enhancing VE-cadherin clustering at the adherens junction, could reduce Endo-MT in SSc. We first tested this hypothesis in normal ECs. Treatment of TGFβ for 72 hours in ECs significantly increased mesenchymal markers at the mRNA level including ACTA2 (encoding for α-smooth muscle actin, αSMA), and S100A4, while significantly downregulating EC markers PECAM1 and CDH5 (encoding for CD31 and VE-cadherin, Figure 3A). In addition, TGFβ also induced SNAI1 (encoding for SNAIL), a transcription factor for Endo-MT. However, TGFβ did not affect FLI1. Co-treatment with lloprost significantly reduced TGFβ- mediated Endo-MT by decreasing the upregulated mesenchymal markers and SNAI1, while increasing the downregulated EC markers. These results suggest that iloprost inhibits Endo-MT induced by TGFβ. This protective effect of iloprost was markedly reduced by the function blocking antibody to VE-cadherin, which enabled restoration of the Endo-MT response to TGFβ (Figure 3A). These results suggest that iloprost inhibits Endo-MT by increasing the clustering of VE-cadherin at endothelial junctions. Similarly, in SSc ECs, iloprost reduced the increased levels of Endo-MT in these cells, significantly reducing expression of mesenchymal markers (ACTA2, COL1A1, S100A4) and SNAI1 (Figure 3B).
Figure 3. Effect of iloprost on Endo-MT in ECs.
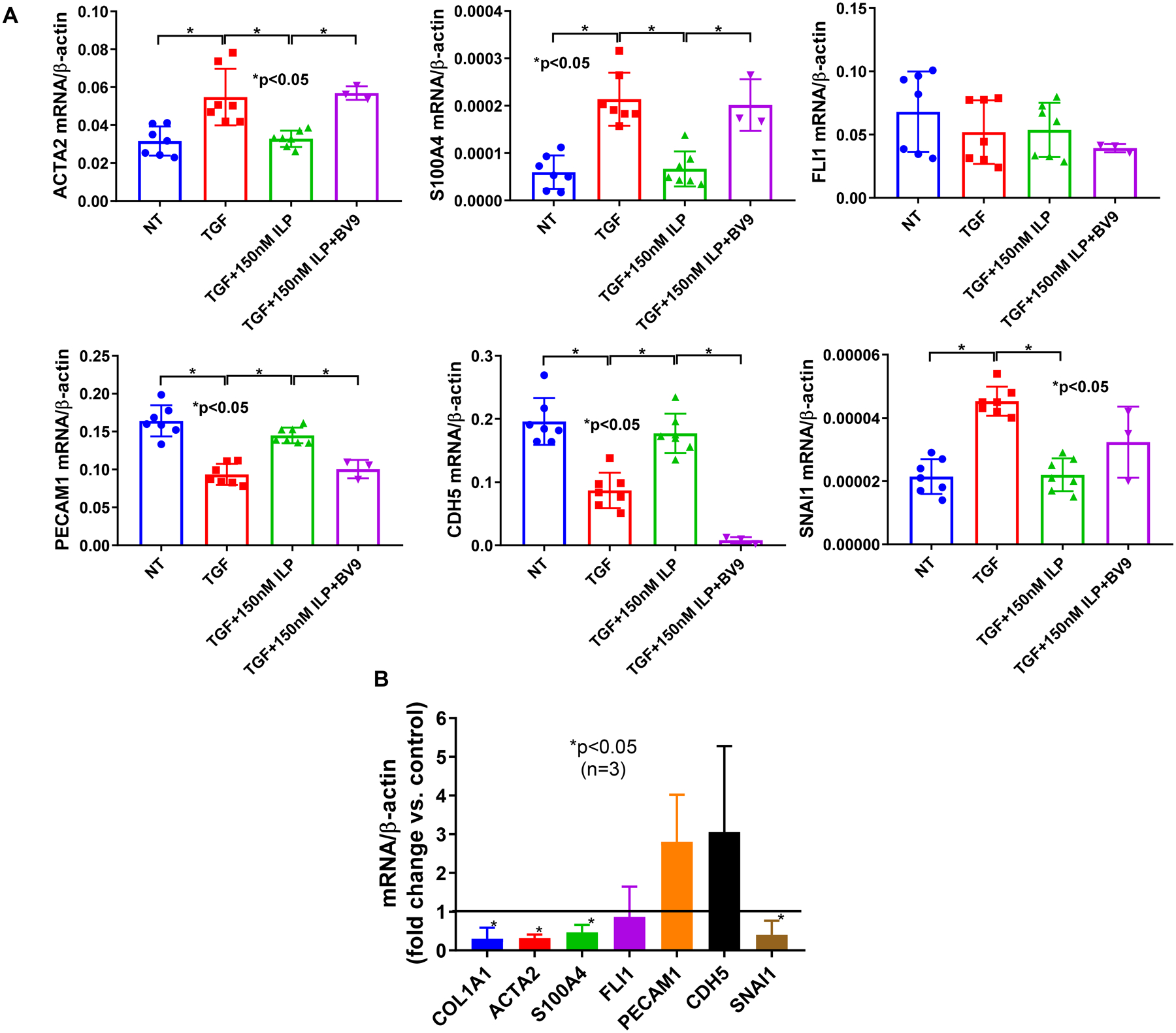
Normal ECs were treated with 10 ng/ml TGFβ and/or 150 nM of iloprost for 3 days. BV9 was added 30 min before the addition of various treatments. Cellular markers for Endo-MT were measured using qPCR. (A) Iloprost inhibited TGFβ-induced Endo-MT in normal ECs, and the effect was blocked by BV9, a VE-cadherin antibody; (B) Iloprost inhibited the Endo-MT phenotype in SSc ECs. Experiments were done with 3–7 subject-derived lines. Results are expressed as mean +/− SD and p<0.05 was considered significant.
Discussion
In this study, we have provided a mechanistic basis for the use of iloprost in treating SSc patients with RP and DU. We have demonstrated that iloprost stabilizes endothelial adherens junctions, increases barrier function, promotes EC tubulogenesis, and inhibits Endo-MT, all of which would be expected to have important therapeutic benefits in SSc (Figure 4). These protective effects of iloprost appear to be dependent on increased clustering of VE-cadherin at endothelial junctions, because they were markedly reduced by a function-blocking antibody to VE-cadherin. Since enhanced VE-cadherin clustering also occurs in response to PGI2 itself and other PGI2 analogues (4, 15), we believe the effects observed in this study are not limited to iloprost itself, but apply to other PGI2 analogues and agonists as well.
Figure 4. Vascular protective effect of iloprost in SSc.
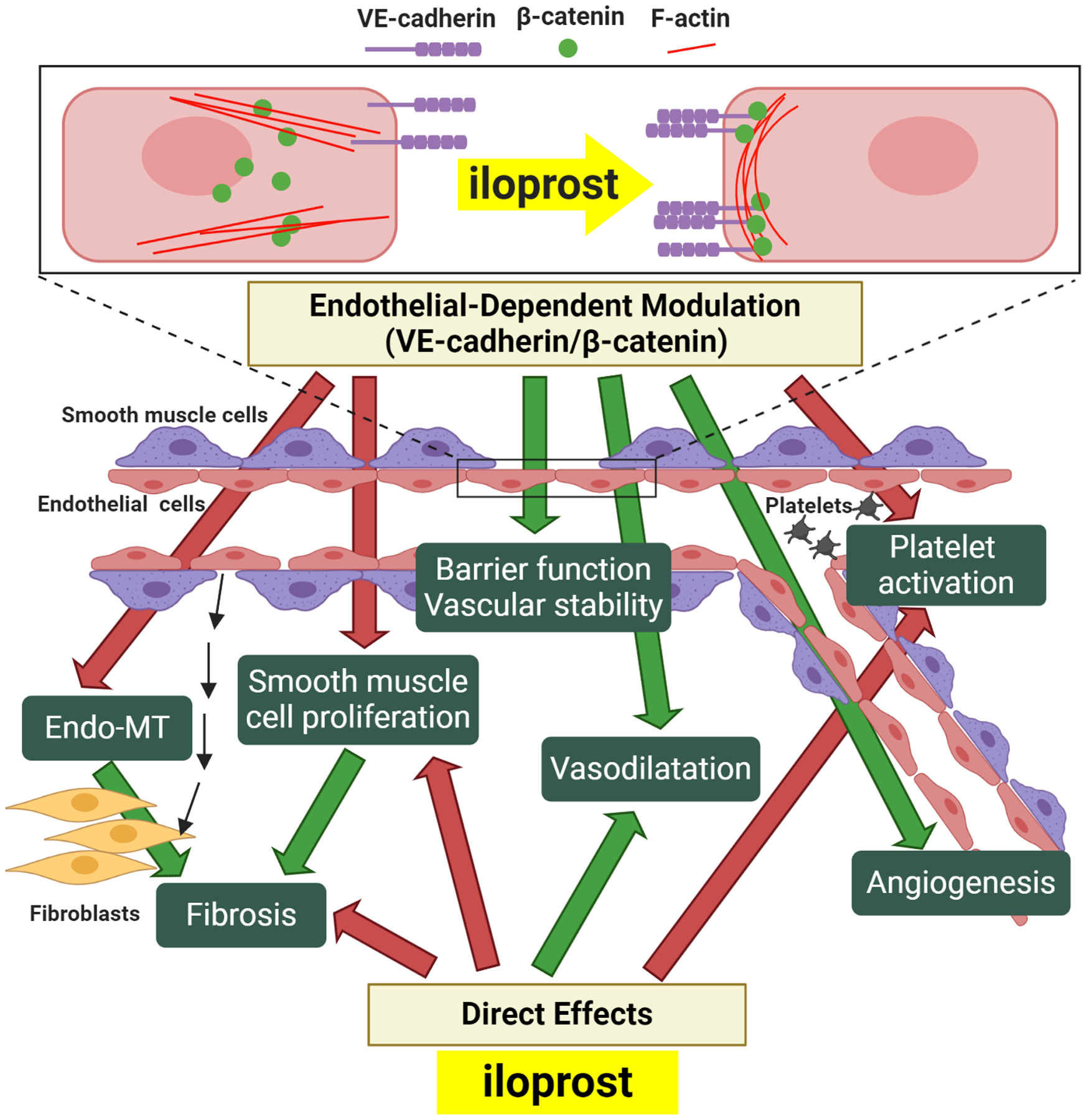
In SSc ECs, Iloprost increases VE-cadherin and β-catenin clustering at the adherens junction, accompanied with accumulation of peripheral F-actin. Increased interaction and signaling of VE-caderin/β-catenin will promote protective endothelial functions and vascular stability including an increase in barrier function, promotion of angiogenesis, inhibition of Endo-MT, and increased NO activity, which will contribute to vasodilation, inhibition of platelet activation, and blockade of smooth muscle cell proliferation. Endo-MT and smooth muscle proliferation can contribute to intravascular and extravascular fibrosis, and to microvascular rarefaction. In addition to these endothelial-dependent modulatory effects, iloprost can also act directly on platelets, fibroblasts, and smooth muscle cells to inhibit platelet activation and fibrosis, and promote vasodilation. Inhibitory effects are highlighted by red arrows, and positive effects are highlighted with green arrows.
Pathologically, endothelial injury is a pivotal initial event in SSc pathogenesis. Persistent activation of SSc ECs by currently unknown sources results in early functional changes and alterations in vasculature including vascular leakage (26). Indeed, electron microscopy of the nailfold in early SSc patients revealed decreased capillary loops with intercellular gaps, associated with interstitial edema (9–11). The regression of the small vessels in SSc is partly due to destabilization of vessels and defect in angiogenesis. As shown by us and others, SSc ECs showed reduced angiogenic properties in in vitro studies (6, 7). These cells also showed intrinsic defect in NO production due to downregulation of endothelial NO synthase (8). Although these endothelial events precede tissue fibrosis, leakage of the blood vessels fuels later tissue fibrosis by involving abnormal ECs, activated inflammatory cells and fibroblasts. In addition, in the presence of TGFβ and immune and pro-fibrotic mediators, SSc ECs acquire a pro-migratory and pro-fibrotic phenotype through Endo-MT, where they differentiate into collagen-producing/αSMA-positive cells that contribute to intravascular and extravascular fibrosis (12). All of these events point to the critical involvement of endothelial dysregulation in SSc pathogenesis. Therapeutic intervention aiming to stabilize ECs and reverse endothelial dysfunction may not only inhibit vascular complications in SSc patients, but also attenuate fibrosis.
Adherens junctions are largely composed of VE-cadherin that binds to several partners, including β-catenin, via its cytoplasmic domain. Junctional clustering of VE-cadherin and β-catenin directly or indirectly modulates various proteins and signaling pathways including Rac-1, tyrosine kinase receptors, protein tyrosine phosphatases, AKT, RhoA/ROCK, Wnt, and Notch signaling (14, 27, 28). These complex signaling events reflect the wide range of biological effects the adherens junctions are involved in. They not only promote endothelial barrier function, they also maintain EC identity by inhibiting Endo-MT, promote NO production, inhibit apoptosis, block leukocyte extravasation and inflammation, and promote endothelial and vascular stability (14). Indeed, when adherens junctions are disrupted, increased permeability, impaired NO production, inflammatory cell infiltration, enhanced transcription of pro-inflammatory mediators, and increased Endo-MT are well documented. Based on these evidences, disruption of adherens junctions could lead to serious pathological consequences in the vasculature, many of which, are characteristic of vascular complications seen in SSc. Interestingly, immunohistochemistry staining of skin biopsies showed that CD31-positive ECs were accompanied with a loss of VE-cadherin expression in SSc patients (13). In vitro experiments, including this study, also confirmed the downregulation of VE-cadherin in SSc ECs (12).
Our results showing that the barrier protective effect of iloprost in SSc ECs was mediated by enhancing VE-cadherin adherens junctions echo the findings by Birukoca et al. (21). This is also demonstrated in pulmonary ECs and human umbilical ECs using PGI2; the barrier-protective effect of PGI2 was mediated by cAMP and downstream pathways, which ultimately led to enhancement of adherens junctions (4, 15). In several follow up studies, the protective effect of iloprost on barrier dysfunction has been further highlighted (29–31). iloprost not only protected mice from ventilator-induced acute lung injury by improving lung endothelial barrier function, it also enhanced barrier function in cultured human lung microvascular ECs (29). The beneficial effect of iloprost in a model of septic lung injury was due, in part, by attenuating barrier dysfunction in the lung (30, 31). In addition, in human pulmonary artery ECs, iloprost attenuated the disruption of the endothelial monolayer, and suppressed the activation of p38 MAPK, NF-κB and Rho signaling after lipopolysaccharide challenge. These studies using PGI2 and its analogues in lungs and pulmonary ECs further support the benefits of using these drugs in patients with pulmonary arterial hypertension.
We postulate that the mechanisms behind the prolonged effect of iloprost on barrier protection in SSc ECs are multifactorial. The cAMP/Epac/Rap1 pathway involved in barrier function might be impaired in SSc ECs. In addition, lower levels of VE-cadherin in SSc ECs might require longer time it to cluster at the junctions. We showed previously that RhoA/Rock expression and activity are elevated in SSc ECs compared to normal ECs (18). Since activation of RhoA/ROCK signaling pathway induced VE-cadherin internalization in ECs (32), it is possible that the activated RhoA/ROCK signaling in SSc ECs reduces VE-cadherin localization at cell junctions. In addition, EZH2, a histone methyltransferase catalyzing repressive H3K27me3 mark, might also play a role. We previously showed that in SSc ECs both EZH2 and H3K27me3 are upregulated compared to normal ECs (33). A study by Morini et al. suggested that clustered VE-cadherin anchors EZH2 at the cell membrane, allowing active gene transcription by impeding the recruitment of EZH2 to the polycomb repressive complex to promoters of genes that strengthen endothelial junctions (34). Downregulation of VE-cadherin at cell junctions and nuclear localization of EZH2 in SSc ECs could both contribute to the vascular leakage and barrier dysfunction in this disease.
In addition to barrier enhancement, we showed that iloprost induced EC angiogenesis on Matrigel. The pro-angiogenic potential of iloprost was also shown in dental organ cultures (35), endothelial progenitor cells (36), and mouse cornea neovascularization models (37, 38). Early studies suggested that the pro-angiogenic properties of iloprost depends on its action on peroxisome proliferator activated receptors (PPARs), which occur via a vascular endothelial growth factor (VEGF)-dependent mechanism (37, 38). Although additional mechanisms may be involved, the results of the present study suggest that the effect of iloprost to increase junctional clustering of VE-cadherin plays a fundamental role in the angiogenic response of this drug.
In SSc, Endo-MT is evident and likely contributes to intravascular and extravascular fibrosis and microvascular rarefaction (12, 39). Both TGFβ and endothelin-1 treatments induced Endo-MT in both normal and SSc ECs in a SMAD-dependent manner (39). In addition, SSc ECs have lower levels of endothelial markers and increased expression of fibroblast markers such as αSMA (12). These cells also show increased ability to contract collagen gels, a major characteristic of myofibroblast function. While the canonical TGFβ/Smad pathway is a major regulator for Endo-MT, TGF-β-induced Endo-MT can also be impacted by crosstalk with other pathways, including Wnt/β-catenin, endothelin-1, and Notch (40). In addition, modulators for TGFβ, such as thrombospondin-1, and proteins regulated by TGFβ, such as connective tissue growth factor (CTGF), can also contribute to Endo-MT directly or indirectly (41, 42). As disruption of adherens junctions is a key initial step of Endo-MT (43) and that many of these aforementioned pathways interact directly or indirectly with adherens junctions (14, 28), the effect of iloprost on Endo-MT inhibition in ECs is not surprising. Indeed, increased clustering of VE-cadherin/β-catenin at adherens junction can inhibit the nuclear localization of β-catenin and block its Endo-MT-promoting effect (14).
PGI2 is synthesized from arachidonic acid by sequential actions of cyclooxygenase and PGI2 synthase. It acts directly on platelets and vascular smooth muscle cells to reduce platelet aggregation and induce vascular relaxation through the cAMP pathway. These effects will be amplified by the effects of PGI2 on ECs. Increased VE-cadherin clustering at adherens junctions can amplify NO production and reverse endothelial NO-dilator dysfunction (14), which is present in SSc (8). Because NO-cGMP signaling acts synergistically with PGI2-cAMP signaling to induce vasodilation as well as inhibition of platelet activation and thrombosis, the ability of PGI2 to stimulate both cGMP and cAMP pathways in the vasculature will provide added benefit for SSc patients. The vascular protective effect of PGI2 analogues therefore likely stems from their combined action on multiple cell types. Likewise, iloprost acts directly on fibroblasts to block CTGF production and collagen synthesis (44). The elevated CTGF levels in skin blister fluid from SSc patients were also reduced by 5 days of iloprost therapy. The anti-fibrotic effect of iloprost was further shown in an animal model of heart failure and pulmonary fibrosis. Figure 4 summarizes the effects of iloprost.
One potential limitation for this study is the lack of recruitment of patients with limited cutaneous SSc. We routinely recruit patients with diffuse cutaneous SSc for EC isolation since this is the most severe form of disease, and ECs from these patients show prominent impairment in endothelial phenotypes (6, 18, 33, 45). It was shown that the vascular effect of iloprost was evident in both diffuse and limited cutaneous SSc patients, with or without DU (46), suggesting that iloprost does not discriminate between these patient groups. Another potential drawback is the variability stemming from patient disease activity and medication differences between patients and controls. Although the overall skin scores ranged from 0 to 28, the skin scores at the site of biopsy ranged from 0 to 2. We randomized patient cells into various experiments, and also controlled the experiments with age-, gender-, and ethnicity-matched healthy subjects. As the cells were cultured for at least 3 passages before they were used in experiments, and that the medications taken at the time of skin biopsy have reversible mechanisms of action, they would have been removed during cell processing. As a result, our experimental findings on endothelial function are unlikely to have been directly affected by the medications. In this study, BV9 was incorporated in all experiments for normal ECs as a validation that the effect of iloprost on VE-cadherin clustering was critical in improving those same functional endpoints. Although we only included BV9 control studies on SSc ECs in the permeability assay (Figure 1E), the effect of VE-cadherin blockade was similar to what was observed in normal ECs. We postulate that the effect of BV9 in tube formation and Endo-MT with SSc ECs would also show similar results performed with normal ECs. We recognize that the iloprost concentration that we used in this study (150 nM) is significantly higher than the steady state concentrations reported in healthy controls receiving iloprost infusion (0.13 and 0.37 nM) (19). Clinically for SSc patients with RP or DU, iloprost is most commonly administered and titrated to the highest tolerated dose between 0.5 and 2 ng/kg/min for 6 to 8 hours of infusion on day 1, and continue for 5 consecutive days (47). It is possible that for short-term iloprost treatments in vitro, a higher dose is needed to achieve therapeutic effects as oppose to the prolonged infusion treatment that the patients receive. In addition, the dose that was used in this study is similar to what was used in in vitro systems published previously (21–23).
In summary, our data provide a novel insight into the vascular effects of iloprost treatment in SSc. Endothelial adherens junctions function as an amplification nexus for protective EC signaling including positive feedback that strengthens the junctions (14). The prolonged clinical endothelial protective effect of PGI2 analogues may therefore stem from their ability to stabilize EC adherens junctions resulting in vasculoprotection, including improved barrier function, normalization of dysregulated angiogenesis, and inhibition of Endo-MT. Together with their anti-platelet, vasodilatory, and potential anti-fibrotic effects, the therapeutic use of PGI2 analogues in treating SSc-associated RP and DU is warranted.
Supplementary Material
Funding info:
Dr. Khanna is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases grant number K24AR063120. Dr. Flavahan is supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development grant number HD078639. Dr. Tsou’s work is supported by the by MICHR grant UL1TR002240, the American Autoimmune Related Disease Foundation, Dr. Donna Shelley and Mr. Lawrence Shelley, and funds from the Edward T. and Ellen K. Dryer Early Career Professorship.
Footnotes
Competing interests: Dr. Khanna is the Chief Medical Officer and stock holder in Eicos Sciences, Inc, which is currently conducting Phase 3 clinical trial for intravenous iloprost in the treatment of digital ischemic episodes due to systemic sclerosis.
References
- 1.Nagaraja V, Cerinic MM, Furst DE, Kuwana M, Allanore Y, Denton CP, et al. Current and future outlook on disease modification and defining low disease activity in systemic sclerosis. Arthritis Rheumatol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olschewski H, Rose F, Schermuly R, Ghofrani HA, Enke B, Olschewski A, et al. Prostacyclin and its analogues in the treatment of pulmonary hypertension. Pharmacology & therapeutics. 2004;102(2):139–53. [DOI] [PubMed] [Google Scholar]
- 3.Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76(8):1327–39. [DOI] [PubMed] [Google Scholar]
- 4.Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, et al. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313(11):2504–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumer Y, Drenckhahn D, Waschke J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem Cell Biol. 2008;129(6):765–78. [DOI] [PubMed] [Google Scholar]
- 6.Tsou PS, Rabquer BJ, Ohara RA, Stinson WA, Campbell PL, Amin MA, et al. Scleroderma dermal microvascular endothelial cells exhibit defective response to pro-angiogenic chemokines. Rheumatology (Oxford). 2016;55(4):745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Alessio S, Fibbi G, Cinelli M, Guiducci S, Del Rosso A, Margheri F, et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50(10):3275–85. [DOI] [PubMed] [Google Scholar]
- 8.Romero LI, Zhang DN, Cooke JP, Ho HK, Avalos E, Herrera R, et al. Differential expression of nitric oxide by dermal microvascular endothelial cells from patients with scleroderma. Vasc Med. 2000;5(3):147–58. [DOI] [PubMed] [Google Scholar]
- 9.Brown GE, O’leary PA. Skin capillaries in scleroderma. Archives of Internal Medicine. 1925;36(1):73–88. [Google Scholar]
- 10.Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol. 1980;2(2):161–70. [DOI] [PubMed] [Google Scholar]
- 11.Frech TM, Revelo MP, Drakos SG, Murtaugh MA, Markewitz BA, Sawitzke AD, et al. Vascular leak is a central feature in the pathogenesis of systemic sclerosis. J Rheumatol. 2012;39(7):1385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. 2017;76(5):924–34. [DOI] [PubMed] [Google Scholar]
- 13.Fleming JN, Nash RA, McLeod DO, Fiorentino DF, Shulman HM, Connolly MK, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS One. 2008;3(1):e1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flavahan NA. In Development-A New Paradigm for Understanding Vascular Disease. J Cardiovasc Pharmacol. 2017;69(5):248–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, et al. Cyclic AMP Potentiates Vascular Endothelial Cadherin-Mediated Cell-Cell Contact To Enhance Endothelial Barrier Function through an Epac-Rap1 Signaling Pathway. Molecular and Cellular Biology. 2005;25(1):136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wigley FM, Wise RA, Seibold JR, McCloskey DA, Kujala G, Medsger TA Jr., et al. Intravenous iloprost infusion in patients with Raynaud phenomenon secondary to systemic sclerosis. A multicenter, placebo-controlled, double-blind study. Ann Intern Med. 1994;120(3):199–206. [DOI] [PubMed] [Google Scholar]
- 17.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–55. [DOI] [PubMed] [Google Scholar]
- 18.Tsou PS, Amin MA, Campbell PL, Zakhem G, Balogh B, Edhayan G, et al. Activation of the Thromboxane A2 Receptor by 8-Isoprostane Inhibits the Pro-Angiogenic Effect of Vascular Endothelial Growth Factor in Scleroderma. J Invest Dermatol. 2015;135(12):3153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krause W, Krais T. Pharmacokinetics and pharmacodynamics of the prostacyclin analogue iloprost in man. Eur J Clin Pharmacol. 1986;30(1):61–8. [DOI] [PubMed] [Google Scholar]
- 20.Janssena MC, Wollersheim H, Kraus C, Hildebrand M, Watson HR, Thien T. Pharmacokinetics of oral iloprost in patients with Raynaud’s phenomenon secondary to systemic sclerosis. Prostaglandins Other Lipid Mediat. 2000;60(4–6):153–60. [DOI] [PubMed] [Google Scholar]
- 21.Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, et al. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol. 2012;227(10):3405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walch L, Labat C, Gascard JP, de Montpreville V, Brink C, Norel X. Prostanoid receptors involved in the relaxation of human pulmonary vessels. Br J Pharmacol. 1999;126(4):859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arner M, Högestätt ED. Endothelium-dependent relaxation and effects of prostacyclin, endothelin and platelet-activating factor in human hand veins and arteries. Acta Physiol Scand. 1991;142(2):165–72. [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Adyshev D, Gorshkov B, Birukov KG, Verin AD. ALK5 and Smad4 are involved in TGF-beta1-induced pulmonary endothelial permeability. FEBS letters. 2005;579(18):4031–7. [DOI] [PubMed] [Google Scholar]
- 25.Machin DR, Gates PE, Vink H, Frech TM, Donato AJ. Automated Measurement of Microvascular Function Reveals Dysfunction in Systemic Sclerosis: A Cross-sectional Study. J Rheumatol. 2017;44(11):1603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruni C, Frech T, Manetti M, Rossi FW, Furst DE, De Paulis A, et al. Vascular Leaking, a Pivotal and Early Pathogenetic Event in Systemic Sclerosis: Should the Door Be Closed? Frontiers in Immunology. 2018;9(2045). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hübner K, Cabochette P, Diéguez-Hurtado R, Wiesner C, Wakayama Y, Grassme KS, et al. Wnt/β-catenin signaling regulates VE-cadherin-mediated anastomosis of brain capillaries by counteracting S1pr1 signaling. Nature Communications. 2018;9(1):4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polacheck WJ, Kutys ML, Yang J, Eyckmans J, Wu Y, Vasavada H, et al. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature. 2017;552(7684):258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birukova AA, Fu P, Xing J, Birukov KG. Rap1 mediates protective effects of iloprost against ventilator-induced lung injury. Journal of Applied Physiology. 2009;107(6):1900–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, et al. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. European Respiratory Journal. 2013;41(1):165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oskolkova O, Sarich N, Tian Y, Gawlak G, Meng F, Bochkov VN, et al. Incorporation of iloprost in phospholipase-resistant phospholipid scaffold enhances its barrier protective effects on pulmonary endothelium. Scientific Reports. 2018;8(1):879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Y, Tan Q, Chen R, Cao B, Li W. Sevoflurane prevents lipopolysaccharide-induced barrier dysfunction in human lung microvascular endothelial cells: Rho-mediated alterations of VE-cadherin. Biochemical and Biophysical Research Communications. 2015;468(1):119–24. [DOI] [PubMed] [Google Scholar]
- 33.Tsou PS, Campbell P, Amin MA, Coit P, Miller S, Fox DA, et al. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc Natl Acad Sci U S A. 2019;116(9):3695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morini MF, Giampietro C, Corada M, Pisati F, Lavarone E, Cunha SI, et al. VE-Cadherin-Mediated Epigenetic Regulation of Endothelial Gene Expression. Circ Res. 2018;122(2):231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seang S, Pavasant P, Limjeerajarus CN. Iloprost Induces Dental Pulp Angiogenesis in a Growth Factor–free 3-Dimensional Organ Culture System. Journal of Endodontics. 2018;44(5):759–64.e2. [DOI] [PubMed] [Google Scholar]
- 36.He T, Lu T, d’Uscio LV, Lam CF, Lee HC, Katusic ZS. Angiogenic function of prostacyclin biosynthesis in human endothelial progenitor cells. Circ Res. 2008;103(1):80–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pola R, Gaetani E, Flex A, Aprahamian TR, Bosch-Marce M, Losordo DW, et al. Comparative analysis of the in vivo angiogenic properties of stable prostacyclin analogs: a possible role for peroxisome proliferator-activated receptors. J Mol Cell Cardiol. 2004;36(3):363–70. [DOI] [PubMed] [Google Scholar]
- 38.Biscetti F, Gaetani E, Flex A, Straface G, Pecorini G, Angelini F, et al. Peroxisome proliferator-activated receptor alpha is crucial for iloprost-induced in vivo angiogenesis and vascular endothelial growth factor upregulation. Journal of vascular research. 2009;46(2):103–8. [DOI] [PubMed] [Google Scholar]
- 39.Cipriani P, Di Benedetto P, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, et al. The Endothelial-mesenchymal Transition in Systemic Sclerosis Is Induced by Endothelin-1 and Transforming Growth Factor-beta and May Be Blocked by Macitentan, a Dual Endothelin-1 Receptor Antagonist. J Rheumatol. 2015;42(10):1808–16. [DOI] [PubMed] [Google Scholar]
- 40.Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC, Ten Dijke P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int J Mol Sci. 2017;18(10):2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SMF, Lawler J, Hynes RO, et al. Thrombospondin-1 Is a Major Activator of TGF-β1 In Vivo. Cell. 1998;93(7):1159–70. [DOI] [PubMed] [Google Scholar]
- 42.He M, Chen Z, Martin M, Zhang J, Sangwung P, Woo B, et al. miR-483 Targeting of CTGF Suppresses Endothelial-to-Mesenchymal Transition: Therapeutic Implications in Kawasaki Disease. Circulation research. 2017;120(2):354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90(11):1189–96. [DOI] [PubMed] [Google Scholar]
- 44.Stratton R, Shiwen X, Martini G, Holmes A, Leask A, Haberberger T, et al. Iloprost suppresses connective tissue growth factor production in fibroblasts and in the skin of scleroderma patients. J Clin Invest. 2001;108(2):241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsou PS, Wren JD, Amin MA, Schiopu E, Fox DA, Khanna D, et al. Histone Deacetylase 5 Is Overexpressed in Scleroderma Endothelial Cells and Impairs Angiogenesis via Repression of Proangiogenic Factors. Arthritis Rheumatol. 2016;68(12):2975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tinazzi E, Dolcino M, Puccetti A, Rigo A, Beri R, Valenti MT, et al. Gene expression profiling in circulating endothelial cells from systemic sclerosis patients shows an altered control of apoptosis and angiogenesis that is modified by iloprost infusion. Arthritis Res Ther. 2010;12(4):R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ingegnoli F, Schioppo T, Allanore Y, Caporali R, Colaci M, Distler O, et al. Practical suggestions on intravenous iloprost in Raynaud’s phenomenon and digital ulcer secondary to systemic sclerosis: Systematic literature review and expert consensus. Semin Arthritis Rheum. 2019;48(4):686–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.