

Abstract
Fanconi anemia (FA) is a genetic disorder due to mutations in any of the 22 FANC genes (FANCA – FANCW) and has high phenotypic variation. Siblings may have similar clinical outcome because they share the same variants, however, such association has not been reported. We present the detailed phenotype and clinical course of 25 sibling sets with FA from two institutions. Haematological progression significantly correlated between siblings, which was confirmed in an additional 55 sibling pairs from the International Fanconi Anemia Registry. Constitutional abnormalities were not concordant, except for a moderate degree of concordance in kidney abnormalities and microcephaly.
INTRODUCTION
Fanconi anemia (FA) is the most common inherited bone marrow failure syndrome.1,2 Patients with FA often present with congenital anomalies and up to 70% of patients develop bone marrow failure (BMF) during the first decade of life, with high risk of progression to myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).3,4 Patients also have high incidence of solid tumors, especially head and neck squamous cell carcinoma.5,6
There are 22 known genes that cause FA, most of which exhibit autosomal recessive inheritance pattern, except FANCB which is X-linked recessive7 and RAD51/FANCR which shows a dominant negative cellular effect.8,9 In case of autosomal recessive inheritance, a sibling has a 25% chance of being affected. As siblings share the same disease-causing variants, it may be expected that a clinical outcome of an affected sibling could be predicted based on that of a proband. Not only is this important information that the parents and family are seeking to allay their anxiety related to the unknown, but in fact it may have implications in treatment decisions for the sibling. However, the published sibling reports suggest that there can be a high degree of discordance.10,11 To date, there has been no systematic evaluation of a large sibling cohort with FA as to whether they have a similar or different clinical course. Here, we show that siblings with FA generally have a similar haematological clinical course as compared to their affected probands.
METHODS
Study participants
We performed a retrospective review of medical records of available FA probands and their affected siblings treated at Memorial Sloan Kettering Cancer Center – MSK Kids (n=9 families) and Cincinnati Children’s Hospital Medical Center (n=16 families) between April 1990 and June 2018. Longitudinal and detailed clinical information was available for this discovery cohort. We collected the onset of neutropenia (absolute neutrophil count (ANC) < 1000 /uL), anemia (hemoglobin (Hb) < 10 g/dL), thrombocytopenia (platelets < 100,000 /uL), MDS or AML, time to hematopoietic stem cell transplantation (HSCT), the presence of any congenital anomalies, radial, thumb or other skeletal abnormalities, kidney or cardiac abnormalities, microcephaly, low birth weight, tracheo-oesophageal fistula, anal atresia, short stature, growth retardation, and learning disability. We then assessed onset of haematological manifestations in an additional 55 sibling pairs from the International Fanconi Anemia Registry (IFAR) as a validation cohort. The IFAR defines onset of haematological manifestations as at least one of the following: ANC < 1000/uL, Hb < 10g/dL, platelets < 100,000/uL, or development of MDS or AML. Institutional Review Boards of each institution approved to perform retrospective chart review and exchange deidentified clinical data for this study. The median age of onset of haematological manifestations of the non-sibling cohort was calculated from 604 subjects from the IFAR who did not have siblings.
Statistical analyses
The Spearman rank correlation coefficient was computed to assess correlation between sibling pair’s clinical manifestations for the discovery cohort. If there were multiple siblings in one family, a proband and the oldest affected sibling were used among available subjects for this analysis. We also calculated the Cohen’s kappa coefficient to determine whether a presence or absence of a particular clinical event was likely to occur in both siblings. For this analysis, we used both a proband vs. an older sibling and a proband vs. a younger sibling pair in cases of triple sibling members. To assess the correlation of onset of haematological manifestations in the validation cohort, the Spearman’s rank correlation was calculated on a total of 55 sibling pairs (excluding sibling pairs with somatic mosaicism or incomplete data). The discovery cohort and validation cohort are mutually exclusive. We excluded known mosaics from the correlation analysis for haematological outcomes.
RESULTS
Demographic characteristics of the discovery cohort is described in the Supplementary Table 1. In the discovery cohort, we examined whether a specific factor or clinical outcome, such as congenital anomaly or development of MDS/AML, was likely to appear in the same sibling pairs or not, by calculating the Cohen’s kappa coefficient (Table 1). We found moderate agreement in kidney abnormalities and microcephaly, weak agreement in other skeletal abnormalities, low birth weight, tracheo-oesophageal fistula, growth retardation and learning disability. Development of MDS or AML were not likely to occur in the same sibling pair.
Table 1.
Cohen’s kappa coefficient test for clinical outcome between sibling pairs (total 28 pairs of siblings from 25 families (MSKCC-CCHMC cohort).
| Variable | Kappa | 95% Confidence Interval |
Level of Agreement |
Frequency of event (%) |
|---|---|---|---|---|
| Any congenital anomalies | 0.14 | (−0.15, 0.43) | None | 73.58 |
| Radial abnormalities | 0.16 | (−0.24, 0.56) | None | 13.21 |
| Thumb abnormalities | 0.32 | (−0.03, 0.67) | Minimal | 37.74 |
| Other skeletal abnormalities | 0.51 | (0.03, 0.99) | Weak | 11.32 |
| Kidney abnormalities | 0.65 | (0.34, 0.96) | Moderate | 28.3 |
| Cardiac abnormalities | 0.35 | (−0.08, 0.78) | Minimal | 15.09 |
| Microcephaly | 0.76 | (0.44, 1.00) | Moderate | 16.98 |
| Low birth weight | 0.48 | (0.12, 0.83) | Weak | 26.42 |
| Tracheo-oesophageal fistula | 0.46 | (−0.17, 1.00) | Weak | 7.55 |
| Anal atresia | 0.34 | (−0.23, 0.92) | Minimal | 9.43 |
| Short stature | 0.23 | (−0.11, 0.57) | Minimal | 43.4 |
| Growth retardation | 0.44 | (0.09, 0.80) | Weak | 22.64 |
| Learning disability | 0.47 | (−0.13, 1.00) | Weak | 5.77 |
| MDS | 0.03 | (−0.34, 0.41) | None | 18.87 |
| AML | −0.05 | (−0.12, 0.02) | None | 5.66 |
A median age of onset of haematological manifestations were 5.69 (95% CI: 4.42 – 8) in the discovery cohort. The most frequent FA complementation group was FA-A, observed in 10 families (40%), followed by FA-C in 6 families (24%) (Fig 1A). The validation cohort had a median age of onset of 6.48 (95% CI: 5.67 – 7.16). The most frequent complementation group was FA-A (63.6%), followed by FA-C (10.9%) and FA-G (9.1%).
Figure 1. The haematological course was similar between probands and affected siblings with Fanconi anemia.
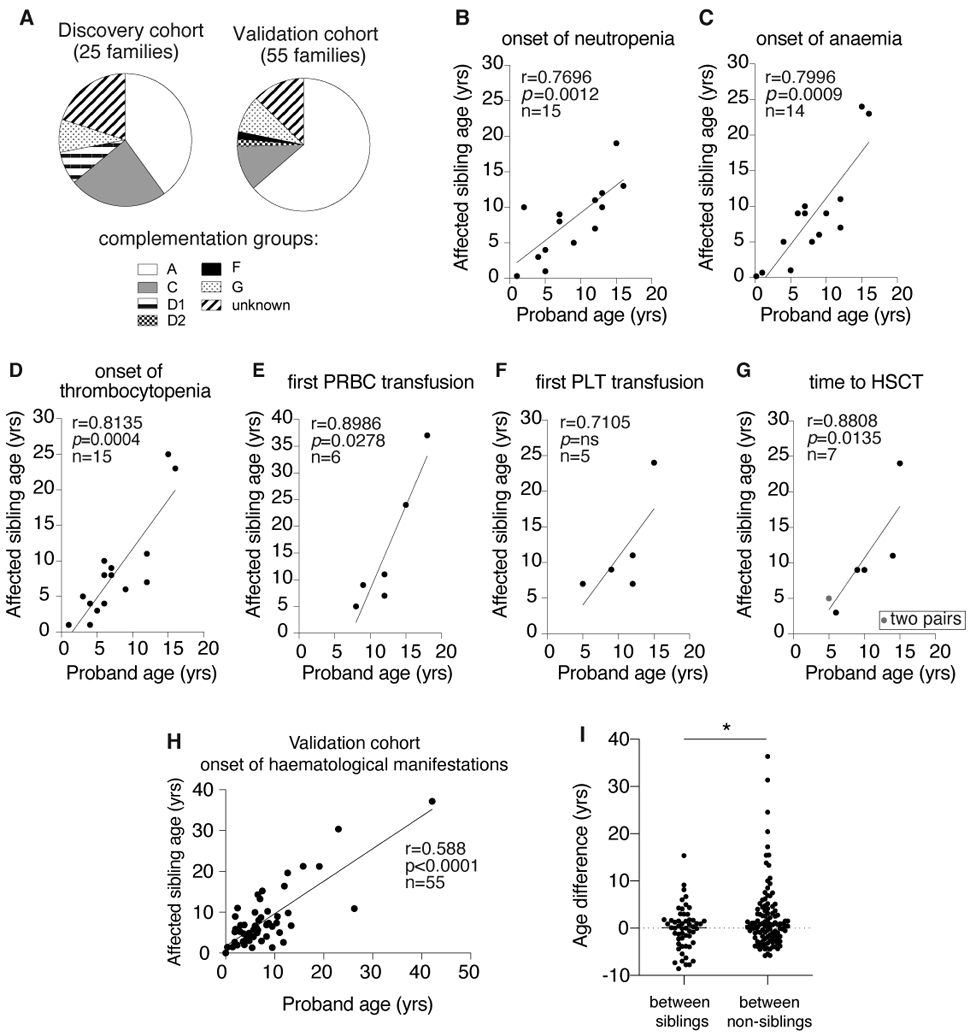
A. The FA complementation group of the discovery cohort and the validation cohort
B. The onset of neutropenia (ANC < 1000 /uL) among the discovery cohort was significantly correlated between sibling pairs.
C. The onset of anemia (Hb < 10 g/dL) among the discovery cohort was significantly correlated between sibling pairs.
D. The onset of thrombocytopenia (platelets < 100,000 /uL) among the discovery cohort was significantly correlated between sibling pairs.
E. The time to first PRCB transfusion among the discovery cohort was significantly correlated between sibling pairs.
F. The time to first platelet transfusion among the discovery cohort did not reach a statistically significant level of correlation between sibling pairs.
G. The time to HSCT among the discovery cohort was significantly correlated between sibling pairs.
H. The onset of haematological manifestations (defined by the presence of any of the following: BMF, MDS or AML) in the validation cohort showed significant correlation between them.
I. The age differences at the onset of haematological manifestations were smaller between siblings than between non-siblings (p = 0.0328 by unpaired t test).
The Spearman rank correlation coefficient calculated from the discovery cohort showed that the onset of neutropenia, anemia and thrombocytopenia were significantly correlated between siblings among evaluable pairs (Fig 1B-D). The ages at the first transfusion of packed red blood cells (PRBC) and of platelets were available for six and five sibling pairs respectively. The age of first transfusion was highly correlated between siblings for PRBC (Fig 1E), but not statistically significant for platelets (Fig 1F). Time to HSCT information was available in seven sibling pairs, which showed a significant correlation between siblings (Fig 1G)
To assess whether the onset of haematological manifestations of a proband are significantly correlated with that of affected siblings in a larger validation sibling cohort, we computed the Spearman rank correlation coefficient on all evaluable sibling pairs in the IFAR. This analysis confirms a significant correlation in the onset of haematological manifestations (see methods for definition) between siblings (r=0.588, p<0.0001, n=55 pairs) (Fig 1H). We also examined whether similar haematological course observed in our sibling study is just due to the nature of the underlying disease, FA. To address that, we tested whether the age differences at the onset of haematological manifestation were smaller between sibling pairs than between non-sibling pairs. We used a median age from 604 subjects from the non-sibling cohort for calculation of age differences with that of 110 subjects in the validation sibling cohort. This analysis showed that there was a median of 0.12-year (95% CI: −1.07 – 1.27) age difference at the onset of haematological manifestation between siblings, while a median of 0.58-year (95% CI: −0.18 – 1.2) age difference was observed between non-siblings (Fig 1I).
DISCUSSION
Siblings with FA share multiple factors that may influence disease course, such as epigenetic factors, causative genetic variants, diet and social environment. Siblings also share not only the FANC genotype, but also may share other genotypes that may influence the FA disease phenotype. For example, FA patients with an ALDH2*2 variant allele have earlier presentation of BMF or MDS/AML12, presumably due to increased DNA damage produced by un-metabolized aldehydes. It is possible that siblings also share such adverse or protective variants outside of the FA pathway, which subsequently either accelerate or delay the onset of haematological manifestations. Therefore, we hypothesized that siblings have similar clinical course because they share same causative variants and/or environmental factors. Indeed, our analyses showed significant correlation in the onset of various haematological manifestations in the discovery cohort. This similarity is confirmed by assessment of the validation cohort. We note that the age at onset of haematological manifestations was slightly younger in the discovery cohort, which may be due to the fact that these subjects were referred to a specialized FA center due to more severe disease and/or more frequent monitoring at these centers.
This is the largest report showing that patients with FA and their affected siblings are likely to have similar haematological course. This is important in counselling of parents with multiple affected children. Additionally it can help in anticipatory guidance and planning of clinical care including timing of transplant for the affected sibling. Being prepared helps parents and patients deal with their clinical course better and also may improve their compliance with the treatment. Congenital anomalies did not occur in the same pattern in siblings except for kidney abnormalities and microcephaly to a moderate degree. The lack of strong correlation in the most developmental abnormalities between sibling pairs is consistent with stochastic events leading to specific stem cell death during development. These findings now formally support the clinical observations that significant difference in size/growth retardation and number of congenital anomalies is observed between affected siblings from the same family. The presence of a proband with MDS or AML was not predictive of development of MDS/AML in affected siblings, however, this analysis is limited by small sample sizes, rare events especially for AML as well as age differences between siblings. Interestingly, common clonal acquisitions (abnormalities of chromosome 1,3 and 7)13,14 observed in most patients with FA point to the possibility of stochastic events leading to clonal evolution, which could be confirmed in future studies in larger number of sibling pairs. Limitations of our study include its retrospective nature resulting in a possible loss of data and limited sample sizes, and descriptive analyses. However, it provides the contextual basis for the future studies addressing underlying mechanisms of clinical similarities for hematological manifestations and dissimilarities for congenital anomalies and clonal evolution between siblings.
Supplementary Material
Table SI. Demographics of the discovery cohort
ACKNOWLEDGEMENTS
We thank the individuals and the families who participated in this study. This work was supported by grants from the National Heart Lung and Blood Institute (R01 HL120922) (A.S.), (K99 HL150628) (M.J.) National Cancer Institute (R01 CA204127) (A.S.), National Center for Advancing Translational Sciences (UL1 TR001866) (M.J., C.S.J., and A.S.), American Society of Hematology Scholar Award (M.J.). A.S. is a HHMI faculty scholar. F.B. acknowledges support from the MSK Cancer Center Support Grant P30 CA008748.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
REFERENCES
- 1.Kutler DI et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101, 1249–1256, doi: 10.1182/blood-2002-07-2170 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Shimamura A & Alter BP Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 24, 101–122, doi: 10.1016/j.blre.2010.03.002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butturini A et al. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood 84, 1650–1655 (1994). [PubMed] [Google Scholar]
- 4.Rosenberg PS, Huang Y & Alter BP Individualized risks of first adverse events in patients with Fanconi anemia. Blood 104, 350–355, doi: 10.1182/blood-2004-01-0083 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Kutler DI et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg 129, 106–112 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Alter BP, Giri N, Savage SA & Rosenberg PS Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39, doi: 10.3324/haematol.2017.178111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jung M et al. Association of clinical severity with FANCB variant type in Fanconi anemia. Blood, doi: 10.1182/blood.2019003249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niraj J, Farkkila A & D'Andrea AD The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478, doi: 10.1146/annurev-cancerbio-030617-050422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang AT et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell 59, 478–490, doi: 10.1016/j.molcel.2015.07.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toraldo R et al. Variable response to the diepoxybutane test in two dizygotic twins with Fanconi's anemia and flow cytometry for diagnosis confirmation. Pediatr Hematol Oncol 15, 45–54 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Koc A, Pronk JC, Alikasifoglu M, Joenje H & Altay C Variable pathogenicity of exon 43del (FAA) in four Fanconi anaemia patients within a consanguineous family. Br J Haematol 104, 127–130, doi: 10.1046/j.1365-2141.1999.01156.x (1999). [DOI] [PubMed] [Google Scholar]
- 12.Hira A et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 122, 3206–3209, doi: 10.1182/blood-2013-06-507962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta PA et al. Numerical chromosomal changes and risk of development of myelodysplastic syndrome--acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet Cytogenet 203, 180–186, doi: 10.1016/j.cancergencyto.2010.07.127 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Tonnies H et al. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood 101, 3872–3874, doi: 10.1182/blood-2002-10-3243 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Demographics of the discovery cohort