

Watch a video presentation of this article
Watch an interview with the author
Abbreviations
- ACP
aceruloplasminemia
- HH
hereditary hemochromatosis
- WD
Wilson’s disease
Wilson’s disease (WD) is a rare liver disease characterized by copper accumulation. Interestingly, iron overload has been observed in patients with WD without a diagnosis of primary hemochromatosis. This association has been recognized in the literature for almost two decades. 1 , 2 , 3 Of the chronic liver diseases known to cause secondary hemochromatosis, WD is classically not listed among them. The prevalence of secondary hemochromatosis in patients with WD is unknown. Despite the rarity of this disease, this knowledge is important because it yields therapeutic and monitoring implications in patients with WD. This article will begin with a review of the etiology and pathophysiology of WD, as well as the iron overload syndromes, followed by an explanation of the mechanism of secondary hemochromatosis in patients with WD. Finally, the authors will discuss the clinical implications of this knowledge with a focus on therapeutics.
Hepatic Metal Storage Disorders: Copper and Iron
The mechanisms of hepatic transport of copper and iron are intimately related. 1 This relatedness can be observed in pathological states. 1 WD is a disorder of copper metabolism that is caused by mutations in the ATP7B gene, which codes for a copper transport protein in the liver. 4 , 5 , 6 The absent or compromised ATP7B protein not only causes copper accumulation in the liver and other organs but also reduces the amount of circulating ceruloplasmin. 4 , 5 , 6 Normally, the ATP7B protein loads copper onto ceruloplasmin, the primary means of copper transport throughout the body (Fig. 1). 1 , 7 In WD, the inability of the ATP7B protein to transport copper and thus facilitate its secretion through the biliary system leads to copper accumulation in the liver. 8 , 9 Copper accumulation and toxicity lead to hepatic, neurological, and psychiatric dysfunction, and if not addressed promptly, it can lead to life‐threatening liver failure. 4 Iron is another essential metal that can accumulate in the liver. Hemochromatosis describes an iron overload syndrome that is classified as either primary (inherited), such as hereditary hemochromatosis (HH), or secondary hemochromatosis, with the latter carrying a broad differential. 10 , 11 HH can be divided into HFE (high Fe2+)‐related HH or non‐HFE‐related HH. 10 HH is an autosomal recessive disease that results from mutations of the HFE gene. 10 , 12 These can be homozygous mutations of C282Y or compound heterozygous mutations involving C282Y and either H63D or S65C. 10 The HFE gene codes the HFE protein, which is believed to increase the expression of hepcidin. 10 , 12 , 13 Mutations of the HFE gene can thus reduce or limit hepcidin, a key iron regulator that leads to iron overload. 13 Secondary causes of iron overload include iron‐loading anemias, parenteral iron overload, and chronic liver diseases. 10 , 11 Rarer miscellaneous inherited forms of hemochromatosis include dysmetabolic iron overload syndrome and aceruloplasminemia (ACP). 10 Iron overload and resulting iron deposition in the liver cause tissue injury and lead to eventual fibrosis and cirrhosis if untreated. 12 Evidence of secondary iron overload can be found in liver biopsies of patients with untreated WD (Fig. 2).
FIG 1.
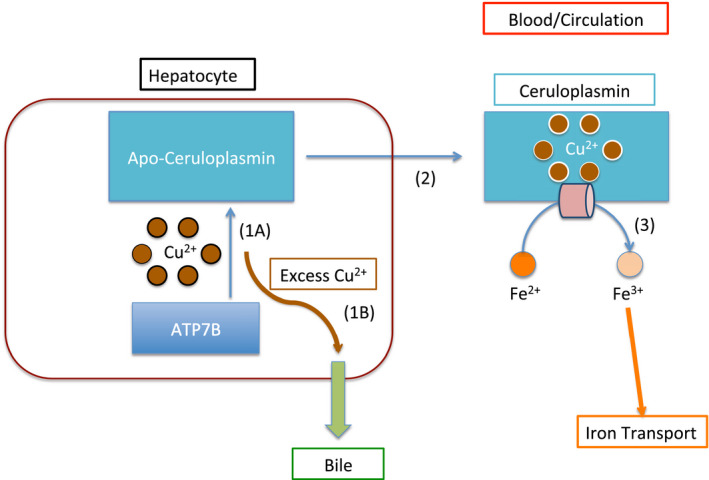
Normal copper and iron transportation. Schematic representation of normal copper transport in the liver. All blue arrows represent normal physiological pathways. (1A) ATP7B loads copper onto Apo‐ceruloplasmin. (2) This transforms it into the copper‐carrying protein Ceruloplasmin. (3) Ceruloplasmin is able to execute its ferroxidation. Ferroxidation is represented by the peach horizontal cylinder. Fe3+ is then able to be transported throughout the body (iron transportation is represented by the orange arrow). (1B) The curved brown arrow represents ATP7B transportation of the excess copper. ATP7B transports excess copper into the bile (thick green arrow) for excretion.
FIG 2.
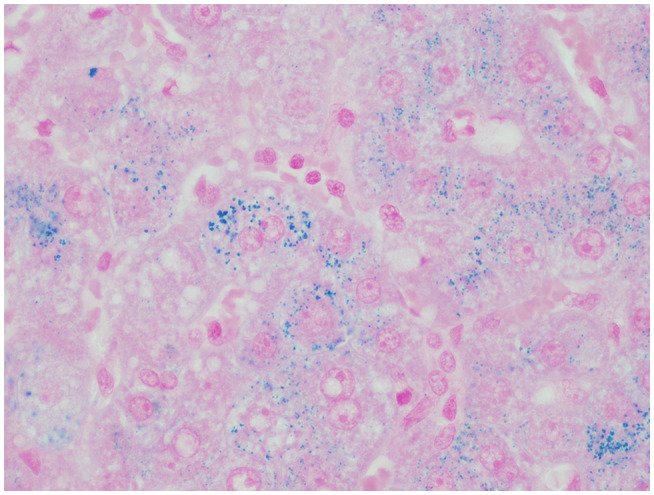
Hepatocellular siderosis in a patient with WD (Prussian blue staining, original magnification ×400).
WD and Iron Overload
WD is a copper overload disorder that can potentially result in secondary hemochromatosis. The pathophysiological mechanism involves the properties of ceruloplasmin, the important link between hepatic copper and iron biochemistry. 14 As aforementioned, ceruloplasmin is the primary means of copper transport throughout the body and plays a key role in copper mobilization and secretion. 7 , 8 , 9 In addition to its copper‐carrying property, ceruloplasmin also has ferroxidase activity. 1 , 14
Copper is an essential cofactor of ceruloplasmin that is required for its function, including ferroxidation. 15 In this context, ferroxidation is the process by which an electron is removed from ferrous iron (Fe2+) to make ferric iron (Fe3+). 16 It is important to note that only ferric iron is loaded onto transferrin, the iron transport protein. 16 , 17 Thus, copper‐containing ceruloplasmin converts iron from Fe2+ to Fe3+, which is needed for iron transportation. 1 In patients with WD whose ceruloplasmin lacks the copper cofactor, iron oxidation and, consequently, iron transportation is disrupted (Fig. 3A). Ultimately, low ceruloplasmin, which is found in WD, leads to decreased amounts of circulating iron, increased iron stores, and eventually iron accumulation in the liver and other organs. 1
FIG 3.
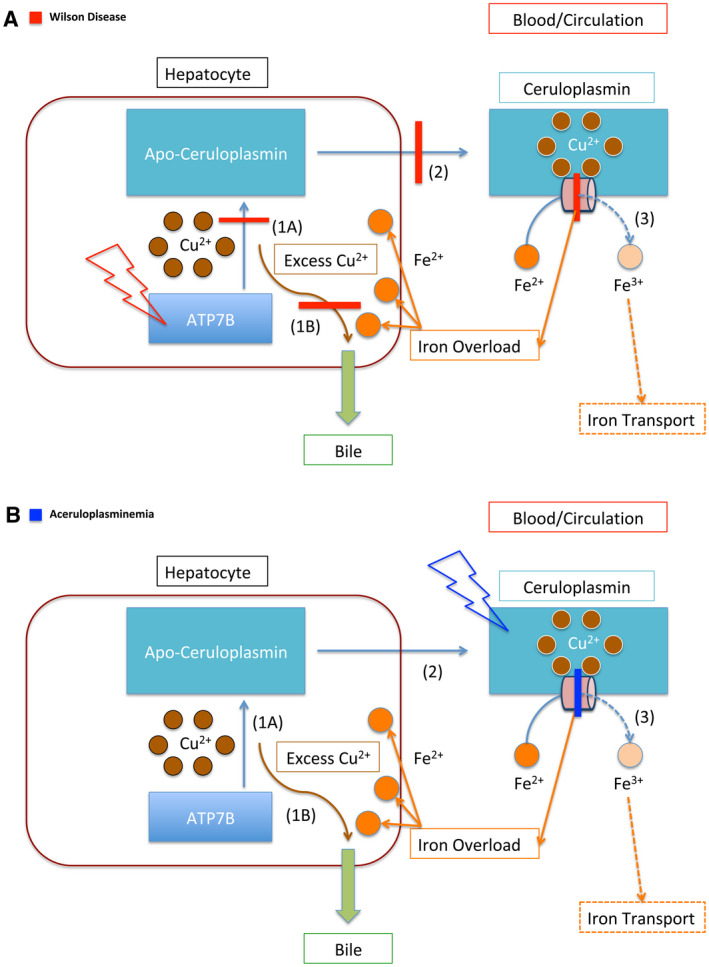
Copper and iron transportation in WD and ACP. (A) Schematic representation of the impaired copper and iron transportation in WD. The red lightning bolt represents mutation. This panel illustrates the mutation of the ATP7B protein, the primary defect in WD. The red bars represent the downstream effects of this protein mutation. In WD, ATP7B is unable to load copper onto Apo‐ceruloplasmin (1A), which prevents copper from being transported throughout the body (2). In addition, ATP7B is unable to facilitate the secretion of copper through the bile, which further causes copper accumulation within the hepatocyte (1B). Furthermore, because Ceruloplasmin lacks copper, it cannot ferroxidize iron (3). The impairment of ferroxidase activity is represented by the red bar across the peach cylinder. The dashed lines represent downstream effects of impaired ferroxidation, leading to iron accumulation in the liver. (B) Schematic representation of the impaired iron transportation in ACP. The blue lightning bolt represents the mutation of the protein Ceruloplasmin. The blue bar represents the impairment of Ceruloplasmin’s ferroxidase activity. As in (A), the dashed lines represent the impaired downstream effects of this, which is the inability of Ceruloplasmin to oxidize iron, which leads to impaired iron transport (3). In contrast with WD (A), the function of the ATP7B protein is not impaired, and it is able to transport copper (1A, 2). It retains the function of facilitating copper excretion through bile (1B). Thus, copper accumulation is not typically associated with ACP, whereas iron accumulation is.
The pathophysiology of ACP, a closely related diagnosis, brings further clarity to the iron accumulation that can be seen in WD. ACP is a rare autosomal recessive disorder that is caused by loss‐of‐function mutations of the ceruloplasmin (CP) gene. 18 , 19 ACP is classically characterized by iron overload, and patients can present with neurological symptoms from iron deposition in the brain. 18 , 19 Diabetes mellitus is usually the first manifestation of this disease, and the median age of onset is 38.5 years. 19 Neurological symptoms signify iron deposition in the brain, and these symptoms typically start in the fifth decade of life. 19 At the biochemical level, WD can be seen as a functional ACP (Fig. 3B).
Understanding iron overload as a second‐order effect of the pathophysiology of WD is an important concept that can influence therapy and disease monitoring.
Therapeutic Implications
WD may be thought of as a primary copper disorder with potential for secondary hemochromatosis. Concurrent pathology may influence treatment options. The mainstay of therapy for patients with WD is lifelong copper chelation with or without zinc. 20 Copper chelators promote urinary copper removal, whereas zinc reduces intestinal absorption of copper. 20 d‐Penicillamine is a copper chelator that promotes urinary excretion of copper but is associated with multiple side effects, making it difficult to tolerate. 6 Trientine, like d‐penicillamine, promotes excretion of copper through the genitourinary system and is generally better tolerated. 20 As previously mentioned, copper is an essential cofactor for ceruloplasmin, which carries an important role in the transportation of iron by its ferroxidase activity. Although removal of copper is the treatment goal for WD, it risks impeding the iron‐related function of ceruloplasmin by reducing the amount of copper, its cofactor. This illustrates why patients treated with d‐penicillamine or trientine can experience hepatic iron accumulation. 1 , 6
Rather than chelation, zinc inhibits intestinal mucosal absorption of copper. 6 , 20 Zinc is mainly used for maintenance therapy instead of induction, although it is often used in conjunction with chelation. 6 , 20 Human data have shown that zinc, in addition to lowering copper levels, reduces iron status. 21 , 22 , 23 This was previously attributed to the competitive interaction between zinc and iron at the divalent metal ion transporter‐1 (DMT1). 24 Newer studies have shown that zinc is not a substrate for DMT1, and the likely site for competitive interaction is the transport protein ZIP14. 21 , 24 , 25 However, recent experiments using enterocytes found that zinc induces the expression of both DMT1 and FPN1, both of which facilitate iron intestinal absorption. 24 , 26 There remain conflicting data regarding the effect of zinc on overall iron status, and further investigations are needed to better understand the complex regulatory mechanisms in place.
Although zinc may induce iron absorption, copper is required for iron export from enterocytes. 17 , 24 Hephaestin is a homologue of ceruloplasmin in enterocytes, and its ferroxidase activity is required for iron export into circulation. 17 , 24 Like ceruloplasmin, hephaestin needs its copper cofactor for ferroxidation. 17 , 24 As discussed previously, zinc blocks enterocyte copper absorption. In addition, the amount of circulating copper‐carrying ceruloplasmin is already low in patients with WD, resulting in a low total serum copper level. 27 Because the enterocytes have reduced copper, the ability of hephaestin to ferroxidize iron for transport may be limited. Duodenal biopsies of patients with WD treated with zinc found increased iron concentrations in the mucosa. 28 The increased iron in the duodenal enterocytes supports the notion that there is impairment in the mobilization of iron due to copper scarcity. Authors of this study also found a considerable increase in metallothionein concentrations, suggesting that metallothionein production was protective against iron accumulation in the enterocytes. 28 The relative containment of iron in these enterocytes (that appear to possess self‐protective processes against iron accumulation) supports the use of zinc in patients with WD despite their induction of iron transporters.
Overall, despite the conflicting data, zinc monotherapy for lifelong maintenance therapy in patients with WD may still be the preferable option over long‐term copper chelation, particularly in those with evidence of iron overload. In patients with considerable iron overload, concurrent phlebotomy with copper chelation therapy should be considered. 1
According to clinical practice guidelines, patients with WD should have their copper and ceruloplasmin levels, as well as liver function tests, checked at least twice annually. 6 Considering the association with secondary hemochromatosis, it is not unreasonable to monitor iron indices regularly, particularly for those undergoing chelation therapy. 1 , 29
Conclusion
Secondary hemochromatosis from WD has been recognized in the literature for almost two decades. 1 , 2 , 3 Because of the rarity of WD, the association between iron overload and WD is not commonly discussed and is absent from practice guidelines as a rare cause of secondary hemochromatosis. Knowing whether a patient with WD has concomitant iron overload is important because it could impact immediate and long‐term therapy. Patients with WD should have iron indices monitored on a regular basis.
Potential conflict of interest: Nothing to report.
References
- 1. Shiono Y, Wakusawa S, Hayashi H, et al. Iron accumulation in the liver of male patients with Wilson's disease. Am J Gastroenterol 2001;96:3147‐3151. [DOI] [PubMed] [Google Scholar]
- 2. Harada M, Miyagawa K, Honma Y, et al. Excess copper chelating therapy for Wilson disease induces anemia and liver dysfunction. Intern Med 2011;50:1461‐1464. [DOI] [PubMed] [Google Scholar]
- 3. Hayashi H, Yano M, Fujita Y, et al. Compound overload of copper and iron in patients with Wilson's disease. Med Mol Morphol 2006;39:121‐126. [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez‐Castro KI, Hevia‐Urrutia FJ, Sturniolo GC. Wilson's disease: a review of what we have learned. World J Hepatol 2015;7:2859‐2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. European Association for Study of Liver . EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol 2012;56:671‐685. [DOI] [PubMed] [Google Scholar]
- 6. Roberts EA, Schilsky ML; American Association for Study of Liver Diseases. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008;47:2089‐2111. [DOI] [PubMed] [Google Scholar]
- 7. Twomey PJ, Viljoen A, House IM, et al. Relationship between serum copper, ceruloplasmin, and non‐ceruloplasmin‐bound copper in routine clinical practice. Clin Chem 2005;51:1558‐1559. [DOI] [PubMed] [Google Scholar]
- 8. Scheiber IF, Bruha R, Dusek P. Pathogenesis of Wilson disease Handb Clin Neurol 2017;142:43‐55. [DOI] [PubMed] [Google Scholar]
- 9. Fatemi N, Sarkar B. Molecular mechanism of copper transport in Wilson disease. Environ Health Perspect 2002;110(Suppl. 5):695‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bacon BR, Adams PC, Kowdley KV, et al.;American Association for the Study of Liver Diseases . Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:328‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kowdley KV, Brown KE, Ahn J, et al. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol 2019;114:1202‐1218. [DOI] [PubMed] [Google Scholar]
- 12. Anderson ER, Shah YM. Iron homeostasis in the liver. Compr Physiol 2013;3:315‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol 2009;122:78‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev 2010;68:133‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr 2002;22:439‐458. [DOI] [PubMed] [Google Scholar]
- 16. Vasilyev VB. Looking for a partner: ceruloplasmin in protein‐protein interactions. Biometals 2019;32:195‐210. [DOI] [PubMed] [Google Scholar]
- 17. MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal 2008;10:997‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kerkhof M, Honkoop P. Never forget aceruloplasminemia in case of highly suggestive Wilson's disease score. Hepatology 2014;59:1645‐1647. [DOI] [PubMed] [Google Scholar]
- 19. Piperno A, Alessio M. Aceruloplasminemia: waiting for an efficient therapy. Front Neurosci 2018;12:903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kathawala M, Hirschfield GM. Insights into the management of Wilson's disease. Therap Adv Gastroenterol 2017;10:889‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Brito NJ, Rocha ED, de Araujo Silva A, et al. Oral zinc supplementation decreases the serum iron concentration in healthy schoolchildren: a pilot study. Nutrients 2014;6:3460‐3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Donangelo CM, Woodhouse LR, King SM, et al. Supplemental zinc lowers measures of iron status in young women with low iron reserves. J Nutr 2002;132:1860‐1864. [DOI] [PubMed] [Google Scholar]
- 23. Rossander‐Hulten L, Brune M, Sandstrom B, et al. Competitive inhibition of iron absorption by manganese and zinc in humans. Am J Clin Nutr 1991;54:152‐156. [DOI] [PubMed] [Google Scholar]
- 24. Kondaiah P, Yaduvanshi PS, Sharp PA, et al. Iron and zinc homeostasis and interactions: does enteric zinc excretion cross‐talk with intestinal iron absorption? Nutrients 2019;11:1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iyengar V, Pullakhandam R, Nair KM. Coordinate expression and localization of iron and zinc transporters explain iron‐zinc interactions during uptake in Caco‐2 cells: implications for iron uptake at the enterocyte. J Nutr Biochem 2012;23:1146‐1154. [DOI] [PubMed] [Google Scholar]
- 26. Kondaiah P, Aslam MF, Mashurabad PC, et al. Zinc induces iron uptake and DMT1 expression in Caco‐2 cells via a PI3K/IRP2 dependent mechanism. Biochem J 2019;476:1573‐1583. [DOI] [PubMed] [Google Scholar]
- 27. Patil M, Sheth KA, Krishnamurthy AC, et al. A review and current perspective on Wilson disease. J Clin Exp Hepatol 2013;3:321‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sturniolo GC, Mestriner C, Irato P, et al. Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson's disease patients. Am J Gastroenterol 1999;94:334‐338. [DOI] [PubMed] [Google Scholar]
- 29. Erhardt A, Hoffmann A, Hefter H, et al. HFE gene mutations and iron metabolism in Wilson's disease. Liver 2002;22:474‐478. [DOI] [PubMed] [Google Scholar]