

Abstract
Immune checkpoints negatively regulate immune cell responses. PD-1:PD-L1 and CTLA-4:B7-1 are among the most important immune checkpoint pathways, and are key targets for immunotherapies which seek to modulate the balance between stimulatory and inhibitory signals and lead to favorable therapeutic outcomes. The current dogma of these two immune checkpoint pathways has regarded them as independent with no interactions. However, the newly characterized PD-L1:B7-1 ligand-ligand cis-interaction and its ability to bind CTLA-4 and CD28, but not PD-1, suggests that these pathways have significant crosstalk. Here, we propose that PD-L1:B7-1 cis-interaction brings novel mechanistic understanding of these pathways, new insights into mechanisms of current immunotherapies, and fresh ideas to develop better treatments in a variety of therapeutic settings.
Keywords: PD-L1, B7–1, cis-interaction, PD-1, CTLA-4, CD28, Immunotherapy
Immune checkpoint blockade
The role of the B7-CD28 family of immune checkpoints (see Glossary) in homeostasis and disease has been increasingly explored over the last three decades. This led to the development of immune checkpoint blockade for which the 2018 Nobel Prize in Physiology or Medicine was awarded to James Allison and Tasuku Honjo for their work targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)/B7–1/B7–2 and programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) pathways in cancer [1]. A similar approach has been extended to autoimmune diseases [2–5] and organ transplants [6, 7]. However, the therapeutic benefits remain variable, working for limited patients [8], diseases [4, 9], or durations [6], suggesting our understanding of these therapies is incomplete.
Recent work demonstrates that the PD-L1:B7–1 ligand-ligand cis-interaction alter trans-interactions with other immune checkpoints [10–15]. Thus, we should reframe the current therapeutic dogma of immune checkpoint pathways (Figure 1A) to include these interactions (Figure 1B) and explore how this contributes to the therapeutic outcomes seen in cancer, autoimmune disease, and organ transplant immunotherapies.
Figure 1. PD-L1 and B7–1 immune checkpoint signaling pathways.
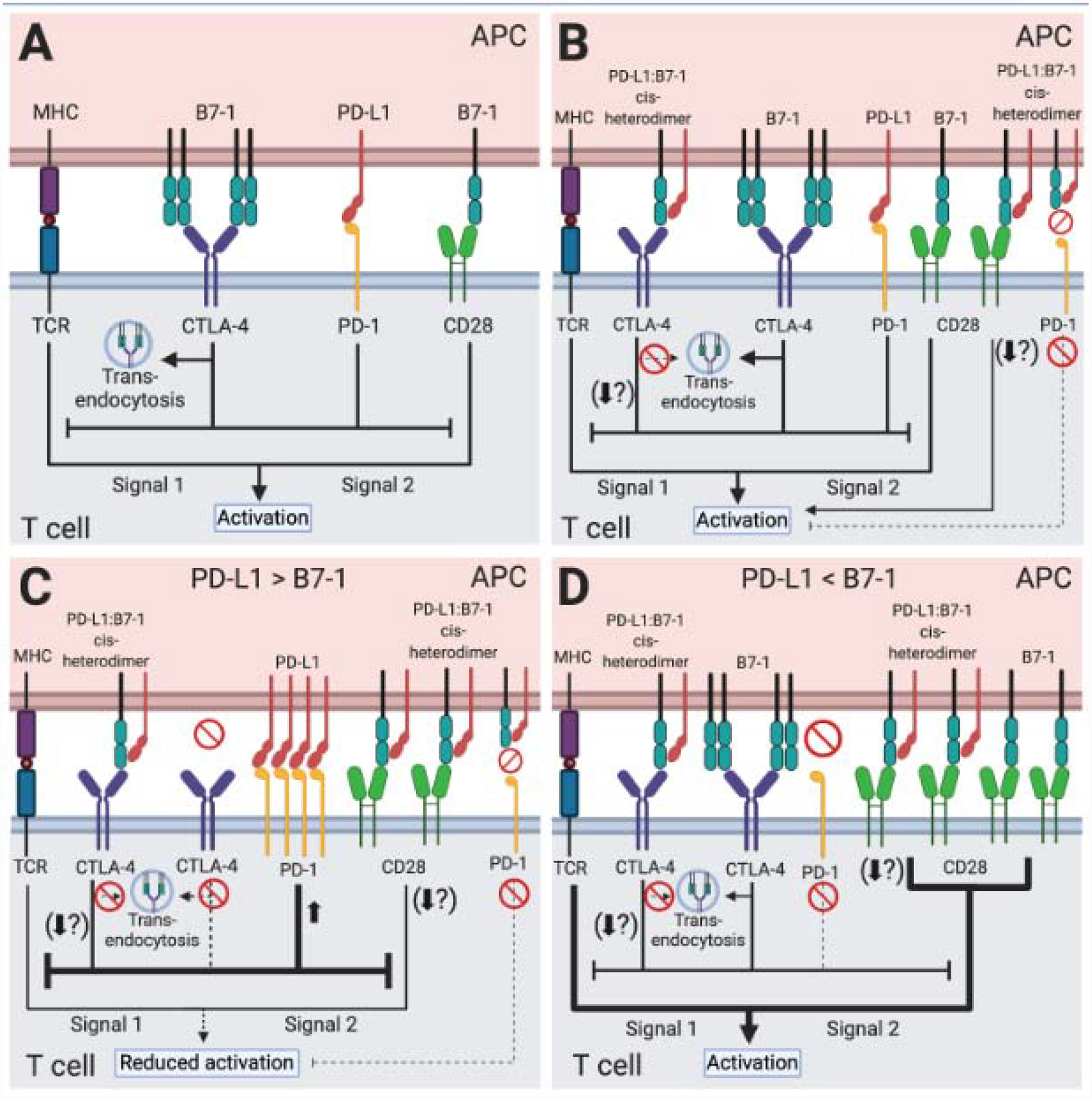
T cell activation requires two signals. Engagement of the peptide-MHC complex on APCs with the TCR complex provides signal one. Costimulatory ligand-receptor interactions such as B7–1:CD28 provide signal two. Immune checkpoint interactions like PD-L1:PD-1 and B7–1:CTLA-4 regulate this response. (A) Although both inhibit T cell activation, originally the PD-L1:PD-1 pathway and the B7–1:CTLA-4 pathway were considered completely independent with no interactions. (B) The PD-L1:B7–1 cis-interaction demonstrates crosstalk between these pathways. The PD-L1:B7–1 cis-heterodimer binds CTLA-4, reduces B7–1 trans-endocytosis, and potentially alters other CTLA-4-mediated cell-intrinsic mechanisms of inhibition. It also binds CD28; while most studies demonstrate no alteration in functional outcomes, one study shows decreased T cell activation. The PD-L1:B7–1 heterodimer does not bind to PD-1. By sequestering B7–1, the PD-L1:B7–1 cis-heterodimer indirectly reduces CTLA-4 inhibition. Although not detailed in this figure, the roles of B7–2 and PD-L2 should be further examined in the context of PD-L1:B7–1 engagement in T cell activation. (C) If PD-L1 outnumbers B7–1, PD-L1:B7–1 cis-heterodimers reduce free B7–1. The PD-L1:B7–1 cis-heterodimer binds to CD28 which most, but not all, studies suggest does not alter functional outcomes as described above. It also binds to CTLA-4, reduces CTLA-4-mediated trans-endocytosis, and possibly other CTLA-4 cell-intrinsic inhibitory mechanisms and outcomes. Finally, excess PD-L1 binds to PD-1, inducing strong inhibitory signals. Altogether, this reduces T cell activation. (D) If B7–1 outnumbers PD-L1, PD-L1:B7–1 cis-heterodimers reduce free PD-L1. The heterodimer prevents PD-1:PD-L1 binding. It reduces CTLA-4:B7–1 trans-endocytosis, possibly other CTLA-4 inhibitory mechanisms. Most studies show that it does not alter B7–1:CD28 functional outcomes, although one study demonstrates that it reduces T cell activation. While B7–1 homodimers may interact with CTLA-4, B7–1 monomers and PD-L1:B7–1 cis-heterodimers continue transducing CD28 costimulatory signals. Collectively, this generates a net-stimulatory effect.
PD-L1 and B7–1: two ligands of the B7 family
PD-L1: protein and cellular characteristics
PD-L1, also called CD274, is a type I transmembrane protein expressed on dendritic cells (DCs), macrophages, epithelial cells, endothelial cells, activated T cells, activated B cells, and others [16–19]. PD-L1 binds to PD-1, recruiting src homology region 2 domain-containing phosphatase (SHP)-1 [20, 21] and SHP-2 [20–23] to PD-1, and dephosphorylates some T cell activation signaling-associated proteins [20–24]. Consequently, the PD-1:PD-L1 pathway inhibits T cells (Figure 1), modulating cytokine release [16, 20, 21], proliferation [16, 25], and affecting cell phenotype [25, 26].
Some tumors express PD-L1 [27], promoting immune evasion by inhibiting cytotoxic responses. In some autoimmune diseases such as systemic lupus erythematous, PD-L1 may be decreased during flares promoting a pro-stimulatory environment [28]. Thus, although important in immune homeostasis, uncoupling PD-L1 equilibrium contributes to the pathogenesis of diseases.
B7–1: protein and cellular characteristics
B7–1, also called CD80, is a type I transmembrane protein that exists as a monomer and homodimer [29]. It can be found on antigen presenting cells (APCs), including DCs, B cells, and monocytes [30–33]. It is upregulated on activated APCs [31–33] and transferred to T cells via trogocytosis [34]. After signal one is induced by T cell receptor (TCR) engagement with a peptide-bound major histocompatibility complex (MHC), B7–1 binds to CD28 on T cells to provide signal two of the activation pathway [35] (Figure 1). It also binds to the inhibitory receptor CTLA-4 [36], inhibiting T cell activation [37] (Figure 1) and proliferation [36, 37].
With some exceptions, most tumors do not express B7–1, thus preventing naïve T cell activation. In autoimmune diseases like rheumatoid arthritis, some cells such as synovial immune cells upregulate B7–1 which may increase T cell stimulation [38]. Thus, abnormal B7–1 expression contributes to the pathogenesis of diseases.
PD-L1:B7–1 cis-interaction: novel ligand-ligand binding
PD-L1:B7–1 cis-interaction in vitro
A PD-L1:B7–1 trans-interaction was initially reported using cell-protein adhesion and surface plasmon resonance assays (SPR) [39, 40]. This interaction decreased T cell proliferation and interferon-γ (IFN-γ), tumor necrosis factor α (TNFα), and interleukin-2 (IL-2) release in CD28/CTLA-4−/− cells, demonstrating an inhibitory effect [39]. Cells from CD28/CTLA-4/PD-L1−/− mice did not respond to B7–1-Ig-mediated inhibition, further supporting the specificity of the PD-L1:B7–1 interaction[39]. Finally, using B7–1 or PD-L1 monoclonal antibodies (mAbs) could disrupt protein-protein interactions. Specifically, one study showed that one B7–1 mAb could block the PD-L1:B7 trans-interaction while another B7–1 mAb could not [39]; the same study showed that one PD-L1 antibody which could block the PD-L1:PD-1 interaction, and another PD-L1 antibody which did not, were both able to block the PD-L1:B7–1 trans-interaction [39]. Another study showed that some PD-L1 mAbs could block both the PD-L1:B7–1 trans-interaction and the PD-L1:PD-1 interaction, while another could only block the PD-L1:PD-1 interaction, and yet another could not block any of the aforementioned interactions [40]. Thus the ability of a B7–1 or PD-L1 antibody to block the PD-L1:B7–1 interaction is unique to each antibody.
Recent studies further dissecting this interaction reveal that PD-L1 and B7–1 interact in cis [10, 12, 13, 15]. PD-L1:B7–1 cis-interaction is detected only with flexible, orientation-permissive PD-L1, suggesting that protein orientation is important [10]. PD-L1:B7–1 cis-interaction is confirmed in multiple systems including proximity-based split luciferase assays [10, 15], fluorescence resonance energy transfer (FRET) assays [12], and protein-cell binding assays [10, 12, 13, 15].
Intriguingly, the PD-L1:B7–1 cis-heterodimer interferes with other immune checkpoints. It impedes PD-1:PD-L1 binding [11–13, 15], inhibits PD-1 microcluster formation [12], alters PD-1-mediated cytokine modulation [11, 13], and thus blocks PD-1-mediated inhibition [10–13, 15]. Mechanistically this is attributed to partially overlapping binding sites of PD-L1:B7–1 and PD-1:PD-L1 [15, 39]. In support of this finding, some PD-L1 mAbs are able to block the PD-L1 cis- and/or trans-interactions [10, 11, 41]. PD-L1 IgV mutagenesis approaches show that specific amino acid mutations (D49K and K124S) may alter both PD-1:PD-L1 and PD-L1:B7–1 binding, while other mutations have a greater effect on PD-L1:B7–1 binding (R113S and M115A) [10]. Additional PD-L1 mutations such as V54E, Y56A, and E58G, and B7–1 mutations such as L96E and L107E, also abrogate PD-L1:B7–1 binding [13]. Another study shows PD-L1 IgV mutations including, but not limited to, D122A, Y123A, and K124A could alter binding to PD-1; other mutations such as Y56A, E72R, and G120D alter B7–1 binding; and L53R, G119R, and A121R alter both PD-1 and B7–1 binding [15]. Together, these data support a model where the binding sites of PD-L1 and B7–1 on B7–1 are in close proximity, and the binding of one ligand to B7–1 prevents the interaction of the other with B7–1.
The PD-L1:B7–1 cis-heterodimer may interfere with the B7–1:CTLA-4 trans-interaction. Structurally the CD28 and CTLA-4 binding sites on B7–1 overlap [15]. The PD-L1 binding site on B7–1 overlaps with the B7–1 dimerization interface [15]. The CTLA-4 and PD-L1 binding sites on B7–1 have been noted to partially overlap [39] or are on opposite sides of B7–1 [15]. These results suggest that the PD-L1:B7–1 cis-interaction not only competes with B7–1 homodimerization, thus limiting the amount of B7–1 homodimers available to bind CTLA-4, but may or may not also affect CTLA-4:B7–1 binding directly. Indeed, one study demonstrates that the PD-L1:B7–1 heterodimer reduces interactions with the CTLA-4 homodimer and reduces CTLA-4-mediated trans-endocytosis of B7–1 [12]. Another study reveals that dimeric CTLA-4-Fc but not monomeric CTLA-4 reduces PD-L1:B7–1 cis-interaction [15]. These results, combined with data from an SPR experiment showing an increase in SPR signal from a chip coated with PD-L1 that was incubated with CTLA-4 and B7–1 [12], suggests that PD-L1 may inhibit the B7–1:CTLA-4 interaction by decreasing the avidity of the B7–1:CTLA-4 interaction rather than by directly blocking it. On the other hand, other data shows that there is no alteration in high avidity CTLA-4 pentamer binding to cells and preserves the CTLA-4 inhibitory effect on IL-2 release [13]; but, this functional, IL-2 release data contradicts other work which showed that CTLA-4+ positive jurkat cells that were co-cultured with CD80+PD-L1+ Raji cells incubated with atezolizumab, an anti-PD-L1 mAb which disrupts PD-L1:B7–1 cis-interaction, released less IL-2 than co-cultured cells without atezolizumab [12]. Given that the relative abundance of immune checkpoints likely determines the stimulatory or inhibitory outcome (e.g. only if PD-L1 outnumbers B7–1 will the PD-L1:B7–1 cis-heterodimer prevent B7–1 homodimerization) [12, 13] (Figure 1C and 1D), additional work is needed for clarification.
Lastly, the effect of the PD-L1:B7–1 cis-heterodimer on the B7–1:CD28 interaction has been contradictory. One study demonstrates that the PD-L1:B7–1 cis-heterodimer partially reduces T cell activation compared to B7–1 alone in a T cell activation reporter cell line, disruption of the PD-L1:B7–1 cis-heterodimer improves T cell priming, and disruption of the PD-L1:B7–1 cis-heterodimer with PD-L1 mAbs could increase B7–1 and CD28-Fc interaction in a Tag-Lite assay [11]. Other studies show that the PD-L1:B7–1 cis-interaction does not alter CD28 protein binding [12, 13], TCR or CD28 microcluster formation [12], cell-cell interface enrichment of CD28 and B7–1 [12], CD28 phosphorylation [12], or IL-2 secretion [12–14]. Given that there were different experimental conditions and technologies between these disparate results, more work will be needed to further solidify if the PD-L1:B7–1 interaction does not interfere with the B7–1:CD28 interaction.
It should be noted that B7–2 is another protein which may also play an important role in this updated paradigm. B7–2 binds to CTLA-4 [42, 43] and CD28 [43]. However, using protein-cell binding assays [13, 39, 40], proximity-based assays [12, 15], or CTLA-4:B7–2 depletion assays [12] it was shown that B7–2 does not bind with PD-L1. In the presence of the PD-L1:B7–1 cis-heterodimer, B7–2 can still interact with CD28 [12]. Using a DC-regulatory T cell B7–1 and B7–2 trans-endocytosis assay, it was shown that administration of the anti-PD-L1 mAb atezolizumab did not alter B7–2 trans-endocytosis, demonstrating that PD-L1 does not affect the B7–2:CTLA-4 interaction [12]. Thus in future studies, it would be interesting to examine the relative role and strength of B7–2 in the context of PD-L1:B7–1 cis-interaction.
PD-L1:B7–1 cis-interaction in vivo
PD-L1:B7–1 cis-interaction shows variable responses in vivo. In an ovalbumin (OVA)-expressing E.G7 T lymphoma murine cancer model, mice engineered to prevent PD-L1:B7–1 cis-interaction via PD-L1 Y56A or CD80 L107E mutations show decreased anti-tumor responses after OVA immunization; these mice also show decreased OVA-induced IL-2 and IFN-γ release [13]. In an autoimmune encephalomyelitis model, these mice exhibit fewer symptoms and have reduced IL-17 production [13]. These results agree with a stimulatory role for the PD-L1:B7–1 cis-heterodimer.
Examining data from patient samples raises intriguing questions about this cis-interaction [11]. Patient tumor-associated conventional DCs express PD-L1 and B7–1 in cis, with PD-L1 approximately 20-fold higher. Re-analyzing RNA-sequencing data from renal cell carcinoma and non-small cell lung cancer clinical trials (NCT01375842 and NCT01903992, respectively) reveals that patients with a high DC signature who received atezolizumab— an anti-PD-L1 mAb which blocks both PD-1:PD-L1 and PD-L1:B7–1 interactions—had improved overall survival. Immune cell PD-L1 expression also predicts atezolizumab response, a result replicated in other studies [44, 45]. Given these results it would be interesting to further examine if the PD-L1:B7–1 cis-interaction in the tumor microenvironment is also associated with anti-PD-L1 immunotherapy responses in future studies.
On the other hand, an inhibitory PD-L1:B7–1 interaction was noted in a non-obese diabetic (NOD) mouse model [41]. While it has been previously established that the PD-1:PD-L1 axis plays a major role in the development of diabetes in NOD mice [46, 47], the contribution of the PD-L1:B7–1 interaction, either in cis or in trans, was unknown. Modulating the binding of PD-L1 and B7–1 via mAb blockade of the PD-L1:B7–1 interaction alone or in combination with PD-1:PD-L1 blockade increases the rate of diabetes when given to 13 week-old mice. When these same mAbs are given to 6–7 week-old mice, blockade of the PD-L1:B7–1 interaction alone precipitates diabetes in a lower percentage of mice than blockade of both the PD-L1:B7–1 and PD-1:PD-L1 pathways. In an adoptive cell transfer model, diabetic donor CD8 T cells given to recipient mice receiving the same mAbs show increased rates of diabetes. Prolonged exposure to these mAbs increases IFN-γ and TNFα double positive cells, as well as spleen localization of T cells. Intriguingly, 22.2%, 4.4%, and 30% of the donor T cells after three days expressed B7–1, PD-1, and PD-L1, respectively. It is unclear if a T cell PD-L1:B7–1 cis-interaction influenced the results, or if a PD-L1:B7–1 trans-interaction was the predominant effector. More work is needed to determine if there is both an inhibitory and stimulatory role for the PD-L1:B7–1 interaction in different experimental contexts, or if there exists a PD-L1:B7–1 trans-interaction in some experimental contexts, as initially described, while in others the cis-interaction predominates.
PD-L1:B7–1 cis-interaction in cancer immunotherapies targeting PD-1/PD-L1
Current principle of anti-PD-1/PD-L1 cancer immunotherapies
Tumor antigen-specific T cells target and eliminate cancer cells [48]. To escape destruction, tumor cells utilize immunosuppressive immune checkpoints such as PD-L1 and B7x to dampen cytotoxic responses [49, 50]. Blocking these immune checkpoints presents an attractive therapeutic avenue. Indeed, anti-PD-1/PD-L1 cancer immunotherapies block PD-1:PD-L1 trans-interaction (referred to as PD-1 binding), thereby preventing PD-1-mediated attenuation of T cell activation [44, 51].
Anti-PD-1 immunotherapy reinvigorates activated T cells regardless of PD-L1 expression in the tumor microenvironment; it also reverses metabolic reprograming in dysfunctional T cells, partially mediating T cell activation [25, 52–54]. Recent studies identified that PD-1 inhibitory pathway preferentially targets the CD28 costimulatory signal which is essential for T cell expansion in anti-PD-1 immunotherapy [22, 54]. Anti-PD-L1 immunotherapies were originally designed to block PD-L1 on tumor cells, thus blocking PD-1 binding. But recent studies demonstrate the involvement of PD-L1:B7–1 cis-interaction in anti-PD-L1 immunotherapies, thereby offering more therapeutic mechanisms in using anti-PD-L1 therapies than anti-PD-1 therapies [10–13, 55].
New considerations of PD-L1:B7–1 interaction in anti-PD-1/PD-L1 cancer immunotherapies
In the tumor microenvironment, the PD-L1:B7–1 cis-interaction mostly exists on APCs since they express both ligands while tumor cells usually do not express B7–1 [11, 56]. PD-L1- and B7–1-expressing DCs are the most important intratumoral APCs in regulating anti-tumor immunity [14]. Given that PD-L1 and B7–1 abundance fluctuates based on DC maturity, we should consider how the PD-L1:B7–1 cis-interaction alters the PD-1, CD28, and CTLA-4 pathways within the tumor microenvironment (Figure 1B).
Intratumoral DCs express more abundant PD-L1 than B7–1 (Figure 1C) [11]. FRET assays examining B7–1 cis-homodimerization in the presence of PD-L1 on cell membranes identified that PD-L1 disrupts B7–1 homodimerization [12]. The smaller population of B7–1 cis-homodimers reduces CTLA-4 binding and CTLA-4-mediated B7–1 trans-endocytosis. The PD-L1:B7–1 cis-heterodimer binds to CD28 and induces a costimulatory signal whose functional outcomes most studies show are comparable to B7–1 alone [12–14], while one study showed that the PD-L1:B7–1 cis-heterodimer results in reduced T cell activation [11]. Additionally, excess PD-L1 increases PD-1 binding [16]. The increased PD-1 binding weakens the CD28 costimulatory signal thereby producing a reduced overall activation signal. If PD-L1 was less abundant than B7–1, the balance of stimulatory and inhibitory signals changes (Figure 1D). In this scenario, the proportion of PD-L1:B7–1 cis-heterodimers decreases while the proportion of free B7–1 increases. Both the free B7–1 and the PD-L1:B7–1 cis-heterodimer bind to CD28 to induce a costimulatory signal [12–14]. In addition, PD-1:PD-L1 binding decreases. The CD28 costimulatory signal and the decreased PD-1 inhibition activates the T cell.
Anti-PD-1/PD-L1 monotherapies alter both PD-1:PD-L1and B7–1:CD28/CTLA-4 pathways through the PD-L1:B7–1 cis-interaction. Anti-PD-1 mAbs block PD-1 on T cells, preventing PD-1 binding and preserving CD28 costimulation (Figure 2A). It also increases PD-L1:B7–1 cis-interaction. This induces CD28 costimulatory signaling that most studies show mediates functional outcomes similar to B7–1 [12–14], although one study showed PD-L1:B7–1 cis-interaction reduced T cell activation [11]. However, CTLA-4 can sequester the PD-L1:B7–1 heterodimer from CD28 since CTLA-4 binds to the PD-L1:B7–1 heterodimer too; this interaction reduces B7–1 trans-endocytosis [12], it may [12] or may not [13] alter other functional outcomes as well. The reduced inhibitory signaling and increased stimulatory signals from these different interactions would lead to increased T cell activation. On one hand, these interactions may provide additional mechanistic insight on current anti-PD-1 immunotherapies by demonstrating continued co-stimulation through CD28 and reduced B7–1 trans-endocytosis. However, if CD28-mediated costimulation is reduced, then this could provide insight into how anti-PD-1 immunotherapies may lose their effectiveness. Future studies should validate these PD-L1:B7–1 cis-heterodimer/CD28 binding outcomes to explore these possibilities.
Figure 2. New considerations of the PD-L1:B7–1 cis-interaction in anti-PD-1/PD-L1 monotherapies to treat cancer.
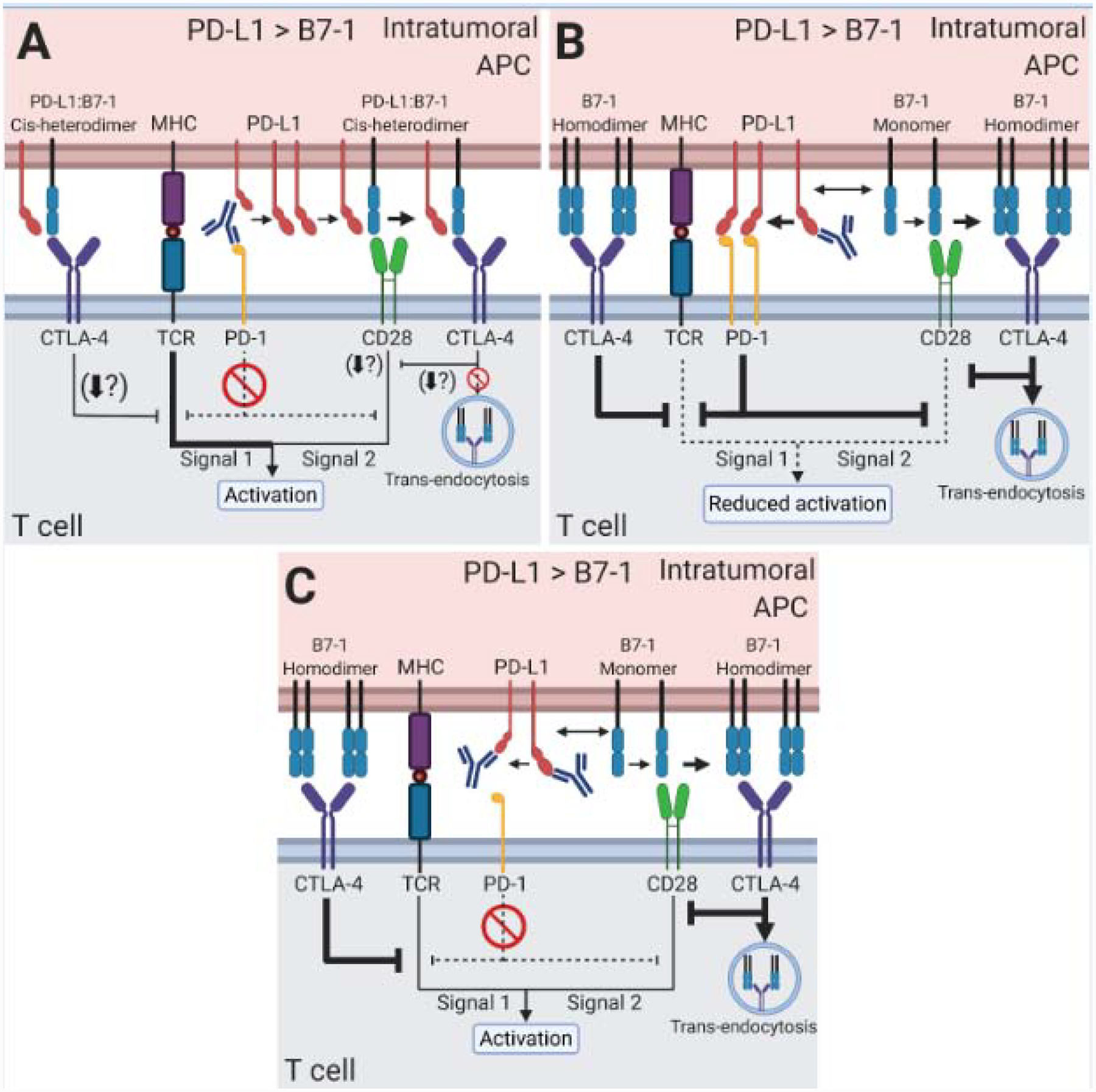
Intratumoral APCs exhibit high PD-L1 expression, outnumbering B7–1. B7–2 and PD-L2 (not shown) likely also play roles in the therapies described below, and their contribution should be explored in future studies. (A) Anti-PD-1 mAb monotherapy blocks PD-1 binding. Excess PD-L1 sequesters B7–1. The PD-L1:B7–1 cis-heterodimer induces CD28 costimulatory signaling. CTLA-4 sequesters the PD-L1:B7–1 cis-heterodimer from CD28, reducing the CD28 costimulatory signal. (B) Anti-PD-L1 mAb monotherapy only targeting the PD-L1:B7–1 cis-interaction frees B7–1 from PD-L1. B7–1 can bind to either CD28 or CTLA-4. B7–1 is sequestered and trans-endocytosed by CTLA-4, reducing its binding to CD28. PD-1 binding increases due to freed PD-L1. The PD-1 pathway inhibits the CD28 costimulatory signal. Together this reduces T cell activation. (C) Anti-PD-L1 mAb monotherapy targeting both PD-1 binding and PD-L1:B7–1 cis-interaction eliminates the PD-1 inhibitory pathway and frees B7–1 from PD-L1. Free B7–1 binds to CD28 to induce a costimulatory signal. Additionally, CTLA-4 may sequester the B7–1 and mediates trans-endocytosis. But the combination of reduced PD-1 and increased CD28 signaling leads to T cell activation. If high CTLA-4 levels are present (not shown), the increased sequestration of B7–1 may reduce T cell activation.
Anti-PD-L1 immunotherapies block PD-1:PD-L1 and/or PD-L1:B7–1 interactions. Theoretically, anti-PD-L1 mAbs that only block PD-1:PD-L1 binding would mimic anti-PD-1 blockade as described above. Anti-PD-L1 mAbs blocking only the PD-L1:B7–1 cis-interaction liberate PD-L1 and B7–1 ligands (Figure 2B). This would increase PD-1 inhibition while also increasing B7–1 interactions with CTLA-4 or CD28. As one study has shown that this increases CTLA-4-mediated B7–1 trans-endocytosis, it would eventually reduce the B7–1 available to interact with CD28 [12]. Thus this immunotherapy reduces the overall T cell activation signal. Anti-PD-L1 mAbs blocking both PD-1:PD-L1 binding and the PD-L1:B7–1 cis-interaction free B7–1 and prevent PD-1 inhibition (Figure 2C). Again, B7–1 could bind to CD28 or CTLA-4. In cases of low CTLA-4 expression, there will be low CTLA-4-mediated inhibition and no PD-1 inhibition, thus activating the T cell (Figure 2C). In cases of high CTLA-4 expression, CTLA-4 reduces free B7–1 capable of binding to CD28 as described above. This will lead to decreased T cell activation. Thus, CTLA-4 expression should be considered in anti-PD-1/PD-L1 monotherapies.
PD-L1:B7–1 cis-interaction in cancer immunotherapy targeting CTLA-4
Current principle of anti-CTLA-4 cancer immunotherapy
One of the major mechanisms of anti-CTLA-4 immunotherapy is to block CTLA-4 from binding B7–1 and B7–2 [57]; this enhances CD28 costimulation, thus promoting T cell activation. As tumor cells do not express B7–1 and B7–2, CTLA-4 blockade is thought to occur in the tumor-draining lymph nodes where DCs present tumor antigens to prime naïve T cells [58]. However, the existence of intratumoral DCs may also imply that CTLA-4 blockade takes place within the tumor [59]. Nevertheless, anti-CTLA-4 immunotherapy leads to the expansion of tumor-specific CD8 T cells and T helper 1 (Th1)-like CD4 effector T cells within the tumor microenvironment [60–62]. New studies suggest that CTLA-4 blockade increases T cell diversity by lowering the activation threshold of T cell clones that recognize poorly immunogenic tumor antigens while maintaining preexisting high-frequency T cell clones [63, 64]. Another emerging mechanism of anti-CTLA-4 immunotherapy is the depletion of intratumoral immunosuppressive regulatory T cells; though, more studies are needed to confirm these findings [65–68]. Additionally, the newly characterized PD-L1:B7–1 cis-interaction may play a role in anti-CTLA-4 immunotherapy [12].
New considerations of PD-L1:B7–1 cis-interaction in anti-CTLA-4 cancer immunotherapy
Anti-CLTA-4 immunotherapy blocks CTLA-4 and frees B7–1 [12, 58]. The free B7–1 alone may interact with CD28, but cannot interact with CTLA-4 due to the presence of the anti-CTLA-4 mAb. Thus, signaling outcomes may heavily rely on PD-L1 expression levels. In situations with normal-to-low PD-L1 expression, the increased B7–1 may bind with PD-L1 and increase PD-L1:B7–1 cis-heterodimer levels. These cis-heterodimers could interact with CD28 but not PD-1 or CTLA-4 due to the anti-CTLA-4 mAb. This would lead to cell activation. However, given a high enough level of PD-L1 to remain able to bind to PD-1 even after new PD-L1:B7–1 cis-heterodimer formation (Figure 3A), the outcome of this therapy may result in sub-optimal activation due to PD-1 signaling. If true, anti-CTLA-4 immunotherapy may be more effective with immune cells expressing low levels of PD-L1.
Figure 3. The PD-L1:B7–1 cis-interaction in anti-CTLA-4 monotherapy and the optimal anti-CTLA-4 and anti-PD-L1 combination therapy to treat cancer.
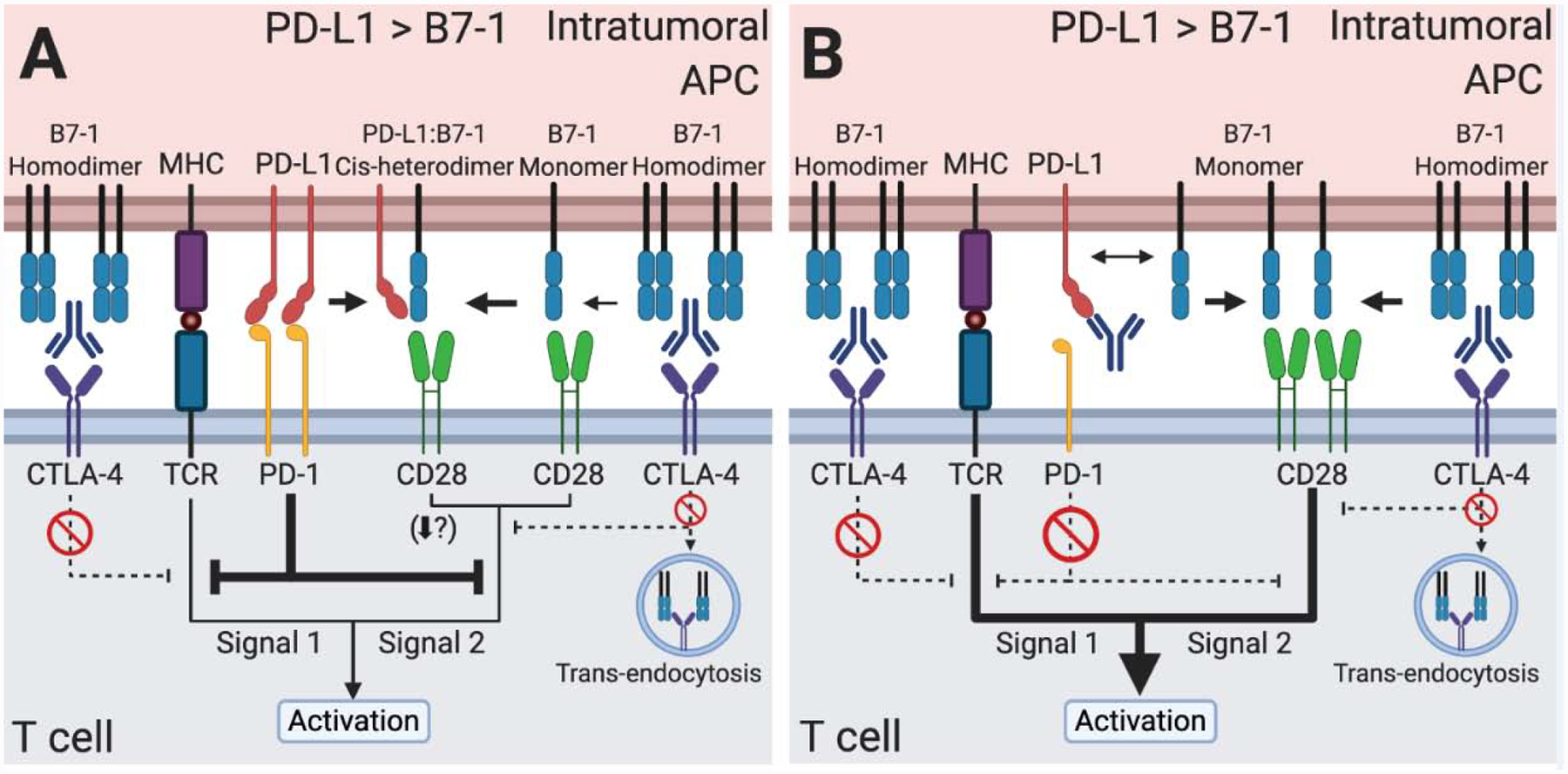
(A) Anti-CTLA-4 mAb monotherapy prevents CTLA-4 sequestration and trans-endocytosis of B7–1. The B7–1 heterodimerizes with PD-L1. The PD-L1:B7–1 cis-heterodimer induces CD28 costimulatory signaling. Given a high enough level of PD-L1 to remain able to bind PD-1, PD-1 signaling may prevent optimal T cell activation. (B) Combination therapy of anti-CTLA-4 mAb and anti-PD-L1 mAb targeting both PD-1 binding and the PD-L1:B7–1 cis-interaction blocks PD-1 inhibitory signaling, CTLA-4 binding of B7–1, and CTLA-4-mediated trans-endocytosis. This combination maximizes B7–1 levels and induces a strong CD28 costimulatory signal, strengthening the overall T cell activation signal. B7–2 and PD-L2 (not shown) may be important in these therapies as well.
Anti-CTLA-4 and anti-PD-1/PD-L1 combination therapies show clinical efficacy [69, 70]. To improve this approach, PD-L1:B7–1 cis-interaction should be considered since anti-PD-L1 immunotherapies can target PD-1:PD-L1 trans-, PD-L1:B7–1 cis-interaction, or both. Combination therapy using anti-CTLA-4 and anti-PD-L1 mAbs blocking only the PD-L1:B7–1 cis-interaction allows for increased B7–1 levels. However, PD-1 binding would simultaneously increase and dampen the overall activation signal. A combination of anti-CTLA-4 mAbs with anti-PD-L1 mAbs blocking only PD-1:PD-L1 binding inhibits both CTLA-4 and PD-1 pathways, but allows for PD-L1:B7–1 cis-heterodimer formation capable of inducing a CD28 costimulatory signal. While most studies show no difference in this signaling [12–14], one study shows that the PD-L1:B7–1 cis-heterodimer induces a weak T cell activation signal compared to B7–1 alone [11]. If the latter finding were true, then this approach would not be optimal, but more studies are needed to confirm this finding. However, combination therapy of anti-CTLA-4 and anti-PD-L1 mAbs blocking both PD-1:PD-L1 binding and PD-L1:B7–1 cis-interaction would circumvent the limitation of the previous approach if the weakened T cell activation induced by PD-L1:B7–1 cis-heterodimer is true. Anti-PD-L1 mAbs blocking both PD-1:PD-L1 binding and PD-L1:B7–1 cis-interaction prevent PD-1 inhibition and PD-L1 sequestration of B7–1 (Figure 3B) [12]. Anti-CTLA-4 mAbs block CTLA-4 from binding to free B7–1. This increases B7–1 levels available to bind to CD28, thereby improving T cell activation and anti-tumor immunity. Thus, including PD-L1:B7–1 cis-interaction into our therapeutic rationale may improve current cancer immunotherapies and guide future therapies.
PD-L1:B7–1 cis-interaction in immunotherapies for autoimmune diseases and organ transplants
Current principle of CTLA-4-Ig immunotherapy in autoimmune diseases
In autoimmune diseases, the immune system targets self-antigens[71]. Dysregulation of immune checkpoints contributes to this maladaptive response. CTLA-4 deficient mice develop fatal lymphoproliferation [72, 73] and humans with CTLA-4 mutations exhibit hyperactivated effector T cells [74]. Medical treatments have sought to leverage the inhibitory properties of CTLA-4. CTLA-4-Ig fusion proteins mimic native CTLA-4 and bind to B7–1 and B7–2 and result in effects such as inhibition of cytokine release and proliferation, though the degree of which depends on the specific fusion protein [75] (Figure 4A). Currently the CTLA-4-Ig abatacept is approved for the treatment of adult rheumatoid arthritis, adult psoriatic arthritis, and juvenile idiopathic arthritisi.
Figure 4. The PD-L1:B7–1 cis-interaction offers new mechanistic insight into CTLA-4-Ig therapy.
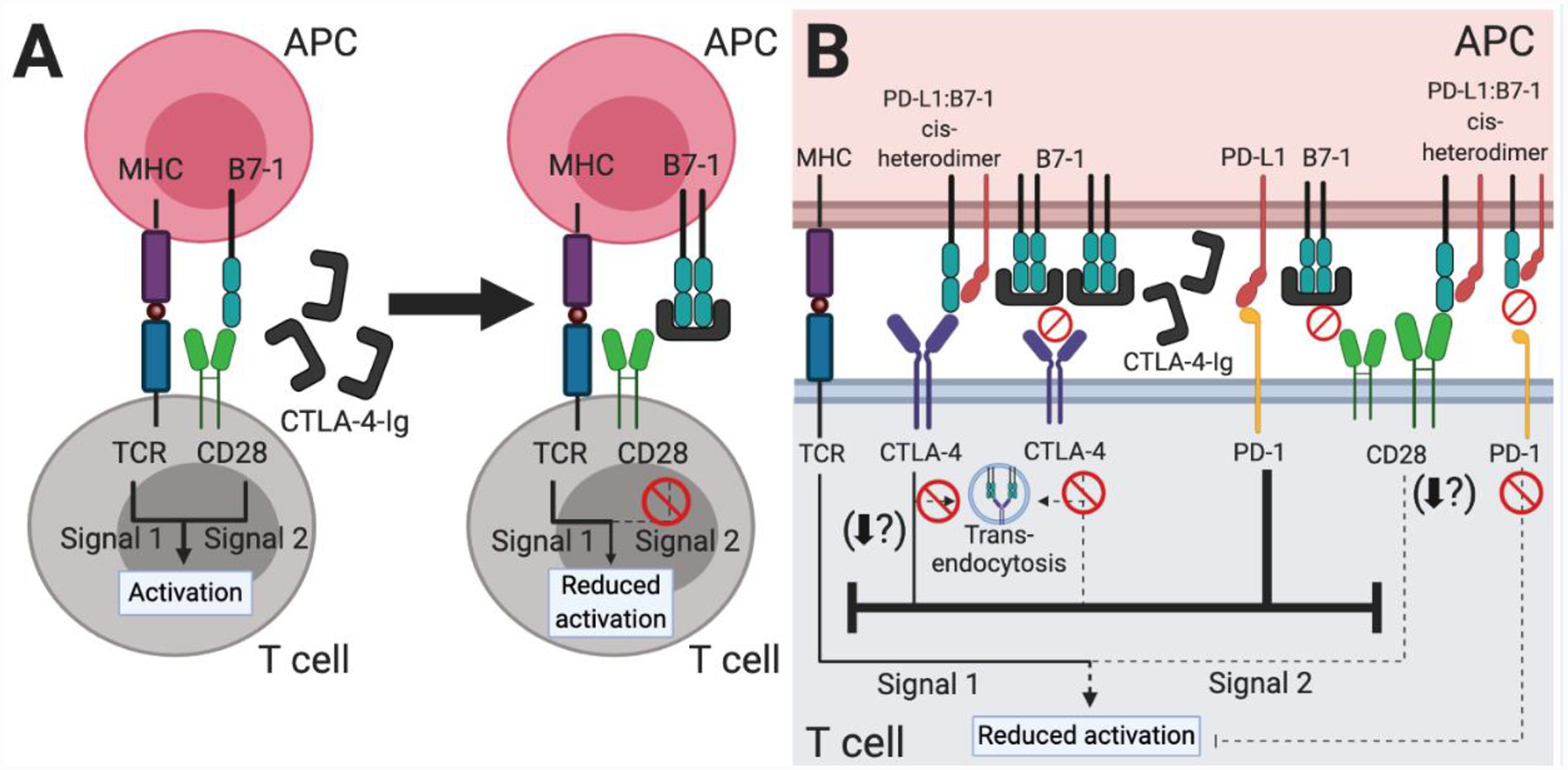
(A) Based on our original understanding of immune checkpoint signaling pathways (Figure 1A), CTLA-4-Ig therapy functions by decreasing CD28-mediated T cell costimulation. The fusion protein mimics the native CTLA-4 protein and binds to B7–1 and B7–2 (not shown) on APCs, thus preventing a CD28:B7–1 or B7–2 interaction. (B) Our new understanding of the PD-L1:B7–1 cis-interaction shows additional immune checkpoint signaling pathways which underlie CTLA-4-Ig therapy. As CTLA-4-Ig sequesters most free B7–1 and B7–2 (not shown), the B7–1 receptors CD28 and CTLA-4 do not transduce activating or inhibitory signals, respectively. PD-L1 is unaffected and may continue to bind PD-1, inhibiting signals one and two of T cell activation. PD-L2 (not shown) may also continue to inhibit T cell activation. Although likely sparse, PD-L1:B7–1 cis-heterodimers may continue to modulate CTLA-4, CD28, and PD-1 signaling. The PD-L1:B7–1:CTLA-4 interaction reduces B7–1 trans-endocytosis and potentially other CTLA-4 functional outcomes as well. Most studies indicate that it mediates continued CD28 costimulation, although one study demonstrated that it reduced T cell activation. As the PD-L1:B7–1 cis-heterodimer cannot bind PD-1, it does not further inhibit T cell activation through this pathway. Taken together, this therapy is highly immunosuppressive, but the preformed PD-L1:B7–1 cis-heterodimer may continue to interact with CD28 and provide enough activation over the long-term to mediate low levels of T cell responses.
Current principle of CTLA-4-Ig immunotherapy in organ transplants
In organ transplantation, organs are transferred from a donor and implanted into a recipient. Inherent genetic differences between the donor and recipient elicit immune responses against the transplanted organ [76]. These reactions are divided into three categories: hyperacute, acute, and chronic rejection which take place within minutes, weeks to months, and months to years, respectively. These rejections are prevented by pre-transplant screenings such as ABO blood type matching, donor-specific human leukocyte antigen (HLA) antibodies, and HLA matching. Additionally, these rejections are also pharmacologically managed.
CTLA-4-Ig is one pharmacological means of rejection management. Currently, the higher affinity CTLA-4-Ig belatacept is approved in the adult kidney transplant settingii [6, 7].This therapy follows the same principle as other CTLA-4-Ig therapies: binding to B7–1 and B7–2 to inhibit T cells [75, 77] (Figure 4A).
New considerations of PD-L1:B7–1 cis-interaction in CTLA-4-Ig therapy
CTLA-4-Ig therapy inconsistently shows benefit across autoimmune diseases [2–5, 9] and fails to permanently maintain transplanted organs[6]. Given our new understanding of PD-L1:B7–1 cis-interaction and its effects on immune checkpoints (Figure 1B), we should reassess these results (Figure 4B).
As CTLA-4-Ig binds B7–1 and B7–2, the expression of these proteins is paramount to therapeutic success. Abatacept-mediated B7–1 downregulation has been noted on B cells[78]; this may occur on other cell types as well. As abatacept likely does not completely disrupt or significantly bind PD-L1:B7–1 heterodimers, these heterodimers may mediate continued costimulation [11–14] and thus disease progression (Figure 4B).
One should also consider CD28 expression. In organs like the colon, not all intraepithelial lymphocytes express CD28 [79] and may instead rely upon other co-stimulatory pathways. Thus, CTLA-4-Ig therapy would be ineffective. Indeed, anti-CTLA-4-Ig failed in double-blind, placebo-controlled, phase III randomized controlled clinical trials (clinicaltrails.gov NCT00406653, NCT00410410) in Crohn’s disease and ulcerative colitis [9].
To improve CTLA-4-Ig therapy we propose to simultaneously manipulate the PD-L1:B7–1 cis-interaction. Decreasing PD-L1:B7–1 cis-heterodimers using PD-L1:B7–1-specific cis-blocking mAbs would increase PD-1:PD-L1 binding, strongly inhibiting T cell activation. In addition, the liberated B7–1 binds to CTLA-4-Ig given proper timing and local concentrations of the immunotherapy. Together, this would lead to decreased activation. Of course, the local abundance of CD28 and dosage of CTLA-4-Ig would be critical factors in this approach as increased levels of B7–1 could increase CD28 signaling. However where appropriate, such a treatment would be beneficial even in cases of low B7–1 or CD28 expression, and represents an untapped therapeutic intervention.
Concluding remarks
The success of immunotherapy in a number of clinical settings, including in cancer, autoimmune disease, and organ transplant has highlighted the importance of the bench-to-bedside research pipeline. New characterizations of immune checkpoints such as the PD-L1:B7–1 cis-interaction and its effects on CD28, CTLA-4, B7–1, PD-L1, and PD-1 demonstrate that a deeper mechanistic understanding helps improve patient outcomes (see Outstanding Questions). To improve our understanding and utilization of immunotherapies, future studies should consider the co-expression, relative concentrations, timing of expression, location, and long-term regulatory changes in expression of these immune checkpoints. Furthermore, newly developed immunotherapies and combination therapies targeting these immune checkpoints should specifically consider effects on the PD-L1:B7–1 cis-interaction to develop more effective strategies in cancer, autoimmunity, and organ transplant (see Clinician’s Corner).
Outstanding Questions.
Do other immune cells that exhibit the PD-L1:B7–1 cis-interaction correlate with clinical responses from anti-CTLA-4 and anti-PD-1/PD-L1 immunotherapy? Does this correlation differ between cancers?
How will the PD-L1:B7–1 cis-interaction behave before and after immune checkpoint blockade on immune cells other than dendritic cells co-expressing PD-L1 and B7–1?
To what degree does CTLA-4 binding to the PD-L1:B7–1 cis-heterodimer disrupt the interaction between CD28 and PD-L1:B7–1 cis-heterodimer?
Does B7–2 play a role in this complex pathway since it can influence both CD28 and CTLA-4?
Do clinical-grade CTLA-4-Ig fusion proteins bind to PD-L1:B7–1 cis-heterodimers?
Does the CD28 costimulatory signal truly vary between the B7–1 and the PD-L1:B7–1 cis-heterodimer?
Does the PD-L1:B7–1 cis-heterodimer induce a CTLA-4-mediated inhibitory signal that attenuates the T cell receptor signaling cascade?
Clinician’s Corner.
PD-L1 antibodies may or may not disrupt co-stimulatory PD-L1:B7–1 cis-interactions. This should be tested for each clinically used antibody as disruption of this cis-interaction may be desirable or undesirable depending on the clinical circumstance.
Combination immunotherapy in cancer may be optimal when inhibiting CTLA-4:B7–1 interaction, PD-1:PD-L1 interaction, and PD-L1:B7–1 cis-interaction.
The use of CTLA-4-Ig in autoimmune diseases should be specifically targeted towards conditions whose pathogenesis involves cells that express B7–1/B7–2 and use the CD28 costimulatory pathway.
Specifically disrupting PD-L1:B7–1 cis-interaction combined with CTLA-4-Ig therapy could provide a novel and improved therapy for autoimmune diseases and organ transplant. The liberated PD-L1 would provide additional inhibition of T cells, and the freed B7–1 would be bound by CTLA-4-Ig. However, the dosage of CTLA-4-Ig in this combination therapy would certainly have to be carefully calculated as the freed B7–1 could conceivably form B7–1 homodimers and interact with CD28 if not first bound by CTLA-4-Ig.
Given that tumor-specific PD-L1 expression alone is a poor predictor of immunotherapy prognosis, additional biomarkers should be pursued. These include the presence or absence of other immune checkpoints, the relative abundance of these proteins in relation to each other, and the immune cell populations on which they are expressed.
Supplementary Material
Highlights.
Dysregulation of immune checkpoints contributes to the pathogenesis of cancer, autoimmunity, and organ transplant rejection by mediating undesired immune responses.
The limited long-term therapeutic efficacy of immunotherapies targeting immune checkpoints underscores the need to better understand the underlying biology of these proteins.
Immune checkpoint receptor-ligand interactions most commonly occur in trans (e.g. the receptor and ligand are expressed on two different cells). The newly characterized PD-L1:B7–1 interaction occurs in cis (e.g. the receptor and ligand are expressed on the same cell).
The PD-L1:B7–1 cis-interaction alters the interactions between CD28, CTLA-4, B7–1, PD-L1, and PD-1, and may underlie the efficacy of immunotherapies in a variety of treatment settings.
Acknowledgements
C.N. and M.P. are supported by National Institutes of Health (NIH) Medical Scientist training grant 5T32GM007288 and PhD in Clinical Investigation training grant 5TL1TR002557 respectively. Research in the Zang lab is supported by NIH R01DK100525 and 2R01CA175495, Department of Defense BC190403, Irma T.Hirschl/Monique Weill-Caulier Trust and Cancer Research Institute.
Glossary
- Antigen presenting cells
immune cells which help T cells recognize target antigens by processing and presenting these proteins on their cell surface
- Autoimmune diseases
diseases in which the immune system attacks self-antigens
- B7–1
an immune checkpoint ligand which can induce stimulatory or inhibitory immune response
- CD28
an immune receptor which transmits costimulatory signal on T cells and natural killer cells
- Cell activation
a process usually described in T cells in which cells gain the ability to exert their mechanistic functions through specific protein interactions
- Cis-interaction
a protein-protein interaction which occurs on the same cell
- CTLA-4
an inhibitory immune checkpoint receptor which inhibits costimulation
- Dysfunction
a phenotypic state describing abnormal cellular responses
- Endocytosis
a highly regulated process in which extracellular material is brought into the cell
- Immune checkpoints
proteins which regulate immune cell responses through inhibition
- Immune checkpoint blockade
a therapeutic strategy in which inhibitory proteins that restrain immune responses are blocked in order to improve immune responses
- Immune evasion
the process by which cells prevent recognition by the body’s immune system
- PD-1
an immune checkpoint receptor that transduces inhibitory signal
- PD-L1
an immune checkpoint ligand which, upon binding to PD-1 receptor, can induce inhibitory signal
- T cell receptor
a protein complex on T cells which recognizes antigens presented by antigen presenting cells
- Trans-interaction
a protein-protein interaction which occurs on different cells
- Trogocytosis
the process by which proteins from one cell membrane are transferred to another cell membrane
- SHP-1/2
proteins that dephosphorylate other proteins by removing bound phosphate groups
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.Ledford H, et al. (2018) Cancer immunologists scoop medicine Nobel prize. Nature 562, 20–21 [DOI] [PubMed] [Google Scholar]
- 2.Mease PJ, et al. (2017) Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann Rheum Dis 76, 1550–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunner HI, et al. (2018) Subcutaneous Abatacept in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase III open-label study. Arthritis Rheumatol 70, 1144–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Nimwegen JF, et al. (2020) Abatacept treatment for patients with early active primary Sjögren’s syndrome: a single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatol 2, e153–e163 [DOI] [PubMed] [Google Scholar]
- 5.Kliwinski C, et al. (2005) Prophylactic administration of abatacept prevents disease and bone destruction in a rat model of collagen-induced arthritis. J Autoimmun 25, 165–171 [DOI] [PubMed] [Google Scholar]
- 6.Durrbach A, et al. (2016) Long-term outcomes in belatacept- versus cyclosporine-treated recipients of extended criteria donor kidneys: final results from BENEFIT-EXT, a phase III randomized study. Am J Transplant 16, 3192–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iasella CJ, et al. (2018) Maintenance belatacept-based immunosuppression in lung transplantation recipients who failed calcineurin inhibitors. Transplantation 102, 171–177 [DOI] [PubMed] [Google Scholar]
- 8.Pons-Tostivint E, et al. (2019) Comparative analysis of durable responses on immune checkpoint inhibitors versus other systemic therapies: a pooled analysis of phase III trials. JCO Precision Oncology, 1–10 [DOI] [PubMed] [Google Scholar]
- 9.Sandborn WJ, et al. (2012) Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology 143, 62–69 e64 [DOI] [PubMed] [Google Scholar]
- 10.Chaudhri A, et al. (2018) PD-L1 binds to B7–1 only in cis on the same cell surface. Cancer Immunol Res 6, 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayoux M, et al. (2020) Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med 12 [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, et al. (2019) PD-L1:CD80 cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 51, 1059–1073 e1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugiura D, et al. (2019) Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 [DOI] [PubMed] [Google Scholar]
- 14.Oh SA, et al. (2020) PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 1, 681–691 [DOI] [PubMed] [Google Scholar]
- 15.Garrett-Thomson SC, et al. (2020) Mechanistic dissection of the PD-L1:B7–1 co-inhibitory immune complex. PLoS One 15, e0233578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman GJ, et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192, 1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamazaki T, et al. (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169, 5538–5545 [DOI] [PubMed] [Google Scholar]
- 18.Rodig N, et al. (2003) Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 33, 3117–3126 [DOI] [PubMed] [Google Scholar]
- 19.Ding H, et al. (2005) PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clin Immunol 115, 184–191 [DOI] [PubMed] [Google Scholar]
- 20.Sheppard KA, et al. (2004) PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett 574, 37–41 [DOI] [PubMed] [Google Scholar]
- 21.Celis-Gutierrez J, et al. (2019) Quantitative Interactomics in primary t cells provides a rationale for concomitant PD-1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Rep 27, 3315–3330 e3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hui E, et al. (2017) T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yokosuka T, et al. (2012) Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med 209, 1201–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plas DR, et al. (1996) Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science 272, 1173–1176 [DOI] [PubMed] [Google Scholar]
- 25.Barber DL, et al. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 [DOI] [PubMed] [Google Scholar]
- 26.Ahn E, et al. (2018) Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A 115, 4749–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pawelczyk K, et al. (2019) Role of PD-L1 expression in non-small cell lung cancer and their prognostic significance according to clinicopathological factors and diagnostic markers. Int J Mol Sci 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mozaffarian N, et al. (2008) Active systemic lupus erythematosus is associated with failure of antigen-presenting cells to express programmed death ligand-1. Rheumatology (Oxford) 47, 1335–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhatia S, et al. (2005) Different cell surface oligomeric states of B7–1 and B7–2: implications for signaling. Proc Natl Acad Sci U S A 102, 15569–15574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young JW, et al. (1992) The B7/BB1 antigen provides one of several costimulatory signals for the activation of CD4+ T lymphocytes by human blood dendritic cells in vitro. J Clin Invest 90, 229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hathcock KS, et al. (1994) Comparative analysis of B7–1 and B7–2 costimulatory ligands: expression and function. J Exp Med 180, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freedman AS, et al. (1987) B7, a B-cell-restricted antigen that identifies preactivated B cells. J Immunol 139, 3260–3267 [PubMed] [Google Scholar]
- 33.Freedman AS, et al. (1991) Selective induction of B7/BB-1 on interferon-gamma stimulated monocytes: a potential mechanism for amplification of T cell activation through the CD28 pathway. Cell Immunol 137, 429–437 [DOI] [PubMed] [Google Scholar]
- 34.Sabzevari H, et al. (2001) Acquisition of CD80 (B7–1) by T cells. J Immunol 166, 2505–2513 [DOI] [PubMed] [Google Scholar]
- 35.Azuma M, et al. (1992) CD28 interaction with B7 costimulates primary allogeneic proliferative responses and cytotoxicity mediated by small, resting T lymphocytes. J Exp Med 175, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linsley PS, et al. (1991) CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 174, 561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krummel MF and Allison JP (1996) CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med 183, 2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranheim EA and Kipps TJ (1994) Elevated expression of CD80 (B7/BB1) and other accessory molecules on synovial fluid mononuclear cell subsets in rheumatoid arthritis. Arthritis Rheum 37, 1637–1646 [DOI] [PubMed] [Google Scholar]
- 39.Butte MJ, et al. (2007) Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butte MJ, et al. (2008) Interaction of human PD-L1 and B7–1. Mol Immunol 45, 3567–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paterson AM, et al. (2011) The programmed death-1 ligand 1:B7–1 pathway restrains diabetogenic effector T cells in vivo. J Immunol 187, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freeman GJ, et al. (1993) Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science 262, 907–909 [DOI] [PubMed] [Google Scholar]
- 43.Freeman GJ, et al. (1993) Cloning of B7–2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 262, 909–911 [DOI] [PubMed] [Google Scholar]
- 44.Herbst RS, et al. (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powles T, et al. (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 [DOI] [PubMed] [Google Scholar]
- 46.Wang J, et al. (2005) Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A 102, 11823–11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keir ME, et al. (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunn GP, et al. (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137–148 [DOI] [PubMed] [Google Scholar]
- 49.John P, et al. (2019) The B7x immune checkpoint pathway: from discovery to clinical trial. Trends Pharmacol Sci 40, 883–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwai Y, et al. (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 99, 12293–12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tumeh PC, et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang AC, et al. (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bengsch B, et al. (2016) Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 45, 358–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamphorst AO, et al. (2017) Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 355, 1423–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haile ST, et al. (2011) Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J Immunol 186, 6822–6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tirapu I, et al. (2006) Low surface expression of B7–1 (CD80) is an immunoescape mechanism of colon carcinoma. Cancer Res 66, 2442–2450 [DOI] [PubMed] [Google Scholar]
- 57.Ramagopal UA, et al. (2017) Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc Natl Acad Sci U S A 114, E4223–E4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei SC, et al. (2018) Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 8, 1069–1086 [DOI] [PubMed] [Google Scholar]
- 59.Engelhard VH, et al. (2018) Immune cell infiltration and tertiary lymphoid structures as determinants of antitumor immunity. J Immunol 200, 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fehlings M, et al. (2017) Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8(+) T cells. Nat Commun 8, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei SC, et al. (2017) Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133 e1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liakou CI, et al. (2008) CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A 105, 14987–14992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cha E, et al. (2014) Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med 6, 238ra270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kvistborg P, et al. (2014) Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 6, 254ra128. [DOI] [PubMed] [Google Scholar]
- 65.Sharma N, et al. (2019) TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc Natl Acad Sci U S A 116, 10453–10462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peggs KS, et al. (2009) Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 206, 1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ha D, et al. (2019) Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A 116, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma A, et al. (2019) Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory t cells (Tregs) in human cancers. Clin Cancer Res 25, 1233–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larkin J, et al. (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Callahan MK, et al. (2014) CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol 4, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Theofilopoulos AN, et al. (2017) The multiple pathways to autoimmunity. Nat Immunol 18, 716–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tivol EA, et al. (1995) Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 [DOI] [PubMed] [Google Scholar]
- 73.Waterhouse P, et al. (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 [DOI] [PubMed] [Google Scholar]
- 74.Kuehn HS, et al. (2014) Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Douthwaite J, et al. (2017) A CD80-biased CTLA4-Ig fusion protein with superior in vivo efficacy by simultaneous engineering of affinity, selectivity, stability, and FcRn binding. J Immunol 198, 528–537 [DOI] [PubMed] [Google Scholar]
- 76.Moreau A, et al. (2013) Effector mechanisms of rejection. Cold Spring Harb Perspect Med 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Larsen CP, et al. (2005) Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant 5, 443–453 [DOI] [PubMed] [Google Scholar]
- 78.Lorenzetti R, et al. (2019) Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun 101, 145–152 [DOI] [PubMed] [Google Scholar]
- 79.Ohteki T and MacDonald HR (1993) Expression of the CD28 costimulatory molecule on subsets of murine intestinal intraepithelial lymphocytes correlates with lineage and responsiveness. Eur J Immunol 23, 1251–1255 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.