

Abstract
The risk factors for prostate cancer include a high-fat diet and obesity, both of which are associated with an altered cell environment including increased inflammation. It has been shown that chronic inflammation due to a high-fat diet or bacterial infection has the potential to accelerate prostate cancer as well as its precursor, prostatic intraepithelial neoplasia (PIN), development. However, the underlying mechanism of how chronic inflammation promotes prostate cancer development, especially PIN, remains unclear. In this study, we showed that more macrophages were present in PIN areas as compared to the normal areas of human prostate. When co-culturing PIN cells with macrophages in 3D, more PIN cells had nuclear localized cyclin D1, indicating that macrophages enhanced PIN cell proliferation. We identified ICAM-1 and CCL2 as chemoattractants expressed by PIN cells to recruit macrophages. Furthermore, we discovered that macrophage-secreted cytokines including C5a, CXCL1, and CCL2 were responsible for increased PIN cell proliferation. These three cytokines activated ERK and JNK signaling in PIN cells through a ligand-receptor interaction. However, only blockade of ERK abolished macrophage cytokines-induced cell proliferation of PIN. Overall, our results provide a mechanistic view on how macrophages activated through chronic inflammation can expedite PIN progression during prostate cancer development. The information from our work can facilitate a comprehensive understanding of prostate cancer development which is required for improvement of current strategies for prostate cancer therapy.
Keywords: macrophage, migration, CXCL1, CCL2, C5a, prostate intraepithelial neoplasia, cell growth, 3D culture
INTRODUCTION
Over the years, prostate cancer has maintained its position as the second most commonly diagnosed form of cancer in men only succeeded by skin cancer. Approximately 175,000 men are diagnosed with prostate cancer annually with the median age of diagnosis coming in at 65 years (1). Fortunately, early detection and isolation of the cancer to the prostate region leads to a five year survival rate of nearly 100%. On the other hand, prostate cancer that relocates to other regions of the body, including bone and lymph node, significantly decreases the chances of survival. Once the cancer begins its migration to other areas of the body, the 5 year survival rate drops to 30% and accounts for the second leading cause of cancer death in men in the United States making early detection critical (1).
Normal prostate glands consist of prostatic ducts lined with epithelium and a stroma consisting of smooth muscle cells. Benign tumor formation begins with increased expression of fibroblasts and myofibroblasts and decreased expression of smooth cell markers within the stroma. The increase in fibroblasts and myofibroblasts causes heavy modification to the extracellular matrix while simultaneous interaction of tumorous epithelial cells creates a safe microenvironment for tumor maturation (2–4). This abnormal neoplastic, precancerous growth of the epithelial cells lining the internal and external surfaces of the prostate gland is known as prostatic intraepithelial neoplasia or PIN, and is considered the precursor to prostate cancer (5, 6).
Although PIN is not considered cancer but rather a precursor lesion, it does require essential features which are conducive for tumor growth, proliferation and maturation. PIN formation is also considered a necessary step in the development of prostate cancer and tumor construction. PIN, based on a diagnostic measure, can be divided into two different categories, low-grade PIN and high-grade PIN (7). As the epithelial cells lining the acini and ducts of the prostate become increasingly abnormal, the transition from low-grade PIN to high-grade PIN, where cells have taken on new characteristics, becomes more visible. Furthermore, high-grade PIN can provide the proper environment for tumor cells to grow and proliferate (7–9).
Cell microenvironment is critical to tumor initiation and development. So far, little is known about the role of inflammation during PIN development. What is known, however, is that immune cells including macrophages secrete cytokines and chemokines which influence the cells surrounding the prostate epithelial cells. Within the prostate cancer area hematopoietic cells are recruited to the area by various signaling factors and are polarized into pro-inflammatory or anti-inflammatory macrophages (10, 11). In return an influx of pro-inflammatory cytokines and chemokines are released from the newly differentiated macrophages creating areas of inflammation (12). Meanwhile, fibroblasts and myofibroblasts within the PIN area cause heavy modification to the extracellular matrix. Consequently, the ECM modification coupled with dense areas of inflammation create an impregnable barrier that prevents cytotoxic T cells from exerting their effects on the tumorous cells. Furthermore, the tumor microenvironment is considered hypoxic (13–15). Lack of oxygen starves the tumor of essential nutrients leading to the recruitment of new blood vessels being formed by growth factors that are secreted by the same pro-inflammatory macrophages in the PIN area.
Altogether, the components of the PIN area work in harmony to provide an environment conducive for tumor growth. The abundance of cytokines, chemokines and growth factors within the PIN area are thought to provide adequate stimulation for tumor progression and enhance cellular proliferation as it relates to cancer. Our current study demonstrates the importance of macrophages within the PIN area and the role they play in tumor development. We show that macrophages were able to increase the numbers of nuclear cyclin D1 positive PIN cells, suggesting macrophages play a role in expediting cell proliferation. Furthermore, later tests showed that PIN cells secreted chemoattractants, notably ICAM-1 and CCL2 cytokines, to attract macrophages to the area. We also demonstrated that macrophage-secreted cytokines including C5a, CXCL1 and CCL2 function as a signaling stimulus which act to enhance or decrease various pathways including JNK and ERK which have been noted for their function in cellular proliferation. However, only blockade of ERK decreased PIN cell proliferation indicating the role of ERK signaling in regulating macrophage-stimulated cell proliferation of PIN. Altogether, our work provided a mechanistic view on how macrophages are recruited by PIN cells to expedite PIN cell proliferation during the prostate tumor progression.
RESULTS
Infiltration of macrophages in human prostate intraepithelial neoplasia and their effect on PIN cell proliferation
Accumulating evidence has indicated a pivotal role for the cell microenvironment in prostate disorders including prostate cancer. The cell microenvironment is influenced by surrounding immune cells which secrete cytokines, chemokines and growth factors that regulate prostate epithelial cells during cancer initiation, development and dissemination. To determine if macrophages are the source of cytokines or chemokines during the development of prostate intraepithelial neoplasia, or PIN, we utilized immunohistochemistry in an attempt to evaluate macrophage density in human tissues of normal prostate and PIN (Fig. 1A). Increased macrophage infiltration was observed in the PIN area as compared to the normal prostate tissue samples, suggesting a potential role for macrophage contribution to prostate cancer development. To test the effect of macrophages on PIN cell proliferation, we plated murine Pr111 PIN cells (16) on matrigel in 3D co-cultured with or without primary macrophages for 72 h. At the endpoint, Pr111 cells were fixed in 3D and then stained for cyclin D1, a surrogate marker for cell proliferation (17–19), for immunocytochemistry analysis (Figs. 1B and S1). As shown in Fig. 1B, co-cultured with primary macrophages, more Pr111 PIN cells were positive for nuclear cyclin D1 as compared to when Pr111 cells were cultured alone. The increased fold of nuclear cyclin D1 of Pr111 cells cultured with primary macrophages was almost exactly same as that of Ki-67 as the cell proliferation indicator (Fig. S1B), indicating that nuclear cyclin D1 is a compatible cell proliferation indicator as to Ki-67. When Raw264.7 macrophages were used to co-culture with Pr111 cells, we detected the same effect of Raw264.7 macrophages on increased numbers of nuclear cyclin D1-positive Pr111 cells (Figs. 1C and S2). Furthermore, the co-cultured Raw264.7 macrophages were also nuclear cyclin D1 positive, suggesting that these macrophages were proliferating under the co-culture conditions (Fig. S3). Altogether, our results suggest that infiltrating macrophages in the PIN area expedite PIN cell proliferation.
Figure 1. Infiltrating macrophages in the prostate intraepithelial neoplasia promote cell proliferation.

(A) Normal prostate and prostate cancer sample containing the area of prostate intraepithelial neoplasia (PIN) of human tissues were immunostained with the antibody of CD68, a marker for macrophages (Top Panel). The tissue samples were also stained with hematoxylin and eosin (H&E) to visualize the morphology (Bottom Panel). Scale bar: 50 μm. The PIN figure shown is representative of five human tissue samples. (B) Murine PIN Pr111 cells were cultured with or without primary peritoneal macrophages on matrigel in 3D. At the endpoint, the cells were fixed, permeabilized and immunostained with cyclin D1 (green), a surrogate marker for cell proliferation. Cell nuclei were stained by DAPI (blue). Cell proliferation index of Pr111 cells under these two conditions was quantified. Scale bar: 20 μm, *: p<0.05 as compared to control (without primary macrophages; student t test), n=3 (C) Murine PIN Pr111 cells were cultured with or without Raw264.7 macrophages on matrigel in 3D. At the endpoint, the cells were fixed, permeabilized and immunostained with cyclin D1 (green), a surrogate marker for cell proliferation. Cell nuclei were stained by DAPI (blue). Cell proliferation index of Pr111 cells under these two conditions was quantified. Scale bar: 20 μm. The data was analyzed by student t test. *: p<0.05 as compared to control (without Raw264.7 macrophages), n=5.
PIN cells expressed chemoattractants that mediate macrophage infiltration
Because macrophages were infiltrating into the PIN areas of human prostate (Fig. 1A), we investigated whether or not PIN cells are able to attract macrophages. We first in vivo labeled murine Pr111 cells and Raw264.7 cells with Vybrant DiI (Red) and Vybrant DiO (green) respectively, and then seeded each type of cells in 3D in the separate compartments of a co-cultivation system (Fig. 2A, left panel). Seventy-two hours post co-culture, we detected macrophage clusters physically attached to PIN cells (Fig. 2A, right panel). Our observation shows that macrophages were attracted to PIN cells but through an unknown mechanism. We determined one possibility may be through chemoattractants expressed by PIN cells resulting in macrophage migration. To identify the potential chemoattractants that mediate macrophage attraction toward Pr111 PIN cells, we subjected the collected conditioned media of our 3D co-culture system and the control media to cytokine profiler arrays. Among 40 tested cytokines, only five cytokines including C5a, ICAM-1, CXCL1, CCL2 and TMP-1 were present in the co-culture media (Fig. 2B). Among these 5 identified cytokines, C5a, ICAM-1 and CCL2 have been reported to function as chemoattractants for immune cells (20–26). To determine if these chemokines were expressed by PIN cells resulting in macrophage attraction, we treated our 3D co-culture system of Pr111 cells and macrophages with the neutralizing antibodies specifically against C5a, ICAM-1 or CCL2 followed by attraction assay (Fig. 2, C and D). Blockade of C5a using the C5a neutralizing antibody had no effect on macrophage attraction (Fig. 2C). Instead, treatment of ICAM-1 neutralizing antibody, CCL2 neutralizing antibody or both significantly decreased macrophage infiltration to PIN cells (Fig. 2D). Furthermore, results from immunohistochemistry analysis indicated that expression of ICAM-1 and CCR2 was increased in PIN cells of the human prostate in comparison to the normal prostate (Fig. 2, E and F).
Figure 2. ICAM-1 and CCR2 expressed by PIN mediate cell attraction between macrophages and PIN cells.
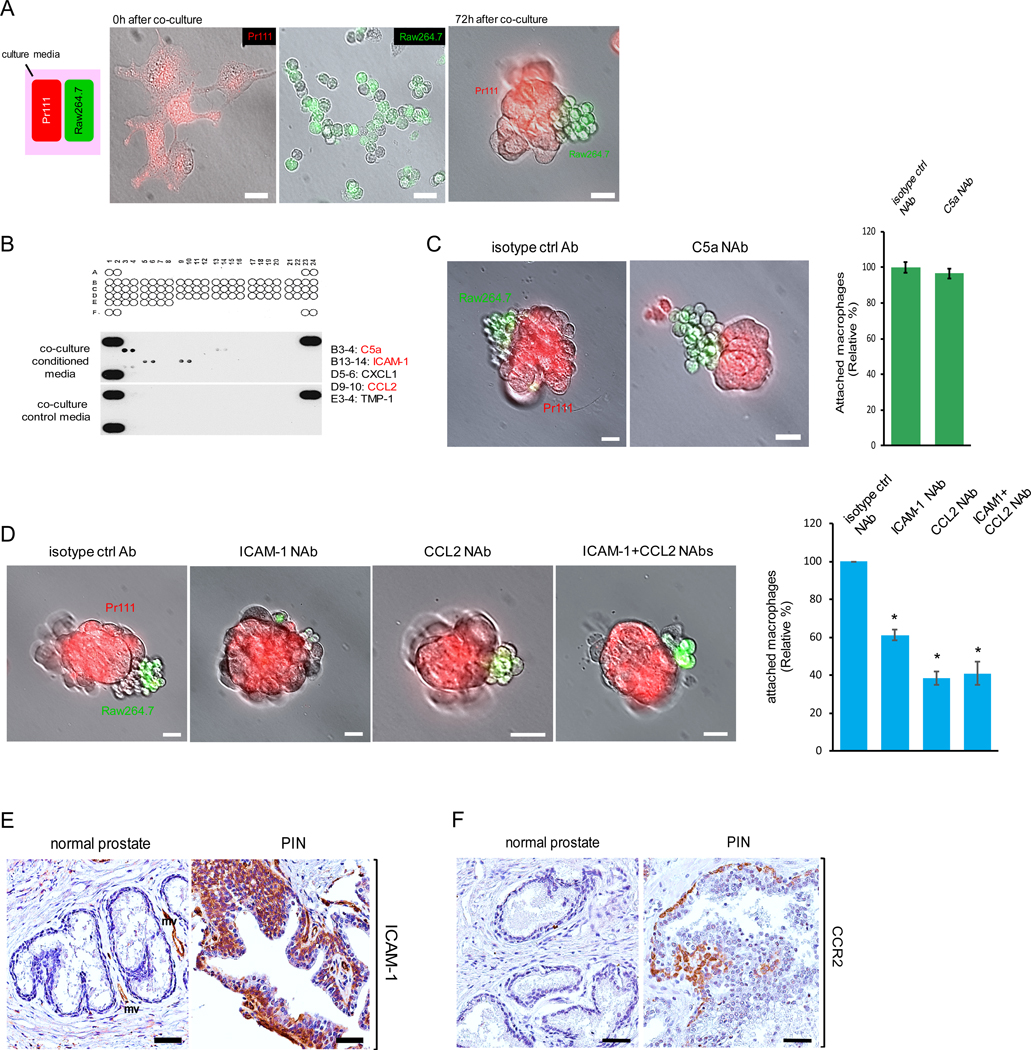
(A) A diagram for demonstrating cell co-cultivation of Pr111 cells and Raw264.7 macrophages that were in vivo labeled with Vybrant DiI (Red) and Vybrant DiO (green) respectively. Cells were plated on matrigel in 3D in μ-Slides (Ibidi) and overlaid with PrEBM complete media. Fluorescent and bright field pictures of Pr111 and Raw264.7 cells were taken at 0 h and 72 h of co-culture. Scale bar: 25 μm. n=3. (B) Raw 264.7-coculture media and control media were subjected to a mouse cytokine array for identifying the cytokines present in Raw 264.7-coculture media. A1/2, A23/24, and F1/2 contain positive controls, and F23/F24 contains negative controls. (C-D) Similar to A, Pr111 cells grown on matrigel in 3D were co-cultured with Raw264.7 cells in the presence of isotype control antibody or the neutralizing antibody against the cytokines identified from Raw 264.7-coculture including C5a (C), ICAM-1 and CCL2 (D). The data shown in C-D is representative of 3 independent experiments. Cell attraction between Pr111 and Raw264.7 cells under these treatments were evaluated 72 h post co-culture and quantified. Scale bar: 25 μm. The data was analyzed by one-way ANOVA with Tukey-Kramer post-hoc test. *: p<0.05 as compared to isotype control NAb, n=3. (E-F) Normal prostate and prostate cancer sample containing PIN of human tissues were immunostained for expression of ICAM-1 (E) and CCR2 (F). The figure shown is representative of three human tissue samples. Scale bar: 50 μm.
Macrophage cytokines promoted PIN cell proliferation without affecting apoptosis
In addition to macrophage attraction, macrophages also potentiated murine PIN Pr111 cell proliferation as shown in Fig 1B. To evaluate if the identified cytokines secreted from macrophages can also regulate cell proliferation of PIN, we treated the Pr111 cells in 3D setting with recombinant C5a. At the endpoint, we utilized immunocytochemistry to assess the nuclear cyclin D1 localization as a readout for cell proliferation to avoid any artificial effects from disrupting the 3D setting. Exogenous addition of recombinant C5a increased Pr111 cell proliferation (Fig. 3A). The same effects were also detected in the cells treated with either recombinant CCL2 or CXCL1 (Fig. 3, B and C). Interestingly, when treating cells with either each of the recombinant C5a, CCL2, CXCL1, in a combination of either one or all three cytokines, all of them were able to promote PIN cell proliferation (Fig. 3D). Notably, there were no additive promoting effects of cell proliferation in the presence of all three cytokines in our 3D co-culture setting, suggesting that these cytokines use similar pathways to regulate cell proliferation of PIN. An increased expression of CD88 and CCR2, the receptor for C5a and CCL2 respectively, was present in the human tissue samples of PIN (Fig. 3E, and Fig. 2F). Furthermore, the cultured Pr111 PIN cells expressed CXCR2, the receptor for CXCL1, and CCR2, the receptor for CCL2 (Fig. S4). Meanwhile, elevated expression of C5a and CCL2 mRNA levels were in Raw264.7 macrophages cultured in PIN media but not in the control media (Fig. S5). It has been reported that CXCL1 is associated with anti-apoptosis in the prostate epithelial cells (27). To test the possibility that these identified macrophage cytokines including C5a, CXCL1 and CCL2 may affect apoptosis of PIN cells, we examined the presence of cleaved poly-(ADP-ribose) polymerase (PARP) which is a product of activated caspases during apoptosis in our system (28–30). As shown in Fig. S6, Pr111 PIN cells treated with C5a, CXCL1 or CCL2 under the 3D culture setting had no effect on apoptosis as compared to control cells, whereas NIH3T3 cells treated with staurosporine (STP), which is well-documented to induce apoptosis (31, 32), underwent apoptosis as indicated by the presence of the cleaved fragment of PARP. Altogether, these results suggested that C5a, CXCL1 and CCL2 secreted by macrophages were capable of regulating PIN cell proliferation without affecting apoptosis through a ligand-receptor interaction.
Figure 3. Macrophage cytokines C5a, CCL2 and CXCL1 upregulate cell proliferation of PIN cells.

(A-C) Murine PIN Pr111 cells grown on matrigel in 3D were treated with the cytokines identified from Raw264.7-coculture media including C5a (A), CCL2 (B) and CXCL1 (C) at the indicated concentrations. 72 h post-treatment, the cells were fixed, permeabilized and immunostained with cyclin D1 (green). The cell nuclei were visualized by DAPI staining (blue). Cell proliferation index of Pr111 cells under these treatments was quantified. Scale bar: 25 μm, *: p<0.05 as compared to control (student t test for C5a (A); one-way ANOVA and multiple comparisons for CXCL1 (B) and CCL2 (C)), n=5. (D) Similar to A, Pr111 cells were treated with either the identified cytokines alone or any combination of the identified cytokines as indicated. Cell proliferation index under these conditions was quantified. The data was analyzed by one-way ANOVA with Tukey-Kramer post-hoc test. *: p<0.05 as compared to the control/vehicle, n=3. (E) Normal prostate and prostate cancer sample containing PIN of human tissues were immunostained for CD88 expression. The figure shown is representative of three human tissue samples. Scale bar: 50 μm.
Blockade of macrophage cytokines attenuated PIN cell proliferation
Next we evaluated the effects of the macrophage-secreted cytokines that were identified from our 3D co-cultured experiments on PIN cell proliferation by blocking each individual cytokine through their specific neutralizing antibody. Murine Pr111 PIN cells were cultured in matrigel in 3D and stimulated with the macrophage conditioned media in the presence of either isotype matched control antibody or C5a neutralizing antibody/C5a NAb (Fig. 4A). Treatment of C5a NAb reduced the number of Pr111 cells that had nuclear cyclin D1, a surrogate marker for cell proliferation, suggesting that blockade of C5a diminished macrophage-induced PIN cell proliferation. Similarly, neutralization of either CXCL1 or CCL2 through the CXCL1 NAb and CCL2 NAb also attenuated macrophage-caused cell proliferation of Pr111 PIN cells (Fig. 4, B and C). However, when treated with all three neutralizing antibodies including C5a, CXCL1 and CCL2, there was no additive inhibitory effect on PIN cell proliferation in comparison to each individual NAb (Fig. 4D). Altogether, these results further bolster our previous conclusion from the experiments which we stimulated PIN cells with recombinant cytokines, C5a, CXCL1 and CCL2 (Fig. 3).
Figure 4. Blockade of C5a, CCL2 or CXCL1 attenuate PIN cell proliferation induced by macrophage conditioned media.

(A-D) Pr111 cells grown on matrigel in 3D were treated with Raw264.7 macrophage conditioned media in the presence of isotype control antibody or the neutralizing antibody of C5a (A), CXCL1 (B), CCL2 (C) or all three (D) for 72 h. The cells were fixed, permeabilized and immunostained with cyclin D1 (green). The cell nuclei were stained and visualized with DAPI (blue). Cell proliferation index of Pr111 cells under these conditions was quantified. Scale bar: 25 μm. The data was analyzed by student t test. *: p<0.05 as compared to isotype control antibody, n=3.
Macrophage cytokines activated ERK and JNK in PIN cells
Several signaling pathways are well-known regulators of cell proliferation including ERK, Akt, NF-κB, JNK and p38 MAP kinase (17, 33–40). To test which signaling pathways are activated by the macrophage cytokines, C5a, CXCL1 and CCL2 which promote PIN cell proliferation, we isolated Pr111 cells that were stimulated by C5a, CXCL1 or CCL2 from our 3D culture system and then subjected them to immunoblotting analysis for the pathways described above. Macrophage cytokine C5a elevated phosphorylated ERK1/2, but had no effect on phosphorylated Akt, suggesting that C5a activates the ERK pathway (Figs. 5A and S7A). Activation of NF-κB transcription factor requires the phosphorylation of its inhibitory suppressor protein IκBα followed by the IκBα degradation through proteasome machinery (41, 42). As judged by the levels of phosphorylated IκBα, which is a readout for activation of NF-κB, treatment of C5a had no effect on NF-κB pathway in Pr111 cells (Fig. 5B). Similarly, C5a did not activate p38 MAP kinase and only slightly increased phosphorylated JNK (Figs. 5C and S7C). Meanwhile, both macrophage cytokines CXCL1 and CCL2 activated ERK but slightly decreased phosphorylated Akt (Figs. 5D and S7B). Neither cytokines, CXCL1 or CCL2, activated NF-κB nor p38 MAP kinase pathways (Fig. 5, E and F). Furthermore, these two cytokines also activated JNK, especially in CCL2-treated Pr111 cells. Among these examined pathways, ERK activation was the strongest one in Pr111 PIN cells in our 3D culture system. We next utilized immunohistochemistry to analyze phospho-ERK levels in the human tissue samples to vindicate our in vitro results. As shown in Fig. 5G, elevated levels of phosphorylated ERK were detected in the PIN areas of prostate cancer patients compared to normal prostate. Meanwhile, little difference was observed among the activated Akt expression between the PIN areas and the normal prostate tissue (Fig. S8). These results further support our in vitro data for ERK activation in PIN cells.
Figure 5. Activation of ERK and JNK in PIN cells stimulated by macrophage cytokines C5a, CCL2 or CXCL1.

(A-C) Pr111 cells grown on matrigel in 3D were treated with either control/ddH2O or recombinant C5a for 72h. Cell lysates were collected and subjected to immunoblotting for examining the protein of interests including p-ERK, ERK, p-AKt and Akt (A); p-IκBα (B); p-JNK, JNK, p-p38 MAPK and p-38 MAPK (C). (D-F) Similar to A-C, Pr111 cells cultured on matrigel in 3D were treated with control, recombinant CXCL1 or CCL2. Cell lysates were collected from 3D culture and subjected to immunoblotting for examining the levels of p-ERK, ERK, p-AKt and Akt (D); p-IκBα (E); p-JNK, JNK, p-p38 MAPK and p-38 MAPK (F). The relative intensity fold change was calculated by the ratio of phosphorylated protein levels over the corresponding total protein levels and set as 1 for control in each case. Each image shown in A-F was representative of 3–5 independent experiments. (G) Normal prostate and prostate cancer sample containing PIN of human tissues (n=3 per group) were immunostained for phosphorylated-ERK expression. Scale bar: 50 μm.
Effect of blockade of ERK or JNK on PIN cell proliferation
Our immunoblotting results suggested differential intensity of ERK and/or JNK activation in Pr111 PIN cells in response to macrophage cytokines, C5a, CXCL1 and CCL2 stimulation in the 3D setting. To test if these two signaling pathways are responsible for PIN cell proliferation, we treated Pr111 PIN cells with either ERK inhibitor U0126 or JNK inhibitor SP600125 in our 3D culture system in the presence of C5a, CXCL1 or CCL2 (Fig. 6, A and B). When treated with recombinant C5a, CXCL1 or CCL2, macrophage cytokines, an increase of Pr111 cell proliferation was detected and confirmed by the nuclear cyclin D1 localization (Fig. 6A). In addition, the increased cell proliferation was abolished by treating cells with U0126, the ERK inhibitor. Interestingly, blockade of JNK activation through SP600125 only impaired C5a-, but not CXCL1- nor CCL2-induced Pr111 cell proliferation (Fig. 6B). Altogether, these results suggested that macrophage-secreted cytokines including C5a, CXCL1 and CCL2 regulate PIN cell proliferation mainly through activation of ERK.
Figure 6. Macrophage-secreted cytokines promote PIN cell proliferation through activation of ERK but not JNK.

(A-B) U0126, a specific ERK inhibitor (A) or SP600125, a specific JNK inhibitor (B) was added to 3D cultures of murine PIN Pr111 cells that were treated with either control/ddH2O, C5a, CXCL1 or CCL2 for 72 h. Vehicle (DMSO) was used as control. Cell proliferation index under these conditions was quantified. The data was analyzed by one-way ANOVA with Tukey-Kramer post-hoc test. *: p<0.05 as compared to the control+vehicle, **: p<0.05 as compared to vehicle+individual cytokine, n=3. (C) A scheme to summarize how macrophages are attracted to PIN cells to promote cell proliferation of PIN.
DISCUSSION
The cell environment plays a critical role in the initiation and development of cancer. With different types of cells such as immune cells, endothelial cells, fibroblasts etc. being nearby, the dynamic interactions between these cells and epithelial cells determine tumor onset and progression. Although there is a discrepancy of clinical reports in regards to the relationship between inflammation and prostate cancer (43–46), several lines of evidence from laboratory bench work and animal studies have indicated an inflammatory environment during prostate cancer development (47–51). Xenografting of the immortalized non-tumorigenic RWPE-1 cells with THP-1 monocytes in nude mice were able to develop tumors due to an epithelial-mesenchymal transition (EMT) of non-tumorigenic prostate epithelial cells by the androgen receptor (AR) of THP-1 monocytes (47). In addition, knockout of AR in macrophages reduced PIN formation in PTEN+/− transgenic mice. Intra-urethral administration of bacteria E.Coli in the C3H/HeJ mouse model induced severe chronic inflammation and subsequent prostate epithelial dysplasia which mimic high grade PIN (48). Furthermore, the dysplastic cells possessed elevated cell proliferation through a decrease of CDK inhibitor p27Kip1, high levels of oxidative DNA damage and loss of AR and PTEN. Our current work demonstrated that Raw264.7 macrophages promoted cell proliferation of murine Pr111 PIN cells that were established from low grade PIN lesions in C3(1)/Tag transgenic model of prostate cancer in a 3D co-culture (Fig 1), thus providing the first direct evidence on the role of inflammation in PIN development and progression. Although there is emerging evidence supporting the notion that chronic inflammation leads to prostate cancer development, whether prostate epithelial cells play a role in modulating this process remains completely unknown. With the live labeling dye technique to distinguish macrophages and PIN cells, we showed that prostate epithelial tumor cells attracted macrophages through ICAM-1 and CCR2 (Fig. 2). This chemoattraction allows more macrophages to be recruited to the areas where PIN cells are and creates a proximity for the macrophages-secreted cytokines including C5a, CXCL1 and CCL2 to activate the cell signaling of PIN cells (Fig. 5).
For the first time, we have identified the cytokines C5a, CXCL1 and CCL2 secreted by macrophage from a 3D co-culture system with PIN Pr111 cells. We also showed that each of these cytokines activated the same signaling pathways in PIN cells to enhance PIN cell proliferation during prostate tumor development and progression. Notably, the specific receptors CD88, CXCR2 and CCR2 for macrophage cytokine C5a, CXCL1 and CCL2 are present in PIN cells (Fig. 2F, 3E and S2), suggesting a ligand-receptor mechanism used by PIN cells to activate the downstream portion of the cell signaling in response to these macrophage cytokines. Among the three identified macrophage cytokines, C5a anaphylatoxin is a cytokine-like polypeptide produced during complement system activation and released at the inflammatory site. Knowledge surrounding cytokine involvement in prostate cancer initiation and precancerous lesion growth has never been fully explained. However, our analysis demonstrates that C5a plays a role in expediting PIN cell proliferation during prostate tumor development. In addition to regulating cancer proliferation, metastasis, angiogenesis and apoptosis, the most well-known function of C5a during cancer development is to attune the microenvironment such as activation of myeloid-derived suppressor cells (MDSC) for cancer cells to prosper (52–55).
Previously, CXCL1 and CCL2 had not been reported to function as macrophage cytokines in the prostate cancer field. However, both cytokines were shown to be upregulated in the prostate cancer cells and controlled cancer cell proliferation and dissemination. Elevated expression of CXCL1 was detected in high-grade prostate cancer (56). It has also been shown that prostate cancer patients who are obese expressed high levels of CXCL1 in their prostate tissues (57). Moreover, blockade of CXCL1 signaling through shRNA or neutralizing antibody impeded the recruitment of adipose stromal cells to the prostate cancer and vascularization in the cancer areas that promote cancer growth. Intriguingly, a high-fat diet not only potentiates prostate cancer growth but also hinders the survival rate of the transgenic adenocarcinoma mouse prostate (TRAMP) and TRAMP-C2 allograft mice (58). CCL2 is upregulated in prostate cancer cells and smooth muscle cells of the prostate gland (59–61). Furthermore, a very large body of evidence has demonstrated its role in promoting prostate cancer cell proliferation, migration, bone metastasis and drug resistance (60, 62–67). This also includes evaluations for the use of CCL2 neutralizing antibody as adjuvant therapy to treat prostate cancer (62, 68).
Our current work showed the activation of ERK and JNK in the PIN cells when stimulated by the macrophage-secreted cytokines C5a, CXCL1 or CCL2 (Fig. 5). However, only blockade of ERK completely impeded macrophage cytokines-induced cell proliferation of PIN (Fig. 6), indicating the function of ERK signaling in promoting PIN cell growth adding a new pathway for modulating PIN progression. In addition to our findings, there are several signaling pathways reported to regulate PIN formation. Overexpression of prostate-specific G-protein-coupled receptor (PSGR) via a probasin promoter in prostate cells induced chronic inflammation as well as subsequent PIN formation in mice (69). In addition, more CD68+ macrophages were detected in the PSGR-induced PIN areas and activation of NF-κB in PIN cells in response to PSGR expression. In addition to elevated expression of cyclooxygenase (COX)-2 and prostaglandin E2 (PGE2) in PIN, treatment of a human PIN cultured cell line with PGE2 caused soluble IL-6 receptor release, gp130 dimerization, STAT3 phosphorylation and eventually accelerated cell proliferation (70). It has been shown that using the morphoproteomic analysis technique to examine the human prostate cancer and high grade PIN in the tissue microarray, activation of mTOR, a protein kinase that regulates protein synthesis and cell growth, was elevated in both cancer and PIN samples, implicating the role of the Akt/mTOR signaling pathway in PIN growth (71). Dysregulation of Notch signaling which controls cell proliferation and differentiation during embryogenesis and adult tissues has been tightly associated with cancer development. Interestingly, it has been reported that higher levels of all members of Notch signaling including Notch1–4, Jagged1–2, Delta, HES1 and HES5 were in high grade PIN of human tissue samples (72). In addition, Notch1, Notch4, HES1 and Jagged1 were the four most highly expressed proteins. Future studies for examining the role of these four Notch signaling molecules in PIN progression are required.
Materials and methods
Cell lines, Antibodies and Reagents
Raw 264.7 macrophage cells (ATCC (Manassas, VA)) were cultured in RPMI 1640 containing 10% FBS and 100 U/ml penicillin/streptomycin. Pr111 cells were cultured on collagen I-coated dishes and maintained as described previously (16). In brief, Pr111 cells were grown in PrEBM™ prostate epithelial cell growth basal media supplemented with MEGM singlequots, 2% FBS, 1 mM sodium Pyruvate, 10 nM dihydro-testosterone and 100 U/ml penicillin/streptomycin. Cells were maintained in a 37°C incubator supplemented with 5% CO2. The passage number of Pr111 cells was less than 10 passages in all experiments. To obtain Raw 264.7-conditioned media, 2.5 × 105 cells per 6-well dish were cultured in Pr111 complete media for 3 days, and supernatant was collected. Conditioned media were freshly prepared for each experiment. All neutralizing antibodies that target murine C5a, CXCL1, and CCL2 as well as the isotype control antibodies were purchased from R&D systems (Minneapolis, MN). Murine ICAM-1 neutralizing antibody and its isotype control antibody were from BioLegend (San Diego, CA). The anti-CD68 antibody was from DAKO (Santa Clara, CA); anti-cyclin D1 antibody was from Thermo Fisher Scientific (Waltham, MA); anti-ICAM-1 antibody was obtained from Abcam; anti-CD88 antibody was from Bio-Rad (Hercules, CA). The anti-CCL2 antibody was from Novus Biologicals; anti-Akt, anti-phospho-Akt (ser473), anti-ERK, anti-JNK, anti-phospho-JNK, anti-p38 MAPK, anti-phospho-p38 MAPK and anti-phospho-IκBα antibodies were purchased from Cell Signaling Technology (Danvers, MA). The anti-phospho-ERK1/2 antibody was from Santa Cruz Biotechnology (Dallas, TX). Mouse recombinant CXCL1 and CCL2 were purchased from PeproTech (Rocky Hill, NJ). Mouse recombinant C5a was from ProSpec (Rehovot, Israel). Matrigel was from Corning (Corning, NY). U0126 and SP600125 were obtained from Cell Signaling Technology and Tocris Bioscience (Bristol, United Kingdom) respectively. Other reagents used are described in the specific experiment sections.
Chemoattraction assays
Pr111 cells and Raw 264.7 macrophages were in vivo labeled with Vybrant DiI (Red) and Vybrant DiO (green) (Invitrogen (Carlsbad, CA)) under the manufacture’s instruction. Cells were separately seeded on top of 50% matrigel of ibid 2×9 well μ chamber slide. The chamber slide does not allow an initial physical contact between two cell types other than sharing the culture media. 2×103 Dil labeled Pr111 cells were seeded in the center of the chamber slide and surrounded by 2.5×103 DiO labeled Raw 264.7 cells in each separate space. The indicated specific neutralizing antibody or isotype control antibody with the final concentrations (C5a NAb: 2 ng/mL; ICAM-1 NAb: 0.625 μg/mL; CCL2 NAb: 18 ng/mL) was immediately added to the shared culture media after cell seeding. The NAbs and their matching isotype antibody were replenished every 24 h. After 72 h, macrophages migrated to the Pr111 cell clusters in the center of the chamber slide were visualized using a Zeiss Axiovert 200M inverted fluorescent microscope. The entire field of the center compartment of the chamber slide where Pr111 cell clusters were located was photographed and analyzed for the interaction of Pr111 and Raw 264.7 cells.
Isolation of Primary Macrophages
Primary murine macrophages were isolated as previously described (73, 74). In brief, mice were intraperitoneally-injected with 2 mL of 3–5% aged thioglycollate solution. On day 5 after injection, peritoneal macrophages were collected through a single injection of 10 mL RPMI-1640 containing 10% FBS into the peritoneal cavity and subsequent withdrawal. The peritoneal exudate was centrifuged and washed with RPMI-1640 media containing 10% FBS before plating onto tissue culture dishes. After 1 h at 37 °C incubator supplemented with 5% CO2, cells were vigorously washed with PBS three times to remove non-adherent macrophages and immediately used to co-culture with Pr111 cells in Pr111 complete media in 3D.
3D culture of Pr111 cells and Treatment
Pr111 single cells were plated on top of 50% matrigel and stimulated with the indicated recombinant proteins with the indicated concentrations or vehicle control. For Raw 264.7 macrophage conditioned media induced cell proliferation, the indicated specific neutralizing antibodies or isotype control antibodies with a final concentration of 1 μg/mL were immediately added to the culture after cell seeding. The NAbs and their matching isotype antibody were replenished every 24 h. For treatment, 2.5 μM U0126, 1M SP600125 or vehicle DMSO was added to the cells simultaneously with or without the indicated recombinant proteins after cells were seeded on matrigel.
Cell proliferation assay
The method has been described in our earlier publication (17). In brief, Pr111 cells grown in 3D matrigel culture with the indicated stimulation/treatment for 72 h were rinsed twice with PBS (140 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4 and 1.5 mM KH2PO4 [pH 7.2]), fixed with 4% paraformaldehyde-PBS for 20 min, permeabilized with 1% TritonX-100 in PBS for 10 min, 0.5% TritonX-100 in PBS for 20 min, 0.1% SDS in PBS for 1 min, and blocked in 10% goat serum 1 h at room temperature. Samples were incubated with anti-cyclin D1 antibody (1:250) overnight at 4 °C. Samples were washed three times with PBS containing 0.05% Tween-20. Secondary antibodies Alexa Fluor 488 goat-anti-rabbit (1:500) from Thermo Fisher Scientific were added for 30 min at room temperature. After another three washes of PBST, DAPI was used to label cell nuclei. Images were visualized in ibidi mounting media. Serial sections of images (Z-stack) were captured by a Zeiss Axiovert 200M inverted fluorescent microscope with a 20x objective lens. Each 3D image were reconstructed from Z-stack images. Numbers of nuclear cyclin D1 were counted in randomly five 3D images per condition. Cell proliferation index was calculated using the ratio: numbers of nuclear cyclin D1/numbers of all cells (DAPI).
Cellular lysates and Immunoblotting
Cells were washed twice with PBS, lysed in buffer A (50 mM Tris/HCl [pH 7.4], 1% TritonX-100, 150 mM NaCl, and 5 mM EDTA [pH 7.4]) or RIPA buffer (25 mM Tris/HCl [pH 7.5], 1% Triton X-100, 140 mM NaCl, 1 mM EDTA, 0.5% SDS) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific), incubated on ice for 30 min and centrifuged at 4 °C, 14000 rpm for 10 min. The supernatants were were subjected to SDS-PAGE, and resolved proteins were transferred onto nitrocellulose membranes. The membranes were blocked in 5% BSA in TBST (50 mM Tris.HCl [pH 7.6], 150 mM NaCl, 0.05% Tween 20) and incubated with the antibodies of interest in 5% BSA in TBST overnight at 4 °C. The appropriate horseradish peroxidase-conjugated 2° antibodies were applied for 30 min at room temperature. Samples were visualized with ECL and X-ray film.
Immunohistochemistry
Human prostate tissue slides were purchased from BioChain (Newark, CA), Origene (Rockville, MD) and Cooperative Human Tissue Network (CHTN). Slides were deparaffinized in 3 changes of xylene and gradually re-hydrated through enthanol to distilled water. The rehydrated slides subjected to 10 mM sodium citrate buffer (pH 6.0) and then rinsed with distilled water. Slides were treated with 3% hydrogen peroxide for 5 min to reduce endogenous peroxidase activity and washed with PBS containing 0.5% Tween-20. Slides were blocked with protein block serum free solution (DAKO) for 10 min at RT. Primary antibody (CD68 1:500 (DAKO); ICAM-1 1:50 (Abcam (Cambridge, United Kingdom)); CCR2 1: 1000 (Novus Biologicals (Littleton, CO)); CD88 1: 500 (Bio-Rad); phospho-ERK 1:150 (Santa Cruz Biotechnology) was incubated in antibody diluent background reducing solution (DAKO) and visualized using the ImmPRESS Polymer Detection Kit (Vector Laboratories (Burlingame, CA)) according to the manufacturer’s instructions. Images were captured using Aperio VERSA tissue scanner with ImageScope software (Aperio (Sausalito, CA)).
Cytokine array
The cytokines secreted by Raw 264.7 macrophages cultured in the Pr111 complete media for 3 days which is the co-culture condition, as previously described in the Cell Lines, Antibodies and Reagents, were examined using the Proteome Profiler Mouse Cytokine Array Kit (R&D Systems) according to the manufacturer’s instructions.
Statistical analysis
Data are presented as means ± SE. P-values were acquired with the Student’s t test for comparing 2 sets of data using Prism (GraphPad Software (San Diego, CA)). For more than 2 sets of data, one-way ANOVA analysis along with multiple comparisons Tukey-Kramer post-hoc test was carried out using Prism (GradPad Software). P < 0.05 is considered statistically significant.
Supplementary Material
Supplemental Figure S1. Increased proliferation of PIN Pr111 cells when co-cultured in 3D with primary macrophages.
Supplemental Figure S2. Pr111 were proliferative when co-cultured in 3D with Raw264.7 macrophage cells.
Supplemental Figure S3. Raw264.7 macrophages were proliferative in 3D co-culture with PIN Pr111 cells.
Supplemental Figure S4. Murine Pr111 cells expressed receptors CXCR2 and CCR2.
Supplemental Figure S5. Raw264.7 macrophages increased expression of C5a and CCL2 as cultured in co-culture media.
Supplemental Figure S6. Macrophage cytokines, C5a, CXCL1 and CCL2 had no effect on apoptosis in murine PIN Pr111 cells.
Supplemental Figure S7. Signaling of ERK and JNK in muirn PIN Pr111 cells stimulated by C5a, CXCL1 or CCL2.
Supplemental Figure S8. The expression of phosphorylated-Akt in the PIN area of human prostate.
ACKNOWLEDGEMENTS
Mikalah Thomas was partially supported by the NIH/NIMHD/RCMI program Grant No. 2U54MD007590–32 and by NIH RISE program Grant No. 5R25G06414–16.
This work was supported in full by the NIH/NIMHD/RCMI program Grant No. 2U54MD007590–32 and partially supported by the NIH RISE program Grant No. 5R25G06414–16. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- AR
androgen receptor
- CCL2
C-C chemokine ligand 2
- COX-2
cyclooxygenase 2
- CXCL1
C-X-C chemokine ligand 1
- ICAM-1
intracellular adhesion molecule-1
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor alpha
- IL-6
interleukin 6
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- ERK
extracellular signal-regulated kinase
- H&E
hematoxylin and eosin
- JNK
c-Jun N-terminal kinase
- MDSC
myeloid-derived suppressor cell
- NAb
neutralizing antibody
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PARP
poly(ADP-ribose) polymerase
- PGE2
prostaglandin E2
- PIN
prostate intraepithelial neoplasia
- PSGR
prostate-specific G-protein-coupled receptor
- PTEN
phosphatase and tensin homolog
- STAT3
signal transducer and activator of transcription 3
- STP
staurosporine
- TIMP-1
tissue inhibitor of metalloproteinases-1
- TRAMP
transgenic adenocarcinoma mouse prostate
Footnotes
Conflicts of Interest
No competing interests declared by the authors regarding the publication of this article.
REFERENCES
- 1.Oncology ASoC. Prostate Cancer: Statistics. 2019. [Google Scholar]
- 2.Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. 2019;18(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otranto M, Sarrazy V, Bonte F, Hinz B, Gabbiani G, Desmouliere A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6(3):203–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clin Cancer Res. 2002;8(9):2912–23. [PubMed] [Google Scholar]
- 5.Brawer MK. Prostatic intraepithelial neoplasia: an overview. Rev Urol. 2005;7 Suppl 3:S11–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Khani F, Robinson BD. Precursor lesions of urologic malignancies. . Arch Pathol Lab Med. 2017;141:1615–32. [DOI] [PubMed] [Google Scholar]
- 7.Brawer MK. Prostatic intraepithelial neoplasia: an overview. Rev Urol. 2005;7 Suppl 3:S11–8. [PMC free article] [PubMed] [Google Scholar]
- 8.De Marzo AM, Haffner MC, Lotan TL, Yegnasubramanian S, Nelson WG. Premalignancy in Prostate Cancer: Rethinking What we Know. Cancer Prev Res (Phila). 2016;9(8):648–56. [DOI] [PubMed] [Google Scholar]
- 9.Goeman L, Joniau S, Ponette D, Van der Aa F, Roskams T, Oyen R, et al. Is low-grade prostatic intraepithelial neoplasia a risk factor for cancer? Prostate Cancer Prostatic Dis. 2003;6(4):305–10. [DOI] [PubMed] [Google Scholar]
- 10.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penet MF, Kakkad S, Pathak AP, Krishnamachary B, Mironchik Y, Raman V, et al. Structure and Function of a Prostate Cancer Dissemination-Permissive Extracellular Matrix. Clin Cancer Res. 2017;23(9):2245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marignol L, Coffey M, Lawler M, Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev. 2008;34(4):313–27. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Schmid T, Schnitzer S, Brune B. Tumor hypoxia and cancer progression. Cancer Lett. 2006;237(1):10–21. [DOI] [PubMed] [Google Scholar]
- 16.Soares CR, Shibata MA, Green JE, Jorcyk CL. Development of PIN and prostate adenocarcinoma cell lines: a model system for multistage tumor progression. Neoplasia. 2002;4(2):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang T, Liou GY. Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt. Sci Rep. 2018;8(1):7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamamori-Adachi M, Ito H, Sumrejkanchanakij P, Adachi S, Hiroe M, Shimizu M, et al. Critical role of cyclin D1 nuclear import in cardiomyocyte proliferation. Circ Res. 2003;92(1):e12–9. [DOI] [PubMed] [Google Scholar]
- 19.Tong W, Pollard JW. Progesterone inhibits estrogen-induced cyclin D1 and cdk4 nuclear translocation, cyclin E- and cyclin A-cdk2 kinase activation, and cell proliferation in uterine epithelial cells in mice. Mol Cell Biol. 1999;19(3):2251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chintakuntlawar AV, Chodosh J. Chemokine CXCL1/KC and its receptor CXCR2 are responsible for neutrophil chemotaxis in adenoviral keratitis. J Interferon Cytokine Res. 2009;29(10):657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–9. [DOI] [PubMed] [Google Scholar]
- 23.DiMartino SJ, Shah AB, Trujillo G, Kew RR. Elastase controls the binding of the vitamin D-binding protein (Gc-globulin) to neutrophils: a potential role in the regulation of C5a co-chemotactic activity. J Immunol. 2001;166(4):2688–94. [DOI] [PubMed] [Google Scholar]
- 24.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. [DOI] [PubMed] [Google Scholar]
- 25.Sawant KV, Poluri KM, Dutta AK, Sepuru KM, Troshkina A, Garofalo RP, et al. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci Rep. 2016;6:33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Son DS, Parl AK, Rice VM, Khabele D. Keratinocyte chemoattractant (KC)/human growth-regulated oncogene (GRO) chemokines and pro-inflammatory chemokine networks in mouse and human ovarian epithelial cancer cells. Cancer Biol Ther. 2007;6(8):1302–12. [DOI] [PubMed] [Google Scholar]
- 27.Killian PH, Kronski E, Michalik KM, Barbieri O, Astigiano S, Sommerhoff CP, et al. Curcumin inhibits prostate cancer metastasis in vivo by targeting the inflammatory cytokines CXCL1 and −2. Carcinogenesis. 2012;33(12):2507–19. [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann SH, Desnoyers S, Ottaviano NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker for chemotherapy-induced apoptosis. Cancer Res. 1993;53(17):3976–85. [PubMed] [Google Scholar]
- 29.Mullen P. PARP cleavage as a means of assessing apoptosis. Methods Mol Med. 2004;88:171–81. [DOI] [PubMed] [Google Scholar]
- 30.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81(5):801–9. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Lee P, Galbiati F, Kitsis RN, Lisanti MP. Caveolin-1 expression sensitizes fibroblastic and epilthelial cells to apoptotic stimulation. Am J Physiol Cell Physiol. 2001;280(4):C823–35. [DOI] [PubMed] [Google Scholar]
- 32.Wu J, Wang Y, Liang S, Ma H. Cytoprotective effect of selective small-molecule caspase inhibitors against staurosporine-induced apoptosis. Drug Des Devel Ther. 2014;8:583–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, et al. Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell. 2001;12(5):1445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am J Pathol. 2001;159(2):387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood. 2005;106(4):1382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. 2009;8(8):1168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saika S, Okada Y, Miyamoto T, Yamanaka O, Ohnishi Y, Ooshima A, et al. Role of p38 MAP kinase in regulation of cell migration and proliferation in healing corneal epithelium. Invest Ophthalmol Vis Sci. 2004;45(1):100–9. [DOI] [PubMed] [Google Scholar]
- 38.Smith A, Ramos-Morales F, Ashworth A, Collins M. A role for JNK/SAPK in proliferation, but not apoptosis, of IL-3-dependent cells. Curr Biol. 1997;7(11):893–6. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35(6):600–4. [DOI] [PubMed] [Google Scholar]
- 40.Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–60. [DOI] [PubMed] [Google Scholar]
- 41.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475–82. [DOI] [PubMed] [Google Scholar]
- 42.Pordanjani SM, Hosseinimehr SJ. The Role of NF-kB Inhibitors in Cell Response to Radiation. Curr Med Chem. 2016;23(34):3951–63. [DOI] [PubMed] [Google Scholar]
- 43.Fujii T, Shimada K, Asai O, Tanaka N, Fujimoto K, Hirao K, et al. Immunohistochemical analysis of inflammatory cells in benign and precancerous lesions and carcinoma of the prostate. Pathobiology. 2013;80(3):119–26. [DOI] [PubMed] [Google Scholar]
- 44.Karakiewicz PI, Benayoun S, Begin LR, Duclos A, Valiquette L, McCormack M, et al. Chronic inflammation is negatively associated with prostate cancer and high-grade prostatic intraepithelial neoplasia on needle biopsy. Int J Clin Pract. 2007;61(3):425–30. [DOI] [PubMed] [Google Scholar]
- 45.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60(1):199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vral A, Magri V, Montanari E, Gazzano G, Gourvas V, Marras E, et al. Topographic and quantitative relationship between prostate inflammation, proliferative inflammatory atrophy and low-grade prostate intraepithelial neoplasia: a biopsy study in chronic prostatitis patients. Int J Oncol. 2012;41(6):1950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang LY, Izumi K, Lai KP, Liang L, Li L, Miyamoto H, et al. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013;73(18):5633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elkahwaji JE, Hauke RJ, Brawner CM. Chronic bacterial inflammation induces prostatic intraepithelial neoplasia in mouse prostate. Br J Cancer. 2009;101(10):1740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A. 2014;111(5):E592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, et al. Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS One. 2013;8(8):e73933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liou GY. Inflammatory Cytokine Signaling during Development of Pancreatic and Prostate Cancers. J Immunol Res. 2017;2017:7979637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. 2017;127(3):780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darling VR, Hauke RJ, Tarantolo S, Agrawal DK. Immunological effects and therapeutic role of C5a in cancer. Expert Rev Clin Immunol. 2015;11(2):255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189(9):4674–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyake M, Lawton A, Goodison S, Urquidi V, Rosser CJ. Chemokine (C-X-C motif) ligand 1 (CXCL1) protein expression is increased in high-grade prostate cancer. Pathol Res Pract. 2014;210(2):74–8. [DOI] [PubMed] [Google Scholar]
- 57.Zhang T, Tseng C, Zhang Y, Sirin O, Corn PG, Li-Ning-Tapia EM, et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat Commun. 2016;7:11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho HJ, Kwon GT, Park H, Song H, Lee KW, Kim JI, et al. A high-fat diet containing lard accelerates prostate cancer progression and reduces survival rate in mice: possible contribution of adipose tissue-derived cytokines. Nutrients. 2015;7(4):2539–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Izhak L, Wildbaum G, Weinberg U, Shaked Y, Alami J, Dumont D, et al. Predominant expression of CCL2 at the tumor site of prostate cancer patients directs a selective loss of immunological tolerance to CCL2 that could be amplified in a beneficial manner. J Immunol. 2010;184(2):1092–101. [DOI] [PubMed] [Google Scholar]
- 60.Loberg RD, Day LL, Harwood J, Ying C, St John LN, Giles R, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8(7):578–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mazzucchelli L, Loetscher P, Kappeler A, Uguccioni M, Baggiolini M, Laissue JA, et al. Monocyte chemoattractant protein-1 gene expression in prostatic hyperplasia and prostate adenocarcinoma. Am J Pathol. 1996;149(2):501–9. [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, et al. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009;69(4):1685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7(19):28697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin TH, Liu HH, Tsai TH, Chen CC, Hsieh TF, Lee SS, et al. CCL2 increases alphavbeta3 integrin expression and subsequently promotes prostate cancer migration. Biochim Biophys Acta. 2013;1830(10):4917–27. [DOI] [PubMed] [Google Scholar]
- 65.Natsagdorj A, Izumi K, Hiratsuka K, Machioka K, Iwamoto H, Naito R, et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2019;110(1):279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qian DZ, Rademacher BL, Pittsenbarger J, Huang CY, Myrthue A, Higano CS, et al. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. Prostate. 2010;70(4):433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Patel L, Pienta KJ. CC chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis and metastasis. Cytokine Growth Factor Rev. 2010;21(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–24. [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez M, Luo W, Weng J, Zeng L, Yi Z, Siwko S, et al. PSGR promotes prostatic intraepithelial neoplasia and prostate cancer xenograft growth through NF-kappaB. Oncogenesis. 2014;3:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu XH, Kirschenbaum A, Lu M, Yao S, Klausner A, Preston C, et al. Prostaglandin E(2) stimulates prostatic intraepithelial neoplasia cell growth through activation of the interleukin-6/GP130/STAT-3 signaling pathway. Biochem Biophys Res Commun. 2002;290(1):249–55. [DOI] [PubMed] [Google Scholar]
- 71.Brown RE, Zotalis G, Zhang PL, Zhao B. Morphoproteomic confirmation of a constitutively activated mTOR pathway in high grade prostatic intraepithelial neoplasia and prostate cancer. Int J Clin Exp Pathol. 2008;1(4):333–42. [PMC free article] [PubMed] [Google Scholar]
- 72.Soylu H, Acar N, Ozbey O, Unal B, Koksal IT, Bassorgun I, et al. Characterization of Notch Signalling Pathway Members in Normal Prostate, Prostatic Intraepithelial Neoplasia (PIN) and Prostatic Adenocarcinoma. Pathol Oncol Res. 2016;22(1):87–94. [DOI] [PubMed] [Google Scholar]
- 73.Liou GY, Doeppler H, Necela B, Edenfiled B, Zhang L, Dawson DW, et al. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015;5:52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Necela BM, Su W, Thompson EA. Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor gamma and nuclear factor-kappaB in macrophages. Immunology. 2008;125:344–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Increased proliferation of PIN Pr111 cells when co-cultured in 3D with primary macrophages.
Supplemental Figure S2. Pr111 were proliferative when co-cultured in 3D with Raw264.7 macrophage cells.
Supplemental Figure S3. Raw264.7 macrophages were proliferative in 3D co-culture with PIN Pr111 cells.
Supplemental Figure S4. Murine Pr111 cells expressed receptors CXCR2 and CCR2.
Supplemental Figure S5. Raw264.7 macrophages increased expression of C5a and CCL2 as cultured in co-culture media.
Supplemental Figure S6. Macrophage cytokines, C5a, CXCL1 and CCL2 had no effect on apoptosis in murine PIN Pr111 cells.
Supplemental Figure S7. Signaling of ERK and JNK in muirn PIN Pr111 cells stimulated by C5a, CXCL1 or CCL2.
Supplemental Figure S8. The expression of phosphorylated-Akt in the PIN area of human prostate.