

Abstract
NUT carcinoma (NC) is an extremely aggressive squamous cancer with no effective therapy. NC is driven, most commonly, by the BRD4-NUT fusion oncoprotein. BRD4-NUT combines the chromatin-binding bromo- and extraterminal domain-containing (BET) protein, BRD4, with an unstructured, poorly understood protein, NUT, which recruits and activates the histone acetyltransferase (HAT) p300. Recruitment of p300 to chromatin by BRD4 is believed to lead to the formation of hyperacetylated nuclear foci, as seen by immunofluorescence. BRD4-NUT nuclear foci correspond with massive contiguous regions of chromatin co-enriched with BRD4-NUT, p300, and acetylated histones, termed ‘megadomains’ (MD). Megadomains stretch for as long as 2 MB. Proteomics has defined a BRD4-NUT chromatin complex in which members that associate with BRD4 also exist as rare NUT-fusion partners. This suggests that the common pathogenic denominator is the presence of both BRD4 and NUT, and that the function of BRD4-NUT may mimic that of wild-type BRD4. If so, then MDs may function as massive super-enhancers, activating transcription in a BET-dependent manner. Common targets of MDs across multiple NCs and tissues are three stem cell-related transcription factors frequently implicated in cancer: MYC, SOX2, and TP63. Recently, MDs were found to form a novel nuclear sub-compartment, called subcompartment M (subM), where MD-MD interactions occur both intra- and inter-chromosomally. Included in subM are MYC, SOX2, and TP63. Here we explore the possibility that if MDs are simply large super-enhancers, subM may exist in other cell systems, with broad implications for how 3D organization of the genome may function in gene regulation and maintenance of cell identity. Finally, we discuss how our knowledge of BRD4-NUT function has been leveraged for the therapeutic development of first-in-class BET inhibitors and other targeted strategies.
Introduction
NUT carcinoma (NC, aka NUT midline carcinoma) is a rare subtype of poorly differentiated squamous cell carcinoma that is defined by rearrangements of the NUTM1 (aka NUT) gene1. In the majority of cases (~75%)4, NUTM1 is fused to BRD4. This results in a chimeric oncoprotein including BRD4, a member of the dual bromodomain and extraterminal domain (BET) family of proteins2, fused to NUT. NUT is a testis-specific protein that facilitates hyperacetylation of chromatin by the histone acetyltransferase (HAT), p300 (aka EP300), in post-meiotic spermatids, a critical step in histone-to-protamine exchange required for chromatin compaction during spermatogenesis3. BRD4-NUT4 is a powerful oncoprotein that, via NUT, recruits p300, and anchors it to chromatin by the acetyl-histone-binding dual bromodomains of BRD42, 5, 6,4, 7(Fig. 1). Variant NUTM1-fusion partners, including BRD3, NSD3, ZNF532, and ZNF592, encode BRD4-interacting proteins that serve to link NUT with BRD4, essentially forming the same oncogenic complex as that of BRD4-NUT8,7, 9, 10. On this basis, and for this review, the oncogenic complexes that drive NC will collectively be referred to as ‘BRD4-NUT’.
Fig.1.
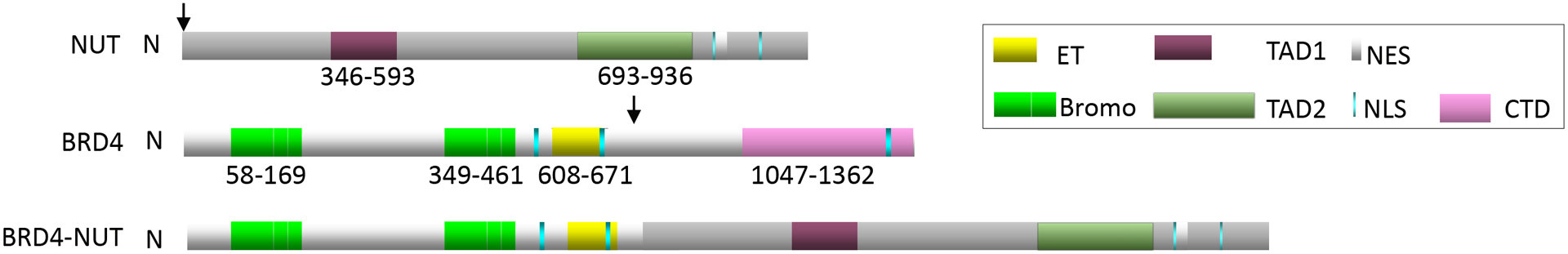
Schematic of BRD4-NUT fusion protein. Arrows denote breakpoints. ET, extra-terminal domain; Bromo, bromodomain; TAD, transcriptional activation domain; NES, nuclear export signal; NLS, nuclear localization signal; CTD, C-terminal domain. TAD1 corresponds with “NUTF1C” fragment that binds p300 described in Reynoird N et al., 2010. Numbers denote amino acid numbers for the indicated domains.
NUT carcinoma is an extremely aggressive cancer. It typically arises in the head and neck (~40%) and thoracic (~50%) regions11 with a reproducible 6.5 month median overall survival (OS) across several large series11, 12. This places NC amongst the most aggressive solid tumors known, surpassing even anaplastic thyroid cancer13(median OS, 9.5 months). Although NC can occur at any age, it affects primarily adolescents and young adults (median age ~2411). Such individuals fall into the so-called ‘AYA gap’, making this a particularly devastating cancer to both patients and loved ones.
The pathogenesis of NC is still in its early years. siRNA-directed depletion of NUT in human NC cell lines has consistently resulted in terminal squamous differentiation and irreversible arrested proliferation7,8, 10,14,15. These findings have led to the accepted concept that BRD4-NUT generally drives NC by blocking differentiation, effectively maintaining the cancer cells in a perpetually undifferentiated, proliferative state. Additionally, these observations indicate that BRD4-NUT, in particular NUT, is a strong therapeutic vulnerability of this cancer. Yet, BRD4-NUT has never been shown, when mis-expressed, to cause malignant transformation, a limitation resulting from the fact that expression of BRD4-NUT is highly toxic to almost all non-NC cells, and from a lack of knowledge of the appropriate host cell, or cell of origin.
We have begun to scratch the surface of how BRD4-NUT may block differentiation, leading to this rapidly progressive, poorly differentiated tumor. First, all evidence indicates that BRD4-NUT is the sole oncoprotein that drives NC growth and dissemination. With the exception of one report that suggests a defect in DNA repair16, whole genome and targeted next-generation sequencing (NGS) have revealed that NCs are genomically stable. Apart from the NUTM1-fusion, NCs lack oncogenic mutations, have low tumor mutation burdens, and have few, if any, copy number alterations17,18,11. Second, like many other high grade malignancies, such as Burkitt’s lymphoma, double-hit lymphoma, and neuroblastoma, NC is a high MYC-expressing tumor15. In fact, the MYC gene is directly upregulated by BRD4-NUT15, 19. Indeed, MYC, which confers pro-growth and anti-differentiative activity in both normal and malignant tissues, is both necessary and partly sufficient for the blockade of differentiation in NC15. Thus NC is properly considered a MYC-driven cancer.
Apart from being a high-grade MYC-driven cancer, the precise mechanism by which fusion of a non-oncogene, BRD4, to another non-oncogene, NUTM1, results in an oncoprotein that subverts the normal function of BRD4 to create one of the most powerful cancer drivers remains elusive. Resolving this question is not only important for understanding disease pathogenesis and to improve treatment, but will also likely uncover insights into normal chromatin and chromosome biology. A major clue may be the observation that NUT recruits p300 to BRD4-bound chromatin6, 8, 19, 20 forming the basis of a model for how BRD4-NUT drives this cancer. This may supercharge the function of BRD4 as a transcription co-activator. In this review, we describe this model and scrutinize data supporting it, and discuss areas in need of further investigation.
Development of a Model
The BRD4-NUT-p300 chromatin complex
In 2010, Dr. N. Reynoird and colleagues6 first demonstrated that NUT interacts with p300 using recombinant proteins as baits in total cell lysates, and by co-immunoprecipitation of ectopically expressed tagged proteins in mammalian cells. An acidic patch of NUT, a domain they termed NUTf1c, interacts with the TAZ2 zinc finger domain of p300 (Fig. 2A). There are two acidic patches on NUT, and for the purposes of this review, we will term this region the transcriptional activation domain 1 (TAD1) of NUT (Fig. 1). Intriguingly, they demonstrated that recombinant TAD1 stimulated the ability of p300 to in vitro acetylate purified H3 histones; and subsequently showed that NUT stimulates acetylation of H3 and H4 histone octamers by p3003. Acetylation of chromatin by NUT-p300 recruitment correlates with in situ activation of a LacO transgene reporter integrated in U2OS 2-6-3 cells20,21.
Fig. 2.
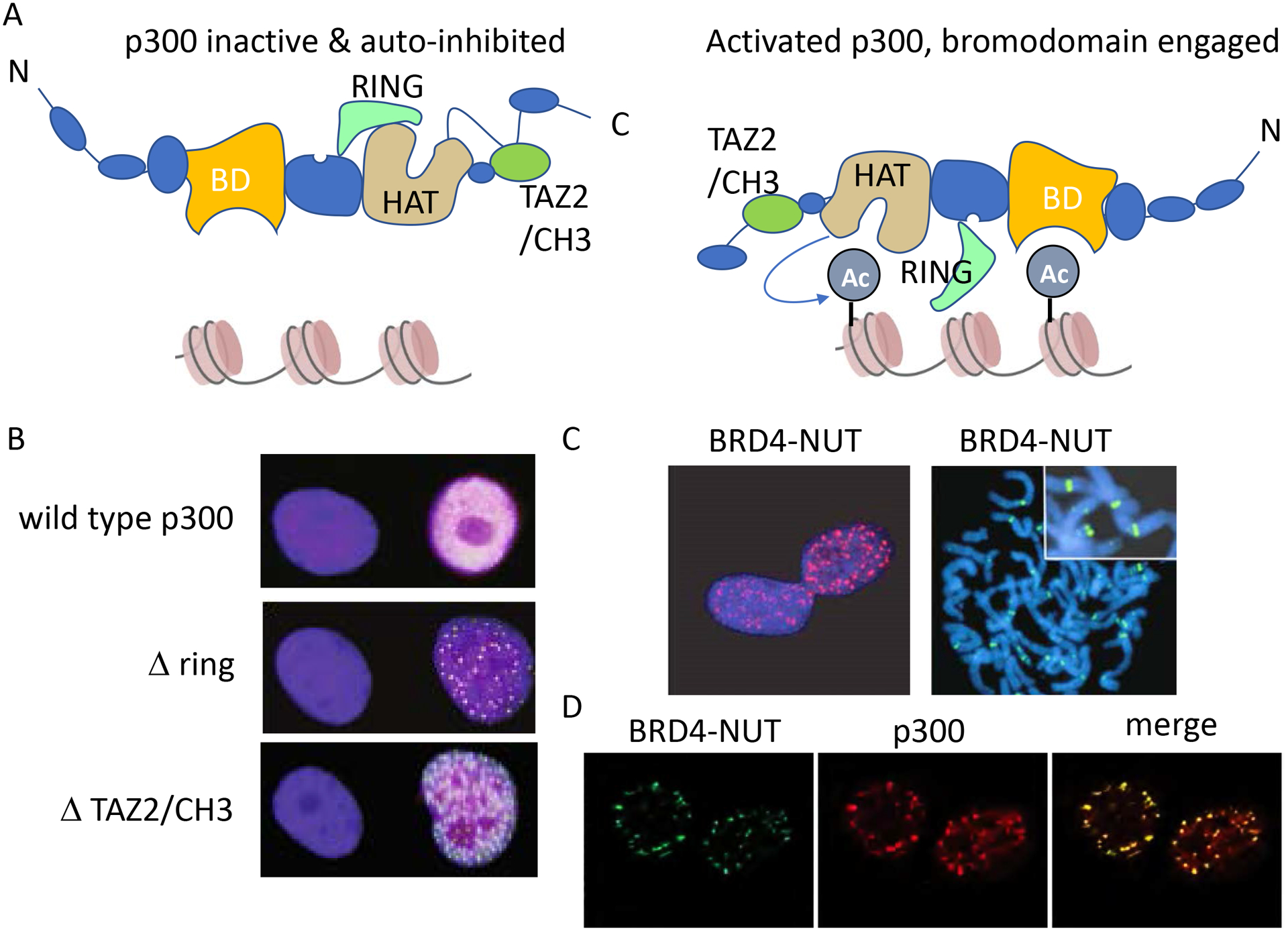
De-repression of p300 causes p300 foci to form, resembling BRD4-NUT. A. Cartoons of inactive and activated conformations of p300. These cartoons derived and simplified by those presented in Ortega E et al., 2018. In this model, p300 activation requires dimerization (not shown), and removal of the auto-inhibitory CH3/TAZ2 and RING domains to allow for HAT activation and engagement of the bromodomain, BD, with acetylated histones. B. Constitutionally activated p300, through deletion of its ring or CH3 domains, results in the formation of nuclear foci. HA-tagged p300 was stained with anti-HA antibody and the images shown are from Ortega E et al., 2018. C. BRD4-NUT immunofluorescence is seen in the NC cell line, TC-797. Left are interphase nuclei, and right are metaphase chromosomes revealing that megadomains cover discrete, chromosomal regions. Immunofluorescence was performed using an antibody to NUT protein (anti-NUT, clone C52B1). Images taken from Alekseyenko A et al., 2015. D. NUT and p300 co-immunofluorescence in the BRD4-NUT+ NC cell line, HCC2429. Taken from Reynoird N et al., 2010.
In 2017, we sought to determine the protein interactors of BRD4-NUT compared with those of the short isoform of BRD4 (BRD4sh); the latter lacks its C-terminal domain (CTD) and is nearly identical to the portion of BRD4 in the BRD4-NUT fusion (Fig. 1). For this, we used a proteomic approach, called BioTAP-XL22, that retains chromatin-bound BRD4-NUT thereby preserving complex integrity and key interactors. Surprisingly, almost all interactions observed with BRD4-NUT were also seen with BRD4sh, indicating that most proteins interact with BRD4-NUT through BRD4 and not NUT. The key difference was that two proteins, p300 and XPO1, appeared to interact only with BRD4-NUT, indicating that they are recruited by NUT8. This observation is substantiated by proteomics data demonstrating interaction of p300 and CBP with endogenous NUT in spermatids3. The findings form the basis of our hypothesis that the recruitment of p300 by NUT is the key difference between BRD4-NUT and BRD4. No evidence thus far suggests a role for XPO1, which likely binds the nuclear export signal of NUT, an obsolete function given that NUT is tethered to chromatin by BRD47. As we will discuss below, the recruitment of p300 by BRD4-NUT may tip the balance between normal levels of transcriptional activation by wild type (wt) BRD4, to higher and sustained levels of transcription of pro-growth genes due to anchoring of p300.
Also notable in our study8 was that the BRD4-NUT complex preserved many known interacting proteins of BRD4, including other BET family members, supporting the idea that BRD4-NUT may maintain some or all of the transcriptional activating/elongation properties of wild-type BRD4. These include Mediator complex members23, Transcription Factor IID (TFIID) complex members23, Nuclear Receptor Binding SET Domain Proteins (NSD1–3)23,10, 24, and Chromodomain Helicase DNA Binding Proteins (CHD4,8)23,24. CREBBP (CBP), a closely related histone acetylatransferase to p300, was also present in the BRD4sh complex, an interaction that could be mediated through acetyl-lysine-bromodomain interaction25,23. The ET domain of BRD4 has been shown to bind to several of the above proteins24. Also present in our BRD4-NUT/BRD4sh pull down were all ‘Z4’ complex members9 (ZNF532, ZNF592, ZNF687, and ZMYND8) known to interact with BRD424,23,26. The function of this complex is largely unknown, and may be repressive to transcription depending on context27, 28. Finally, other BET proteins, including BRD2, BRD3, and BRD4 (long isoform, BRD4l) were also present, in keeping with previous observations23,24, and likely because they bind the same chromatin template. The presence of BRD4l in our pull down may explain the presence of P-TEFb (CDK9/Cyclin T1) in the BRD4-NUT complex. P-TEFb binds the CTD of BRD4l29, 30, which is absent from BRD4-NUT, thus its presence in the BRD4-NUT pull down is likely due to its binding wt BRD4l, and provides further support that the transcriptional co-activator function of wt BRD4 may be maintained.
Many of the above BRD4-NUT complex members, including BRD3, NSD3, ZNF532, and ZNF592 encode novel and rare NUTM1-fusion partners in NC7–10. Knockdown of their wild-type counterparts in BRD4-NUT+ NCs results in either differentiation and/or growth arrest thereby validating their functional importance to NC. Because most of the functional domains of the above variant partners are lost (except BRD3), their oncogenic role may primarily be their interaction with wt BRD4. For example, the NSD3 fusion partner lacks its methyltransferase SET domain, ZNF532 lacks 9 of 11 of its zinc fingers8, and ZNF592 lacks two of its thirteen zinc fingers9. Thus, these variant fusions may be alternative genomic means to recapitulate the BRD4-NUT complex.
Taken together, the above findings indicate that the BRD4-NUT chromatin complex closely mimics that of BRD4, with the added ability to recruit p300. P300 is a known transcriptional co-activator31. One might logically surmise that its recruitment and activation by NUT might increase the ability of BRD4 to activate transcription. Indeed, when p300 HAT is constitutively activated through deletion of its negative regulatory RING domain or the CH3 domain, and while tethered to chromatin through its own bromodomain, it forms nuclear foci (Fig. 2A–B)32. Intriguingly, deletion of the CH3 domain also removes the TAZ2 domain that binds NUT. This suggests that the role of NUT is to alleviate autoinhibition of p300 by the CH3 domain32.
Discovery of the megadomain
It has long been observed that BRD4-NUT and p300 co-localize in hyperacetylated foci in NC cells (Fig. 2C–D), resembling the foci seen in cells expressing constitutively activated p300 (Fig. 2B). We thus hypothesized that these foci might represent transcriptionally active centers, despite initial data indicating otherwise6. Chromatin immunoprecipitation coupled with NGS (ChIP-seq) revealed that BRD4-NUT nuclear foci correspond with massive genomic regions (100kb – 2MB) contiguously co-enriched with BRD4-NUT (Fig. 3), wild-type BRD4, p300, and active histone marks, including H3K27Ac, and exclusive of repressive (H3K27Me3) marks19. We termed these regions ‘megadomains’ (MD). In support of the idea that BRD4-NUT foci represent transcriptional centers, MDs were associated with increased transcription of associated coding and non-coding DNA in BRD4-NUT gain and loss experiments19.
Fig. 3.
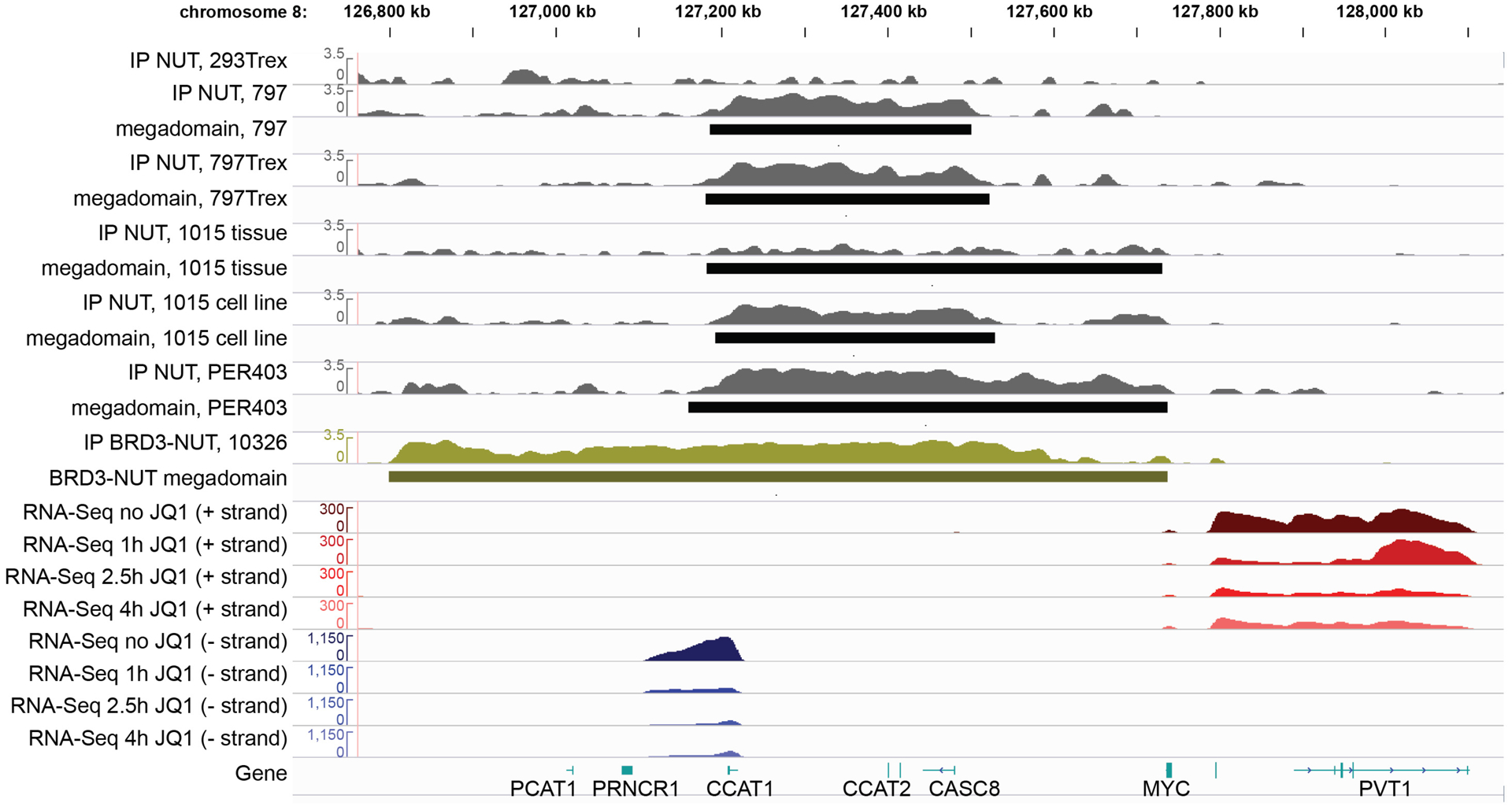
BRD-NUT forms megadomains over the MYC locus in five NMCs, but not in 293TRex cells. Shown is anti-NUT ChIP-seq and RNA-seq over a time course of JQ1. 293TRex and 797TRex cells were induced to express BRD4-NUT. TC-797, 797TRex, PER-403, and 1015 (rust) express endogenous BRD4-NUT. 10326 NMC cells (pea soup green) express BRD3-NUT. Figure taken from Alekseyenko A et al., 2015.
Are megadomains just large super-enhancers?
The above data support our central hypothesis that the BRD4-NUT complex(s) is essentially a super-active BRD4 complex, and raise the possibility that MDs may be aberrant, extremely large super-enhancers. Super-enhancers (aka stretch enhancers or locus control regions), originally discovered at the beta-globin locus control region33, are clusters of large, cell-type-specific enhancers. They are enriched for cell-type specific transcription factors as well as transcriptional co-activators and activators such as p300, the p300/CBP histone post-translational modification (PTM), H3K27Ac, Mediator, and BRD4. Binding of these factors at super-enhancers maintains transcriptional programs that dictate cell phenotypes both in normal and disease/cancerous states34–36. Like regular enhancers, super-enhancers activate RNA polymerase II by linking transcription factors to promoters through these transcriptional co-activators. This likely occurs through long distance looping contacts between the enhancers and promoters. The presence of BRD4l at super-enhancers enables transcriptional elongation by facilitating phosphorylation of RNA pol II by P-TEFb29, 30, 37, 38. All of these features are conserved with MDs. MDs and super-enhancers are also detected using identical bioinformatics algorithms19. Super-enhancer activity can be detected through the production of various non-coding RNAs, including short enhancer RNAs (eRNA)39 and long-non-coding RNAs (lncRNA)40. Indeed, much of the upregulated transcription associated with MDs is from enhancer lncRNAs, such as CCAT1 and PVT119, 40–42 (Fig. 3).
While MDs possess many overlapping features with super-enhancers, there are some differences, the importance of which is unknown, including: 1. on average, MDs are an order of magnitude larger than super-enhancers, 2. MDs have a profile that is much more contiguous and plateau-like than super-enhancers, which are comprised of clusters of peaks, and 3. MDs arise not from super-enhancers, but regular enhancers19. The significance of these differences is unknown, and for the purposes of this review, our working hypothesis is that MDs are very large super-enhancers.
Megadomains upregulate oncogenic target genes
If MDs function as large super-enhancers, then they would be predicted to upregulate cell-type-specific transcriptional programs that confer the malignant properties of NC, as is the case for other cancers whose super-enhancers have been characterized34, 36, 43–45. While the former property has not yet been demonstrated, data do support the latter19. Locations of the vast majority of MDs vary across different NC cell lines and tissues, as do super-enhancers. Variation among NC cells is as strong as variation between NC and non-NC cells induced to express BRD4-NUT (note: in this assay, non-NC cells are lysed prior to the onset of toxic effects of BRD4-NUT expression)19. Across five different cell line and tissues we have identified a very limited set of NC-specific, common MDs: those associated with the MYC, TP63, and SOX2 loci. MYC, TP63, and SOX2 all encode transcription factors implicated in cancer pathogenesis. Expression of all of these genes is directly driven by BRD4-NUT MDs14, 15, 19.
MYC is constitutively upregulated in a large number of cancers, and when amplified or translocated is a driver of a subset of high-grade malignancies, such as Burkitt lymphoma and double-hit lymphoma. MDs upregulate centromeric and telomeric lncRNAs known to upregulate MYC, including CCAT140, 41, 46and PVT142, 47–49. Indeed, knockdown of PVT1 or MYC leads to differentiation of NC cells19, and ectopic MYC overexpression can nearly completely rescue loss of BRD4-NUT in NC15. MYC is well known to promote cancer growth through the upregulation and activation of CDK4/650–54 and cyclin D155, 56. Upregulation of cyclin D1 has been shown by the Elledge lab as a means by which NC can bypass growth arrest induced by BET inhibition57.
TP63 is a squamous cell stem cell factor and tumor suppressor related to TP5358. A truncated isoform, ΔNp63, has been implicated in driving oncogenic transcriptional programs in multiple cancers through complex, poorly understood mechanisms59–63. TP63 is expressed in the majority of NCs64, and knockdown of TP63 potently induces death of NC cells19.
SOX2 is a TF involved in maintaining stemness that when overexpressed by amplification has been implicated a squamous cell carcinoma-driving oncoprotein65–67. As with MYC, knockdown of SOX2 leads to differentiation of NC cells14.
How MYC, SOX2, and p63 cooperate to drive NC is unclear, but it is notable that all of them are involved in maintenance of the stem cell state68, 69. Two, MYC and SOX2, are members of the four Yamanaka factors that are necessary and sufficient of reprograming lineage-committed somatic cells to multi-potent stem cells70. SOX2 and ΔNp63 may work together to drive squamous-specific programs that promote NC growth, as they do in conventional squamous cancer71. ΔNp63 may be important in maintaining MYC levels68 and bypassing cell cycle checkpoint and apoptosis though inhibition of p5358, 72, 73. MYC and SOX2 may collaborate to block differentiation70 and maintain a stem-like state of NC.
Are megadomains p300 sinks?
Although there is substantial evidence that MDs upregulate transcription of associated genes, there is essentially no data on their effect on transcription of non-associated genes. Given that, compared with TFs and co-repressors, p300 is present in limiting quantities in nuclei74, it is possible that p300 recruitment to MDs may lower its abundance at other loci. If so, MDs may have a ‘yin-yang’ function to increase transcription of target regions while simultaneously repressing transcription of non-target regions. Such repression could affect differentiation transcriptional programs in a manner that cooperates with the anti-differentiative effects of MYC and SOX2.
Formation of megadomains
To determine how MDs form, we performed ChIP-seq at multiple early time points following tet-inducible expression of BRD4-NUT in a 293T derivative cell line, 293TRex19. BRD4-NUT first bound to regular enhancers, and then spread contiguously and uni- or bi-directionally over time (7h) to form mature MDs. These findings indicate a process whereby MDs form from processive expansion from a small seed locus, rather than simultaneous occupation of an entire region. This observation supports the feed-forward model proposed by Dr. S. Khochbin, where BRD4-NUT binds a small region of acetylated histones, recruits p300 to locally acetylate nearby histones, resulting in further recruitment of BRD4-NUT and p300. This feed forward loop leads to the massive MDs we observe. Size limitation of MDs may be determined by boundaries that control genome folding and/or by limiting quantities of p30074. Additional potential factors, yet to be determined, that could impact MD size may include histone deacetylase (HDAC) complexes, such as NuRD, recruited by Z427.
Megadomains form a novel nuclear subcompartment
Based on genome-wide chromosome conformation capture analysis (i.e. Hi-C), mammalian genomes are spatially organized into compartments and subcompartments. Compartments represent the preferential association of chromatin of similar epigenetic state and, conversely, the segregation of chromatin of opposite epigenetic state. Initially, an active, euchromatic ‘A’ compartment and inactive, heterochromatic ‘B’ compartment were identified75. These are further sub-divided into subcompartments A1, A2, B1, B2, B3, based on Hi-C data from the GM12878 lymphoblastoid cell line76. Recently, the Eagen lab discovered that endogenous MDs form a novel sixth subcompartment, termed subcompartment M (subM)77. SubM arises from intrachromosomal interactions between MDs separated by extremely long distances, such as opposite chromosome arms, as well as interactions between different chromosomes. These interactions are fundamentally different than shorter-range (<1 Mb) looping events characteristic of TADs. Discovery of subcompartment M revealed that the previously identified subcompartments, though a useful conceptual framework for globally understanding folding of the human genome, do not apply to all cells nor account for the whole genome. This also highlights how studying a rare disease, NC, illuminates aspects of the genome that are understudied, and, until recently, unseen. SubM interactions are BET protein-, presumably BRD4-NUT, dependent, because they disappear upon BET-degrader treatment. Yet, they are transcription-independent as they are stable when RNA pol II transcription is inhibited by triptolide. A limitation is that these studies were performed in one NC cell line, and in our inducible 293TRex cell line, so whether subM is generalizable to all NCs will need to be confirmed.
Does subcompartment M facilitate auto-regulation of core regulatory circuitry
A key observation by the Eagen group was that the common MD-associated TFs, MYC, SOX2, and TP63, cluster within subM77. The finding raises the possibility that spatial clustering of these key NC-factors may upregulate their own and each other’s expression to create an auto-regulatory, feed-forward loop. This may provide a window into the mechanism by which super-enhancers establish a cell’s core regulatory circuitry (CRC). As described above, we believe that MDs function similarly to, and even behave like super large super-enhancers, with their size enabling facile investigations into subcompartment contacts that may also exist for super-enhancers. Super-enhancers are known to drive expression of a small subset (5–10) of cell type specific transcription factors (TFs) that define cell identity. The group of unique and conserved TFs for a given cell type are called core regulatory circuitry (CRC) TFs because they, as master regulators of transcriptional programs that define cell lineage, auto-regulate each other’s transcription in a feed-forward loop of mutual super-enhancer and promoter binding78, 79. We postulate that spatial clustering of the regulatory regions of the genes that encode these TFs (e.g. SOX2, MYC, TP63) and clustering of the encoded TF proteins themselves within subM maintains this feed-forward loop that defines cell identity. This model would explain physically why mutual contacts are coordinated across large distances and between different chromosomes, and how CRC TFs, both in NC and other cell types, auto-regulate in an exquisitely sensitive and coordinated fashion, with broad implications for normal and abnormal cell biology.
The BRD4-NUT mechanistic model
Our conceptualization of how BRD4-NUT drives growth and blocks differentiation is centered on the idea that the pro-transcriptional function of BRD4 is maintained, but overamplified. A single change to BRD4, recruitment and stimulation of p300, leads to a feed-forward process of acetylation and further recruitment that transforms enhancers into ultra-large super-enhancers (MDs). MDs stabilize expression of pro-growth target genes (Fig. 4) through at least two reinforcements, including 1. a broad region of acetylation, increasing the number and strength of enhancer activation, and 2. an auto-regulatory loop enforced by MD-MD interactions within subM. With these reinforcements, expression of MD-target genes is predicted to be less plastic and responsive to intrinsic or extrinsic regulation, such as those mediated by co-repressor complexes, thereby ensuring their continued expression. The lack of plasticity conferred by MDs is predicted to limit the cell’s ability to change or adapt, for example to exit the cell cycle and/or differentiate, keeping it in a perpetually undifferentiated, proliferative state.
Fig. 4.
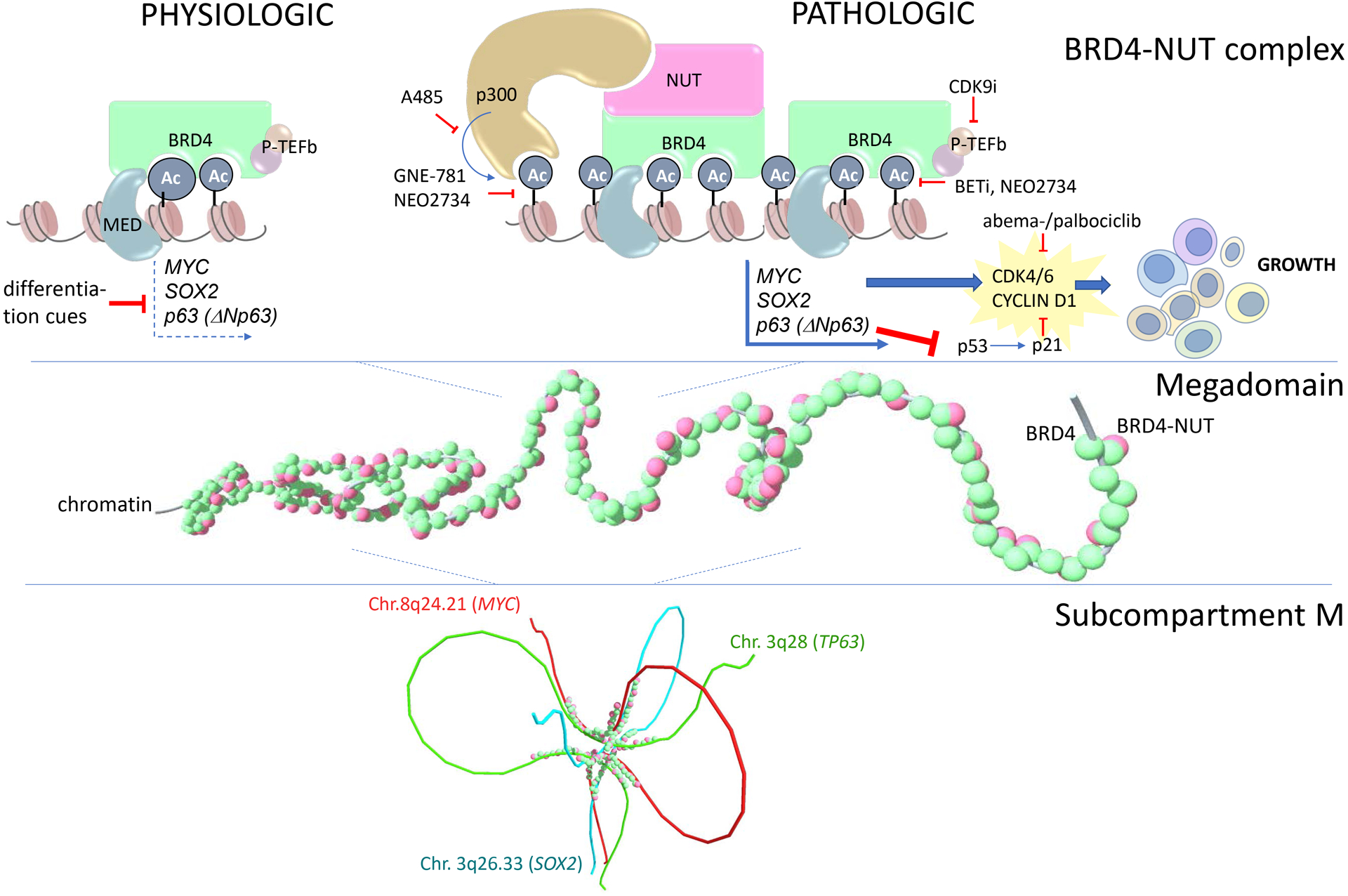
Mechanistic model of how BRD4-NUT drives growth and blocks differentiation in NUT carcinoma. Ac, acetyl-lysine; MED, mediator complex.
What remains unclear in this model is how NC cell proliferation remains unchecked. NGS studies have revealed no inactivating p53 mutations or deletions11, 16–18. We propose that MD-mediated upregulation of p63 (ΔNp63) counteracts p53 by inhibiting its transcriptional targets and opposing apoptosis, as indicated in other cancer models58, 73. There is additional data that suggests that stimulation of p300 by BRD4-NUT may also inhibit p536 by sequestration of its acetylated, activated form6.
The other paradox is that BRD4 has consistently been shown to interact with CBP, likely through its bromodomains80, which probably can replace the function of p300, so how is the recruitment of p300 any different? Likely, the difference is NUT’s ability to stimulate p300 by alleviating autoinhibition from p300’s CH3 domain (Fig. 2A–B). It is unclear if interaction of BRD4 with CBP alters CBP’s activity. It could also be that the affinity of interaction between p300 and NUT is much higher than that between BRD4 and CBP. In fact it is not clear whether the BRD4-CBP interaction is direct or dependent upon the presence of other factors.
Targeted inhibitors of the BRD4-NUT pathway
BET inhibition
Acetyl-lysine mimetics that competitively inhibit binding of BET protein bromodomains to acetylated histones made their debut in cancer in the treatment of NUT carcinoma18, 81. These compounds evict BRD4-NUT from chromatin by directly binding the dual bromodomains of BRD4, resulting in decreased expression of MD target genes, including MYC15, 19, TP6319, and SOX214, followed by rapid differentiation, growth arrest, and differentiation of NC cells81. The development of this drug class, pioneered by Dr. J. Bradner, has resulted in an explosion of studies in other cancer models34, 43, 82–89, and in the very least provided an invaluable tool compound to explore the oncogenic role of mutated and non-mutated BET proteins in cancer35, 44, 80–87. A number of clinical trials have also been completed using BET inhibitors for treatment of liquid and solid cancers. Amongst solid tumors, the only responses seen have been in NC90, 91. Given that NC is the only known cancer to harbor a BRD4 oncogene (BRD4 is wild type in other ‘BRD4-dependent’ cancers), the findings are not surprising, but point to the on-target activity of this class of small molecule inhibitors.
The results of the clinical trials so far have shown that the efficacy of BET inhibitors in treating NC is limited by a narrow therapeutic window, the most common toxicity being thrombocytopenia. No cures were seen, and most responses were not durable, the median duration of response ranging from 2–3 months. We believe the limited efficacy of BET inhibitors in NC is due to two major factors: 1. BET inhibitors do not fully evict BRD4-NUT from chromatin, and 2. the target, BET proteins, is ubiquitously expressed and essential for many normal cellular activities4. Even with sustained exposure to the tool BET inhibitor, JQ181, we observed only partial (~60%) eviction of BRD4-NUT from MDs globally in NC cells19. Essentially complete removal was observed when using a version of JQ1 that degrades, rather than simply evicts BET proteins77. Such small-molecule proteolysis targeting chimeras (PROTACs) therefore have enormous therapeutic potential, but at the same time may be limited by their large size and thus limited bioavailability. Moreover, an important aspect of all BET inhibitors used in clinical trials, is that they inhibit not only BRD4, but all BET proteins, including BRD2, BRD3, and BRDT92–94.
As more selective BET inhibitors emerge, for example the second bromodomain (BD2)-selective ABBV-74495, 96, toxicity and tumor selectivity may improve and provide a path for therapeutic use of this compound class.
p300 inhibition
If p300 plays a critical role in BRD4-NUT function, it is logical that its inhibition will affect NC cell growth and viability, and in fact it does. Both selective p300 HAT46 or bromodomain inhibition97 using A-48598 or GNE-78199, respectively, inhibits NC growth, but are not very effective as single agents. Remarkably, these inhibitors have overlapping transcriptional effects with those of BET inhibition. When combined with a BET inhibitor, both p300 HAT and p300 bromodomain inhibitors are synergistic46, though with our cell lines the HAT inhibitor was less effective.
Recently, a new compound, NEO2734, which inhibits both BET and p300 bromodomains at low nM IC50s in cell-free assays was described97, 100. Because this compound is predicted to synergistically inhibit both BRD4-NUT and p300, we tested its activity in NC. In vitro, growth inhibition and induction of differentiation of NC cells approximately matched that of selective BET inhibitors, however in vivo, it was significantly superior in prolonging survival of mouse xenograft models, demonstrating tumor regression in two of three models97. Genes affected by NEO2734 resembled those affected by a BET inhibitor, though NEO2734 induced markedly greater global changes in expression levels, suggesting that the addition of p300 bromodomain inhibition cooperatively inhibits the same transcriptional programs as BET inhibition alone97. The results are encouraging and provide a strong rationale for clinical development of NEO2734.
HDAC inhibition
HDAC inhibitors, like BET inhibitors, induce differentiation and growth arrest of NC cells, and they have even shown activity in the clinic101, 102. However, the mechanism by which HDAC inhibitors induce differentiation is unclear. Based on our model (Fig. 4), inhibition of HDAC activity in NC cells would be predicted to spread acetylation across the genome, including in regions not occupied by MDs. BRD4-NUT, which binds to acetylated histones in a largely DNA sequence-independent manner, might be expected to spread to areas of newly acetylated chromatin, and p300 would follow. Alternatively, given that there are limiting quantities of p300 and that MDs might act as p300 sinks, one might expect its relative depletion in MDs, and repletion in non-MDs, to effectively short-circuit the function of the MD. This could result in loss of expression of pro-growth MD target genes, and gain of expression of pro-differentiation genes. Indeed, this transcriptional effect is observed in NC cells, however epigenetic analysis of changes in chromatin have yet to be performed101.
CDK9 inhibition
P-TEFb is a heterodimeric complex comprised of cyclin T1 and CDK9, and is required for BRD4-mediated transcriptional elongation, as explained above. Evidence that CDK9 inhibition in a large panel (N = 80) of cancer cell lines selectively kills NC cells is further evidence that wtBRD4 function is required for BRD4-NUT function, as BRD4-NUT lacks the CTD of BRD4 that binds P-TEFb103. In the work by Bragelmann and colleagues103, it was shown that NC cell death occurred as a result of MCL1 downregulation, and provided intriguing evidence that CDK9 inhibition should be considered for treatment of NC. This idea is tempered with the fact that very large concentrations (10μM) of the tool CDK9 inhibitor, LDC67, were used in the study, therefore follow-on studies are required with more potent selective molecules.
CDK4/6 inhibition
The first evidence that NC may be sensitive to inhibition of CDKs was provided by Beesley et al.104, who showed highly potent inhibitory activity of NC cells, in vitro and in vivo, by the pan-CDK inhibitor, flavopiridol. In that work, the authors attributed the effect to CDK9 inhibition, however, flavopiridol is not selective, and inhibits many other CDKs with low nM IC50s (20–100nM), including CDK1, CDK2, CDK4, CDK6 and CDK9. In an effort to identify why NC is ultimately resistant to BET inhibitors clinically, the Elledge lab performed a CRISPR ORF screen to identify what genetic factors can confer resistance of NC cells to JQ157. They identified that cyclin D1 upregulation or Rb loss could bypass cell cycle arrest induced by JQ1. In keeping with this result, they demonstrated that CDK4/6 inhibitors were synergistic in vitro and in vivo with BET inhibitors in inhibiting NC growth. These findings provide a powerful rationale for combining CDK4/6 inhibitors in the next phase of clinical trials that use a combination approach to BET inhibitor therapy of NC.
Unresolved questions
Despite progress in characterizing epigenetic aberrations induced by BRD4-NUT, two fundamental questions remain. First, are MDs really necessary or even sufficient for the maintenance of target gene expression, or are they an epiphenomenon with no intrinsic function? The fact that MDs are characteristic of all NC cell lines (N = 5) and primary tissues (N=1) tested indicates that they are a conserved feature unique to NC cells. Moreover, NUT immunohistochemistry performed clinically to diagnose NC demonstrates nuclear foci (Fig. 5) in the majority of cases105–107, supporting the idea that MDs are not an artifact of cell culture and are conserved in vivo as well. Of course, the conserved nature of MDs does not prove their necessity. Gain or loss of MDs would need to be performed experimentally to establish their function. However, it is technically challenging to remove or create MDs without perturbing the biology of a cell in undesired ways. The same question and experimental challenges pertain to subM. Is subM needed to reinforce and stabilize expression of MD target genes?
Fig. 5.
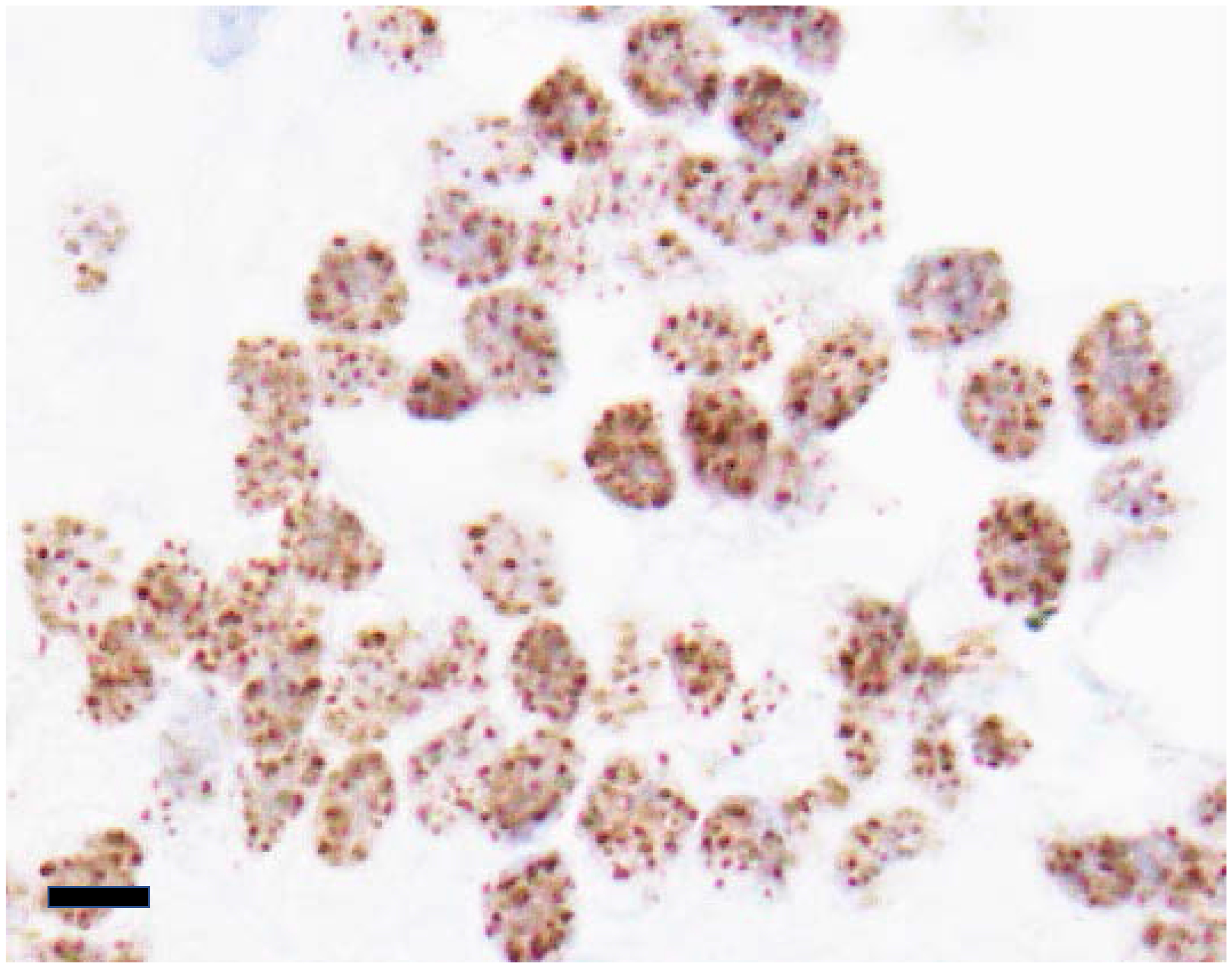
The nuclear foci of BRD4-NUT seen in this biopsy of a NUT carcinoma correspond with megadomains and subM. Immunohistochemistry was performed using an antibody to NUT (clone C52B1, Cell Signaling Technologies, Danvers, MA). Scale bar is 15μm.
Perhaps more addressable and compelling questions with broad relevance are 1. is subM a mechanism by which CRC TFs can auto-regulate in NC?, and 2. do super-enhancers in other, non-NC cell types also exist in subM or, analogously, a ‘super-enhancer subcompartment’?; if so, 3. is subM a general mechanism by which CRC TFs auto-regulate in many if not all cell types? Super-enhancers have previously been reported by Hi-C to interact with each other, but only after removal of cohesin108. The lack of detection of super-enhancer-super-enhancer interactions is likely because Hi-C is typically not sensitive enough to detect such interactions. This points to the value of MDs, which allow for the detection of these ultra-long-range interactions, likely due to enhanced sensitivity afforded by the breadth of MDs. It is possible that structural mechanisms of CRC auto-regulation can be explored in the NC model, with wide-reaching implications in normal and cancer cell biology.
Future directions
In summary, much has been learned of NC pathogenesis, but these insights have yet to substantially impact therapy of this disease. Much more is needed to make this a treatable disease, but we can build upon our current knowledge, and recent technological advances to make progress. For example, a new generation of bromodomain-selective BET inhibitors is emerging with more selective cell type inhibition. As referenced above, ABBV-744 selectively inhibits BD2 of BET proteins with ~300-fold greater affinity than the first bromodomain (BD1). Initial reports demonstrate that a much smaller subset of cancer subtypes, particularly prostate cancer and acute leukemia, respond to this inhibitor compared with the pan-inhibitor, ABBV-07595, 96. Moreover, ABBV-744 was tolerated much more than ABBV-075, with markedly less thrombocytopenia, no observed gastro-intestinal toxicity, and no observed histopathologic effects on testes using 8–16-fold higher relative doses of ABBV-744 than −07596.
Unfortunately, many oncologists believe that BET inhibitor monotherapy may never cure or even have a major impact on NC outcomes. Combinations, as has historically been needed for most cancer therapies with either chemotherapy or targeted approaches109, are likely needed to achieve meaningful responses in NC. NEO2734, which possesses combined targeting of p300 and BET BDs in a single compound, holds promise. However, there is concern that combining epigenetic inhibitors may be too toxic, as these in general are not cell type selective. Rational combinations, such as that with CDK4/6 and BET inhibition, may have a larger therapeutic window. Genetic screens designed to identify genes whose loss sensitizes NC cells to BETi, for example a CRISPR sensitizer screen110, is a powerful approach that can be used to identify further rational combinations with BET inhibitors.
Rather than focusing only on BET inhibitors, we may need to consider other therapeutic strategies. For example, we know that the most specific approach to targeting NC is through direct inhibition of NUT, whose expression, in contrast to BRD4, is restricted to testes4. While this is unachievable currently with small molecule inhibitors due to the untargetable nature of this unstructured protein, efficient genetic downregulation of BRD4-NUT is accomplished through siRNA knockdown of NUT7, 10. Until recently, this was not a viable approach in vivo, but the development of lipid nanoparticles (LNP) has led to the FDA approval of an siRNA-based approach to treating hereditary ATTR amyloidosis111. This technological breakthrough may provide the groundwork for this approach in NC. Another, though still unproven, approach is to use RNA-targeted CRISPR systems such as CasRx112.
Finally, some NC patients respond to immune therapy (unpublished observations), however very little is known about the immune micro-environment of NC. Thus, biomarkers predictive of response and a mechanistic understanding of these therapies are lacking. A major limitation to characterizing the immune micro-environment of NC has been the lack of an animal model that recapitulates that of the natural disease. The development of a genetically engineered mouse model (GEMM) could make this possible. While we wait for a NC GEMM, single cell RNA sequencing is a powerful approach that can be leveraged to characterize the immune infiltrate of human NC when coupled with multiplex immunofluorescence to provide the spatial distribution of the immune subsets identified by RNAseq. This approach has led to recent paradigm-changing discoveries of novel predictive biomarkers of response to immune checkpoint blockade in several other cancer types113, 114.
Acknowledgments
Competing Interests statement: C.A.F. receives consultation and research funding from Boehringer-Ingelheim. K.P.E. declares no competing interests.
References
- 1.French CA, Kutok JL, Faquin WC, Toretsky JA, Antonescu CR, Griffin CA, et al. Midline carcinoma of children and young adults with nut rearrangement. J Clin Oncol. 2004;22(20):4135–9. [DOI] [PubMed] [Google Scholar]
- 2.Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, et al. A bromodomain protein, mcap, associates with mitotic chromosomes and affects g(2)-to-m transition. Mol Cell Biol. 2000;20(17):6537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiota H, Barral S, Buchou T, Tan M, Coute Y, Charbonnier G, et al. Nut directs p300-dependent, genome-wide h4 hyperacetylation in male germ cells. Cell Rep. 2018;24(13):3477–87 e6. [DOI] [PubMed] [Google Scholar]
- 4.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. Brd4-nut fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003;63(2):304–7. [PubMed] [Google Scholar]
- 5.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A. 2003;100(15):8758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reynoird N, Schwartz BE, Delvecchio M, Sadoul K, Meyers D, Mukherjee C, et al. Oncogenesis by sequestration of cbp/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010;29(17):2943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, et al. Brd-nut oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27(15):2237–42. [DOI] [PubMed] [Google Scholar]
- 8.Alekseyenko AA, Walsh EM, Zee BM, Pakozdi T, Hsi P, Lemieux ME, et al. Ectopic protein interactions within brd4-chromatin complexes drive oncogenic megadomain formation in nut midline carcinoma. Proc Natl Acad Sci U S A. 2017;114(21):E4184–E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiota H, Elya JE, Alekseyenko A, Chou PM, Gorman SA, Barbash O, et al. ‘Z4’ complex member fusions in nut carcinoma: Implications for a novel oncogenic mechanism. Mol Cancer Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.French CA, Rahman S, Walsh EM, Kuhnle S, Grayson AR, Lemieux ME, et al. Nsd3-nut fusion oncoprotein in nut midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014;4(8):928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chau NG, Ma C, Danga K, Al-Sayegh H, Nardi V, Barrette R, et al. An anatomical site and genetic based prognostic model for patients with nut midline carcinoma: Analysis of 124 patients. JNCI Cancer Spectrum. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer DE, Mitchell CM, Strait KM, Lathan CS, Stelow EB, Luer SC, et al. Clinicopathologic features and long-term outcomes of nut midline carcinoma. Clin Cancer Res. 2012;18(20):5773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maniakas A, Dadu R, Busaidy NL, Wang JR, Ferrarotto R, Lu C, et al. Evaluation of overall survival in patients with anaplastic thyroid carcinoma, 2000–2019. JAMA Oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang R, Liu W, Helfer CM, Bradner JE, Hornick JL, Janicki SM, et al. Activation of sox2 expression by brd4-nut oncogenic fusion drives neoplastic transformation in nut midline carcinoma. Cancer Res. 2014;74(12):3332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grayson AR, Walsh EM, Cameron MJ, Godec J, Ashworth T, Ambrose JM, et al. Myc, a downstream target of brd-nut, is necessary and sufficient for the blockade of differentiation in nut midline carcinoma. Oncogene. 2014;33(13):1736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stirnweiss A, Oommen J, Kotecha RS, Kees UR, Beesley AH. Molecular-genetic profiling and high-throughput in vitro drug screening in nut midline carcinoma-an aggressive and fatal disease. Oncotarget. 2017;8(68):112313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JK, Louzada S, An Y, Kim SY, Kim S, Youk J, et al. Complex chromosomal rearrangements by single catastrophic pathogenesis in nut midline carcinoma. Ann Oncol. 2017;28(4):890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord JP, de La Motte Rouge T, et al. Clinical response of carcinomas harboring the brd4-nut oncoprotein to the targeted bromodomain inhibitor otx015/mk-8628. Cancer Discov. 2016;6(5):492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alekseyenko AA, Walsh EM, Wang X, Grayson AR, Hsi PT, Kharchenko PV, et al. The oncogenic brd4-nut chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015;29(14):1507–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, You J. Mechanistic analysis of the role of bromodomain-containing protein 4 (brd4) in brd4-nut oncoprotein-induced transcriptional activation. J Biol Chem. 2015;290(5):2744–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, et al. From silencing to gene expression: Real-time analysis in single cells. Cell. 2004;116(5):683–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alekseyenko AA, McElroy KA, Kang H, Zee BM, Kharchenko PV, Kuroda MI. Biotap-xl: Cross-linking/tandem affinity purification to study DNA targets, rna, and protein components of chromatin-associated complexes. Curr Protoc Mol Biol. 2015;109:21 30 1–21 30 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, et al. Bet inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The brd4 extraterminal domain confers transcription activation independent of ptefb by recruiting multiple proteins, including nsd3. Mol Cell Biol. 2011;31(13):2641–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. Bet bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol Cell. 2015;58(6):1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145(5):787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spruijt CG, Luijsterburg MS, Menafra R, Lindeboom RG, Jansen PW, Edupuganti RR, et al. Zmynd8 co-localizes with nurd on target genes and regulates poly(adp-ribose)-dependent recruitment of gatad2a/nurd to sites of DNA damage. Cell Rep. 2016;17(3):783–98. [DOI] [PubMed] [Google Scholar]
- 28.Li N, Li Y, Lv J, Zheng X, Wen H, Shen H, et al. Zmynd8 reads the dual histone mark h3k4me1-h3k14ac to antagonize the expression of metastasis-linked genes. Mol Cell. 2016;63(3):470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, et al. Recruitment of p-tefb for stimulation of transcriptional elongation by the bromodomain protein brd4. Mol Cell. 2005;19(4):535–45. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z, He N, Zhou Q. Brd4 recruits p-tefb to chromosomes at late mitosis to promote g1 gene expression and cell cycle progression. Mol Cell Biol. 2008;28(3):967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, et al. Molecular cloning and functional analysis of the adenovirus e1a-associated 300-kd protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8(8):869–84. [DOI] [PubMed] [Google Scholar]
- 32.Ortega E, Rengachari S, Ibrahim Z, Hoghoughi N, Gaucher J, Holehouse AS, et al. Transcription factor dimerization activates the p300 acetyltransferase. Nature. 2018;562(7728):538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talbot D, Collis P, Antoniou M, Vidal M, Grosveld F, Greaves DR. A dominant control region from the human beta-globin locus conferring integration site-independent gene expression. Nature. 1989;338(6213):352–5. [DOI] [PubMed] [Google Scholar]
- 34.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein brd4 is a positive regulatory component of p-tefb and stimulates rna polymerase ii-dependent transcription. Mol Cell. 2005;19(4):523–34. [DOI] [PubMed] [Google Scholar]
- 38.Di Micco R, Fontanals-Cirera B, Low V, Ntziachristos P, Yuen SK, Lovell CD, et al. Control of embryonic stem cell identity by brd4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014;9(1):234–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by erna. Nature. 2011;474(7351):390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCleland ML, Mesh K, Lorenzana E, Chopra VS, Segal E, Watanabe C, et al. Ccat1 is an enhancer-templated rna that predicts bet sensitivity in colorectal cancer. J Clin Invest. 2016;126(2):639–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiang JF, Yin QF, Chen T, Zhang Y, Zhang XO, Wu Z, et al. Human colorectal cancer-specific ccat1-l lncrna regulates long-range chromatin interactions at the myc locus. Cell Res. 2014;24(5):513–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. Pvt1 dependence in cancer with myc copy-number increase. Nature. 2014;512(7512):82–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large b cell lymphoma. Cancer Cell. 2013;24(6):777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, et al. Notch1-rbpj complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A. 2014;111(2):705–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A notch1-driven myc enhancer promotes t cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20(10):1130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Zegar T, Lucas A, Morrison-Smith C, Knox T, French CA, et al. Therapeutic targeting of p300/cbp hat domain for the treatment of nut midline carcinoma. Oncogene. 2020;39(24):4770–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo L, Li J, Zeng H, Guzman AG, Li T, Lee M, et al. A combination strategy targeting enhancer plasticity exerts synergistic lethality against beti-resistant leukemia cells. Nat Commun. 2020;11(1):740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vito D, Eriksen JC, Skjodt C, Weilguny D, Rasmussen SK, Smales CM. Defining lncrnas correlated with cho cell growth and igg productivity by rna-seq. iScience. 2020;23(1):100785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riquelme E, Suraokar MB, Rodriguez J, Mino B, Lin HY, Rice DC, et al. Frequent coamplification and cooperation between c-myc and pvt1 oncogenes promote malignant pleural mesothelioma. J Thorac Oncol. 2014;9(7):998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, et al. Direct induction of cyclin d2 by myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18(19):5321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warner BJ, Blain SW, Seoane J, Massague J. Myc downregulation by transforming growth factor beta required for activation of the p15(ink4b) g(1) arrest pathway. Mol Cell Biol. 1999;19(9):5913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mateyak MK, Obaya AJ, Sedivy JM. C-myc regulates cyclin d-cdk4 and -cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999;19(7):4672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, et al. Identification of cdk4 as a target of c-myc. Proc Natl Acad Sci U S A. 2000;97(5):2229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miliani de Marval PL, Macias E, Rounbehler R, Sicinski P, Kiyokawa H, Johnson DG, et al. Lack of cyclin-dependent kinase 4 inhibits c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol. 2004;24(17):7538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ. Myc induces cyclin d1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene. 1994;9(12):3635–45. [PubMed] [Google Scholar]
- 56.Steiner P, Philipp A, Lukas J, Godden-Kent D, Pagano M, Mittnacht S, et al. Identification of a myc-dependent step during the formation of active g1 cyclin-cdk complexes. EMBO J. 1995;14(19):4814–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao S, Maertens O, Cichowski K, Elledge SJ. Genetic modifiers of the brd4-nut dependency of nut midline carcinoma uncovers a synergism between betis and cdk4/6is. Genes Dev. 2018;32(17–18):1188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. P63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2(3):305–16. [DOI] [PubMed] [Google Scholar]
- 59.Giacobbe A, Compagnone M, Bongiorno-Borbone L, Antonov A, Markert EK, Zhou JH, et al. P63 controls cell migration and invasion by transcriptional regulation of mtss1. Oncogene. 2016;35(12):1602–8. [DOI] [PubMed] [Google Scholar]
- 60.Latina A, Viticchie G, Lena AM, Piro MC, Annicchiarico-Petruzzelli M, Melino G, et al. Deltanp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene. 2016;35(12):1493–503. [DOI] [PubMed] [Google Scholar]
- 61.Hamdan FH, Johnsen SA. Deltanp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci U S A. 2018;115(52):E12343–E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abbas HA, Bui NHB, Rajapakshe K, Wong J, Gunaratne P, Tsai KY, et al. Distinct tp63 isoform-driven transcriptional signatures predict tumor progression and clinical outcomes. Cancer Res. 2018;78(2):451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westcott JM, Camacho S, Nasir A, Huysman ME, Rahhal R, Dang TT, et al. Deltanp63-regulated epithelial-to-mesenchymal transition state heterogeneity confers a leader-follower relationship that drives collective invasion. Cancer Res. 2020;80(18):3933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tilson MP, Bishop JA. Utility of p40 in the differential diagnosis of small round blue cell tumors of the sinonasal tract. Head Neck Pathol. 2014;8(2):141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. Sox2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D, et al. Sox2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One. 2010;5(1):e8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukhopadhyay A, Berrett KC, Kc U, Clair PM, Pop SM, Carr SR, et al. Sox2 cooperates with lkb1 loss in a mouse model of squamous cell lung cancer. Cell Rep. 2014;8(1):40–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y, Li Y, Peng Y, Zheng X, Fan S, Yi Y, et al. Deltanp63alpha down-regulates c-myc modulator mm1 via e3 ligase herc3 in the regulation of cell senescence. Cell Death Differ. 2018;25(12):2118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alexandrova EM, Petrenko O, Nemajerova A, Romano RA, Sinha S, Moll UM. Deltanp63 regulates select routes of reprogramming via multiple mechanisms. Cell Death Differ. 2013;20(12):1698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, et al. Sox2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124(4):1636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chiang CT, Chu WK, Chow SE, Chen JK. Overexpression of delta np63 in a human nasopharyngeal carcinoma cell line downregulates ckis and enhances cell proliferation. J Cell Physiol. 2009;219(1):117–22. [DOI] [PubMed] [Google Scholar]
- 73.Lanza M, Marinari B, Papoutsaki M, Giustizieri ML, D’Alessandra Y, Chimenti S, et al. Cross-talks in the p53 family: Deltanp63 is an anti-apoptotic target for deltanp73alpha and p53 gain-of-function mutants. Cell Cycle. 2006;5(17):1996–2004. [DOI] [PubMed] [Google Scholar]
- 74.Gillespie MA, Palii CG, Sanchez-Taltavull D, Shannon P, Longabaugh WJR, Downes DJ, et al. Absolute quantification of transcription factors reveals principles of gene regulation in erythropoiesis. Mol Cell. 2020;78(5):960–74 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3d map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosencrance CD, Ammouri HN, Yu Q, Ge T, Rendleman EJ, Marshall SA, et al. Chromatin hyperacetylation impacts chromosome folding by forming a nuclear subcompartment. Mol Cell. 2020;78(1):112–26 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res. 2016;26(3):385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu T, Kamikawa YF, Donohoe ME. Brd4’s bromodomains mediate histone h3 acetylation and chromatin remodeling in pluripotent cells through p300 and brg1. Cell Rep. 2018;25(7):1756–71. [DOI] [PubMed] [Google Scholar]
- 81.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of bet bromodomains. Nature. 2010;468(7327):1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell. 2011;146(6):904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting myc dependence in cancer by inhibiting bet bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of bet recruitment to chromatin as an effective treatment for mll-fusion leukaemia. Nature. 2011;478(7370):529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, et al. Bet inhibition silences expression of mycn and bcl2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8(8):e72967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, et al. Bet bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget. 2013;4(11):2080–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting mycn in neuroblastoma by bet bromodomain inhibition. Cancer Discov. 2013;3(3):308–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qiu H, Jackson AL, Kilgore JE, Zhong Y, Chan LL, Gehrig PA, et al. Jq1 suppresses tumor growth through downregulating ldha in ovarian cancer. Oncotarget. 2015;6(9):6915–30. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of bet bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510(7504):278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol. 2018:JCO2018782292. [DOI] [PubMed] [Google Scholar]
- 91.Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 study of molibresib (gsk525762), a bromodomain and extra-terminal domain protein inhibitor, in nut carcinoma and other solid tumors. JNCI Cancer Spectrum. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. Bet inhibitor otx015 targets brd2 and brd4 and decreases c-myc in acute leukemia cells. Oncotarget. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, et al. Small-molecule inhibition of brdt for male contraception. Cell. 2012;150(4):673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sheppard GS, Wang L, Fidanze SD, Hasvold LA, Liu D, Pratt JK, et al. Discovery of n-ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]- 6-methyl-7-oxo-1h-pyrrolo[2,3-c]pyridine-2-carboxamide (abbv-744), a bet bromodomain inhibitor with selectivity for the second bromodomain. J Med Chem. 2020;63(10):5585–623. [DOI] [PubMed] [Google Scholar]
- 96.Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the bd2 bromodomain of bet proteins in prostate cancer. Nature. 2020;578(7794):306–10. [DOI] [PubMed] [Google Scholar]
- 97.Morrison-Smith CD, Knox TM, Filic I, Soroko KM, Eschle BK, Wilkens MK, et al. Combined targeting of the brd4-nut-p300 axis in nut midline carcinoma by dual selective bromodomain inhibitor, neo2734. Mol Cancer Ther. 2020;19(7):1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/cbp inhibitor that targets lineage-specific tumours. Nature. 2017;550(7674):128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L, et al. Gne-781, a highly advanced potent and selective bromodomain inhibitor of cyclic adenosine monophosphate response element binding protein, binding protein (cbp). J Med Chem. 2017;60(22):9162–83. [DOI] [PubMed] [Google Scholar]
- 100.Yan Y, Ma J, Wang D, Lin D, Pang X, Wang S, et al. The novel bet-cbp/p300 dual inhibitor neo2734 is active in spop mutant and wild-type prostate cancer. EMBO Mol Med. 2019;11(11):e10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schwartz BE, Hofer MD, Lemieux ME, Bauer DE, Cameron MJ, West NH, et al. Differentiation of nut midline carcinoma by epigenomic reprogramming. Cancer Res. 2011;71(7):2686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maher OM, Christensen AM, Yedururi S, Bell D, Tarek N. Histone deacetylase inhibitor for nut midline carcinoma. Pediatr Blood Cancer. 2015;62(4):715–7. [DOI] [PubMed] [Google Scholar]
- 103.Bragelmann J, Dammert MA, Dietlein F, Heuckmann JM, Choidas A, Bohm S, et al. Systematic kinase inhibitor profiling identifies cdk9 as a synthetic lethal target in nut midline carcinoma. Cell Rep. 2017;20(12):2833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beesley AH, Stirnweiss A, Ferrari E, Endersby R, Howlett M, Failes TW, et al. Comparative drug screening in nut midline carcinoma. Br J Cancer. 2014;110(5):1189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Haack H, Johnson LA, Fry CJ, Crosby K, Polakiewicz RD, Stelow EB, et al. Diagnosis of nut midline carcinoma using a nut-specific monoclonal antibody. Am J Surg Pathol. 2009;33(7):984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.French CA, Bakker MAD. Who classification of head and neck tumours. 4th ed. El-Naggar A, Chan JKC, Grandis JR, Takata T, Slootweg P, editors. Lyon: International Agency for Research on Cancer (IARC); 2017. [Google Scholar]
- 107.French CA. Nut carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol Int. 2018;68(11):583–95. [DOI] [PubMed] [Google Scholar]
- 108.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171(2):305–20 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. 2017;171(7):1678–91 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hinze L, Pfirrmann M, Karim S, Degar J, McGuckin C, Vinjamur D, et al. Synthetic lethality of wnt pathway activation and asparaginase in drug-resistant acute leukemias. Cancer Cell. 2019;35(4):664–76 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rizk M, Tuzmen S. Patisiran for the treatment of patients with familial amyloid polyneuropathy. Drugs Today (Barc). 2019;55(5):315–27. [DOI] [PubMed] [Google Scholar]
- 112.Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. Transcriptome engineering with rna-targeting type vi-d crispr effectors. Cell. 2018;173(3):665–76 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5. [DOI] [PubMed] [Google Scholar]