

Abstract
The nitric oxide (NO)–cyclic guanosine monophosphate (cGMP)–protein kinase G (PKG) pathway plays a critical role in skeletal homeostasis. Pre-clinical data using NO and its donors and genetically modified mice demonstrated that NO was required in bone remodeling and partly mediated the anabolic effects of mechanical stimuli and estrogen. However, the off-target effects and tachyphylaxis of NO limit the long-term use, and previous clinical trials using organic nitrates for osteoporosis have been disappointing. Among the other components in the downstream pathway, targeting cGMP-specific phosphodiesterase to promote the NO–cGMP–PKG signal is a viable option. There are growing in vitro and in vivo data that, among many other PDE families, PDE5A is highly expressed in skeletal tissue, and inhibiting PDE5A using currently available PDE5A inhibitors might increase the osteoanabolic signal and protect skeleton. These pre-clinical data open the possibility of repurposing PDE5A inhibitors for treating osteoporosis. Further research is needed to address the primary target bone cell of PDE5A inhibition, the contribution of direct and indirect effects of PDE5A inhibition, and the pathophysiological changes in skeletal PDE5A expression in aging and hypogonadal animal models.
Keywords: nitric oxide, soluble guanylate cyclase, cGMP, PDE5A, PKG, bone
Graphical abstract
Over the decades, researchers have learned that nitric oxide (NO) and its downstream pathway play a critical role in skeletal homeostasis. We summarize findings from preclinical studies on the skeletal effect of NO and its downstream components and revisit the idea of taking advantage of this pathway for treating osteoporosis.
Introduction
Osteoporosis is a major public health problem affecting almost 54 million U.S. women and men aged 50 and older.1 Anti-resorptive such as bisphosphonates and denosumab are used for treating primary and secondary osteoporosis to reduce fracture risk.2 For a high-bone turnover disease such as postmenopausal osteoporosis, anti-resorptive suppresses bone remodeling, and increased bone densities but not necessarily bone quality. The prolonged use of an anti-resorptive, especially the bisphosphonate, increases the risk of unwanted complications such as osteonecrosis of the jaw and atypical femur fracture. Conversely, anabolic agents are shown to improve bone quality and quantity with new bone formation.3 Unfortunately, only a few anabolics are available currently, and the use of the anabolics in the clinical practice is very limited. PTH and PTHrP analog are approved for a maximum of two years by the U.S. Food and Drug Administration (FDA) due to the theoretical concern of the risk of osteosarcoma based on an animal study using very high doses (30 to 4500 μg/day for humans) of teriparatide for a prolonged period.4 Romosozumab, anti-sclerostin antibody, was recently approved with a black box warning because of an increased signal of serious cardiovascular events compared with alendronate treatment.5 There are many conditions with a low born turnover disease such as diabetes, aging, or adynamic bone disease from CKD where anabolics might work better than anti-resorptive. Also, PTH 1–34 and PTH 1–84 have been investigated to improve fracture healing; however, the results are conflicting.6–8
Therefore, new anabolic agents with anti-fracture efficacy and long-term safety for treating chronic diseases such as osteoporosis are desperately needed. The nitric oxide (NO)–cyclic guanosine monophosphate (cGMP)–protein kinase G (PKG) pathway might provide a new potential treatment target. Over the decades, researchers have learned that NO and its downstream pathway play a critical role in skeletal homeostasis. Although initial attempts to use organic nitrates as therapeutics in osteoporosis were disappointing,9, 10 their potential should be revisited in light of new research findings. Therefore, in our review we summarize findings from preclinical studies on the skeletal effect of NO and its downstream components and revisit the idea of taking advantage of this pathway for treating osteoporosis.
NO–cGMP–PKG signaling and bone
Nitric oxide is known to play important functional roles in a variety of physiological and pathological pathways. It is a free radical with a very short half-life (less than 30 seconds). Three isoforms of nitric oxide synthases have been identified, namely, neuronal (nNOS or NOS1), inducible (iNOS or NOS2), and endothelial (eNOS or NOS3) that produce NO using L-arginine as a substrate. While it is well-known that NO diffuses freely across cell membranes, wherein the vascular system acts as a potent vasodilator, importantly NO also a critical signal for skeletal remodeling. All three NOS isoforms were found in whole bone tissue or bone-derived cells such as osteoblast or osteoclast by PCR and immunohistochemistry. eNOS expression was reported in osteoblasts, stromal cells, and osteoclasts.11–15 eNOS is calcium- and calmodulin-dependent and constitutively releases of picomolar concentrations of nitric oxide.16, 17 iNOS was also found in osteoblast and osteocytes,18 as well as in osteoclast-like cells, bone marrow, and osteoclast precursor cells. However, basal iNOS expression was minimal, if any, and triggered by inflammatory cytokines such as tumor necrosis factor (TNF) α, interferon (IFN) γ, or interleukin 1.19–21 iNOS can generate much larger quantities of NO (nanomolar range) in response to inflammatory cytokines.16, 17 nNOS mRNA was also detected in whole bone, but the expression in a specific bone-derived cell is not known.14
Although different types of bone-cells are shown to express NOS isoforms, osteoblasts seem to be a dominant player in terms of generating NO. Co-culture of osteoblast and hematopoietic stem cells showed that the majority of NO was produced from osteoblasts compared with osteoclast precursors (100 fold greater in osteoblasts) in response to cytokines treatment.22 The effect of NO on osteoblast is well-described from studies using genetically modified mice. Osteoblasts derived from eNOS–/– (Nos3–/–) mice showed slow growth, poor differentiation, and decreased alkaline phosphatase activity in response to PTH treatment.11, 23 However, the effects of NO are not restricted to osteoblasts. In the absence of NO, the activity and mobility of osteoclasts significantly decreased, which suggests NO is an important signal in healthy bone remodeling.19 Furthermore, RANKL secreted from osteoblasts promotes iNOS expression and generates NO in osteoclast, which, in turn, negatively affects RANK-induced osteoclastogenesis, suggesting the role of NO in osteoblast-osteoclast crosstalk and bone remodeling.20
The skeletal phenotype of NOS-deficient mice further demonstrated the skeletal effect of NOS and NO production. The basal constitutive activation of eNOS and resultant low-grade NO production seem to be required in skeletal remodeling. eNOS−/− mice showed impaired bone density and bone microarchitecture.11, 23 These mice showed an exaggerated bone loss after ovariectomy and a blunted response to high-dose estrogen treatment.11, 24 Histomorphometry analysis of eNOS−/− mice showed reduced bone volume/tissue volume (BV/TV), trabecular number, osteoblast number and surfaces, bone formation rate (BFR) and mineral apposition rate (MAR).11, 23
On the other hand, the iNOS–/– (Nos2–/–) mice showed normal skeletal phenotype at the baseline, although iNOS mediates the effect of mechanical stimuli. In mice with a tail suspension model, which mimick mechanical un-loading, iNOS-deficient mice did not reverse bone loss after mechanical re-loading, suggesting the role of iNOS and NO production in mechanical-loading induced anabolic stimuli.18 More importantly, iNOS seem to mediate cytokine-induced bone remodeling in an inflammatory condition. iNOS–/– mice showed decreased NF-κB activation in bone marrow cells in the inflammatory state and did not lose bone in response to cytokine treatment.25 Also, TNF-Tg mice with inflammatory arthritis did not show increased bone resorption or osteoclast formation in the absence of NOS (TNF-Tg × NOS–/–).26 But this skeletal phenotype of global eNOS–/– and iNOS–/– mice needs to be interpreted with caution considering the role of NO in broad physiological processes. Bone-specific eNOS or iNOS gene knockout mice will provide a better understanding of skeletal specific effects.
In terms of nNOS, although the cell-specific nNOS expression has not known in skeletal tissue, nNOS deficient mice showed a high bone mass. The osteosclerotic phenotype was accompanied by reduced bone remodeling. Histomorphometry showed decreased osteoblast and osteoclast numbers with a significant reduction in bone formation.27 Considering abundant expression of nNOS in neurons and vascular tissues,28 this skeletal phenotype of global knockout nNOS–/– mice could be indirect, which needs to be further studied.
The skeletal effect of NO through mechanical loading and estrogen is partly transcriptional: the regulatory 5′ flanking region of bovine eNOS gene includes an estrogen binding motif, shear stress response, and tumor necrosis factor response elements.29 Thus, 17-β estradiol stimulates eNOS expression and activity in human osteoblast-like cells and endothelial cells of the cultured human umbilical vein.30, 31 The anti-apoptotic effect of estrogen on osteocyte requires cGMP and cGMP-dependent PKG activation. 17β estradiol or cGMP analog treatment prevented osteocyte-like cell (MLO-Y4) from undergoing apoptosis, but not in the presence of inhibitors for NOS, sGC, or PKGs.32 PKG1 and PKG2 function independently, where PKG1 isoform PKG1α directly phosphorylates BAD on Ser155 and prevents apoptosis, while PKG2 mediates estrogen-induced Akt/ERK activation, which ultimately phosphorylates BAD on Ser136 and Ser122.32
Mechanical loading also requires the NO–cGMP–PKG signaling pathway. Mechanical shear stress promoted eNOS expression and increased NO production.13, 33 Like fluid shear stress, NO donor, or cGMP induced Src activation in primary human osteoblast and murine osteoblast-like cells (MC3T3).34 NOS, soluble guanylate cyclase (sGC) or PKG inhibitors abolished fluid stress-induced Src activation. siRNA-mediated knockdown of PKG1 and 2 showed membrane-bound PKG2 is required for fluid stress and cGMP-induced Src activation.34 Also, ERK/Akt activation through NO-mediated cGMP elevation did not occur in the absence of PKG2.35 However, PKG2 did not activate Src directly but through phosphorylation of SHP-1 and −2, which, in turn, dephosphorylate and activate Src.34
PKG-deficient mice further demonstrated the vital role of the NO–cGMP–PKG pathway in bone remodeling. Global Prkg2–/– mice caused dwarfism because of impaired endochondral ossification from defective chondroblast differentiation.36 Primary osteoblast from Prkg2–/– mice showed decreased c-fos and Fra2 expression, which involve in osteoblast cell cycle regulation.34 On the other hand, osteoblast-specific PKG2 gain-of-function mutation (Col1a1-Prkg2RQ) showed significantly increased bone formation parameters such as trabecular mineralizing surface, MAR, and BFR compared with littermates.37 Interestingly, this skeletal phenotype was more evident in male mice, suggesting the possible interaction of sex hormone and the NO–cGMP–PKG2 signal. Moreover, Col1a1-Prkg2RQ transgenic mice were protected from diabetes-induced bone loss caused by streptozocin injection.37
Global PKG1-deficient mice showed high mortality where ~80% of mice died within 2 months as loss of PKG1 result in severe vascular and intestinal dysfunctions.38 Osteoblast-specific Prkg1 knockout (Col1a1-CRETg/+ Prkg1fl/fl) showed decreased osteoblast activities; decreased osteoblastic gene expression (Alpl, Bglap, and Cola1a) and decreased MS/BS, MAR and BFR in histomorphometry. However, it did not affect osteoblast proliferation as the number of osteoblasts remained unchanged on the bone surface.39 The number of osteoclasts was also unchanged.39 In the fracture model using monocortical defect model, Prkg1 OB-KO mice showed impaired bone regeneration. Alongside decreased VEGF and VEGFR1 expression and capillary density in bone, BMP-2 and −4 signaling in osteoblast were also significantly decreased in Prkg1 OB-KO, suggesting PKG1 play a vital role in angiogenesis and osteoblast differentiation.39
Complicating the understanding of the effects of NO in bone is its biphasic nature. Many studies have reported opposite effects of NO depending upon low and high concentrations achieved by using different NO donors. Although the dose, the study drug, and study methods are heterogeneous, NO at higher concentrations consistently decreases cellular activity in osteoblasts and osteoclasts thereby suppressing bone remodeling. An in vitro study, NO generating agents such as nitroprusside at higher concentrations (30 μM vs 9 μM), which decreased the osteoclast spread area.40 NO donor (S-nitroso-acetyl penicillamine (SNAP)) increased bone turnover at lower concentrations (1–100 μM); yet at much higher concentration (500 μM), bone turnover became significantly suppressed.41 The dramatic increase in NO production responding to a combination of applied cytokines, including IL-1β, TNF-α, and IFN-γ, led to arrested osteoclast differentiation and activity.22 These effects of NO at high concentration generated in response to high cytokine levels caused an apoptotic change not only in osteoclast precursor22 but also decreased osteoblast (MC3T3-E1) cell viability.42 The biphasic effects of NO need to be further clarified with future studies. The definition of physiological and pathological concentration of NO thus remains unclear. More importantly, whether and how NO production or NOS expression in the skeleton is changed and responsible for primary and secondary osteoporosis in both males and females needs to be further studied.
NO donors and bone
With all the available physiologic data, NO and its donors were studied as a potential therapeutic for osteoporosis. In an in vivo study using ovariectomized-mice, nitroglycerin prevented bone loss, and estradiol failed to protect bone loss when NOS inhibitors were given simultaneously.43 Also, nitroglycerin (0.2 mg/kg of 2% nitroglycerin ointment daily) restored bone mass after ovariectomy to levels comparable to estradiol treatment.44 However, organic nitrates require mitochondrial biotransformation, and during the process reactive oxygen species (ROS) are generated.45, 46 For this reason, long-term use of organic nitrate is limited due to intolerance, tachyphylaxis, and off-target effects. A newer NO-releasing agent, nitrosyl-cobinamide (NO-Cbi), that releases NO directly without biotransformation was studied; NO-Cbi increased intracellular cGMP, osteoblast proliferation, and survival.35 NO-Cbi treatment (10 mg/kg/d, i.p) increased osteoblastic bone formation while inhibiting osteoclast differentiation in ovariectomized mice.35
Epidemiology studies support the protective skeletal effects of NO donors as well. A Danish population-based study using 123,655 subjects reported a ~15% reduced risk of fracture in users of organic nitrates.47 A case-control study using the Dutch PHARMO Record Linkage System showed a decreased risk of hip fracture in patients with the current use of nitrates. In this study, intermittent users had a lower risk of fracture compared with daily users.48 The US-population based cohort from the Study of Osteoporotic Fractures also showed that adjusted hip BMD was significantly higher in daily users of organic nitrates, especially intermittent users, compared with non-users.49
However, the results of clinical trials have been very disappointing. Two randomized clinical trials using nitroglycerin ointment and isosorbide mononitrate in postmenopausal women with low bone densities were retracted because of scientific misconduct.9, 10 More recently, a 1-year double-blind, randomized, placebo-controlled trial of three different nitrate preparations (short-acting isosorbide mononitrate, long-acting isosorbide mononitrate, and nitroglycerin) at two different doses examined the change of BMD and bone turnover markers. In this study of 240 postmenopausal women with low bone densities, no significant BMD change was noted. Of note, the withdrawal rate was quite high (~ 20%) in the active treatment arm mainly due to headaches.50
Soluble guanylate cyclase (sGC) and bone
Soluble guanylate cyclase (sGC) converts guanosine triphosphate (GTP) to secondary messenger cGMP and inorganic pyrophosphate and increases the NO–cGMP–PKG signal. The soluble guanylate cyclases (Gucy1a2 and Gucy1a3) were expressed in mouse skeletal tissue.15 And sGC agonist, cinaciguate, increased intracellular cGMP concentration in murine primary osteoblast.51
In vivo studies using different heme-dependent and heme-independent sGC agonists increased bone formation and resorption, resulting in increased bone turnover.52 sGC agonists also increased bone remodeling in parathyroidectomized rats, which suggested the effect was independent of PTH action.52 The skeletal effect of sGC agonist seems to be osteoblast-driven. Cinaciguat enhanced osteoblast proliferation and differentiation and reversed the trabecular bone loss in ovariectomized mice whereas, it did not suppress the osteoclast differentiation or osteoclastic gene expression in ovariectomized mice51 Cinaciguate was also studied in the streptozocin-induced type 1 diabetes mice model. Cinaciguate treatment group showed significantly improved BMD and micro-skeletal architecture as well as an increased number of osteoblasts and collagen content.39 Importantly, cinaciguate also increased Vegfa expression in skeletal tissue, suggesting that cinaciguate-induced cGMP-PKG signal not only works as an osteogenic stimulus but also improves angiogenesis in diabetes-related bone disease.39
Currently, sGC agonist, riociguat, is approved by the FDA for treating pulmonary hypertension. It has shown a favorable safety profile and well-tolerated with hypotension and syncope as the most frequent side effect.53 Randomized, controlled, phase IIb trial of cinaciguat use for acute heart failure syndromes showed short-term use of intravenous cinaciguat caused frequent hypotension without beneficial effect on respiratory and cardiac index.54
Phosphodiesterase (PDE) 5 and Bone
cGMP-specific phosphodiesterase is another potential target to promote the NO-sGC-cGMP-PKG signal in the skeleton. Among 11 known PDE families, PDE5A, 6, and 9A are known to specifically bind to and degrade cGMP. Taqman assays using mRNA from mouse skeletal tissue evaluating the expression of 11 PDEs showed the high levels of Pde5a expression, which was comparable to expression of Tnfrsf11a and Tnfsf11.15 Immunohistochemistry confirmed the co-localization of RUNX2 and PDE5A in osteoblast precursors; however, PDE5A was not stained in osteoclast precursors. Microarray analysis of human osteoblast and osteoclast also showed the presence of PDE5A only in osteoblasts.15 Furthermore, PDE5 inhibitors sildenafil and vardenafil increased cGMP concentration significantly in calvarial cells from neonatal mice.55
PDE5A is an attractive therapeutic target; PDE5A inhibitors are readily available and have been widely and safely used in clinical practice. Currently, four different PDE5A inhibitors, namely sildenafil, tadalafil, vardenafil, and avanafil are approved for treating erectile dysfunction, which is the most common sexual dysfunction worldwide.56 Almost 70% of elderly men (70 years of age and older) and more than half of the patients with diabetes suffer from erectile dysfunction; this group also has a high risk of osteoporosis and fractures.56 Among them, tadalafil was approved for daily chronic use given its long-term safety profile, and low-dose daily use of tadalafil significantly improved erectile dysfunction and lower urinary tract symptoms in diabetes patients.57 A large epidemiology study from Taiwan suggested that the two multifactorial diseases erectile dysfunction and osteoporosis are closely associated. Patients with erectile dysfunction had a significantly higher risk of low bone mass and, more importantly, hip fracture.58 Therefore, repurposing PDE5 inhibitors, which have been safely used for decades, for osteoporosis may be a viable therapeutic option.
In vivo studies showed protective skeletal effects of PDE5A inhibition, which are summarized in Table 1. In wild-type mice, tadalafil and vardenafil significantly increased areal and volumetric BMD, including better microarchitecture, compared with vehicle-treated mice.15 Tadalafil and vardenafil increased serum NO, cGMP, and PKG expression, as well as BMD, in ovariectomized rats.59 Also, sildenafil and vardenafil promoted new bone formation in mice with femur osteotomy and completely restored bone mass and trabecular microarchitecture parameters in ovariectomized mice.55 Avanafil and zaprinast also showed protective effects in rats with glucocorticoid-induced bone loss.60 PDE5 inhibitors were also studied in mice fracture model and showed fastened fracture healing and better biomechanical properties.61–64
Table 1.
PDE5 inhibitors and skeleton effect
| Medication | Intervention | Skeletal changes | Other findings | |
|---|---|---|---|---|
| Alp et al.59 | Tadalafil Vardenfail Udenafil (10 mg/kg/d, OG), 2 mon |
OVX (8 mon-old rats) | BMD↑ tadalafil and vardenafil (+12%) udenafil (+14%) P1NP↑ CTX↓ |
|
| Huyut et al.60 | Avanafil Zaprinast (10mg/kg/d, OG), 1mon |
Dexamethasone (120ug/kg) (8 mon-old rat) |
BMD↑ avanafil (+8%) and zaprinast (+2%) |
|
| Pal et al.55 | Sildenafil (6 mg/kg/d) Vardenafil (2.5 mg/kg/d), OG, 6 wks |
OVX (5 mon-old Balb/c mice) |
BMD↑ sildenafil and vardenafil (L5,+45%) BV/TV↑, MS/BS↑, MAR↑, BFR↑ P1NP ↑, CTX ↔ |
VEGF and VEGFR2 expression ↑ In-vivo vascularization (FITC) ↑ |
| Kim et al.15 | Tadalafil (2 mg/kg/d) and vardenafil (10 mg/kg/d) OG, 6 wks | No intervention 4 mon-old C57Bl.6J mice | BMD↑ tadalafil (L4–6, 7%), vardenafil (~2%) BV/TV↑, MS/BS↑, MAR↑, BFR↑ and N.Oc/BS ↓, Cfu-Ob ↑ and ACP5+ OC ↓ |
Negative CNS-mediated effect |
| Histing et al.61 | Sildenafil (5mg/kg/d), OG, 5wks | Femur fracture | Fracture healing (Radiologic and histologic finding)↑ Improved bending stiffness ↑ |
VEGF/CYR61 expression in callus ↑ |
| Togral et al.63 | Tadalafil (1mg/kg/d) Sildenafil (5mg/kg/d), OG, 6wks | Femur fracture | Fracture healing (Radiologic and histologic finding)↑ | In-vivo vascularization (FITC) ↑ |
| Cakir-Ozkan et al.62 | Sildenafil (10mg/kg/d), OG, 4wks | Mandibular fracture | Fracture healing (Radiologic and histologic finding)↑ | BMP2 and Col1 expression ↑ in callus |
| Dincel64 | Sildenafil (10mg/kg/d), OG, 2 wks | Femur fracture | Fracture healing (Radiologic and histologic finding)↑ |
OG, oral gavage; MS/BS, mineralized surface/bone surface; MAR, mineral apposition rate; BFR, bone formation rate; IVIS: in vivo imaging system; FITC, fluorescein isothiocyanate-dextran
The effect of PDE5A inhibitors seems to primarily come from osteoblasts. PDE5A inhibitor treatment promoted alkaline phosphatase (ALP) activity, increased bone formation parameters (BV/TV, MAR, and BFR), and upregulated osteoblastic gene expression.55 Bone formation markers (P1NP) increased without any change in bone resorption marker (CTX),55 yet there is evidence that tadalafil and vardenafil suppressed osteoclast differentiation and osteoclastic bone resorption.15 In this study, PDE5 expression was exclusively noted in osteoblasts, suggesting anti-osteoclastic effect might be driven by osteoblast.15 Although the negative effect of NO on RANK/RANKL-induced osteoclastogenesis was studied,20 the effect of PDE5 inhibition on osteoblast-osteoclast interaction is not known.
While the skeletal effects of PDE5A inhibition may predominantly be caused by direct actions on osteoblasts, there are also indirect effects that may contribute. PDE5A inhibitors are potent vasodilators and the resultant increased blood flow might promote anabolic signal and affect bone remodeling positively. In fractured bone, sildenafil upregulated cysteine-rich 61 (CYR61) in callus, which might increase endothelial cell migration.61 Also, PDE5 inhibitors increased VEGF and VEGFR expression in osteoblasts,55 which is consistent with upregulated VEGF and VEGFR gene expression with cGMP analog or sGC agonist treatment.39 In vivo imaging using fluorescein isothiocyanate-dextran (FITC dextran) demonstrated increased bone vasculature after sildenafil or vardenafil treatment.55 PDE5 inhibitors also were shown to modulate lymphatic function. Sildenafil and tadalafil reduce lymphatic contraction and dilated collateral lymphatic vessels,65 which might play a role in bone remodeling indirectly through the inflammatory cell or osteoclast progenitor recruitment.
In addition, PDE5A is relatively ubiquitous and also expressed in the central nervous system and PDE5 inhibitors were studied for vascular dementia and Alzheimer’s disease.66, 67 As a CNS–bone relay through the sympathetic nervous system is well-documented,68 another possible indirect action on the skeleton is via CNS-mediated effects of PDE5A. Co-localization of PDE5A and dopamine β-hydroxylase (DBH) in specific parts of the brain was confirmed by using co-staining. The connection between bone and DBH/PDE5A positive neurons was confirmed by tracing retrograde sympathetic neuron-specific PRV152 virus after injecting it to the bone.15 Sympathetic nervous system (SNS)-related clock genes expression profiles including Myc and Ccnd in osteoblasts were down-regulated, which suggested PDE5A inhibition might work negatively on osteoblast proliferation via the SNS.15
Further studies are needed before repurposing those agents in treating osteoporosis. Although serum cGMP levels were noted to be significantly lower in hypogonadal ovariectomized mice or streptozocin-induced type 1 diabetes mice,35, 37, 51 the pathological change of PDE5A expression in the skeletal tissues with hypogonadism, aging, type 1 diabetes or fracture has not been studied. Genetically modified mice with upregulated or downregulated PDE5A, particularly a bone cell-specific genetic modification, will give a better understanding of the role of PDE5A in skeletal remodeling and definitive answer regarding the primary target cell.
Summary
There is growing evidence that suggests the NO–cGMP–PKG pathway will provide a new therapeutic target for osteoporosis, as summarized in Figure 1. Organic nitrates have been previously tried; however, the intolerance, tachyphylaxis, and off-target side effects prevented their long-term therapeutic use. More recently, a soluble guanylate agonist and PDE5A inhibitors have been approved by the FDA and used in men and women for long-term treatment of pulmonary hypertension and erectile dysfunction in clinical practice. PDE5A inhibitors are well-tolerated with minimal side effects, and there are promising preliminary pre-clinical data on skeletal protection via osteoblast-driven anabolic effects. Further research is needed to repurpose these agents for osteoporosis therapy. The primary target cell of PDE5A inhibition in bone remains to be definitively demonstrated, and the contribution of direct versus indirect effects of PDE5A inhibition on skeletal remodeling remains unclear. Last, the pathophysiological changes in skeletal PDE5A expression in aging and hypogonadal osteoporosis animal models need to be studied.
Figure 1.
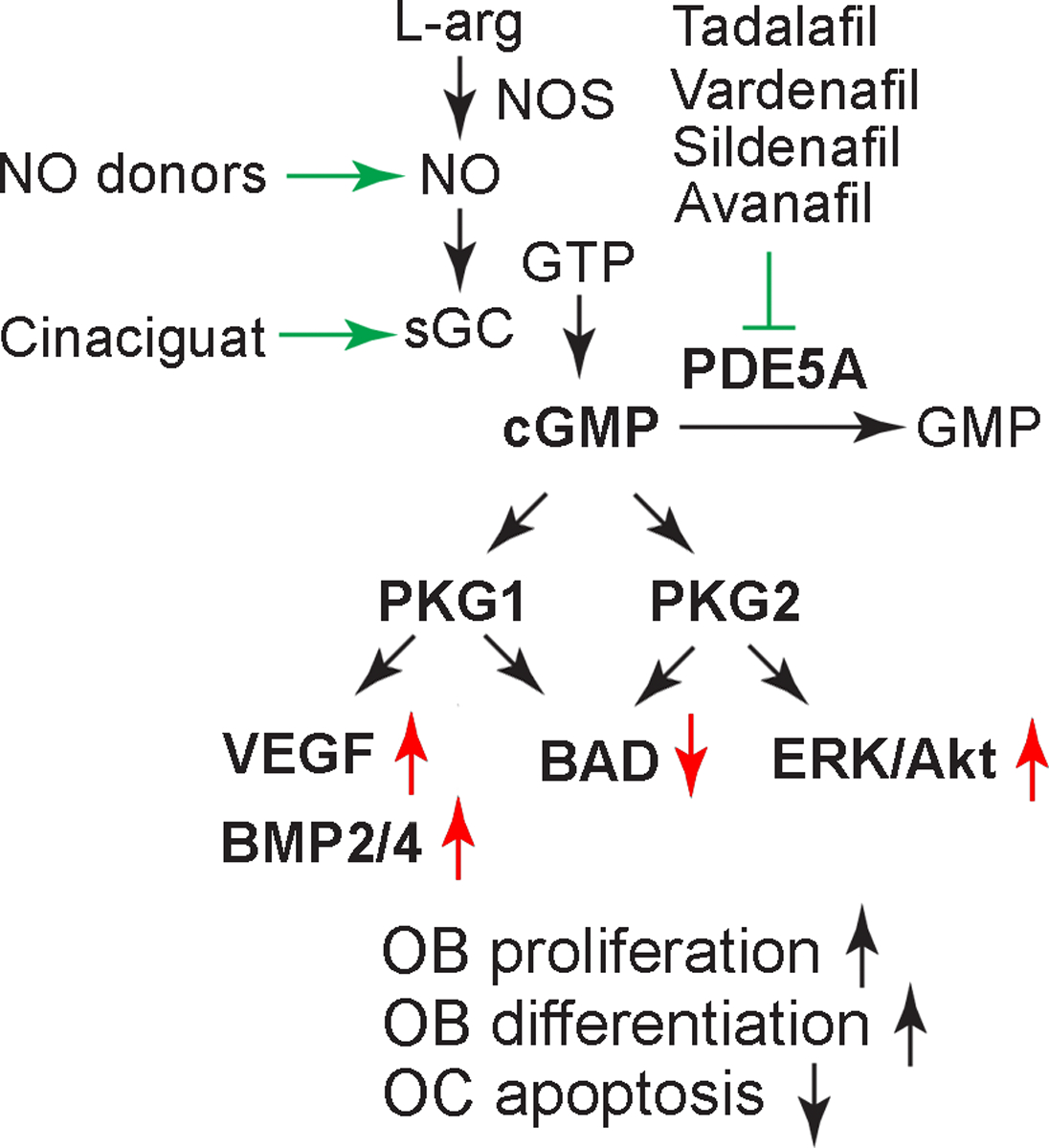
The NO–cGMP–PKG1/2 Axis. The PDE5A inhibitors tadalafil, vardenafil, sildenafil, and avanafil prevent cGMP degradation and thus stimulate PKG1/2.
Acknowledgments
S-M.K., J.I., T.Y. and M.Z. are supported by funding from the NIH (U19 AG60917).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Wright NC, Looker AC, Saag KG, et al. 2014. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res. 29: 2520–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iqbal J, Sun L & Zaidi M. 2010. Denosumab for the treatment of osteoporosis. Curr Osteoporos Rep. 8: 163–167. [DOI] [PubMed] [Google Scholar]
- 3.Weiss AJ, Iqbal J, Zaidi N, et al. 2010. The skeletal subsystem as an integrative physiology paradigm. Curr Osteoporos Rep. 8: 168–177. [DOI] [PubMed] [Google Scholar]
- 4.Vahle JL, Sato M, Long GG, et al. 2002. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1–34) for 2 years and relevance to human safety. Toxicol Pathol. 30: 312–321. [DOI] [PubMed] [Google Scholar]
- 5.Saag KG, Petersen J, Brandi ML, et al. 2017. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N Engl J Med. 377: 1417–1427. [DOI] [PubMed] [Google Scholar]
- 6.Aspenberg P, Genant HK, Johansson T, et al. 2010. Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J Bone Miner Res. 25: 404–414. [DOI] [PubMed] [Google Scholar]
- 7.Johansson T 2016. PTH 1–34 (teriparatide) may not improve healing in proximal humerus fractures. A randomized, controlled study of 40 patients. Acta Orthop. 87: 79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peichl P, Holzer LA, Maier R, et al. 2011. Parathyroid hormone 1–84 accelerates fracture-healing in pubic bones of elderly osteoporotic women. J Bone Joint Surg Am. 93: 1583–1587. [DOI] [PubMed] [Google Scholar]
- 9.Eastell R, Hamilton CJ & Cummings SR. 2016. Notice of Retraction: Jamal SA, et al. Effect of Nitroglycerin Ointment on Bone Density and Strength in Postmenopausal Women: A Randomized Trial. JAMA. 2011;305(8):800–807. JAMA. 315: 418–419. [DOI] [PubMed] [Google Scholar]
- 10.Nabhan AF 2006. A randomized clinical trial of the effects of isosorbide mononitrate on bone formation and resorption in post-menopausal women: a pilot study. Hum Reprod. 21: 1320–1324. [DOI] [PubMed] [Google Scholar]
- 11.Armour KE, Armour KJ, Gallagher ME, et al. 2001. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology. 142: 760–766. [DOI] [PubMed] [Google Scholar]
- 12.Lowik CW, Nibbering PH, van de Ruit M, et al. 1994. Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J Clin Invest. 93: 1465–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zaman G, Pitsillides AA, Rawlinson SC, et al. 1999. Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res. 14: 1123–1131. [DOI] [PubMed] [Google Scholar]
- 14.Helfrich MH, Evans DE, Grabowski PS, et al. 1997. Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J Bone Miner Res. 12: 1108–1115. [DOI] [PubMed] [Google Scholar]
- 15.Kim SM, Taneja C, Perez-Pena H, et al. 2020. Repurposing erectile dysfunction drugs tadalafil and vardenafil to increase bone mass. Proc Natl Acad Sci U S A. 117: 14386–14394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van’t Hof RJ & Ralston SH. 2001. Nitric oxide and bone. Immunology. 103: 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moncada S & Higgs A. 1993. The L-arginine-nitric oxide pathway. N Engl J Med. 329: 2002–2012. [DOI] [PubMed] [Google Scholar]
- 18.Watanuki M, Sakai A, Sakata T, et al. 2002. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Miner Res. 17: 1015–1025. [DOI] [PubMed] [Google Scholar]
- 19.Brandi ML, Hukkanen M, Umeda T, et al. 1995. Bidirectional regulation of osteoclast function by nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 92: 2954–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng H, Yu X, Collin-Osdoby P, et al. 2006. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem. 281: 15809–15820. [DOI] [PubMed] [Google Scholar]
- 21.Armour KE, Van THRJ, Grabowski PS, et al. 1999. Evidence for a pathogenic role of nitric oxide in inflammation-induced osteoporosis. J Bone Miner Res. 14: 2137–2142. [DOI] [PubMed] [Google Scholar]
- 22.van’t Hof RJ & Ralston SH. 1997. Cytokine-induced nitric oxide inhibits bone resorption by inducing apoptosis of osteoclast progenitors and suppressing osteoclast activity. J Bone Miner Res. 12: 1797–1804. [DOI] [PubMed] [Google Scholar]
- 23.Aguirre J, Buttery L, O’Shaughnessy M, et al. 2001. Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol. 158: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grassi F, Fan X, Rahnert J, et al. 2006. Bone re/modeling is more dynamic in the endothelial nitric oxide synthase(−/−) mouse. Endocrinology. 147: 4392–4399. [DOI] [PubMed] [Google Scholar]
- 25.van’t Hof RJ, Armour KJ, Smith LM, et al. 2000. Requirement of the inducible nitric oxide synthase pathway for IL-1-induced osteoclastic bone resorption. Proc Natl Acad Sci U S A. 97: 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell RD, Slattery PN, Wu EK, et al. 2019. iNOS dependent and independent phases of lymph node expansion in mice with TNF-induced inflammatory-erosive arthritis. Arthritis Res Ther. 21: 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van’t Hof RJ, Macphee J, Libouban H, et al. 2004. Regulation of bone mass and bone turnover by neuronal nitric oxide synthase. Endocrinology. 145: 5068–5074. [DOI] [PubMed] [Google Scholar]
- 28.Costa ED, Rezende BA, Cortes SF, et al. 2016. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front Physiol. 7: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venema RC, Nishida K, Alexander RW, et al. 1994. Organization of the bovine gene encoding the endothelial nitric oxide synthase. Biochim Biophys Acta. 1218: 413–420. [DOI] [PubMed] [Google Scholar]
- 30.Armour KE & Ralston SH. 1998. Estrogen upregulates endothelial constitutive nitric oxide synthase expression in human osteoblast-like cells. Endocrinology. 139: 799–802.9449657 [Google Scholar]
- 31.Hayashi T, Yamada K, Esaki T, et al. 1995. Estrogen increases endothelial nitric oxide by a receptor-mediated system. Biochem Biophys Res Commun. 214: 847–855. [DOI] [PubMed] [Google Scholar]
- 32.Marathe N, Rangaswami H, Zhuang S, et al. 2012. Pro-survival effects of 17beta-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J Biol Chem. 287: 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein-Nulend J, Helfrich MH, Sterck JG, et al. 1998. Nitric oxide response to shear stress by human bone cell cultures is endothelial nitric oxide synthase dependent. Biochem Biophys Res Commun. 250: 108–114. [DOI] [PubMed] [Google Scholar]
- 34.Rangaswami H, Schwappacher R, Marathe N, et al. 2010. Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci Signal. 3: ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalyanaraman H, Ramdani G, Joshua J, et al. 2017. A Novel, Direct NO Donor Regulates Osteoblast and Osteoclast Functions and Increases Bone Mass in Ovariectomized Mice. J Bone Miner Res. 32: 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfeifer A, Aszodi A, Seidler U, et al. 1996. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science. 274: 2082–2086. [DOI] [PubMed] [Google Scholar]
- 37.Ramdani G, Schall N, Kalyanaraman H, et al. 2018. cGMP-dependent protein kinase-2 regulates bone mass and prevents diabetic bone loss. J Endocrinol. 238: 203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfeifer A, Klatt P, Massberg S, et al. 1998. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 17: 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schall N, Garcia JJ, Kalyanaraman H, et al. 2020. Protein kinase G1 regulates bone regeneration and rescues diabetic fracture healing. JCI Insight. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacIntyre I, Zaidi M, Alam AS, et al. 1991. Osteoclastic inhibition: an action of nitric oxide not mediated by cyclic GMP. Proc Natl Acad Sci U S A. 88: 2936–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ralston SH, Ho LP, Helfrich MH, et al. 1995. Nitric oxide: a cytokine-induced regulator of bone resorption. J Bone Miner Res. 10: 1040–1049. [DOI] [PubMed] [Google Scholar]
- 42.Damoulis PD & Hauschka PV. 1997. Nitric oxide acts in conjunction with proinflammatory cytokines to promote cell death in osteoblasts. J Bone Miner Res. 12: 412–422. [DOI] [PubMed] [Google Scholar]
- 43.Wimalawansa SJ, De Marco G, Gangula P, et al. 1996. Nitric oxide donor alleviates ovariectomy-induced bone loss. Bone. 18: 301–304. [DOI] [PubMed] [Google Scholar]
- 44.Wimalawansa SJ 2000. Restoration of ovariectomy-induced osteopenia by nitroglycerin. Calcif Tissue Int. 66: 56–60. [DOI] [PubMed] [Google Scholar]
- 45.Bennett BM, McDonald BJ, Nigam R, et al. 1994. Biotransformation of organic nitrates and vascular smooth muscle cell function. Trends Pharmacol Sci. 15: 245–249. [DOI] [PubMed] [Google Scholar]
- 46.Sydow K, Daiber A, Oelze M, et al. 2004. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 113: 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rejnmark L, Vestergaard P & Mosekilde L. 2006. Decreased fracture risk in users of organic nitrates: a nationwide case-control study. J Bone Miner Res. 21: 1811–1817. [DOI] [PubMed] [Google Scholar]
- 48.Pouwels S, Lalmohamed A, van Staa T, et al. 2010. Use of organic nitrates and the risk of hip fracture: a population-based case-control study. J Clin Endocrinol Metab. 95: 1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jamal SA, Browner WS, Bauer DC, et al. 1998. Intermittent use of nitrates increases bone mineral density: the study of osteoporotic fractures. J Bone Miner Res. 13: 1755–1759. [DOI] [PubMed] [Google Scholar]
- 50.Bolland MJ, House ME, Horne AM, et al. 2020. Nitrates Do Not Affect Bone Density or Bone Turnover in Postmenopausal Women: A Randomized Controlled Trial. J Bone Miner Res. [DOI] [PubMed] [Google Scholar]
- 51.Joshua J, Schwaerzer GK, Kalyanaraman H, et al. 2014. Soluble guanylate cyclase as a novel treatment target for osteoporosis. Endocrinology. 155: 4720–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Homer BL, Morton D, Bagi CM, et al. 2015. Oral administration of soluble guanylate cyclase agonists to rats results in osteoclastic bone resorption and remodeling with new bone formation in the appendicular and axial skeleton. Toxicol Pathol. 43: 411–423. [DOI] [PubMed] [Google Scholar]
- 53.Sawabe T, Chiba T, Kobayashi A, et al. 2019. A novel soluble guanylate cyclase activator with reduced risk of hypotension by short-acting vasodilation. Pharmacol Res Perspect. 7: e00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gheorghiade M, Greene SJ, Filippatos G, et al. 2012. Cinaciguat, a soluble guanylate cyclase activator: results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur J Heart Fail. 14: 1056–1066. [DOI] [PubMed] [Google Scholar]
- 55.Pal S, Rashid M, Singh SK, et al. 2020. Skeletal restoration by phosphodiesterase 5 inhibitors in osteopenic mice: Evidence of osteoanabolic and osteoangiogenic effects of the drugs. Bone. 135: 115305. [DOI] [PubMed] [Google Scholar]
- 56.Selvin E, Burnett AL & Platz EA. 2007. Prevalence and risk factors for erectile dysfunction in the US. Am J Med. 120: 151–157. [DOI] [PubMed] [Google Scholar]
- 57.Bolat MS, Cinar O, Akdeniz E, et al. 2018. Low dose daily versus on-demand high dose tadalafil in diabetic patients with erectile and ejaculatory dysfunction. Int J Impot Res. 30: 102–107. [DOI] [PubMed] [Google Scholar]
- 58.Wu CH, Tung YC, Lin TK, et al. 2016. Hip Fracture in People with Erectile Dysfunction: A Nationwide Population-Based Cohort Study. PLoS One. 11: e0153467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alp HH, Huyut Z, Yildirim S, et al. 2017. The effect of PDE5 inhibitors on bone and oxidative damage in ovariectomy-induced osteoporosis. Exp Biol Med (Maywood). 242: 1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huyut Z, Bakan N, Yildirim S, et al. 2018. Effects of the Phosphodiesterase-5 (PDE-5) Inhibitors, Avanafil and Zaprinast, on Bone Remodeling and Oxidative Damage in a Rat Model of Glucocorticoid-Induced Osteoporosis. Med Sci Monit Basic Res. 24: 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Histing T, Marciniak K, Scheuer C, et al. 2011. Sildenafil accelerates fracture healing in mice. J Orthop Res. 29: 867–873. [DOI] [PubMed] [Google Scholar]
- 62.Cakir-Ozkan N, Bereket C, Sener I, et al. 2016. Therapeutic Effects of Sildenafil on Experimental Mandibular Fractures. J Craniofac Surg. 27: 615–620. [DOI] [PubMed] [Google Scholar]
- 63.Togral G, Arikan M, Korkusuz P, et al. 2015. Positive effect of tadalafil, a phosphodiesterase-5 inhibitor, on fracture healing in rat femur. Eklem Hastalik Cerrahisi. 26: 137–144. [DOI] [PubMed] [Google Scholar]
- 64.Dincel YM, Alagoz E, Arikan Y, et al. 2018. Biomechanical, histological, and radiological effects of different phosphodiesterase inhibitors on femoral fracture healing in rats. J Orthop Surg (Hong Kong). 26: 2309499018777885. [DOI] [PubMed] [Google Scholar]
- 65.Bouta EM, Bell RD, Rahimi H, et al. 2018. Targeting lymphatic function as a novel therapeutic intervention for rheumatoid arthritis. Nat Rev Rheumatol. 14: 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puzzo D, Loreto C, Giunta S, et al. 2014. Effect of phosphodiesterase-5 inhibition on apoptosis and beta amyloid load in aged mice. Neurobiol Aging. 35: 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puzzo D, Staniszewski A, Deng SX, et al. 2009. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model. J Neurosci. 29: 8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elefteriou F, Ahn JD, Takeda S, et al. 2005. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 434: 514–520. [DOI] [PubMed] [Google Scholar]