

Abstract
Hypotension and changes in fluid-electrolyte balance pose immediate threats to survival. Juxtaglomerular cells respond to such threats by increasing the synthesis and secretion of renin. In addition, smooth muscle cells along the renal arterioles transform into renin cells until homeostasis has been regained. However, chronic unrelenting stimulation of renin cells leads to severe kidney damage. Here, we discuss the origin, distribution, function, and plasticity of renin cells within the kidney and immune compartments and the consequences of distorting the renin programme. Understanding how chronic stimulation of these cells in the context of hypertension may lead to vascular pathology will serve as a foundation for targeted molecular therapies.
Keywords: Cell Fate, Cell Memory, Chromatin, RAS Inhibition, Transdifferentiation
Graphical Abstract
The Renin Angiotensin System and the Diversity of Renin Cell Functions
Regulation of blood pressure and fluid electrolyte homeostasis is crucial for survival [1,2]. The Renin-Angiotensin-System (RAS) (Figure 1) was refined throughout evolution to maintain normal blood pressure and the constancy of the extracellular fluid volume and composition [2]. Proper functioning of the RAS results in normal tissue perfusion and delivery of oxygen and nutrients to tissues. The RAS enzymatic cascade is described in Box 1. The synthesis and release of renin, a hormone enzyme catalyzing the rate-limiting step of the RAS, by juxtaglomerular (JG) cells is tightly regulated by physiological stimuli. JG cells are sensors that respond to signals conveying the status of the extracellular fluid volume and blood pressure. Thus, a reduction in extracellular volume or blood pressure which occurs during dehydration, blood loss, heart failure, or treatment with RAS inhibitors, result in increased renin synthesis and release which ultimately leads to increased Angiotensin II generation and reestablishment of homeostasis [Box1]. When the threat to homeostasis becomes protracted or severe, smooth muscle cells along the kidney arterioles adopt the renin phenotype enhancing the production of renin to ensure survival [3].
Figure 1: The-Renin Angiotensin-System.

The rate-limiting step of this cascade involves the secretion of renin from juxtaglomerular cells in response to sympathetic stimulation, changes in perfusion pressure and salt levels in the urinary system. Renin, a hormone enzyme, cleaves angiotensinogen into angiotensin I, an inactive deca-peptide. Angiotensin I is further processed into angiotensin II by angiotensin-converting-enzyme (ACE) to produce angiotensin II. Angiotensin has a critical role in restoring homeostasis namely by inducing vasoconstriction to increase blood pressure and stimulating the adrenal gland to produce aldosterone which promotes sodium re-uptake in the kidney. Together, these actions regulate blood pressure and fluid-electrolyte balance, thus restoring homeostasis.
Text Box 1: The renin-angiotensin system enzymatic cascade.
The RAS is an enzymatic cascade involving multiple organs and peptides. Angiotensinogen, a 485-amino acid long peptide is released from the liver and circulates in an inactive form. Renin, secreted from juxtaglomerular cells (JG cells) of the kidney, cleaves Angiotensinogen into Angiotensin I which is further processed to Angiotensin II by Angiotensin-Converting-Enzyme produced in the lungs, endothelial cells, and other tissues [2]. Angiotensin II, an octapeptide, acts on Angiotensin receptors inducing vasoconstriction and increasing heart rate. In addition, Angiotensin II stimulates NaCl tubular reabsorption by the kidney. These actions result in elevation of blood pressure and maintenance of fluid electrolyte balance [2].
In addition to the endocrine control of fluid-electrolyte balance, renin cells are involved in kidney morphogenesis, regeneration of injured glomeruli and erythropoietin production [4-9]. In extrarenal locations such as in hematopoietic organs, specialized renin producing cells may play a role in innate immunity to fight infections.
In this review, we discuss how renin cells differentiate and acquire their identity, how they change fate in response to disease and the chromatin states that characterize and govern the identity and plasticity of renin cells. We also discuss the heterogeneity of renin-expressing cells, their diverse functions, and the molecular pathways whereby renin cells are controlled. Finally, we describe the evolutionary history of these cells and the clinical consequences of their perturbation.
Differentiation of renin-expressing cells in the kidney.
The differentiation of the renin cell precursors is intimately linked to the development of nephrons and the vasculature that irrigates them. In humans, nephrogenesis of the definitive kidney or metanephros starts around the 5th week of gestation culminating with the formation of approximately 1 million (400,000 to 2.4 million) nephrons. In mice, nephrogenesis starts around E10.5 of gestation resulting in 20-30 thousand nephrons. Whereas in humans, no new nephrons are formed after 36-38 weeks of gestation, in mice nephrogenesis and vascular development continues for a week after birth.
Nephrogenesis results from the reciprocal co-induction of two embryonic structures: the metanephric mesenchyme and the primitive ureteric bud [10]. The mesenchyme induces the ureteric bud to grow and divide into multiple ureteric tips (Figure 2A, B). The mesenchyme condenses around each ureteric tip and undergoes mesenchymal to epithelial transformation (MET) differentiating into a vesicle, then a comma, and an S-shaped body which in turn will give rise to a mature nephron. At around E11.5, the embryonic kidney is composed of three main compartments from which all mature kidney structures derive: 1) the ureteric bud (UB), characterized by the expression of Hoxb7; gives rise to the collecting ducts and ureter, 2) the cap mesenchyme adjacent to the ureteric bud tips is characterized by the expression of Six2. It undergoes the MET described above and ultimately gives rise to the Bowman’s capsule, podocytes, proximal tubules, loops of Henle, and distal tubules, 3) the stromal compartment composed of cells that express the Forkhead Box protein 1 (Foxd1) which give rise to mural cells of the kidney arterioles including smooth muscle cells, pericytes, renin precursors, and mesangial cells [11], and SCL+ progenitors that give rise to all endothelial cells of the kidney vasculature (Figure 2B) [12].
Figure 2: Nephrogenesis and Renin Expression.

A) Illustration of the two structures that give rise to the nephron in adult animals: the metanephric mesenchyme (MM) and the ureteric bud (UB) at E11. B) Reciprocal interaction of the MM and UB results in the growth of the UB towards the MM which begins to coalesce and continues through a series of progressive structures – namely a vesicle, comma and s-shaped body – before forming the tubular system and stromal compartment. Expression of key marker genes such as Foxd1 in the stromal cells and Hoxb7 in the collecting duct progenitors of the UB becomes detectable. C) By E19, the glomerulus is fully formed, and renin expression can be seen widely in the afferent arterioles and broader vasculature. D) During adulthood, expression of renin recedes to the JGA and those cells which expressed renin differentiate into vascular smooth muscle cells, pericytes and mesangial cells.
Renin precursor cells which are derived from the stromal compartment are crucial in the formation and branching of nascent kidney vessels [10,11]. As the arterioles are forming, renin cells coalesce to form a macula which turns into a bulge, further elongating to form an arteriole completely covered by renin-expressing cells (Figure 2C) [7]. This process repeats multiple times, in a fractal pattern so that from E11.5 to 7-10 days after birth there is constant branching, elongation and maturation of the kidney arterioles. As development is completed, renin-expressing cells differentiate into smooth muscle, pericytes, mesangial cells and mature renin-expressing JG cells (Figure 2D) [3]. During this process and until vascular development is complete (from E11.5 to P 15), the distribution of renin-expressing cells along the arterioles is extensive and heterogenous. A classification of the diversity of renin expression patterns observed throughout development of the vascular tree can be found in [7]. Thus, renin cells play a fundamental role in the assembly, elongation and branching of the kidney vasculature. Whether this angiogenic activity is due to a direct action of renin, or angiotensin produced locally, or other growth factors produced by renin cells remains to be investigated. The angiogenic properties of renin cells during embryonic life also manifests -pathologically- during the development of concentric arterial hypertrophy ensuing from chronic stimulation of renin cells as discussed below [see Clinician’s Corner].
Clinician’s Corner:
Conditions associated with a drop in blood pressure or extracellular fluid loss (dehydration, heart failure, hemorrhage) result in continuous stimulation of renin cells until balance is achieved. This mechanism, so beneficial throughout our evolution and everyday life, can turn deleterious when the renin cells are chronically and persistently challenged.
Thus, in animals and humans with mutations of the RAS genes or in rodents treated chronically with inhibitors of the RAS, overstimulated renin cells occupy the walls of the renal arterioles and cause the development of concentric vascular hypertrophy and focal renal ischemia.
Given the critical role that renin cells play not only in homeostasis and the negative consequences associated with their perturbation, it is important to examine the kidney structural effects of chronic RAS inhibition in hypertensive patients.
Renin progenitor cells participate directly in kidney vascular development. In the adult they are involved in glomerular regeneration.
Renin-expressing lymphocytes (B1 cells) may participate in innate immunity and defense against bacterial infections.
Perturbation of renin expression or its downstream effectors lead to severe defects in the cardiovascular and renal systems, particularly during development.
Renin cells are diverse and multifunctional
Although the classical notion of renin-expressing cells places them at the JG apparatus in the kidney, it is now known that renin cells can be found in a variety of different tissues during normal development and disease [3,13].
Even within the kidney, renin expression is not limited to the vasculature and has been detected in other cell types [1,3,14-18]. Cells from the renin lineage have been observed in a subset of proximal tubules, cells of the Bowman’s capsule and principal cells of the cortical collecting ducts [4]. Of these cells, proximal tubules have been found to express low levels of renin (approx. 1/100th of the mRNA levels found in native JG cells) and those levels are increased by hypotension [1]. In addition, mice subjected to deletion of Angiotensinogen, resulting in a chronic and severe model of hypotension, also display renin expression in tubules and pericytes [14]. Whether renin expressed in these cells plays a role as part of a local RAS in the kidney is not clear. Collecting duct cells and cells of the Bowman’s capsule also constitute a subset of the renin lineage, yet, no conclusive studies have demonstrated renin transcripts in these subpopulations during adulthood. Indeed, deletion of renin in collecting duct cells elicited no changes in circulating renin levels and in the renin response to a homeostatic threat [18]. In contrast, deletion of renin in the kidney vasculature resulted in a phenotype similar to the global deletion of renin [18]. These findings indicate that vascular renin is the primary source of circulating renin and is solely responsible for the normal development and maintenance of the kidney.
Lineage tracing studies also demonstrated renin expression outside the kidney including hematopoietic tissues, testis, adrenal glands, Muller cells, central nervous system, and skin [3,19]. The vast heterogeneity in renin-expressing cells transcending organs and embryonic layers raises a fascinating question: what role does renin expression play in these distinct cell types? Is the production/secretion of renin involved in the classical RAS to regulate blood pressure or is renin used in other non-canonical functions related to the cell types expressing it? These exciting questions remain unanswered and constitute a new opportunity for investigation. However, it should be noted that lineage tracing using the Cre-recombinase/loxP system has the potential to label cells that may not be related by lineage to a unique precursor. All that is required is that a cell expresses the gene in question driving Cre-recombinase -even transiently- independently of its origin and the cell and its descendants will be labeled permanently and erroneously classified. For example, all the collecting ducts derive from the ureteric bud which expresses Hoxb7, however, later during embryonic development a subset of collecting duct cells turn on transiently the renin gene. Therefore, when using a constitutive renin cre mouse crossed to a reporter mouse some collecting duct cells will be labeled and would erroneously appear to be directly derived from the stromal (Foxd1) renin precursor. Therefore, lineage relationships should be verified using multiple techniques. This does not negate that collecting duct renin expression may become relevant in hitherto unknown and untested physiological threats/conditions. Furthermore, single-cell RNA-seq studies demonstrated that progenitor cells may exhibit expression of markers from multiple lineages by virtue of their permissive chromatin [20]. As these cells differentiate, they undergo progressive, epigenetic repression of unrelated genes and positive chromatin modifications at specific enhancers that strengthen the identity of such cell. Because lineage tracing and labelling can occur as a result of even one transcriptional event (due to efficiency of the Cre-recombinase), it is possible that collecting ducts may label as a result of this “stochastic” and “pulsatile” expression of the renin gene in Hoxb7 progenitors. The exact function of renin in these subpopulations remains to be determined.
Renin cells and the innate immune system
As mentioned above, renin-producing hematopoietic cells may be involved in immune responses. These renin-expressing, hematopoietic progenitors first appear in the primitive yolk sac followed by their occurrence in the spleen, liver, bone marrow and peripheral blood [19,21]. The cells are immature progenitor B lymphocytes. Transplant experiments revealed that they give rise to B1 cells which colonize the liver and spleen. Preliminary data suggests that these B1-cells produce cable-like extensions that entrap bacteria which they subsequently phagocytose. When incubated with Salmonella typhimurium, renin-expressing B1 lymphocytes decreased the number of bacterial colony-forming-units (CFUs). This inhibition of bacterial growth is diminished if the B1 cells are derived from renin null mice. Although preliminary, these results suggest not only that these cells have the ability to decrease bacterial growth but also a potential novel role for renin in the immune system. Previous hypotheses had centered around the potential ability of a local RAS through renin-expressing lymphocytes causing localized inflammation and vasoconstriction; thus, enhancing resistance to infections and improving wound healing [22,23]. These data suggest that the enzyme renin, may participate directly in immunity potentially by digesting peptides found in the bacterial cell wall. However, given that Ren1c-KO mice have renal failure it is also possible that their B1-cells may be impaired rather than having an innate loss of anti-microbial activity as a result of absence of renin. Further studies will be needed to answer this question. The existence of renin-expressing lymphocytes adds a new dimension to our understanding of the defensive roles of renin cells linking the immune system with the endocrine control of blood pressure and fluid-electrolyte homeostasis.
Increasing the number of renin cells: a strategy for survival.
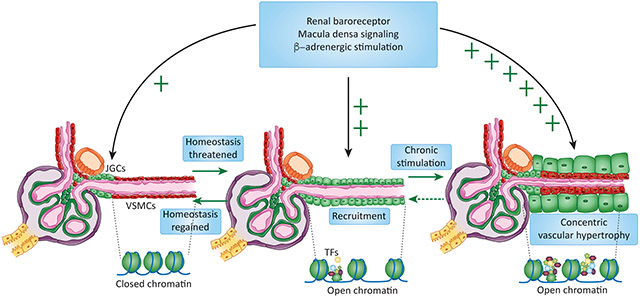
In adult mammals, JG cells comprise 0.01 % of the total kidney cells [24]. Under normal circumstances, renin release from those few cells usually suffice to maintain blood pressure homeostasis. However under intense and prolonged threats to homeostasis (such as dehydration, blood loss, hypotension) requiring that more renin be available, the kidney arterioles adopt an endocrine phenotype: renin cell descendants (smooth muscle cells, pericytes, mesangial cells) are transformed to synthesize renin, resulting in an increase in the number of cells releasing renin to the bloodstream. This “recruitment” occurs by a re-enactment of the embryonic pattern of renin expression [3]. The process is believed to be reversible and it is evident when there is a chronic threat to homeostasis [1,3,14,25-29]. The ability to produce large quantities of renin allows animals to restore their blood pressure and fluid volume and maintain blood supply/oxygen delivery to critical organs thus ensuring survival. Contrary to what has been suggested, the recruitment of renin cells does not involve proliferation [30] or migration of cells but occurs by de-differentiation or re-transformation of smooth muscle cells to the renin phenotype [3,31]. A small percentage of renin-expressing cells may be generated by neogenesis, de novo expression of renin by cells that never expressed it [4]. Thus, the smooth muscle cells retain the memory of the renin phenotype. Such memory resides in the cells’ chromatin and is mediated by a set of epigenetic marks at the locus of the renin gene and other loci throughout the genome [31]. It is possible that during early development the early expression of renin throughout the renal arteriolar smooth muscle cells may imprint these cells – epigenetically – for renin expression during critical threats to homeostasis. The ability to respond to homeostatic threats appears early in evolution [32-34] and is retained throughout phylogeny. Whether this plasticity to transdifferentiate confers an evolutionary advantage remains to be investigated.
The ability to re-express renin is not limited to the kidney and has been observed also in the adrenal gland. In embryonic life, the fetal zone of the adrenal cortex expresses a non-secreted form of renin [35] before renin cells appear in the kidney [36]. As the cells from the adrenal gland differentiate, they cease to express renin but the organ retains radial stripes of cells derived from the renin lineage [3,28]. As in the kidney, those cells maintain the ability to re-express renin, notably seen in mice subjected to global deletion of the aldosterone synthase gene [37]. Similarly, mice with bilaterally nephrectomy and excision of the submandibular glands exhibit strong renin expression in the inner cortex of the adrenal gland [28,38]. This activation of renin expression is molded by endocrine signaling cues; the release of adrenocorticotropic hormone appears to stimulate the production of renin in the zona glomerulosa and capsular cells while it is inhibited by circulating Angiotensin II levels [39]. It would be interesting to understand whether recruitment of adrenal cells to synthesize renin is controlled by the same epigenetic and transcriptomic mechanisms that control the recruitment of renal arteriolar smooth muscle cells. In addition to the kidney and adrenal glands, the recruitment of cells to perform specific homeostatic functions has been observed in other organs including insulin producing beta cells, atrial natriuretic peptide-producing myocardial cells, follicular cells of the thyroid, and albumin-producing hepatocytes [40-43]. Thus, recruitment of cells to perform a specific function in response to a physiological challenge is a phenomenon of major biological importance conserved among different cell types and species as a mechanism to preserve homeostasis [see clinician’s corner].
Where does the memory of the renin phenotype reside?
Establishment, preservation and reenactment of cell identity rests on the ability to maintain expression of key genes and repression of those associated with alternative cell fates [44]. This is achieved in part via deposition of activating or repressing epigenetic marks at specific regulatory regions of genes that are crucial for the identity and function of the cell. Strong enrichment of activating marks such as acetylation of histone 3 at lysine 27 (H3K27Ac), deposition of p300 and/or BRD4 combined with an open chromatin configuration of gene regulatory regions denote the presence of enhancers and super-enhancers which have a fundamental role in governing the identity of cells [45-48]. Super-enhancers are typically associated with transcription factors and genes that play key roles in the cell [49]. Their importance can be seen through the impact of the combined deletion of histone acetyl transferases CBP/p300 which have dual roles as partners for cyclic AMP-response element binding protein (CREB), a transcription factor, and also serve as epigenetic writers of activating epigenetic marks such as H3K27Ac. In whole body knock-out animals, the absence of these epigenetic remodelers is embryonically lethal [50,51]. Their conditional ablation in the renin lineage using the Cre-recombinase/loxP system results in a profound phenotype including a near absence of renin expression in JG cells and the inability of cells of the renin lineage to re-express the renin gene during homeostatic threats [52]. In addition, mutant kidneys have structural and physiological abnormalities resembling those found in patients with end stage renal disease (ESRD) or Chronic Kidney Disease (CKD) [52]. Adding another layer of complexity, the 3D organization of chromatin may be equally important in the control of renin cell identity. Within the nucleus, the chromatin is arranged in territories which are further compartmentalized in topological associated domains (TADs). TADs are delimitated by CCCTC-binding factor (Ctcf), a prominent chromatin organizer with binding sites in the vicinity of the renin gene, in renin cells [53]. The impact of chromatin architecture on renin gene expression, was evaluated in mice with conditional deletion of CTCF in renin cells. Loss of CTCF resulted in decreased endowment of renin-expressing cells accompanied by reduced circulating renin, hypotension, severe kidney abnormalities, and renal failure [53]. In renin cells, CTCF and the mediator complex -mentioned below- may regulate the formation of chromatin loops bringing into proximity crucial super-enhancers with their promoters. Understanding chromatin architecture represents a fascinating opportunity to understand renin cell fate.
Because perturbation of H3K27Ac levels and the associated changes in chromatin configuration abolish the ability to express renin, it can be inferred that renin cells have a unique and essential epigenetic landscape which serves as the foundation for their cell identity. Indeed, recent work indicates that renin cells possess unique epigenetic states including open chromatin, deposition of Med 1 and H3K27Ac at loci crucial for the renin cell programme, including the renin locus itself [31]. In addition, in a renin KO model which has chronic and persistent activation of the renin cell programme due to their inability to raise blood pressure [26,54], establishment of super-enhancers at key cell identity genes was observed including at the renin gene and the transcription factors which may regulate its transcriptional activity [31]. These results suggest that the chromatin may act as a sensor of physiological conditions that can be changed accordingly to facilitate re-establishment of homeostasis. The resting epigenetic state of renin cell descendants has not yet been ascertained. It would be exciting to ascertain whether these cells have a vastly different epigenetic profile including closed chromatin at the renin gene. This would suggest that remodeling of their epigenetic configuration may be necessary to allow SMCs to re-express the renin cell program (Figure 3). In fact, treatment of SMCs from the renin lineage with cAMP or HDAC inhibitors resulted in re-expression of renin suggesting that the cells retain the memory of the renin phenotype. To define whether this memory can be elicited epigenetically, a mutated form of Cas9 harboring the catalytic unit of p300 was targeted to the super-enhancer region of the renin gene. Upon stimulation with p300, a fraction of the cells re-expressed the renin gene indicating that the memory of the renin phenotype can be brought up by changing the acetylation of chromatin at the renin locus [31]. These exciting experiments indicate that it is possible to change the fate of renin cell descendants and other cells using epigenetic rewriting.
Figure 3: Model of Epigenetic Landscape of Renin Lineage Cells.

A) Cells actively expressing renin have a distinct epigenetic signature at the renin gene (and others) characterized by the presence of open chromatin, activating histone marks such as H3K27Ac and binding of key transcription factors (TF) such as CREB and Med1. Together, these features constitute a super-enhancer which drives strong expression of renin and other key genes. B) Vascular smooth muscle cells which activate the renin program under stress may have poised epigenetic features such as H3K4me1 and low transcription of the renin gene. The absence of open chromatin and activating marks prevents these cells from ectopically expressing renin until the body is challenged by physiological threats requiring increased renin output. C) Tubular cells (and other cell types) may possess silencing epigenetic features including closed chromatin, repressive epigenetic features such as H3K27me3 and restricted transcription factor binding. As a result, these cells lack the ability to activate the renin cell programme.
Clinical and pathological consequences of excessive inhibition of the renin-angiotensin system: Experimental lessons
As mentioned above, the ability of renin cells to “recruit” establishes an additional layer of defense to deal with protracted challenges to homeostasis and meet the demands for circulating renin that cannot be met by a few JG cells. This ability has been demonstrated in several species including amphibians [55], fish [32], mice [56,57] and humans [1,58]. It can be speculated that the ability to generate a large number of renin cells when subjected to potentially lethal threats to homeostasis constitutes a strong evolutionary adaptation to enable survival. Whereas this ability to recruit cells is crucial for the organism, the cells have been designed to continue their attempt to make renin for as long the challenge exists. Unfortunately, under conditions of unrelenting stimulation, renin cells may contribute to concentric arteriolar hypertrophy and focal renal ischemia as described below [see Clinician’s corner].
Given the importance of the RAS and that renin cells are essential for survival across taxa, it is unsurprising that mutations or pharmacological inhibition of the RAS has severe consequences. Thus, in early life, RAS inhibitors are contraindicated during pregnancy as their use result in renal abnormalities, calvaria defects and lung hypoplasia in the developing fetus [59] [Clinician’s corner]. Mice with deletion of any of the of the RAS genes display systemic abnormalities, including hypotension, and inability to concentrate their urine [25-27,54,55,58,60-62]. Furthermore, their renal anatomy is compromised with papillary atrophy, an underdeveloped renal medulla and disorganized, hypertrophic arterioles. Similarly, mutations of the RAS genes in humans lead to renal tubular dysgenesis and arteriolar thickening [59]. Furthermore, mutations which result in a decline in Angiotensin II signaling activity (including deletion of renin or Angiotensin II receptors) also diminish the endowment of smooth muscle cells in the ureter and cause impaired peristalsis as a result [63]. The consequent buildup of pressure in the renal parenchyma causes inflammation, fibrosis and hydronephrosis in the renal medulla [26,63]. Interestingly, treatment of adult hypertensive rats with RAS inhibitors results in marked improvement in blood pressure and proteinuria. However, these animals develop significant kidney damage, including tubulo-interstitial fibrosis and concentric arteriolar hypertrophy [55,64]. These results are mirrored by vascular abnormalities found in Rana catesbiana tadpoles subjected to ACE inhibition [55]. Chronic stimulation of the renin cells and their unrelenting continuous recruitment perturbs tissue homeostasis: the kidneys develop concentric arteriolar hypertrophy accompanied by signs of focal renal ischemia and fibrosis [Clinician’s corner]. The concentric arteriolar hypertrophy is not caused by the renin protein itself but rather by the activities of the renin cells themselves. In fact, in animals with ablation of renin cells using Diphtheria toxin [65] or in mice with conditional deletion of Integrin β1 [66] [Box 2] the vascular hypertrophy does not occur suggesting that during chronic stimulation, renin cells contribute physically by inserting themselves into the thickening vessel and/or by releasing angiogenic /growth factors that stimulate the accumulation of concentric layers of smooth muscle underneath the renin cells [54].
Text Box 2: Beta-1 Integrin and Renin Cell Vitality.
Because survival of animals is predicated upon the ability to regulate renin expression in a rapid and precise fashion, it is unsurprising that renin cells are highly sensitive to their environment and extracellular milieu. A historical challenge in studying renin cells has been that they lose the ability to express renin in culture where they transdifferentiate into other cell types due to the absence of physiological input [69]. Recent studies in our lab on integrins have uncovered some of the underlying molecular mediators which facilitate the survival of JG cells. Integrins are cell adhesion molecules that interact with extracellular matrix proteins, notably proteins containing the RGD amino acid motif, to mediate attachment as well as signaling pathways that promote survival [70,71]. β1-integrin is expressed by renin cells throughout development which led us to hypothesize that it may play a critical role in safeguarding renin cell identity and maintaining survival. Deleting β1-integrin in renin lineage cells of mice results in a severe phenotype characterized by kidney failure and a drastic reduction in the endowment of renin cells in the adult kidney as seen through immunostaining and lineage tracing experiments [66]. The vasculature is significantly thinner with fewer cells; consistent with the reduction seen in renin lineage cells. Additionally, mutant animals had lower mitotic indices in neonatal life and have JG areas positive for TUNEL staining indicating apoptosis of renin cells. The reduction in proliferation together with the increased death of renin cells explains the near absence of renin staining in adult animals. How does loss of β1-integrin result in cell death/apoptosis? One hypothesis is that these cells undergo apoptosis through anoikis – a term referring to removal of cells detached from their extracellular matrix to prevent ectopic displacement of cells as commonly seen in metastatic tumors which escape this checkpoint [72,73]. Additionally, these mice suffer from various symptoms of renal failure and present compromised renal and vascular architecture. Because deletion of β1-integrin occurred in various cell types which resulted in a rapid deterioration of the kidney with markers of damage such as interstitial fibrosis and perivascular infiltration, the compromised architecture of the kidney may have negatively affected JG cells and, eventually, resulted in their death. However, the clear overlap of apoptotic bodies in the JG area early in development before progression of the phenotype suggests that this it is more likely that the renin cells underwent apoptosis due to the direct absence of β1-integrin in a cell-autonomous fashion rather than alterations at the tissue / organ level. These experiments not only emphasize the importance of the structural molecular components of the renin cells, they also provide a window into the potential regulators/targets which may support maintaining JG cell identity in culture.
Considering the widespread use of RAS inhibitors to treat essential hypertension and their proven efficacy in preventing fatal cardiovascular events [67], it would be important to define whether treatment of hypertension with RAS blockers in humans leads to the same kidney abnormalities found in animals as described above [68]. It should also be noted that expression of ACE is vital for fighting infections and promoting the immune function of macrophages [22,23]. Therefore, it would be prudent to evaluate whether prolonged use of RAS inhibitors in humans is associated with diminished immune capabilities [Clinician’s corner]. At present, the long-term consequences of using RAS inhibitors in normal and hypertensive animals is being evaluated to determine whether their protracted use results in vascular and interstitial damage. The studies may encourage the design of novel drugs which can play the same role as current RAS inhibitors while leaving renin cells and the vasculature unperturbed.
Concluding Remarks and Future Perspectives
In short, renin-expressing cells manifest in various forms and play a diverse set of functions. A subset of these cells plays the classical roles associated with the RAS to regulate fluid-electrolyte and blood pressure homeostasis. The cells have a remarkable plasticity – termed recruitment – which enables them to respond to threats to ensure survival. Renin-expressing cells also exist outside the kidney and protect the body from infections through innate immunity. The conserved function of these cells across taxa highlights their critical role in survival and the loss/inhibition of renin expression results in severe physiological consequences making it essential to understand downstream effects of RAS inhibitors on renin cells in the clinical setting.
Discoveries that remain to be made (see Outstanding Questions) include the epigenetic landscape of the cells which adopt the renin cell identity, the exact role of the different renin-expressing cells detected by lineage tracing and the molecular and physiological consequences of RAS inhibition as a clinical treatment. Answering these questions will require the use of advanced, emerging technologies, but will result in a more nuanced insight into cell fate and ultimately facilitate the development of life-saving treatments while preventing complications associated with systems regulated by renin and its partner molecules.
Outstanding Questions.
Given the findings in experimental or spontaneous models of RAS deletion and/or inhibition, It will be important to define whether patients treated chronically with RAS blockers develop structural kidney abnormalities.
What are the chromatin events, gene–gene interactions, and transcriptional factories that allow cells of the renin lineage – and not other cell types – to adopt the renin cell identity during homeostatic threats?
How do renin-expressing immune cells compare with their non-renin-expressing counterparts?
Do changes in the epigenetic landscape of renin lineage cells represent the causal event in their transdifferentiation into renin cells or are they facilitating events to reinforce new expression patterns?
What is the exact role of renin expression in adult renin lineage cells which only exhibit very low levels of renin expression during development?
Figure 4: Renin Cell Programme.
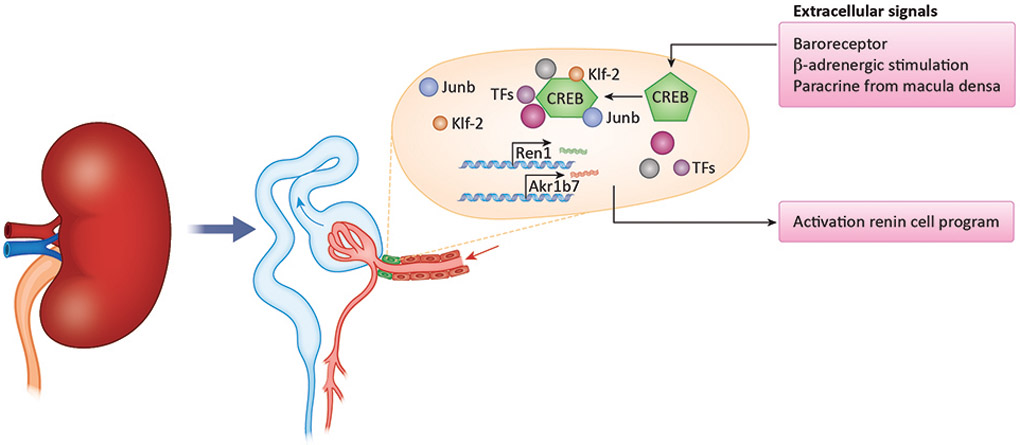
Our tentative hypothesis regarding the renin cell programme. In response to changes in perfusion pressure, sympathetic innervation and fluid-electrolyte balance, transcription factors and epigenetic remodelers are mobilized to activate the renin programme. We hypothesize that these signaling pathways result in the expression of various transcription factors such as Klf-2, JunB and others. Together with epigenetic remodeling, these molecular factors both maintain the resting JG cell identity as well as induce transformation of smooth muscle cells under stress by activating genes central to the renin cell identity such as renin and Akr1b7.
Acknowledgments
Studies were funded by National Institutes of Health grants P50 DK-096373 and R01 DK-116718 to RAG, DK-116196, DK-096373 and HL-148044 to MLSSL and American Heart Association Pre-Doctoral Fellowship 18PRE34020090 to OG.
Glossary
- Metanephric Mesenchyme:
An embryonic structure derived from the mesoderm which in conjunction with the ureteric bud, develops into the kidney proper during nephrogenesis. The metanephric mesenchyme specifically gives rise to nephrons, the functional units of the kidney and their blood vessels. These nephrovascular units are responsible for filtering blood and producing urine
- Ureteric Bud:
An embryonic structure developing from the intermediate mesoderm which contributes to kidney development via its interactions with the metanephric blastema. The ureteric bud then develops into the collecting ducts and ureter
- Forkhead Box Protein1 (Foxd1):
A transcription factor which characterizes a diverse lineage of cells found in eyes, brain, lungs, skin among others and in the kidney. During nephrogenesis, it is expressed in the stromal cells which give rise to the mural cells of the kidney vasculature including renin cells
- Homeobox Protein 7 (HoxB7):
A transcription factor belonging to the homeobox gene family which are critical for development. It is characteristically expressed in collecting ducts and cells which develop from the ureteric bud during kidney development
- Transdifferentiation / Recruitment:
A process by which cells undergo a change in cell identity from one cell type to another without progressing through a less differentiated intermediate. The initial description of transdifferentiation in renin lineage cells into renin cells was described as “recruitment” in reference to the functional recruitment of the cells to produce more renin during homeostatic threat. The term “recruitment” is used in a functional sense; it does not refer to spatial recruitment with migration or production of new cells via proliferation
- Epigenetics:
A compound word of epi + genetics meaning “above genetics” or “above sequence”. The term refers to heritable changes affecting gene expression without changing the genetic sequence of DNA in a cell and represents a critical factor in regulating cell identity and enabling cells with the same DNA to adopt markedly different identities
- ChIP-seq:
Chromatin immunoprecipitation followed by high-throughput sequencing is a technique used to identify the DNA sequences where in the genome binding of a particular protein such as a transcription factor or epigenetic modification occurs
- Lineage Tracing:
Lineage tracing refers to techniques which attempt to identify the ancestry of a cell. Various methods have been used including direct observation, inference using expression markers, a cre-lox approach linked to the expression of an unerasable reporter, and computationally through barcoding
- Cre-recombinase/LoxP:
A protein reconstituted from the P1 bacteriophage which has the catalytic capacity to recombine DNA sequences which are flanked by 34 bp specific sequences consisting of two 13 bp inverted and palindromic repeats and 8 bp core sequences termed LoxP sites. This protein has been used to recombine genetic sequences to allow expression of reporters such as B-galactosidase or fluorescent proteins to identify cells which express the Cre-recombinase driven by particular genes; thus, achieving lineage/fate tracing
References
- 1.Taugner R et al. (2013) The Juxtaglomerular Apparatus: Structure and Function. Springer Verlag [Google Scholar]
- 2.Fournier D et al. (2012) Emergence and evolution of the renin-angiotensin-aldosterone system. , Journal of Molecular Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sequeira López MLS et al. (2004) Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev. Cell 6, 719–728 [DOI] [PubMed] [Google Scholar]
- 4.Hickmann L et al. (2017) Persistent and inducible neogenesis repopulates progenitor renin lineage cells in the kidney. Kidney Int. 92, 1419–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurt B et al. (2013) Deletion of von Hippel-Lindau protein converts renin-producing cells into erythropoietin-producing cells. J. Am. Soc. Nephrol 24, 433–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurt B et al. (2015) Chronic Hypoxia-Inducible Transcription Factor-2 Activation Stably Transforms Juxtaglomerular Renin Cells into Fibroblast-Like Cells In Vivo. J. Am. Soc. Nephrol 26, 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddi V et al. (1998) Renin-expressing cells are associated with branching of the developing kidney vasculature. J. Am. Soc. Nephrol 9, 63–71 [DOI] [PubMed] [Google Scholar]
- 8.Starke C et al. (2015) Renin lineage cells repopulate the glomerular mesangium after injury. J. Am. Soc. Nephrol 26, 48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pippin JW et al. (2013) Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am. J. Pathol 183, 542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sequeira Lopez MLS and Gomez RA (2011) Development of the renal arterioles. J. Am. Soc. Nephrol 22, 2156–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sequeira-Lopez MLS et al. (2015) The earliest metanephric arteriolar progenitors and their role in kidney vascular development. Am. J. Physiol. - Regul. Integr. Comp. Physiol 308, R138–R149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Y et al. (2016) Hemovascular progenitors in the kidney require sphingosine-1-phosphate receptor 1 for vascular development. J. Am. Soc. Nephrol 27, 1984–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez RA and Sequeira-Lopez MLS (2018) Renin cells in homeostasis, regeneration and immune defence mechanisms. Nat. Rev. Nephrol 14, 231–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berg AC et al. (2013) Pericytes synthesize renin. World J. Nephrol 2, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohrwasser A et al. (2003) Renin and kallikrein in connecting tubule of mouse. Kidney Int. 64, 2155–2162 [DOI] [PubMed] [Google Scholar]
- 16.Prieto-Carrasquero MC et al. (2008) Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hpertensive rats. Hypertension 51, 1590–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang JJ et al. (2008) The collecting duct is the major source of prorenin in diabetes. Hypertension 51, 1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sequeira-Lopez MLS et al. (2015) Vascular versus tubular renin: Role in kidney development. Am. J. Physiol. - Regul. Integr. Comp. Physiol 309, R650–R657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belyea BC et al. (2014) Identification of renin progenitors in the mouse bone marrow that give rise to B-cell leukaemia. Nat. Commun 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magella B et al. (2018) Cross-platform single cell analysis of kidney development shows stromal cells express Gdnf. Dev. Biol 434, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belyea BC et al. (2019) A primitive type of renin-expressing lymphocyte protects the organism against infections. bioRxiv DOI: 10.1101/770511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen XZ et al. (2007) Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol 170, 2122–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okwan-Duodu D et al. (2010) Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J. Biol. Chem 285, 39051–39060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellanos Rivera RM et al. (2011) Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol. Genomics 43, 1021–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilgers KF et al. (1997) Aberrant renal vascular morphology and renin expression in mutant mice lacking angiotensin-converting enzyme. Hypertension 29, 216–221 [DOI] [PubMed] [Google Scholar]
- 26.Takahashi N et al. (2005) Ren1c homozygous null mice are hypotensive and polyuric, but heterozygotes are indistinguishable from wild-type. J. Am. Soc. Nephrol 16, 125–32 [DOI] [PubMed] [Google Scholar]
- 27.Tsuchida S et al. (1998) Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J. Clin. Invest 101, 755–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naruse M et al. (1984) Immunoreactive renin in mouse adrenal gland: localization in the inner cortical region. Hypertension 6, 275–280 [PubMed] [Google Scholar]
- 29.Makhanova N et al. (2006) Kidney function in mice lacking aldosterone. Am. J. Physiol. - Ren. Physiol 290, 61–69 [DOI] [PubMed] [Google Scholar]
- 30.Guessoum O et al. (2020) Proliferation does not Contribute to Murine Models of Renin Cell Recruitment. Acta Physiol. DOI: DOI: 10.1111/apha.13532 [DOI] [Google Scholar]
- 31.Martinez MF et al. (2018) Super-enhancers maintain renin-expressing cell identity and memory to preserve multi-system homeostasis. J. Clin. Invest 128, 4787–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rider SA et al. (2015) Renin expression in developing zebrafish is associated with angiogenesis and requires the notch pathway and endothelium. Am. J. Physiol. - Ren. Physiol 309, F531–F539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rider SA et al. (2017) Zebrafish mesonephric renin cells are functionally conserved and comprise two distinct morphological populations. Am. J. Physiol. - Ren. Physiol 312, F778–F790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann S et al. (2018) Investigating the RAS can be a fishy business: Interdisciplinary opportunities using Zebrafish. , Clinical Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steckelings UM et al. (2004) Human skin: Source of and target organ for angiotensin II. Exp. Dermatol 13, 148–154 [DOI] [PubMed] [Google Scholar]
- 36.Peters J et al. (2008) A renin transcript lacking exon 1 encodes for a non-secretory intracellular renin that increases aldosterone production in transgenic rats. J. Cell. Mol. Med DOI: 10.1111/j.1582-4934.2008.00132.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee G et al. (2005) Homeostatic responses in the adrenal cortex to the absence of aldosterone in mice. Endocrinology 146, 2650–2656 [DOI] [PubMed] [Google Scholar]
- 38.Naruse M and Inagami T (1982) Markedly elevated specific renin levels in the adrenal in genetically hypertensive rats. Proc. Natl. Acad. Sci. U. S. A 79, 3295–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baba K et al. (1986) Mechanisms by which nephrectomy stimulates adrenal renin. Hypertension 8, 997–1002 [DOI] [PubMed] [Google Scholar]
- 40.Thorel F et al. (2010) Conversion of adult pancreatic α-cells to B-cells after extreme B-cell loss. Nature DOI: 10.1038/nature08894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lattion AL et al. (1986) Myocardial recruitment during ANF mRNA increase with volume overload in the rat. Am. J. Physiol. - Hear. Circ. Physiol 251, H890–H896 [DOI] [PubMed] [Google Scholar]
- 42.Lin CT et al. (1986) Estrogen induction of very low density apolipoprotein ii synthesis, a major avian liver yolk protein, involves the recruitment of hepatocytes. Endocrinology 118, 538–544 [DOI] [PubMed] [Google Scholar]
- 43.Gerber H et al. (1987) Progressive recruitment of follicular cells with graded secretory responsiveness during stimulation of the thyroid gland by thyrotropin. Endocrinology 120, 91–96 [DOI] [PubMed] [Google Scholar]
- 44.Alberts B et al. (2014) Molecular Biology of the Cell 6e, 6 [Google Scholar]
- 45.Hnisz D et al. (2013) Super-enhancers in the control of cell identity and disease. Cell 155, 934–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pott S and Lieb JD (2014) What are super-enhancers? Nat. Genet 47, 8–12 [DOI] [PubMed] [Google Scholar]
- 47.Buenrostro JD et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buenrostro JD et al. (2015) ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol DOI: 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niederriter AR et al. (2015) Super enhancers in cancers, complex disease, and developmental disorders. , Genes, 6. 1183–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka Y et al. (2000) Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech. Dev 95, 133–145 [DOI] [PubMed] [Google Scholar]
- 51.Lipinski M et al. (2020) KAT3-dependent acetylation of cell type-specific genes maintains neuronal identity in the adult mouse brain. Nat. Commun DOI: 10.1038/s41467-020-16246-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomez RA et al. (2009) CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. AJP Hear. Circ. Physiol 296, H1255–H1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martinez MF et al. (2020) Ctcf is Required for Renin Expression and Maintenance of the Structural Integrity of the Kidney. Clin. Sci DOI: 10.1042/CS20200184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oka M et al. (2017) Chronic Stimulation of Renin Cells Leads to Vascular Pathology. Hypertension 70, 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tufro-McRreddie A et al. (1995) Angiotensin II regulates nephrogenesis and renal vascular development. Am. J. Physiol. - Ren. Fluid Electrolyte Physiol 269, F110–F115 [DOI] [PubMed] [Google Scholar]
- 56.Cantin M et al. (1979) Metaplastic and mitotic activity of the ischemic (endocrine) kidney in experimental renal hypertension. Am. J. Pathol 96, 545–565 [PMC free article] [PubMed] [Google Scholar]
- 57.Everett AD et al. (1990) Renin release and gene expression in intact rat kidney microvessels and single cells. J. Clin. Invest 86, 169–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gribouval O et al. (2012) Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat 33, 316–326 [DOI] [PubMed] [Google Scholar]
- 59.Bullo M et al. (2012) Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: A systematic review. , Hypertension. [DOI] [PubMed] [Google Scholar]
- 60.Okubo S et al. (1998) Angiotensinogen gene null-mutant mice lack homeostatic regulation of glomerular filtration and tubular reabsorption. Kidney Int. 53, 617–625 [DOI] [PubMed] [Google Scholar]
- 61.Tanimoto K et al. (1994) Angiotensinogen-deficient mice with hypotension. J. Biol. Chem 269, 31334–31337 [PubMed] [Google Scholar]
- 62.Esther CR et al. (1996) Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab. Investig 74, 953–965 [PubMed] [Google Scholar]
- 63.Miyazaki Y et al. (1998) Angiotensin induces the urinary peristaltic machinery during the perinatal period. J. Clin. Invest DOI: 10.1172/JCI4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moniwa N et al. (2013) Hemodynamic and hormonal changes to dual renin-angiotensin system inhibition in experimental hypertension. Hypertension 61, 417–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pentz ES et al. (2004) Ablation of renin-expressing juxtaglomerular cells results in a distinct kidney phenotype. Am. J. Physiol. - Regul. Integr. Comp. Physiol 286, R474–483 [DOI] [PubMed] [Google Scholar]
- 66.Mohamed T et al. (2020) Renin-expressing cells require β1-integrin for survival and for development and maintenance of the renal vasculature. Hypertension DOI: 10.1161/HYPERTENSIONAHA.120.14959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whelton PK et al. (2017) 2017 Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults A Report of the American College of Cardiology / American Heart Association T [DOI] [PubMed]
- 68.Sequeira-Lopez MLS and Gomez RA (2018) Preserving kidney health during intensive blood pressure control. Nat. Rev. Nephrol 14, 537–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karginova EA et al. (1997) Zis: A developmentally regulated gene expressed in juxtaglomerular cells. Am. J. Physiol. - Ren. Physiol 273, F731–738 [DOI] [PubMed] [Google Scholar]
- 70.Jalali S (2001) Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc. Natl. Acad. Sci DOI: 10.1073/pnas.031562998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pinkse GGM et al. (2006) Integrin signaling via RGD peptides and anti-β1 antibodies confers resistance to apoptosis in islets of Langerhans. Diabetes DOI: 10.2337/diabetes.55.02.06.db04-0195 [DOI] [PubMed] [Google Scholar]
- 72.Alanko J et al. (2015) Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol DOI: 10.1038/ncb3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Silginer M et al. (2014) Integrin inhibition promotes atypical anoikis in glioma cells. Cell Death Dis. DOI: 10.1038/cddis.2013.543 [DOI] [PMC free article] [PubMed] [Google Scholar]