

Abstract
Although the activities of many signaling pathways are dysregulated during the progression of neurodegenerative and muscle degeneration disorders, the precise sequence of cellular events leading to degeneration have not been fully elucidated. Two kinases of particular interest, the growth-promoting Tor kinase and the energy sensor AMPK, appear to show reciprocal changes in activity during degeneration, with increased Tor activity and decreased AMPK activity reported. These changes in activity have been predicted to cause degeneration by attenuating autophagy, leading to accumulation of unfolded protein aggregates and dysfunctional mitochondria, the consequent increased production of reactive oxygen species (ROS), and ultimately oxidative damage. Here we propose that this increased ROS production not only causes oxidative damage but also ultimately induces an oxidative stress response that reactivates the redox-sensitive AMPK and activates the redox-sensitive stress kinase JNK. Activation of these kinases re-activates autophagy. Because at this late stage, cells have become filled with dysfunctional mitochondria and protein aggregates, which are autophagy targets, this autophagy reactivation induces degeneration. The mechanism proposed here emphasizes that the process of degeneration is dynamic, that dysregulated signaling pathways change over time and can transition from deleterious to beneficial and vice versa as degeneration progresses.
Similar signaling pathway disruptions are observed in neurodegenerative and muscle degenerative disorders
Although the neurodegenerative and muscle degenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and muscle disuse/denervation atrophy (MDA) are distinct entities with unique properties, these various disorders display similarities in the types of signaling pathways and cellular processes that are disrupted. Here we describe how activities of two kinases, the growth-promoting kinase Tor (reviewed in Schmelzle and Hall1) and the AMP-activated kinase AMPK (reviewed in Hardie, 20112), as well as the transcription factor Foxo change during degeneration (Figure 1A). We also describe evidence that these changes in activity have causal roles in promoting degeneration (Figure 1B).
Figure 1:
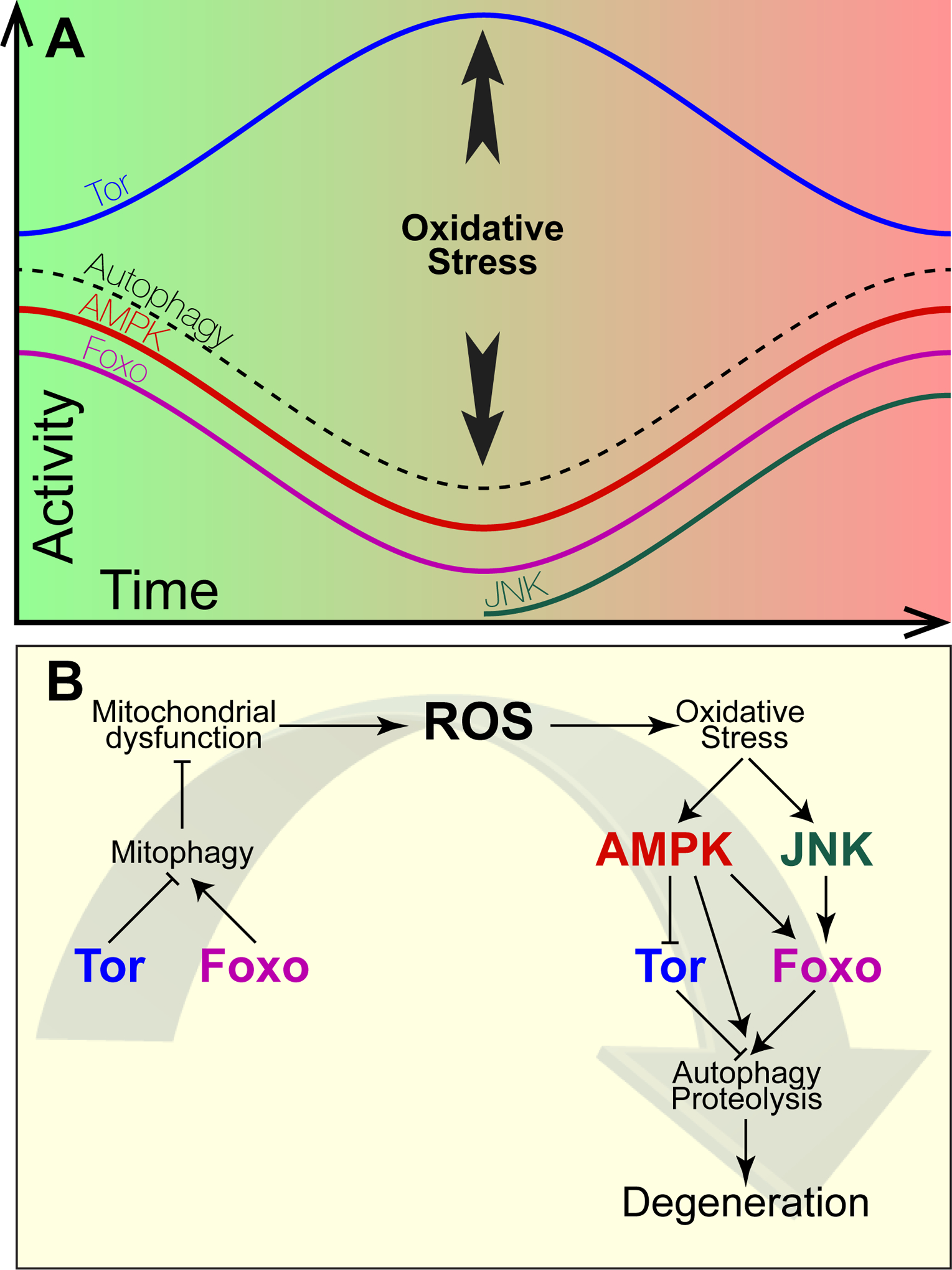
Mechanisms regulating the changes in signaling pathway activities during progression of degeneration. Upper panel: Changes in activity of the indicated signaling molecules or processes as a function of time during the progression of degeneration. Green shading represents a reduced cytoplasm, red shading represents an oxidized cytoplasm. The time at which the appearance of the oxidative stress response, which activates AMPK and JNK, is indicated. Lower panel: Causal relationships among observed changes in signaling pathway activities. Bars indicate repression, arrows indicate activation.
a). Increasing Tor kinase activity, decreasing AMPK and Foxo activities, in degeneration
The Tor kinase is activated under conditions of rapid growth, and when active, increases growth rate and decreases stress resistance. Several investigators have reported the progressive activation of Tor during the progression of AD3–5, PD6–8, and MDA9. In contrast, activity of AMPK, which is activated in cells under energy stress and tends to inhibit growth, has been reported to decrease during degeneration in both AD and MDA10, 11. AMPK activity also decreases during aging12, 13, which is a strong risk factor in all degenerative disorders. The transcription factor Foxo is also implicated in development and progression of degenerative disorders. Foxo is encoded by single genes in C. elegans and Drosophila, but in mammals comprises a family of related factors encoded by distinct genes. Foxo family members tend to be co-expressed within cells and exhibit functional redundancy14, although discrete roles of specific family members are sometimes observed15. The effects of Foxo activity, which are growth inhibition, tumor suppression, and stress resistance16, tend to be similar to those of AMPK but opposite to those of Tor. Although dynamics of Foxo activity during progression of most degenerative disorders have not been intensely studied, expression of the pro-apoptotic gene beclin, which is induced transcriptionally by Foxo17, declines during AD development in the mouse model18, which is consistent with a loss of Foxo activity in AD.
b). Effects of altered Tor, AMPK, and Foxo activities on degeneration
Several reports indicate that these changes in Tor, AMPK and Foxo activities have functional consequences, specifically that activating Tor or inhibiting either AMPK or Foxo promotes degeneration. Thus, therapeutic effects have been reported in rodent or Drosophila models of applying the Tor inhibitor rapamycin or of inhibiting Tor targets such as S6 Kinase. These observations suggest that Tor activation contributes to the pathology in these disorders6, 9, 19–29. In contrast, chemical or genetic activation of AMPK can be therapeutic in rodent or Drosophila degeneration models, whereas chemical inhibition of AMPK, or expression of a dominant negative AMPK transgene (AMPKDN) can exacerbate degeneration in a PD cell culture model30–33. Similarly, many reports indicate that Foxo, like AMPK, has a protective role in degenerative disorders and that blocking Foxo can be deleterious. In particular, blocking Foxo activity by expressing a FoxoDN transgene induces ROS production and loss of dopaminergic neurons in the rat. Similarly, in Drosophila, the Foxo null mutation confers degeneration in a manner similar to loss of the PD gene PINK134, 35, whereas Foxo+ overexpression protects dopaminergic neurons from degeneration caused by PINK1 mutations. Taken together, these results support the notion that the increasing Tor and the decreasing AMPK and Foxo activities observed in degeneration actively promote the degeneration process.
c). Cross-talk among Tor, AMPK and Foxo signaling molecules.
The opposing nature of AMPK and Foxo activities versus Tor is also revealed through cross-talk among these signaling molecules. AMPK inhibits Tor, both through direct Tor phosphorylation36 and by phosphorylation and activation of the Tor inhibitor Tsc237. Furthermore, AMPK directly phosphorylates each Foxo isoform38 and activates Foxo1 and Foxo339, 40. Thus, decreasing AMPK activity during degeneration might be responsible, at least in part, for the loss of Foxo activity and increased Tor activity observed during degeneration.
Disrupted autophagy is critical for driving degeneration forward
How might Tor activation, or AMPK or Foxo inhibition, contribute to progression of degenerative disorders? One pathway regulated in common by Tor, AMPK and Foxo is autophagy. Tor inhibits autophagy, whereas AMPK and Foxo each promote autophagy. In particular, Tor phosphorylates and inhibits the autophagy inducer ATG1 as well as ATG1341, 42, whereas AMPK phosphorylates and activates ATG136, 43. Foxo, in turn, activates autophagy by inducing transcription of critical autophagy genes such as LC3 (also known as ATG8) and ATG1244. These effects of Tor, AMPK and Foxo likely contribute to the defective autophagy observed in AD45, 46 and PD7, 26, 47. This autophagy inhibition, in turn, likely contributes to the observed accumulation of protein aggregates containing poly-ubiquitin in AD48 and α-synuclein in PD, as such aggregates are normally degraded by autophagy49–55. Given the toxic nature of these protein aggregates56, 57 it is possible that the loss of autophagy and consequent accumulation of these aggregates contributes to degeneration.
A critical role for mitochondrial autophagy (mitophagy) in degeneration
However, some evidence indicates that deficits in one specific form of autophagy, mitochondrial autophagy (mitophagy) is particularly important for progression of degeneration. Mitophagy is a critical mitochondrial quality control process and disruptions of mitophagy increase the persistence of dysfunctional mitochondria58. In fact, both disrupted mitophagy and accumulation of dysfunctional mitochondria are widely observed in tissues from degenerating samples such as postmortem samples from AD patients, and in mouse AD models59, 60. These observations have led several investigators to suggest that disrupted mitophagy might be causal for AD pathology61, 62. Accumulation of dysfunctional mitochondria also plays a key role in the initiation of MDA as well63, 64. For example, Trevino et al., (2019)65 reported that mitochondrial dysfunction was observed within three days of initiating muscle disuse in mice, and prior to onset of degeneration.
The importance of mitophagy in degeneration is most directly demonstrated for PD. At least two PD disease genes, PINK1 and Parkin, encode proteins specifically required for mitophagy66–68, and deficits in these genes are likely responsible for the attenuated mitophagy and mitochondrial dysfunction observed both in PD patients and animal models30, 69–71. Given that Foxo directly induces transcription of PINK172, 73, the loss of Foxo activity that occurs during degeneration is predicted to attenuate PINK1 transcription and hence attenuate mitophagy.
Oxidative damage in degeneration
How might an accumulation of dysfunctional mitochondria cause degeneration? In addition to decreasing ATP production, dysfunctional mitochondria also increase production of reactive oxygen species (ROS)74–77, which can lead to oxidative damage to proteins, lipids and nucleic acids. Such oxidative damage is widely observed in several different degenerative disorders, and the accumulation of damaged macromolecules has been implicated as causal for degeneration63, 69, 70, 74–85.
A preliminary model for degeneration: loss of autophagy causes accumulation of protein aggregates and increases ROS production and oxidative damage.
These data support, as a first approximation, a model in which degeneration develops as a consequence of long-term Tor activation, and Foxo and AMPK inhibition. The consequent mitophagy inhibition causes accumulation of dysfunctional mitochondria that overproduce ROS and increase oxidative damage of essential cellular structures. Combining this increase in oxidative damage with inhibited general autophagy, which prevents the clearance of damaged molecules, leads to accumulation of aggregates containing damaged proteins. These various pathologies and stressors ultimately lead to cell death.
This model predicts that administering autophagy inducers such as the Tor inhibitor rapamycin would be beneficial for degenerative disorders4, 86–88. Such therapies have been administered in both animal models and human patients, albeit with variable results.
Not so fast……..
Despite its appeal, the simple model described above fails to explain several features of degenerative disorders and therefore requires modification. First, decreased mitophagy is not always observed in degenerating tissues, as increased mitophagy has been reported in some human AD postmortem samples89, 90. Second, deleterious effects of Tor on degeneration have not always been observed. For example, it was reported that Tor activation, via Cre-mediated deletion of the Tor repressor PTEN, protects dopaminergic neurons from degeneration in a mouse PD model91. Similarly, Tor inhibition was reported to exacerbate degeneration, rather than protect from degeneration, in certain animal PD models92. In addition, some reports have indicated that amyloid-β oligomers decrease, rather than increase Tor activity93. Third, several reports investigating a variety of cell types indicate that autophagy is not always protective in degenerative disorders, but rather can contribute to cell death47, 94–99. Finally, Foxo activity is not always beneficial in degenerative conditions, but rather shows a complex relationship with cell survival in degeneration models. For example, Foxo is activated late in the MDA process and directly triggers degeneration by inducing transcription of the genes encoding the ubiquitin ligases atrogin and MuRF19, while expressing a FoxoDN transgene attenuates degeneration in MDA100. Foxo expression has also been reported to be up-regulated in both PD and AD brains101, 102 and some evidence indicates that this Foxo activation promotes apoptosis102, thereby conferring deleterious effects on afflicted cells.
To resolve these apparent contradictions, several investigators have noted that changes in activity of Tor, AMPK and Foxo are not static, but rather can change as degeneration progresses. For example, Tor is activated early in MDA, but Foxo is activated late in this process9. In contrast, AMPK activity decreases early in MDA, but increases late in this process11, 103.
These activity changes during disease progression were also reported to have important consequences on degeneration. As described above, the late stage Foxo activation during MDA is the proximate trigger for degeneration9, 104. In addition, it was proposed that both AMPK and Foxo transition from protective to deleterious during AD progression33, 105. Finally, it was suggested that administration of rapamycin might be beneficial early in degeneration, but might become ineffective late, when protein aggregates have stabilized and become too large for autophagy4. Taken together, these data indicate that the activities of Tor, AMPK and Foxo might confer distinct phenotypic consequences on degeneration dependent on timing, with AMPK and Foxo activities beneficial early in degeneration but deleterious late, and Tor demonstrating reciprocal effects. However, neither the timing nor the cellular changes underlying such an early to late transition have been identified.
Central hypothesis: induction of an oxidative stress response triggers an early to late transition during progression of degenerative disorders.
Taken together, the results described above suggest that degeneration comprises an early stage and a late stage. During the early stage, Tor activity increases while AMPK and Foxo activities decrease. As a result of impaired autophagy and mitophagy, protein aggregates and dysfunctional mitochondria accumulate. The increase in mitochondrial ROS generation caused by dysfunctional mitochondria progressively oxidizes the cytoplasm. We propose that this cytoplasmic oxidation ultimately becomes sufficient to induce an oxidative stress response. We hypothesize that the induction of this oxidative stress response marks the switch from the early to the late, degenerative, stage of degeneration. We suggest that oxidative stress induces the late, degenerative, stage by reactivating the redox-sensitive AMPK as well as by activating a second kinase, the stress- and redox-activated kinase JNK, which is described in more detail below. AMPK and JNK, in turn, together reactivate Foxo and inhibit Tor. We suggest that the consequent reactivation of autophagy and the proteasome during this late stage is the direct cause of degeneration (Figure 1). Below we describe the experimental data supporting this hypothesis.
Evidence supporting a role for oxidative stress in the transition to degeneration
a). Role of JNK in degeneration
Several reports indicate that JNK is activated in degenerative disorders, and that this activation participates in degeneration. In particular, JNK activation is observed both in postmortem brain samples from AD patients106–108 and in a transgenic mouse AD model109. Furthermore, the extent of JNK activity is correlated with the extent of cognitive decline as well as the levels of Aβ4 protein. Finally, inhibiting JNK in a mouse AD model restores normal synaptic function. These observations suggest a causal role for activated JNK in AD.
Similarly, studies in PD show that JNK inhibitors are protective in mouse MPTP models110–112 and that JNK activation is required for degeneration in a PC12 PD model113, 114.
Finally, a role for JNK activation is reported for MDA. JNK is activated in a rat disuse atrophy model115. In addition, it was reported that disuse atrophy activates JNK, which then activates Foxo indirectly, via phosphorylation of IRS-1, which detaches the insulin receptor from PI3K, thus preventing activation of the Foxo inhibitor Akt116.
b). Activation of AMPK and JNK by oxidative stress.
Oxidative stress activates AMPK either via the upstream activating kinase ATM36, 117, 118 or via direct oxidation119. Similarly, oxidative stress activates JNK120 by relieving the JNK-activating kinase ASK1 from inhibition by reduced thioredoxin121 or via the Ral GTPase122.
c). Foxo activation and Tor inhibition by AMPK and JNK
As described above, AMPK inhibits Tor via direct phosphorylation or by activating the Tor inhibitor Tsc1/Tsc2 complex, whereas AMPK activates Foxo by direct phosphorylation. Furthermore, JNK activates Foxo4 by direct phosphorylation122 and also activates Foxo1 and Foxo3123, 124, possibly by phosphorylating the 14-3-3 scaffold to prevent Foxo binding125. Either phosphorylation event activates Foxo by causing its nuclear translocation, enabling transcription of target genes. This Foxo activation, combined with AMPK activation and Tor inhibition, is anticipated to re-activate autophagy, mitophagy, and the proteasome122, 125–128,44–46, 104.
The two faces of Foxo and AMPK: benevolent protectors and merciless executioners.
AMPK and Foxo are generally considered protective for organismal viability, longevity and stress resistance, whereas Tor is thought to promote growth at the expense of longevity and stress resistance. In particular, increasing either AMPK or Foxo activity, or inhibiting Tor, increases lifespan in model organisms129–135. Furthermore, administering the AMPK activator metformin or the Tor inhibitor rapamycin has likewise been reported to increase lifespan in both invertebrate and mammalian species136–139. Thus, it is well established that AMPK and Foxo, or inhibited Tor, are beneficial to organismal survival. How can the activation of these beneficial pathways be the proximate cause for the degeneration observed in degenerative conditions?
We propose that cellular context determines whether these AMPK, Tor, Foxo and autophagy activities are beneficial or deleterious (Figure 2). In particular, we propose that in young cells, few dysfunctional mitochondria and few oxidized, unfolded or aggregated proteins are present. In this context, activating the AMPK/Foxo pathway increases lifespan and stress resistance by increasing the timely, autophagy-dependent removal of the few dysfunctional structures present (Figure 2, left panel). However, as cells age, progressively attenuating autophagy enables accumulation of aggregates of unfolded proteins and dysfunctional mitochondria (Figure 2, center panel); these mitochondria, albeit damaged, are generating ATP necessary for survival. Ultimately, the ROS generated by these dysfunctional mitochondria oxidize the cytoplasm sufficiently to induce an oxidative stress response. The consequent reactivation of autophagy and the proteasome now triggers removal of aggregates of unfolded proteins, other damaged cellular structures, and most importantly, the removal of damaged, but essential mitochondria (Figure 2, right panel). The result of this overzealous removal of these damaged structures is a precipitous decline in ATP production, degeneration and loss of cell viability.
Figure 2:
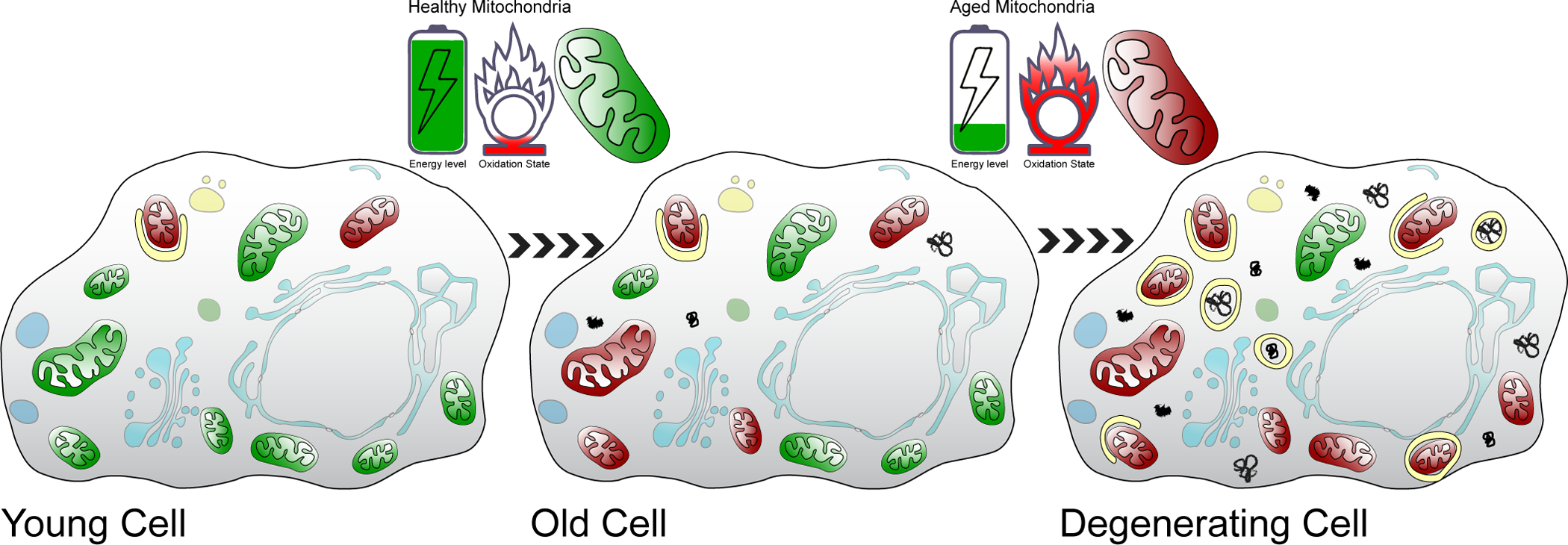
Accumulation of damaged structures during cell aging. Cells at three different stages (young, old and degenerating) are shown. Green mitochondria: healthy, polarized, non-oxidatively damaged. Red mitochondria: dysfunctional, depolarized, oxidatively damaged (see upper panels). Black shapes indicate protein aggregates. Yellow circles or semi-circles indicated autophagic vesicles. Shaded blue indicates other cellular structures.
This proposed mechanism supports the notion that decreased ATP production might underlie degeneration140. Despite intense investigation, the role of diminished energy production in degeneration has been difficult to demonstrate unambiguously, due in large part to lack of reporters with sufficient spatial and temporal precision. However, several indirect observations support the possibility that decreased energy utilization correlates with degeneration140, 141. These observations include reduced glucose uptake in degenerating neurons142, 143, increased lactate accumulation in Huntington’s Disease (HD) brains144, low ATP levels in PD brains145, and decreased ATP levels in PINK1 knockout mice146. In addition, using a novel ATP reporter, Pathak et al.147 showed decreased ATP production in the Leigh’s neurodegenerative disorder. Taken together, these data are consistent with the possibility shown in Figure 2 that declining energy production at least partially underlies degeneration.
Differential sensitivity of various cell types to degeneration
It is not known with certainty why specific neuronal or muscle subtypes show differential sensitivity to degeneration. However, one possible explanation could be cell type specific expression of pro-degeneration genes. For example low expression of PGC-1α, which inhibits Foxo, could explain the increased sensitivity of 2B muscle fibers to degeneration148. In addition, neurons particularly sensitive to degeneration could have increased energy requirements, leading to increased sensitivity to mitochondrial stressors141, 149. In this regard, neurons exhibiting functional plasticity might be more sensitive to degeneration than others150 because continuous retraction and regrowth of synapses could increase demand for ATP.
Oxidative stress-triggered degeneration induced by various causes
Although the mechanism shown in Figure 2 most directly relates to neurodegeneration caused by aging, other factors have been reported to advance the onset of neurodegenerative disorders. These include genetic factors, metabolic factors such as insulin resistance, or physiological factors such as neuroinflammation or accumulation of senescent cells151–157. In some cases, a direct link to altered Tor and Foxo signaling is apparent. For example, insulin resistance arises as a consequence of long-term increases in insulin signaling, which activates Tor and inhibits Foxo. Such long-term increases in Tor activity and decreases in Foxo activity are predicted to accelerate progression through the early stage of degeneration (see Figure 1) and advance the onset of the late, degenerative stage. Likewise, results from a transgenic mouse model, in which mutant Amyloid Precursor Protein and Presenilin-1 genes are co-overexpressed, support the notion that AD comprises an early stage of Tor activation and Foxo inhibition, followed by a late, degenerative stage of Tor inhibition and Foxo activation158. Thus, accelerating the changes in Tor and Foxo activities during the early stage might be a general property of factors that advance the onset of neurodegeneration. For other factors, although no obvious mechanisms directly link to Tor/Foxo/JNK signaling, many are predicted to induce oxidative stress, which would likewise advance the onset of the late, degenerative stage159.
Oxidative stress-triggered degeneration in other neurodegenerative or muscle degeneration disorders
A similar involvement of oxidative stress, mitochondrial dysfunction and autophagy impairment contributes to the development and progression of other neurodegenerative disorders, such as HD and Amyotrophic Lateral Sclerosis (ALS)160–169. Furthermore, alterations in activities of Tor, AMPK and JNK have been observed in these disorders, with therapeutic effects of Tor, AMPK or JNK inhibition reported170–173. Consistent with the model presented in Figure 2 and with what was described above for MDA, AMPK activity suppresses HD phenotypes, in a Foxo-dependent manner, early in disease progression174, while enhancing HD phenotypes late in disease progression175. These results suggest that transitions of signaling pathways from beneficial to deleterious or vice versa during disease progression might be a common property of neurodegenerative disorders.
ROS-induced oxidative stress and mitochondrial dysfunction have likewise been implicated in two additional muscle degeneration disorders, cachexia and sarcopenia176–184. In addition, previous reports indicate that both JNK and Foxo are activated during cancer cachexia, and this activation is required for the observed loss of muscle tissue185–187. These results are consistent with the role described above for JNK and Foxo in AD, PD and MDA. However, in cachexia, the transcription factor NF-κB is also activated, via ROS or TNF-α. This pathway appears to play a major role in degeneration via induced transcription of proteolysis genes188–190. This pathway might operate in parallel to a JNK-Foxo pathway in inducing degeneration.
Relevance to treatment of degenerative disorders
We emphasize that the progression of degeneration is a dynamic process; activities of specific signaling pathways change over time, and specific signaling pathways can transition from beneficial to deleterious, or vice versa, as degeneration progresses. Thus, for example, rapamycin or metformin administered early in disease progression might be beneficial, but deleterious when administered in the late stage. We anticipate that considering these timing issues will help clarify the roles of altered signaling molecule activity in progression of degenerative disorders.
Acknowledgements:
We are grateful to Drs. Saurabh Srivastav and Kevin van der Graaf for comments on the manuscript. This work was funded by NIH grant R01 NS102676 awarded to MS and JAM.
Footnotes
Conflicts of Interest: The authors have no conflicts to disclose.
References
- 1.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell 2000; 103(2): 253–262. [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25(18): 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med 2008; 12(6B): 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011; 12(8): 437–452. [DOI] [PubMed] [Google Scholar]
- 5.Wong M Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J 2013; 36(2): 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lan AP, Chen J, Zhao Y, Chai Z, Hu Y. mTOR Signaling in Parkinson’s Disease. Neuromolecular Med 2017; 19(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 7.Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010; 5(2): e9313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Zhu Z, Yang C, Iyaswamy A, Krishnamoorthi S, Sreenivasmurthy SG, Liu J et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int J Mol Sci 2019; 20(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang H, Inoki K, Lee M, Wright E, Khuong A, Khuong A et al. mTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci Signal 2014; 7(314): ra18. [DOI] [PubMed] [Google Scholar]
- 10.Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med 2012; 14(1): 1–14. [DOI] [PubMed] [Google Scholar]
- 11.Thomson DM. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int J Mol Sci 2018; 19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma D, Chen Y, Sun Y, Yang B, Cheng J, Huang M et al. Molecular analysis of the CYP21A2 gene in Chinese patients with steroid 21-hydroxylase deficiency. Clin Biochem 2014; 47(6): 455–463. [DOI] [PubMed] [Google Scholar]
- 13.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 2007; 5(2): 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt-Ney M The FOXO’s Advantages of Being a Family: Considerations on Function and Evolution. Cells 2020; 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 2003; 301(5630): 215–218. [DOI] [PubMed] [Google Scholar]
- 16.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005; 24(50): 7410–7425. [DOI] [PubMed] [Google Scholar]
- 17.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 2014; 39(4): 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118(6): 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36(6): 585–595. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J 2005; 272(16): 4211–4220. [DOI] [PubMed] [Google Scholar]
- 21.Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 2007; 3(6): 620–622. [DOI] [PubMed] [Google Scholar]
- 22.Caccamo A, Branca C, Talboom JS, Shaw DM, Turner D, Ma L et al. Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer’s Disease. J Neurosci 2015; 35(41): 14042–14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011; 6(9): e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC. Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol 2010; 21(7): 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Talboom JS, Velazquez R, Oddo S. The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease. NPJ Aging Mech Dis 2015; 1: 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 2010; 30(37): 12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci 2010; 30(3): 1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci 2009; 12(9): 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu S, Stern M, McNew JA. Beneficial effects of rapamycin in a Drosophila model for hereditary spastic paraplegia. J Cell Sci 2017; 130(2): 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK et al. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J Neurosci 2012; 32(41): 14311–14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curry DW, Stutz B, Andrews ZB, Elsworth JD. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J Parkinsons Dis 2018; 8(2): 161–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun 2010; 391(1): 147–151. [DOI] [PubMed] [Google Scholar]
- 33.Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer’s disease. J Neurochem 2011; 118(4): 460–474. [DOI] [PubMed] [Google Scholar]
- 34.Pino E, Amamoto R, Zheng L, Cacquevel M, Sarria JC, Knott GW et al. FOXO3 determines the accumulation of alpha-synuclein and controls the fate of dopaminergic neurons in the substantia nigra. Hum Mol Genet 2014; 23(6): 1435–1452. [DOI] [PubMed] [Google Scholar]
- 35.Koh H, Kim H, Kim MJ, Park J, Lee HJ, Chung J. Silent information regulator 2 (Sir2) and Forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in Drosophila PTEN-induced kinase 1 (PINK1) null mutant. J Biol Chem 2012; 287(16): 12750–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kjobsted R, Hingst JR, Fentz J, Foretz M, Sanz MN, Pehmoller C et al. AMPK in skeletal muscle function and metabolism. FASEB J 2018; 32(4): 1741–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115(5): 577–590. [DOI] [PubMed] [Google Scholar]
- 38.Greer EL, Banko MR, Brunet A. AMP-activated protein kinase and FoxO transcription factors in dietary restriction-induced longevity. Ann N Y Acad Sci 2009; 1170: 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H et al. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem 2012; 113(2): 695–710. [DOI] [PubMed] [Google Scholar]
- 40.Yun H, Park S, Kim MJ, Yang WK, Im DU, Yang KR et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J 2014; 281(19): 4421–4438. [DOI] [PubMed] [Google Scholar]
- 41.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584(7): 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13(2): 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331(6016): 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem 2009; 284(41): 28319–28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Li L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front Mol Neurosci 2019; 12: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N. Autophagy in Alzheimer’s disease. Rev Neurosci 2015; 26(4): 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170(1): 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 2015; 16(6): 345–357. [DOI] [PubMed] [Google Scholar]
- 49.Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest 2015; 125(1): 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34(3): 259–269. [DOI] [PubMed] [Google Scholar]
- 51.Madeo F, Eisenberg T, Kroemer G. Autophagy for the avoidance of neurodegeneration. Genes Dev 2009; 23(19): 2253–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gundersen V Protein aggregation in Parkinson’s disease. Acta Neurol Scand Suppl 2010; (190): 82–87. [DOI] [PubMed] [Google Scholar]
- 53.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278(27): 25009–25013. [DOI] [PubMed] [Google Scholar]
- 54.Xilouri M, Brekk OR, Stefanis L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov Disord 2016; 31(2): 178–192. [DOI] [PubMed] [Google Scholar]
- 55.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M et al. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol 1988; 155(1): 9–15. [DOI] [PubMed] [Google Scholar]
- 56.Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med 2008; 14(7–8): 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stefanis L alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2012; 2(2): a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018; 20(9): 1013–1022. [DOI] [PubMed] [Google Scholar]
- 59.Selfridge JE, E L, Lu J, Swerdlow RH. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol Dis 2013; 51: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 2004; 63(1): 8–20. [DOI] [PubMed] [Google Scholar]
- 61.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA et al. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci 2017; 40(3): 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakravorty A, Jetto CT, Manjithaya R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front Aging Neurosci 2019; 11: 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Powers SK, Wiggs MP, Duarte JA, Zergeroglu AM, Demirel HA. Mitochondrial signaling contributes to disuse muscle atrophy. Am J Physiol Endocrinol Metab 2012; 303(1): E31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calvani R, Joseph AM, Adhihetty PJ, Miccheli A, Bossola M, Leeuwenburgh C et al. Mitochondrial pathways in sarcopenia of aging and disuse muscle atrophy. Biol Chem 2013; 394(3): 393–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trevino MB, Zhang X, Standley RA, Wang M, Han X, Reis FCG et al. Loss of mitochondrial energetics is associated with poor recovery of muscle function but not mass following disuse atrophy. Am J Physiol Endocrinol Metab 2019; 317(5): E899–E910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392(6676): 605–608. [DOI] [PubMed] [Google Scholar]
- 67.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004; 304(5674): 1158–1160. [DOI] [PubMed] [Google Scholar]
- 68.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107(1): 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 2013; 3(4): 461–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaki GS, Papavassiliou AG. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromolecular Med 2014; 16(2): 217–230. [DOI] [PubMed] [Google Scholar]
- 71.Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010; 16(11): 1313–1320. [DOI] [PubMed] [Google Scholar]
- 72.Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci U S A 2009; 106(13): 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001; 20(33): 4457–4465. [DOI] [PubMed] [Google Scholar]
- 74.Bin-Umer MA, McLaughlin JE, Butterly MS, McCormick S, Tumer NE. Elimination of damaged mitochondria through mitophagy reduces mitochondrial oxidative stress and increases tolerance to trichothecenes. Proc Natl Acad Sci U S A 2014; 111(32): 11798–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T et al. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287(5): 3265–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang CH, Wu SB, Wu YT, Wei YH. Oxidative stress response elicited by mitochondrial dysfunction: implication in the pathophysiology of aging. Exp Biol Med (Maywood) 2013; 238(5): 450–460. [DOI] [PubMed] [Google Scholar]
- 77.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469(7329): 221–225. [DOI] [PubMed] [Google Scholar]
- 78.Giordano S, Lee J, Darley-Usmar VM, Zhang J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One 2012; 7(9): e44610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hwang O Role of oxidative stress in Parkinson’s disease. Exp Neurobiol 2013; 22(1): 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord 1998; 13 Suppl 1: 35–38. [PubMed] [Google Scholar]
- 81.Stojkovska I, Wagner BM, Morrison BE. Parkinson’s disease and enhanced inflammatory response. Exp Biol Med (Maywood) 2015; 240(11): 1387–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A et al. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 2007; 293(3): R1159–1168. [DOI] [PubMed] [Google Scholar]
- 83.Qiu J, Fang Q, Xu T, Wu C, Xu L, Wang L et al. Mechanistic Role of Reactive Oxygen Species and Therapeutic Potential of Antioxidants in Denervation- or Fasting-Induced Skeletal Muscle Atrophy. Front Physiol 2018; 9: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Domenico F, Barone E, Perluigi M, Butterfield DA. The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid Redox Signal 2017; 26(8): 364–387. [DOI] [PubMed] [Google Scholar]
- 85.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 2001; 60(8): 759–767. [DOI] [PubMed] [Google Scholar]
- 86.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110(19): E1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2012; 2(4): a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11(9): 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 2001; 21(9): 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M et al. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 2007; 66(6): 525–532. [DOI] [PubMed] [Google Scholar]
- 91.Domanskyi A, Geissler C, Vinnikov IA, Alter H, Schober A, Vogt MA et al. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J 2011; 25(9): 2898–2910. [DOI] [PubMed] [Google Scholar]
- 92.Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal 2014; 26(8): 1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem 2005; 94(1): 215–225. [DOI] [PubMed] [Google Scholar]
- 94.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 2001; 21(24): 9549–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 2003; 73(3): 341–350. [DOI] [PubMed] [Google Scholar]
- 96.Choi KC, Kim SH, Ha JY, Kim ST, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem 2010; 112(2): 366–376. [DOI] [PubMed] [Google Scholar]
- 97.Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286(12): 10814–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 2009; 4(5): e5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng HC, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M et al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci 2011; 31(6): 2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol 2010; 298(1): C38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dumitriu A, Latourelle JC, Hadzi TC, Pankratz N, Garza D, Miller JP et al. Gene expression profiles in Parkinson disease prefrontal cortex implicate FOXO1 and genes under its transcriptional regulation. PLoS Genet 2012; 8(6): e1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wong HK, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C et al. De-repression of FOXO3a death axis by microRNA-132 and −212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet 2013; 22(15): 3077–3092. [DOI] [PubMed] [Google Scholar]
- 103.Stouth DW, Manta A, Ljubicic V. Protein arginine methyltransferase expression, localization, and activity during disuse-induced skeletal muscle plasticity. Am J Physiol Cell Physiol 2018; 314(2): C177–C190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117(3): 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shi C, Viccaro K, Lee HG, Shah K. Cdk5-Foxo3 axis: initially neuroprotective, eventually neurodegenerative in Alzheimer’s disease models. J Cell Sci 2016; 129(9): 1815–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H et al. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem 2001; 76(2): 435–441. [DOI] [PubMed] [Google Scholar]
- 107.Gourmaud S, Paquet C, Dumurgier J, Pace C, Bouras C, Gray F et al. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J Psychiatry Neurosci 2015; 40(3): 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front Pharmacol 2015; 6: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sclip A, Tozzi A, Abaza A, Cardinetti D, Colombo I, Calabresi P et al. c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis 2014; 5: e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang Y, Zhang Y, Wei Z, Li H, Zhou H, Zhang Z et al. JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J Neurol Sci 2009; 285(1–2): 172–177. [DOI] [PubMed] [Google Scholar]
- 111.Wang W, Ma C, Mao Z, Li M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect 2004; 17(10): 646–654. [DOI] [PubMed] [Google Scholar]
- 112.Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 2004; 101(2): 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wilhelm M, Xu Z, Kukekov NV, Gire S, Greene LA. Proapoptotic Nix activates the JNK pathway by interacting with POSH and mediates death in a Parkinson disease model. J Biol Chem 2007; 282(2): 1288–1295. [DOI] [PubMed] [Google Scholar]
- 114.Kuan CY, Burke RE. Targeting the JNK signaling pathway for stroke and Parkinson’s diseases therapy. Curr Drug Targets CNS Neurol Disord 2005; 4(1): 63–67. [DOI] [PubMed] [Google Scholar]
- 115.Kim JK, HKim B. Differential Regulation of MAPK Isoforms during Cast-Immobilization —Induced Atrophy in Rat Gastrocnemius Muscle. J Phys Ther Sci 2010; 22: 217–222. [Google Scholar]
- 116.Hilder TL, Tou JC, Grindeland RE, Wade CE, Graves LM. Phosphorylation of insulin receptor substrate-1 serine 307 correlates with JNK activity in atrophic skeletal muscle. FEBS Lett 2003; 553(1–2): 63–67. [DOI] [PubMed] [Google Scholar]
- 117.Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol 2013; 15(10): 1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A 2013; 110(32): E2950–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 2010; 285(43): 33154–33164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem 1996; 271(26): 15703–15707. [DOI] [PubMed] [Google Scholar]
- 121.Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol 2000; 20(6): 2198–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 2004; 23(24): 4802–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shen B, Chao L, Chao J. Pivotal role of JNK-dependent FOXO1 activation in downregulation of kallistatin expression by oxidative stress. Am J Physiol Heart Circ Physiol 2010; 298(3): H1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE et al. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis 2012; 3: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol 2005; 170(2): 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luo Y, Umegaki H, Wang X, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem 1998; 273(6): 3756–3764. [DOI] [PubMed] [Google Scholar]
- 127.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 2011; 1813(11): 1938–1945. [DOI] [PubMed] [Google Scholar]
- 128.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 2005; 121(1): 115–125. [DOI] [PubMed] [Google Scholar]
- 129.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 2003; 426(6967): 620. [DOI] [PubMed] [Google Scholar]
- 130.Stenesen D, Suh JM, Seo J, Yu K, Lee KS, Kim JS et al. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab 2013; 17(1): 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Payne S, Shivaprasad HL, Mirhosseini N, Gray P, Hoppes S, Weissenbock H et al. Unusual and severe lesions of proventricular dilatation disease in cockatiels (Nymphicus hollandicus) acting as healthy carriers of avian bornavirus (ABV) and subsequently infected with a virulent strain of ABV. Avian Pathol 2011; 40(1): 15–22. [DOI] [PubMed] [Google Scholar]
- 132.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 1997; 278(5341): 1319–1322. [DOI] [PubMed] [Google Scholar]
- 133.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004; 14(10): 885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004; 429(6991): 562–566. [DOI] [PubMed] [Google Scholar]
- 135.Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science 2004; 305(5682): 361. [DOI] [PubMed] [Google Scholar]
- 136.Chen J, Ou Y, Li Y, Hu S, Shao LW, Liu Y. Metformin extends C. elegans lifespan through lysosomal pathway. Elife 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M et al. Metformin improves healthspan and lifespan in mice. Nat Commun 2013; 4: 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 2010; 11(1): 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460(7253): 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pathak D, Berthet A, Nakamura K. Energy failure: does it contribute to neurodegeneration? Ann Neurol 2013; 74(4): 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Haddad D, Nakamura K. Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett 2015; 589(24 Pt A): 3702–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Feigin A, Leenders KL, Moeller JR, Missimer J, Kuenig G, Spetsieris P et al. Metabolic network abnormalities in early Huntington’s disease: an [(18)F]FDG PET study. J Nucl Med 2001; 42(11): 1591–1595. [PubMed] [Google Scholar]
- 143.Petrie EC, Cross DJ, Galasko D, Schellenberg GD, Raskind MA, Peskind ER et al. Preclinical evidence of Alzheimer changes: convergent cerebrospinal fluid biomarker and fluorodeoxyglucose positron emission tomography findings. Arch Neurol 2009; 66(5): 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, MacDonald M et al. 1H NMR spectroscopy studies of Huntington’s disease: correlations with CAG repeat numbers. Neurology 1998; 50(5): 1357–1365. [DOI] [PubMed] [Google Scholar]
- 145.Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C, Steinmetz H et al. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 2009; 132(Pt 12): 3285–3297. [DOI] [PubMed] [Google Scholar]
- 146.Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci 2011; 124(Pt 7): 1115–1125. [DOI] [PubMed] [Google Scholar]
- 147.Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA et al. The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem 2015; 290(37): 22325–22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 2013; 45(10): 2191–2199. [DOI] [PubMed] [Google Scholar]
- 149.Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 2011; 31(48): 17649–17658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roussarie JP, Yao V, Rodriguez-Rodriguez P, Oughtred R, Rust J, Plautz Z et al. Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2008; 2(6): 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wan L, Nie G, Zhang J, Luo Y, Zhang P, Zhang Z et al. beta-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med 2011; 50(1): 122–129. [DOI] [PubMed] [Google Scholar]
- 153.Ill-Raga G, Ramos-Fernandez E, Guix FX, Tajes M, Bosch-Morato M, Palomer E et al. Amyloid-beta peptide fibrils induce nitro-oxidative stress in neuronal cells. J Alzheimers Dis 2010; 22(2): 641–652. [DOI] [PubMed] [Google Scholar]
- 154.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1–42). Neurobiol Aging 1999; 20(3): 325–330; discussion 339–342. [DOI] [PubMed] [Google Scholar]
- 155.Butterfield DA, Swomley AM, Sultana R. Amyloid beta-peptide (1–42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal 2013; 19(8): 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res 2007; 85(4): 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Castillo X, Castro-Obregon S, Gutierrez-Becker B, Gutierrez-Ospina G, Karalis N, Khalil AA et al. Re-thinking the Etiological Framework of Neurodegeneration. Front Neurosci 2019; 13: 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jimenez S, Torres M, Vizuete M, Sanchez-Varo R, Sanchez-Mejias E, Trujillo-Estrada L et al. Age-dependent accumulation of soluble amyloid beta (Abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J Biol Chem 2011; 286(21): 18414–18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rekatsina M, Paladini A, Piroli A, Zis P, Pergolizzi JV, Varrassi G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv Ther 2020; 37(1): 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pollari E, Goldsteins G, Bart G, Koistinaho J, Giniatullin R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front Cell Neurosci 2014; 8: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Jaronen M, Arens E, Akerman K et al. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J Biol Chem 2008; 283(13): 8446–8452. [DOI] [PubMed] [Google Scholar]
- 162.Jaiswal MK, Zech WD, Goos M, Leutbecher C, Ferri A, Zippelius A et al. Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci 2009; 10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014; 10(4): 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Song CY, Guo JF, Liu Y, Tang BS. Autophagy and Its Comprehensive Impact on ALS. Int J Neurosci 2012; 122(12): 695–703. [DOI] [PubMed] [Google Scholar]
- 165.Cipolat Mis MS, Brajkovic S, Frattini E, Di Fonzo A, Corti S. Autophagy in motor neuron disease: Key pathogenetic mechanisms and therapeutic targets. Mol Cell Neurosci 2016; 72: 84–90. [DOI] [PubMed] [Google Scholar]
- 166.Kumar A, Ratan RR. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J Huntingtons Dis 2016; 5(3): 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Acevedo-Torres K, Berrios L, Rosario N, Dufault V, Skatchkov S, Eaton MJ et al. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst) 2009; 8(1): 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington’s disease: focusing on huntingtin and the striatum. J Neurochem 2010; 114(1): 1–12. [DOI] [PubMed] [Google Scholar]
- 169.Quintanilla RA, Johnson GV. Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull 2009; 80(4–5): 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chaplot K, Pimpale L, Ramalingam B, Deivasigamani S, Kamat SS, Ratnaparkhi GS. SOD1 activity threshold and TOR signalling modulate VAP(P58S) aggregation via reactive oxygen species-induced proteasomal degradation in a Drosophila model of amyotrophic lateral sclerosis. Dis Model Mech 2019; 12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC et al. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci 2012; 32(3): 1123–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schellino R, Boido M, Vercelli A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019; 8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci Transl Med 2017; 9(403). [DOI] [PubMed] [Google Scholar]
- 174.Vazquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millan JM et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum Mol Genet 2016; 25(6): 1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol 2011; 194(2): 209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 1992; 267(8): 5317–5323. [PubMed] [Google Scholar]
- 177.Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J 1998; 12(10): 871–880. [DOI] [PubMed] [Google Scholar]
- 178.Li YP, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J 2003; 17(9): 1048–1057. [DOI] [PubMed] [Google Scholar]
- 179.Carson JA, Hardee JP, VanderVeen BN. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin Cell Dev Biol 2016; 54: 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Abrigo J, Elorza AA, Riedel CA, Vilos C, Simon F, Cabrera D et al. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxid Med Cell Longev 2018; 2018: 2063179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Coen PM, Musci RV, Hinkley JM, Miller BF. Mitochondria as a Target for Mitigating Sarcopenia. Front Physiol 2018; 9: 1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gonzalez-Freire M, Scalzo P, D’Agostino J, Moore ZA, Diaz-Ruiz A, Fabbri E et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018; 17(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Migliavacca E, Tay SKH, Patel HP, Sonntag T, Civiletto G, McFarlane C et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun 2019; 10(1): 5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Andreux PA, van Diemen MPJ, Heezen MR, Auwerx J, Rinsch C, Groeneveld GJ et al. Mitochondrial function is impaired in the skeletal muscle of pre-frail elderly. Sci Rep 2018; 8(1): 8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A et al. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol 2006; 291(3): R674–683. [DOI] [PubMed] [Google Scholar]
- 186.Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J 2012; 26(3): 987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mulder SE, Dasgupta A, King RJ, Abrego J, Attri KS, Murthy D et al. JNK signaling contributes to skeletal muscle wasting and protein turnover in pancreatic cancer cachexia. Cancer Lett 2020; 491: 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cai D, Frantz JD, Tawa NE Jr., Melendez PA, Oh BC, Lidov HG et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004; 119(2): 285–298. [DOI] [PubMed] [Google Scholar]
- 189.Barreiro E, de la Puente B, Busquets S, Lopez-Soriano FJ, Gea J, Argiles JM. Both oxidative and nitrosative stress are associated with muscle wasting in tumour-bearing rats. FEBS Lett 2005; 579(7): 1646–1652. [DOI] [PubMed] [Google Scholar]
- 190.Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 2003; 285(4): C806–812. [DOI] [PubMed] [Google Scholar]