

Abstract
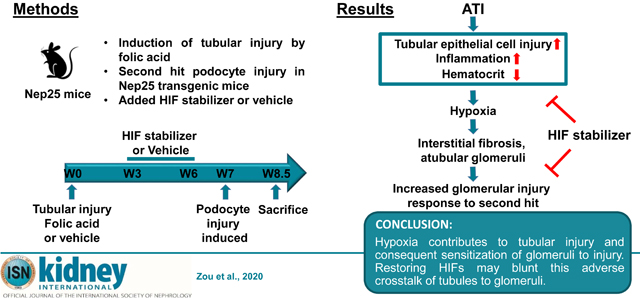
Previously, we found that mild tubulointerstitial injury sensitizes glomeruli to subsequent injury. Here, we evaluated whether stabilization of hypoxia-inducible factor-α (HIF-α), a key regulator of tissue response to hypoxia, ameliorates tubulointerstitial injury and impact on subsequent glomerular injury. Nep25 mice, which express the human CD25 receptor on podocytes under control of the nephrin promotor and develop glomerulosclerosis when a specific toxin is administered were used. Tubulointerstitial injury, evident by week two, was induced by folic acid, and mice were treated with an HIF stabilizer, dimethyloxalylglycine or vehicle from week three to six. Uninephrectomy at week six assessed tubulointerstitial fibrosis. Glomerular injury was induced by podocyte toxin at week seven, and mice were sacrificed ten days later. At week six tubular injury markers normalized but with patchy collagen I and interstitial fibrosis. Pimonidazole staining, a hypoxia marker, was increased by folic acid treatment compared to vehicle while dimethyloxalylglycine stimulated HIF-2α expression and attenuated tubulointerstitial hypoxia. The hematocrit was increased by dimethyloxalylglycine along with downstream effectors of HIF. Tubular epithelial cell injury, inflammation and interstitial fibrosis were improved after dimethyloxalylglycine, with further reduced mortality, interstitial fibrosis, and glomerulosclerosis induced by specific podocyte injury. Thus, our findings indicate that hypoxia contributes to tubular injury and consequent sensitization of glomeruli to injury. Hence, restoring HIFs may blunt this adverse crosstalk of tubules to glomeruli.
Keywords: HIF, Dimethyloxalylglycine, interstitial fibrosis, glomerulosclerosis
Graphical Abstract
Introduction
Numerous mechanisms have been proposed for chronic kidney disease (CKD) progression, including the “overload hypothesis”, which emphasizes glomerular injury, and the “fibrosis hypothesis”, which focuses on the tubulointerstitium. 1 Tubulointerstitial fibrosis (TIF) is uniformly observed in CKD, and is the best morphological predictor for progression. 2 Acute kidney injury (AKI) episodes correlate with development of CKD. 3–5 Our recent research indicated that even mild preexisting tubulointerstitial injury with clinical recovery, sensitizes glomeruli to subsequent podocyte-specific injury. 6 However, the mechanism of the crosstalk between the tubulointerstitium and glomeruli is uncertain. The kidney is susceptible to hypoxic injury. 7, 8 Peritubular capillary (PTC) loss and subsequent hypoxia caused by tubulointerstitial injury may be important mechanisms promoting tubulointerstitial fibrosis. 9–11
Hypoxia-inducible factors (HIFs) are oxygen-sensitive transcription factors, which regulate oxygen delivery and cellular adaptation to oxygen deprivation. 12, 13 HIFs are heterodimers composed of oxygen-sensitive α subunits (HIF-1α, HIF-2α, or HIF-3α), regulated by hypoxia that modulate HIF activity, and a constitutively expressed β subunit. With normal oxygen, HIF-α is hydroxylated and rapidly degraded by the VHL/E3-ubiquitin ligase. 12–14 In hypoxia, HIF-α is stabilized and either dimerizes with HIF-β in the nucleus to form transcriptionally active HIF or interacts with non-HIF proteins. HIFs regulate genes involved in energy metabolism, angiogenesis, erythropoiesis and other biological processes, resulting in renal protection in many settings. 13, 15–17 Pharmacologic inhibition of HIF-α degradation could potentially improve tubulointerstitial injury, which shows PTC loss and subsequent hypoxia, and ameliorate oxidative stress. 18 Dimethyloxalylglycine (DMOG), a competitive antagonist of α-ketoglutarate, can inhibit hydroxylation and induce HIF-dependent transcription. 19
We established a mouse model to study the interaction between tubulointerstitial injury and glomerular injury and to assess the impact of HIFs on these injuries. First, to assess fibrotic mechanisms, we used a Col I-luciferase mouse, in which the collagen I promoter drives luciferase, visualized by bioluminescence imaging in living animals. We mated these mice with Nep25 transgenic mice, which express human CD25 on podocytes under the nephrin promoter, and develop primary podocyte injury with proteinuria and progressive glomerulosclerosis after injection of the LMB2 toxin that binds this receptor. 20 We induced tubulointerstitial injury in these mice by injection of folic acid, and waited six weeks, when they had recovered functionally from AKI. Glomerular injury was then induced by LMB2. We found that hypoxia and HIFs are involved in the pathogenesis of acute to chronic tubulointerstitial injury transition, and that even mild and focal existing chronic tubulointerstitial injury then sensitized glomeruli to subsequent injury. These changes were partially reversed after administration of the HIF activator, DMOG. Our findings indicate that hypoxia-induced tubulointerstitial injury contributes to glomerular sensitization to injury, and HIFs are amongst the pivotal molecules that mediate this interaction.
Results
Folic Acid-induced Hypoxia and Tubulointerstitial Fibrosis
Mice underwent tubular injury induced by high dose folic acid (Figure 1). Intratubular crystal formation and acute tubular injury, with significantly increased urinary kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) were detected at week 2 (Figure 2A). Acute tubular injury was manifest by increased blood urea nitrogen (BUN) at day 1 (217.8±12.0 mg/dl) versus baseline (30.1±1.4 mg/dl), then returned to near baseline (33.7±2.0 mg/dl) at week 6, with urinary KIM-1 and NGAL also returning to baseline (Figure 2A), indicating functional recovery after tubular injury. Despite this functional recovery, patchy interstitial fibrosis was observed at week 6 in the folic acid group (Figure 2B). Collagen I transcription activity in kidney, analyzed by bioluminescence imaging, increased in the folic acid versus vehicle group at week 6 (Figure 2C). Folic acid induced more collagen I accumulation assessed by immunostaining than vehicle (Figure 2D), with patchy striped fibrosis.
Figure 1. Experimental design.

Mice received folic acid or vehicle at week 0. DMOG was given from week 3 till 6, and uninephrectomy was performed at week 6. All mice then received LMB2 injection to induce podocyte injury at week 7 and were sacrificed at week 8.5.
Figure 2. Folic acid-induced acute tubular injury in the acute phase and tubulointerstitial fibrosis in the chronic phase.

Urinary kidney injury molecules, KIM-1 and NGAL, excretion increased at week 2 and recovered at week 6 after folic acid injection (A). At week 6, folic acid-treated mice showed patchy tubulointerstitial fibrosis (PAS, ×100) (B). Quantitation assessed by region of interest (ROI) signal intensity of bioluminescence in each representative area of kidneys increased in folic acid versus vehicle (C). Representative images and quantitation of collagen I immunostaining positive area (percentage of the whole kidney cortex) showed increased collagen I production in the tubulointerstitium (×200) (D).
Pimonidazole, which is reduced to an adduct under conditions of low oxygen availability and thus is a tissue marker of hypoxia, accumulated mainly in tubular epithelial cells in folic acid mice, compared to faint staining in the vehicle group (Figure 3A), with increased pimonidazole confirmed by Western blot (Figure 3B). Nuclear HIF-2α expression, but not HIF-1 α, was decreased in folic acid versus vehicle mice (Figure 3C). Peritubular capillary (PTC) density, assessed by CD31 staining, was not different between folic acid and vehicle group (5.96±0.84 vs. 5.46±0.74 %, pNS). However, PTC permeability, measured by extravasated Evans blue dye in the kidney, was significantly increased in folic acid versus vehicle group (Figure 3D).
Figure 3. Hypoxia and hypoxia inducible factors were involved in the pathogenesis of folic acid-induced tubulointerstitial fibrosis.

Pimonizadole adducts staining was increased in tubular epithelial cells in folic acid versus vehicle (×100) (A). Pimonizadole adducts were increased in folic acid versus vehicle (B). HIF-2α, but not HIF-1 α, corrected for histone 3, decreased in the folic acid versus vehicle group (C). Extravasated Evans blue dye, a marker of peritubular capillary permeability, increased in the folic acid group (D).
DMOG Treatment Improved Hypoxia, Tubular injury, Inflammation and Decreased Interstitial Fibrosis and Atubular Glomeruli
After three weeks’ of DMOG treatment, kidney nuclear HIF-2α protein, but not HIF-1 α, was elevated in folic acid+DMOG compared to folic acid (Figure 4A). HIF1 and HIF2 gene expression level also increased after DMOG treatment (Figure 4B). Hematocrit significantly increased in DMOG-treated groups (Figure 4C). Carbonic anhydrase IX (CA IX) mRNA, an endogenous hypoxia marker regulated by HIFs, also significantly increased in folic acid +DMOG versus folic acid (Figure 4 D).
Figure 4. DMOG intervention stabilized hypoxia inducible factors and increased hematocrit.

DMOG in folic acid-treated mice increased nuclear expression of HIF-2α in kidney cortex (A). HIF1 and HIF2 gene expression increased after DMOG treatment (B). Hematocrit increased in response to DMOG (C). Carbonic anhydrase IX mRNA increased after DMOG intervention (D).
Pimonidazole immunostaining density was substantially attenuated in kidney at week 6 in folic acid+DMOG compared to folic acid (Figure 5A), which suggests tubulointerstitial hypoxia improvement after DMOG administration. Vascular endothelial growth factor A (VEGF-A) mRNA in kidney cortex, a dominant inducer of angiogenesis, significantly increased in folic acid +DMOG versus folic acid (Figure 5B). Although CD31 mRNA in kidney cortex was numerically increased in folic acid+DMOG versus folic acid group (3.12±1.03 vs. 0.88±0.20, P=0.07, Figure 5C), CD31 immunostaining did not differ. The peritubular endothelial cell marker (plvap) and glomerular endothelial cell marker (hecw2) were also similar in folic acid+DMOG and folic acid groups (Table 1). eNOS and VCAM, endothelial function markers, were not altered by DMOG (Table 1). Evans blue dye extravasation, a marker of capillary integrity, was similar in folic acid+DMOG vs. folic acid (Figure 5D). These data show that in spite of hypoxia improvement, endothelial cell number and function were not changed by DMOG.
Figure 5. DMOG intervention improved renal hypoxia but did not change endothelial cell number and function.

Pimonizadole adducts immunostaining in tubular epithelial cells was less when DMOG was added after folic acid injury (×100) (A). Expression of VEGF-A mRNA in kidney cortex increased in response to DMOG after folic acid injury (B). CD31 mRNA was not different in response to DMOG after folic acid injury (C). Extravasated Evans blue dye did not change in the DMOG vs. vehicle treatment group (D).
Table 1.
Endothelial related genes relative expression at UniNx
| Vehicle | Folic acid | Vehicle+DMOG | Folic acid+DMOG | |
|---|---|---|---|---|
| CD31 | 1.10±0.28 | 0.88±0.20 | 1.85±0.62 | 3.12±1.03 |
| PLVAP | 1.08±0.12 | 0.91±0.09 | 0.68±0.11* | 0.71±0.09 |
| HECW2 | 1.12±0.15 | 0.85±0.08 | 0.71±0.08 | 1.08±0.19 |
| eNOS | 1.04±0.07 | 1.14±0.11 | 1.15±0.22 | 0.96±0.12 |
| VCAM-1 | 1.05±0.09 | 11.05±2.18* | 1.08±0.30 | 9.70±1.88*# |
| ICAM-1 | 1.06±0.10 | 1.80±0.21* | 1.21±0.27 | 2.09±0.27*# |
| SELE | 1.16±0.17 | 1.48±0.18* | 1.67±0.52 | 1.72±0.31* |
Abbreviations: UniNx- uninephrectomy; PLVAP, plasmalemmavesicle vesicle-associated protein; HECW2, C2 and WW domain containing E3 ubiquitin protein ligase 2; eNOS, endothelial nitric oxide synthase 3; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion aolecule-1; 34.17±5.53; SELE, selectin E.
Data are expressed as mean±SEM
p<0.05 vs Vehicle
p<0.05 vs Vehicle+DMOG
Epithelial cadherin (E-cadherin) mRNA, a marker of tubular epithelial differentiation, was significantly increased in folic acid +DMOG versus folic acid (Figure 6A). The non-phosphorylated, active form of β-catenin translocates from cytoplasm to the nucleus and promotes tubular epithelial cell transdifferentiation and interstitial fibrosis. 21 Folic acid stimulated active β-catenin expression, which was attenuated by DMOG (Figure 6B). MMP-7, a downstream target of β-catenin previously shown to originate from tubules and affect podocyte injury, was upregulated by folic acid (folic acid 36.36±7.08 vs. Vehicle 1.06±0.12, p<0.05), but this induction was not blunted by DMOG (folic acid+DMOG 34.17±5.53). 22
Figure 6. DMOG intervention improved tubular injury and inflammation.

In response to DMOG after folic acid injury, epithelial cadherin (E-cadherin) mRNA in kidney cortex, corrected for 18s and normalized to vehicle group, increased (A), active β-catenin was attenuated (B) and F4/80 immunostaining was decreased (C).
Interstitial macrophage infiltration, shown by F4/80 immunostaining, increased in folic acid compared to Vehicle, and was lessened by DMOG (Figure 6C). ICAM-1 and selectin E (SELE) are expressed on endothelial cells and can recruit leukocytes to sites of injury. Both ICAM-1 and SELE were upregulated by folic acid, but not affected by DMOG (Table 1). Interstitial leukocyte infiltration, detected by CD45, was increased by folic acid (vehicle 5.8±0.6 vs. folic acid 25.6±1.5 /HPF, p<0.05), but not affected by DMOG (24.2±1.8 /HPF).
BUN and Scr were significantly increased in folic acid versus vehicle. BUN was similar in folic acid+DMOG versus folic acid (31.9±1.5 vs. 37.2±3.6 mg/dl, P=0.096, Figure 7A). At week 6, total collagen content in kidney cortex was not reduced by DMOG (vehicle 18.45±2.36, folic acid 32.76±4.03, vehicle+DMOG 12.62±0.98, folic acid+DMOG 30.57±2.87 mg/g). However, collagen I transcriptional activation was increased in the folic acid versus vehicle group, then lessened in folic acid+DMOG versus folic acid (Figure 7B). Collagen I immunostaining was focally increased in the kidney cortex of folic acid versus vehicle group, but was decreased in folic acid+DMOG versus folic acid (Figure 7C). Tubular epithelial cell injury and interstitial fibrosis may also obliterate the connection of glomeruli to proximal tubules, resulting in so-called atubular glomeruli. After folic acid, atubular glomeruli increased, especially in areas of striped interstitial fibrosis. Atubular glomeruli were decreased significantly by DMOG added to folic acid-injured mice, even in fibrotic areas (Figure 7D). No glomerulosclerosis was detected in the mice after folic acid before podocyte injury was induced. Differentiated podocyte density, assessed by Wilms tumor-1 (WT-1) staining, was not changed by tubular injury alone and/or DMOG (Figure 7E) before additional podocyte injury was added.
Figure 7. DMOG reduced tubulointerstitial injury but did not directly affect glomerular cells.

Renal function, assessed by BUN and Scr, was worsened in folic acid versus vehicle mice, and improved only numerically after DMOG intervention (A). Ex vivo bioluminescence images. Quantitation of collagen I signal in folic acid+DMOG versus folic acid mice (B). In response to DMOG after folic acid injury, collagen I immunostaining in whole kidney cortex was decreased (C), the proportion of atubular glomeruli was reduced (D), and glomerular WT-1 density was not changed (E).
Improvement of Tubulointerstitial Injury Ameliorated Glomerular Sensitivity to Injury
We next tested whether decreased tubulointerstitial injury could reduce glomerular sensitivity to injury. All these NEP25 mice received LMB2 injection to injure podocytes, at week 7 after preceding folic acid or vehicle as above. Ten days later, body weight, a reflection of nephrotic syndrome-related edema, increased similarly in vehicle +LMB2 and vehicle+DMOG+LMB2, but was ameliorated by added DMOG (Figure 8A). None of the mice without tubulointerstitial injury, those without folic acid injury, died after LMB2 injection. In contrast, mice with tubulointerstitial injury and added podocyte injury showed significant mortality, decreased by DMOG (folic acid+LMB2 55.6% mortality rate vs folic added DMOG 14.3%). BUN was markedly increased, as expected, in folic acid+LMB2 compared to vehicle+LMB2 mice, and was less when DMOG was added, with similar change in creatinine (Figure 8B). Albuminuria and proteinuria (urinary albumin or protein to creatinine ratio) in these mice with combined tubular and glomerular injury was not changed by added DMOG (data not shown).
Figure 8. Tubulointerstitial injury improvement lessened subsequent renal response to LMB2 injury.

Body weight, a measure of edema related to marked proteinuria, was less at day 10 after podocyte injury in when DMOG was added (A). Renal function, assessed by BUN and Scr, was worse in folic acid versus vehicle mice, and BUN was decreased after DMOG intervention (B). Ex vivo bioluminescence images confirmed decreased collagen I mRNA signal in folic acid +DMOG+LMB2 versus folic acid+LMB2 mice (C). In vivo bioluminescence images showed lessened collagen I mRNA signal when DMOG was added to mice with combined folic acid and podocyte injury (D).
Ten days after LMB2 injection, collagen I transcriptional activation further increased in folic acid+LMB2 versus vehicle+LMB2 mice, and was decreased by added DMOG (Figure 8C). In vivo bioluminescence imaging data were confirmed by ex vivo data from isolated kidneys, with less collagen I signal with added DMOG (Figure 8D).
Of note, overall average glomerular injury showed no differences among groups. However, in this folic acid model, the areas of patchy interstitial fibrosis showed more atubular glomeruli compared to preserved cortical areas. These atubular glomeruli showed less podocyte injury and sclerosis after LMB2 injection, probably due to less filtration, thus minimizing toxin effects, as we have shown previously.23 We then compared glomerular morphology in areas of cortex without fibrosis. Glomerulosclerosis was worse at day 10 after podocyte injury even in these nonfibrotic areas in mice with preexisting folic acid-induced injury, and was ameliorated by DMOG (Figure 9A), supporting an impact of preexisting tubulointerstitial injury on subsequent glomerular injury, even in area without extensive fibrosis. Similarly, glomerular collagen IV immunostaining in these areas was also increased in mice with preceding folic acid-induced injury, and was decreased in glomeruli in areas of intact cortex after DMOG (Figure 9B). Glomerular WT-1 density and nephrin expression were also higher after podocyte injury in mice with preexisting tubulointerstitial injury that also received DMOG, comparing glomeruli in areas without fibrosis (Figure 9C and 9D).
Figure 9. Tubulointerstitial injury improvement resulted in less severe subsequent glomerular Injury.

Ten days after podocyte injury by LMB2 injection, glomerulosclerosis was decreased in the non-fibrotic areas in when DMOG was added to mice with folic acid +podocyte injury (A). Glomerular collagen IV staining was also decreased in glomeruli in non-fibrotic areas by added DMOG (B), the glomerular density of WT-1 positive cells was higher (C) and glomerular nephrin expression was increased (D).
Discussion
In this study, we investigated potential mechanisms of the effect of preexisting tubulointerstitial fibrosis on subsequent glomerular injury. Acute tubular injury and glomerular podocyte-specific injury were induced serially.20, 24 Folic acid injection induces acute folic acid intratubular crystals with tubular necrosis and patchy interstitial fibrosis in the chronic phase.24–26 Acute toxic injury in rodent models is accompanied by subsequent hypoxia, in which peritubular capillary number and/or integrity may be involved.25, 27 Our data showed patchy tubulointerstitial fibrosis at week 6 after folic acid accompanied by tissue hypoxia, and associated with decreased nuclear HIF-2α. In this model, the fibrosis is striped and focal, unlike the diffuse fibrosis in our previous model.6 We found that tissue hypoxia was increased after folic acid-induced injury, and that existing chronic tubulointerstitial injury with apparent clinical recovery then sensitized glomeruli to subsequent injury. The resulting subsequent glomerular injury, induced by injection of a separate podocyte-specific toxin, was more severe in the folic acid-treated mice. However, these changes were partially reversed after administration of the HIF stabilizer DMOG after tubular injury and before glomerular injury. Our findings indicate that HIF-related hypoxia plays a role in tubular epithelial cell injury, interstitial inflammation and fibrosis, but not endothelial cell functional change, and finally contributes to sensitizing glomeruli to subsequent injury.
AKI is now recognized as a contributor to CKD. 3–5 Cohort studies show that AKI pooled adjusted hazard ratios were 8.8 and 3.1 for CKD and end stage renal disease, respectively.3 Various mechanisms, including tubulointerstitial hypoxia, have been proposed for AKI to CKD transition.28, 29 Peritubular capillary rarefaction in foci of tubulointerstitial fibrosis is thought to contribute to AKI to CKD transition by giving rise to hypoxia-signaling pathways. These pathways then augment interstitial inflammation and fibrosis. Capillary rarefaction, hypoxic signaling, and diverse hypoxia targets in the tubulointerstitium mutually reinforce each other in a final common pathway to CKD. Patients with CKD studied by blood oxygenation level-dependent MRI (BOLD-MRI, show loss of kidney oxygen.30, 31 In our study, 6 weeks after folic acid injury, significant tissue hypoxia was detected without reduced glomerular and peritubular capillary number. We observed increased PTC permeability, VCAM-1, ICAM-1 and SELE suggesting functional change of endothelial cells. HIFs are expressed on endothelial cells and endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. 32 We therefore hypothesized that DMOG, a HIF stabilizer, would preserve hypoxia by targeting endothelial cells. However, although tissue hypoxia was decreased in response to DMOG, no endothelial benefit was detected. DMOG increased hematocrit in our study, an effect mediated by stabilizing HIF2 in interstitial cells and upregulating EPO production. Increased hematocrit results in increased renal oxygen delivery.33 DMOG also has increased renal blood flow in the rodent subtotal nephrectomy model. 34 In addition, increased mitochondrial oxygen consumption and superoxide production occur with tubular injury. 35 HIF-1α stabilization lowers mitochondrial oxygen consumption o, which could also contribute to improvement of hypoxia by DMOG. 34 These mechanisms could have mediated less hypoxia in response to DMOG even without change in endothelial cell number or endothelial function.
Hypoxia also activates other pathways in which HIFs are key mediators. In the normal kidney, HIF1 is expressed on tubular epithelial cells and endothelial cells, while HIF2 is expressed on interstitial cells, endothelial cells and glomerular cells, with cell-specific pathways observed by microarray studies of cultured tubular cells and podocytes. 36 Endothelial HIF2, but not HIF1, reduced inflammation and protected against kidney injury in the ischemia-reperfusion model. Stabilization of HIF-2 in renal interstitial cells stimulates EPO production, while HIF activation in proximal tubular epithelial cells negatively modulates EPO. 37 Systemic HIF stabilization in experimental models of ischemia-reperfusion injury or acute renal failure showed beneficial effects.38–40 However, in CKD, the role of HIF stabilization is controversial. HIFs could exacerbate fibrosis by either epithelial-to-mesenchymal transition-triggering pathways or interactions with transforming growth factor-β signaling.41–43 HIF stabilization could also lessen oxidative stress in settings of tubulointerstitial injury.10, 18, 44 In our study, DMOG treatment was initiated after tubular injury and administered for only 3 weeks. We showed reduced tubular injury in response to DMOG, with less E-cadherin loss and less active β-catenin expression. DMOG also decreased infiltration by macrophages but not leukocytes. DMOG may also affect macrophage polarization with less M1 phenotype, as shown to downregulate nuclear factor-kappa B. 45
Importantly, HIF stabilization short term after tubular injury also decreased subsequent glomerular injury in response to the second hit. In our previous study, we found that atubular glomeruli are one contributor to adverse tubular-glomerular crosstalk.6 In the current study, atubular glomeruli were increased, especially in the patchy fibrotic areas. When we induced glomerular injury by LMB2, glomerulosclerosis was increased, mostly in non-fibrotic areas. Our previous study showed that decreasing GFR by performing UUO after podocyte toxin injection in Nep25 mice, resulted in complete protection of the non-filtering obstructed kidney to sclerosis, contrasting marked sclerosis in the non-obstructed kidney.23 In the current study, DMOG decreased interstitial fibrosis and atubular glomeruli, and also decreased glomerular injury after LMB2 induction in connected glomeruli. Glomerular effects of HIF are complex. Global HIF-1α knockout resulted in less glomerulosclerosis in the NEP25 model with decreased TGF-β activation. 46 In contrast, DMOG activated HIFs and attenuated glomerular injury in the rat remnant kidney model when DMOG was given starting 2 weeks after nephrectomy. 47 Considering the brief time course of DMOG exposure in our study, the results are most consistent with effects to reduce tubulointerstitial injuries rather than direct glomerular protection. MMP-7, a factor secreted from tubular epithelial cell after injury, has been shown to have direct effects on podocytes. 22 In our study, MMP-7 was increased after folic acid injury, but not further affected by DMOG. However, we cannot exclude that HIF stabilizing could affect other factors originating from tubules or interstitium, such as exosomes or miRNAs, that finally could target the glomerulus. Our study shows that even a short period of HIF stabilizer reduced tubulointerstitial injury and lessened subsequent glomerular injury response to a second hit.
HIF-α degradation is mediated by the pVHL-E3-ubiquitin ligase complex and requires oxygen- and iron-dependent prolyl hydroxylase domain (PHD) enzymes. In addition, the hydroxylation of the HIF-α asparagine-residue by factor inhibiting HIF (FIH) attenuates HIF’s transactivation activity. Roxadustat, Vadadustat, Daprodustat are PHD selective, while DMOG targets both PHD and FIH. 48 DMOG increased HIF activity in an in vitro study 7.8–11.7 folds, vs 1.2–2 fold and 3.1–4.6 folds for Vadadustat and Daprodustat, respectively. 48 In our study, DMOG preserved HIF2-α protein level after folic acid injury. Although hypoxia and exposure to certain hypoxia-mimetics such as CoCl2 can result in increased HIF-α transcription in certain cell types, PHD inhibition is expected to regulate HIF activity on the post-translational level. 49 However, we found HIF1-α and HIF2-α mRNA levels were also increased after DMOG treatment, which most likely involves cross-talk between multiple signaling pathways that may or may not be HIF-dependent. 50–52 Several more specific prolyl-hydroxylase inhibitors have been investigated or are undergoing clinical trials. Comparison of those treatments with DMOG could be of interest in future experiments to further dissect specific PHD vs other mechanisms of potential benefit in AKI to CKD progression.
In conclusion, preexisting folic acid-induced tubulointerstitial injury sensitized to subsequent glomerular podocyte-specific injury, and restoring HIFs blunted this adverse crosstalk of tubules to glomeruli. PHD inhibitors to stabilize HIFs are a promising new generation alternative treatment to oral erythropoietin. Our study suggests that these drugs may play roles beyond erythropoiesis.53 Our findings have important clinical implications for blocking AKI to CKD transition in native kidneys as well as in transplant settings. Moreover, since the tubulointerstitium may actively participate in the progression of glomerular injury, our findings suggest that the progression of glomerular diseases might also benefit from interventions to stabilize HIFs and blunt adverse tubular-glomerular crosstalk.54
CONCISE METHODS
Animals
Heterozygous Col I-luciferase mice (C57BL6), with luciferase in the collagen I promoter, were mated with heterozygous Nep25 mice (C57BL6), with human CD25 receptor on podocytes, and develop glomerulosclerosis when LMB2 immunotoxin is administered. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Vanderbilt University Medical Center.
Experimental protocol
Twelve weeks old Nep25+/Col I+ male mice were randomized to folic acid (240 μg/g, i.p., Sigma Aldrich, St Louis, MO, USA) or vehicle. At week 3 (age 15 weeks), half of the mice in each group, vehicle (n=8) or folic acid (n=10), were randomized to also receive dimethyloxaloylglycine (DMOG, 8 mg/mouse, i.p. qod, Cayman Chemical, Ann Arbor, MI, USA) for three weeks. Uninephrectomy was then performed. One week later, all mice received LMB2 injection (8 ng/g Bwt i.v.). Mice were sacrificed at age 20.5 weeks, 8.5 weeks after folic acid injection and 10 days after LMB2 injection (Figure 1).
Renal Function and Urinalysis
Body weight was measured before and after LMB2 injection. Renal function was assessed by serum creatinine (LC-MS/MS, UAB/UCSD O’Brien Center Core C Resource), blood urea nitrogen concentration (Bioassay Systems, Hayward, CA, USA), urinary total protein (Bio-Rad Protein Assay, ThermoFisher, Rockford, IL, USA) and albumin (Exocell, Philadelphia, PA, USA). Tubular injury was monitored by urinary NGAL and KIM-1 excretion (ELISA kits, R&D Systems, Minneapolis, MN, USA).
Western Blot
Frozen kidney tissue samples were transferred in RIPA buffer to extract total protein, while nuclear protein was extracted using Nuclear Extraction Kit (Abcam, Cambridge, MA, USA). HIF-1α (1:61000; Novus, Littleton, CO, USA), HIF-2α (1:4000; Novus), pimonidazole adducts (1:500; Hypoxyprobe, Burlington, MA, USA), and Histone 3 (1:6000; Abcam) and β-actin (1:5000; Sigma Aldrich) were detected by using the corresponding antibody overnight at 4°C. Protein bands on western blots were visualized by ECL Plus (Amersham, Arlington Heights, IL, USA). Pimonidazole adducts were expressed relative to β-actin. HIF-1 α and HIF-2 α were expressed relative to Histone 3.
Collagen I Bioluminescence Analysis
For in vivo bioluminescence imaging, mice received luciferin (150 μg/g, i.p., Promega, Madison, WI, USA). Bioluminescence light emissions were measured and integrated over 10 to 20 min, and the image with highest intensity was analyzed. For ex vivo bioluminescence imaging, the uninephrectomized or sacrificed fresh kidneys were immersed in luciferin/phosphate-buffered saline solution (150 μg/ml). Bioluminescence light emission was measured and integrated at 15 min. Region of interest (ROI) signal intensity was measured in each representative area and expressed as radiance (p/sec/cm2/sr).
Determination of Total Collagen Content
The total collagen content was determined by QuickZyme Collagen Assay (QuickZyme BioSciences, Leiden, Netherlands) in frozen kidney cortex, and corrected by total protein content (QuickZyme BioSciences).
Morphological Assessment
Glomerulosclerosis was evaluated by scoring each glomerulus 0–4, 0 no lesion; 1+ sclerosis of <25% of tuft; 2+ for 25−<50%; 3+ for 50–75% of the glomerulus; 4+ >75%. Average score was calculated from all glomeruli on PAS-stained section for each mouse. All sections for all morphologic studies were examined without knowledge of the treatment protocol.
Atubular glomeruli
Atubular glomeruli counting was performed by using serially sectioned slides of kidney (7 μm). All glomeruli identified in a midsection (section #6 or 7 of total 12 sections) were examined and traced. Glomeruli with connected proximal tubules were identified by lotus lectin staining. Atubular glomeruli were those that lay entirely within the section series and showed no connection to a proximal tubule. A total of 60 to 90 glomeruli were scored in each slide and noted whether in areas of patchy interstitial fibrosis vs. nonfibrotic areas.
Immunohistochemistry
3μm sections from kidneys fixed in paraformaldehyde stained. For antigen retrieval, sections were either incubated with 0.1% trypsin (Sigma Aldrich), 0.8% collagenase or microwaved in citrate buffer (pH 6.0). Primary antibodies were incubated overnight at 4°C by adding rabbit anti-mouse collagen IV antibody (1:400, Millipore, Darmstadt, Germany), rat anti-mouse CD31 (1:50, Dianova GmbH, Hamburg, Germany), rabbit anti-mouse active β-catenin (1:600, Cell signaling, Danvers, MA, USA), goat anti-mouse nephrin (1:500, R&D), rabbit anti-mouse WT-1 (1:200, Novus), rabbit anti-mouse collagen I (1:500 dilution, Abcam), rabbit anti-mouse CD45 (1:400, Abcam) and rat anti-mouse F4/80 (1:600, Biorad, Hercules, CA, USA). Slides treated with nonspecific antisera instead of primary antibody were used as negative control.
Collagen I, CD31, active β-catenin, F4/80, nephrin and collagen IV stained slides were scanned using the Aperio CSO system (Leica Biosystems, Buffalo Grove, IL, USA). The positive area was analyzed using Aperio Imagescope software. WT1-positive nuclei per glomerulus and CD45 positive cells per high power field were counted.
Endothelial function and tissue hypoxia
Vascular permeability was determined by Evans blue dye extravasation (Sigma Aldrich), after i.v. injection (20μg/g BW), washing out remaining dye 30 mins later by perfusing with normal saline via the renal artery. The mice were then euthanized immediately and the kidney was removed. The dye remaining in the tissue was extracted with pure formamide and measured by spectrophotometry (absorbance at 620 nm).
Pimonidazole (60 μg/g i.p., Hypoxyprobe) was used to detect tissue hypoxia. Kidneys were harvested 1 hour after pimonidazole injection and pimonidazole adducts were detected by immunohistochemistry (rabbit anti-pimonidazole antibody 1:400, Hypoxyprobe).
Quantitative Real-Time PCR
Reverse transcription was performed using the TaqMan Reverse Transcription Kit (Applied Biosystems, Branchburg, NJ, USA). Quantitative real-time PCR was performed in a total reaction volume of 25 μl using iQ SYBR Green Supermix (Bio-rad, Hercules, CA, USA), 0.5 μl cDNA, and 500 nM forward and reverse primers. VEGF-A, CA IX, E-Cadherin, MMP-7, CD31, PLVAP, HECW2, eNOS, VCAM-1, ICAM-1, SELE, Collagen I α1, and 18s primers were from ThermoFisher Scientific (Waltham, MA, USA). Experimental cycle threshold (Ct) values were normalized to 18s measured on the same plate, and fold differences in gene expression were determined by using the 2−△△Ct method.
Statistical Analysis
All data are presented as mean ± standard error of the mean. Statistical difference was evaluated by one-way analysis of variance (ANOVA), followed by post-hoc Bonferroni correction for multiple comparisons between the groups. Nonparametric data were compared by Mann-Whitney U test. All tests were two-tailed and p<0.05 was considered statistically significant.
Translational Statement.
Previous data show that acute tubular injury can sensitize glomeruli to a second hit and this mechanism can promote CKD progression. Our studies reveal that HIF activation reduced hypoxia after tubular injury, decreased tubular epithelial cell injury, interstitial inflammation and interstitial fibrosis. Critically, even a short period of treatment to stabilize HIF after acute tubular injury was induced ameliorated the glomerular sensitization to subsequent injury. These results have important clinical implications for PHD inhibitors to potentially decrease AKI to CKD transition in native kidneys as well as in transplant settings.
Acknowledgements.
The work was supported in part by NIH NIDDK DK56942-09 (ABF).
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Hodgkins KS, Schnaper HW Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol 2012; 27: 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nath KA Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis 1992; 20: 1–17. [DOI] [PubMed] [Google Scholar]
- 3.Coca SG, Singanamala S, Parikh CR Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012; 81: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chawla LS, Eggers PW, Star RA, et al. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med 2014; 371: 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palant CE, Amdur RL, Chawla LS Long-term consequences of acute kidney injury in the perioperative setting. Curr Opin Anaesthesiol 2017; 30: 100–104. [DOI] [PubMed] [Google Scholar]
- 6.Lim BJ, Yang JW, Zou J, et al. Tubulointerstitial fibrosis can sensitize the kidney to subsequent glomerular injury. Kidney Int 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lübbers DW, Baumg?rtl H. Heterogeneities and profiles of oxygen pressure in brain and kidney as examples of the pO2 distribution in the living tissue. Kidney Int. 1997; 51: 372–380. [DOI] [PubMed] [Google Scholar]
- 8.Brezis M, Rosen S Hypoxia of the renal medulla--its implications for disease. N. Engl. J. Med 1995; 332: 647–655. [DOI] [PubMed] [Google Scholar]
- 9.Fine LG, Norman JT Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008; 74: 867–872. [DOI] [PubMed] [Google Scholar]
- 10.Nangaku M Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J. Am. Soc. Nephrol 2006; 17: 17–25. [DOI] [PubMed] [Google Scholar]
- 11.Norman JT, Clark IM, Garcia PL Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000; 58: 2351–2366. [DOI] [PubMed] [Google Scholar]
- 12.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292: 468–472. [DOI] [PubMed] [Google Scholar]
- 13.Haase VH Hypoxia-inducible factors in the kidney. Am. J. Physiol. Renal Physiol 2006; 291: F271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG, Ratcliffe PJ Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 2008; 30: 393–402. [DOI] [PubMed] [Google Scholar]
- 15.Majmundar AJ, Wong WJ, Simon MC Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010; 40: 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haase VH Pathophysiological Consequences of HIF Activation: HIF as a modulator of fibrosis. Ann. N. Y. Acad. Sci 2009; 1177: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapitsinou PP, Sano H, Michael M, et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest 2014; 124: 2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nordquist L, Friederich-Persson M, Fasching A, et al. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J. Am. Soc. Nephrol 2015; 26: 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn JM, You SJ, Lee YM, et al. Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS One 2012; 7: e48952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J. Am. Soc. Nephrol 2005; 16: 1013–1023. [DOI] [PubMed] [Google Scholar]
- 21.Tan RJ, Zhou D, Zhou L, et al. Wnt/beta-catenin signaling and kidney fibrosis. Kidney Int Suppl (2011) 2014; 4: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan RJ, Li Y, Rush BM, et al. Tubular injury triggers podocyte dysfunction by beta-catenin-driven release of MMP-7. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsusaka T, Kobayashi K, Kon V, et al. Glomerular sclerosis is prevented during urinary tract obstruction due to podocyte protection. Am. J. Physiol. Renal Physiol 2011; 300: F792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim BJ, Yang HC, Fogo AB Animal models of regression/progression of kidney disease. Drug Discov. Today Dis. Models 2014; 11: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan HT, Li XZ, Pitera JE, et al. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am. J. Pathol 2003; 163: 2289–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Long DA, Woolf AS, Suda T, et al. Increased renal angiopoietin-1 expression in folic acid-induced nephrotoxicity in mice. J. Am. Soc. Nephrol 2001; 12: 2721–2731. [DOI] [PubMed] [Google Scholar]
- 27.Basile DP, Donohoe DL, Roethe K, et al. Chronic renal hypoxia after acute ischemic injury: effects of L-arginine on hypoxia and secondary damage. Am. J. Physiol. Renal Physiol 2003; 284: F338–348. [DOI] [PubMed] [Google Scholar]
- 28.Ferenbach DA, Bonventre JV Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol 2015; 11: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferenbach DA, Bonventre JV Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther 2016; 12 Suppl 1: S41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milani B, Ansaloni A, Sousa-Guimaraes S, et al. Reduction of cortical oxygenation in chronic kidney disease: evidence obtained with a new analysis method of blood oxygenation level-dependent magnetic resonance imaging. Nephrol. Dial. Transplant 2016. [DOI] [PubMed] [Google Scholar]
- 31.Pruijm M, Milani B, Burnier M Blood Oxygenation Level-Dependent MRI to Assess Renal Oxygenation in Renal Diseases: Progresses and Challenges. Front. Physiol 2016; 7: 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapitsinou PP, Sano H, Michael M, et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest 2014; 124: 2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marini CP, Russo GC, Nathan IM, et al. Effect of hematocrit on regional oxygen delivery and extraction in an adult respiratory distress syndrome animal model. Am. J. Surg 2000; 180: 108–114. [DOI] [PubMed] [Google Scholar]
- 34.Thomas JL, Pham H, Li Y, et al. Hypoxia-inducible factor-1alpha activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am. J. Physiol. Renal Physiol 2017; 313: F282–F290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitaker RM, Wills LP, Stallons LJ, et al. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J. Pharmacol. Exp. Ther 2013; 347: 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberger C, Mandriota S, Jürgensen JS, et al. Expression of hypoxia-inducible factor-1alpha and −2alpha in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol 2002; 13: 1721–1732. [DOI] [PubMed] [Google Scholar]
- 37.Farsijani NM, Liu Q, Kobayashi H, et al. Renal epithelium regulates erythropoiesis via HIF-dependent suppression of erythropoietin. J. Clin. Invest 2016; 126: 1425–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kapitsinou PP, Jaffe J, Michael M, et al. Preischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am. J. Physiol. Renal Physiol 2012; 302: F1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernhardt WM, Campean V, Kany S, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol 2006; 17: 1970–1978. [DOI] [PubMed] [Google Scholar]
- 40.Hill P, Shukla D, Tran MG, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol 2008; 19: 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanna C, Hubchak SC, Liang X, et al. Hypoxia-inducible factor-2α and TGF-β signaling interact to promote normoxic glomerular fibrogenesis. Am. J. Physiol. Renal Physiol 2013; 305: F1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest 2007; 117: 3810–3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumann B, Hayashida T, Liang X, et al. Hypoxia-inducible factor-1α promotes glomerulosclerosis and regulates COL1A2 expression through interactions with Smad3. Kidney Int. 2016; 90: 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eardley KS, Kubal C, Zehnder D, et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008; 74: 495–504. [DOI] [PubMed] [Google Scholar]
- 45.Hirai K, Furusho H, Hirota K, et al. Activation of hypoxia-inducible factor 1 attenuates periapical inflammation and bone loss. Int J Oral Sci 2018; 10: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumann B, Hayashida T, Liang X, et al. Hypoxia-inducible factor-1alpha promotes glomerulosclerosis and regulates COL1A2 expression through interactions with Smad3. Kidney Int. 2016; 90: 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song YR, You SJ, Lee YM, et al. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol. Dial. Transplant 2010; 25: 77–85. [DOI] [PubMed] [Google Scholar]
- 48.Sulser P, Pickel C, Gunter J, et al. HIF hydroxylase inhibitors decrease cellular oxygen consumption depending on their selectivity. FASEB J. 2020; 34: 2344–2358. [DOI] [PubMed] [Google Scholar]
- 49.Wang GL, Jiang BH, Rue EA, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A 1995; 92: 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmer LA, Semenza GL, Stoler MH, et al. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am. J. Physiol 1998; 274: L212–219. [DOI] [PubMed] [Google Scholar]
- 51.Belaiba RS, Bonello S, Zahringer C, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007; 18: 4691–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008; 453: 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maxwell PH, Eckardt KU HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond. Nat. Rev. Nephrol 2016; 12: 157–168. [DOI] [PubMed] [Google Scholar]
- 54.Lee PT, Chou KJ, Fang HC Are tubular cells not only victims but also perpetrators in renal fibrosis? Kidney Int. 2012; 82: 128–130. [DOI] [PubMed] [Google Scholar]