

Abstract
Multiple approaches to QSAR modeling using various statistical or machine learning techniques and different types of chemical descriptors have been developed over the years. Oftentimes models are used in consensus to make more accurate predictions at the expense of model interpretation. We propose a simple, fast, and reliable method termed Multi-Descriptor Read-Across (MuDRA) for developing both accurate and interpretable models. The method is conceptually related to the well-known kNN approach, but uses different types of chemical descriptors simultaneously for similarity assessment. To benchmark the new method, we have built MuDRA models for six different endpoints (Ames mutagenicity, aquatic toxicity, hepatotoxicity, hERG liability, skin sensitization, and endocrine disruption) and compared the results with those generated with conventional consensus QSAR modeling. We find that models built with MuDRA show consistently high external accuracy similar to that of conventional QSAR models. However, MuDRA models excel in terms of transparency, interpretability, and computational efficiency. We posit that due to its methodological simplicity and reliable predictive accuracy, MuDRA provides a powerful alternative to a much more complex consensus QSAR modeling. MuDRA is implemented and freely available at the Chembench web portal (https://chembench.mml.unc.edu/mudra).
Introduction
The Occam’s razor principle states, in its most popular version, that “Entities are not to be multiplied without necessity”.1 Many industries have followed this rule. A great example is provided by consumer electronics, where the iPhone’s simplistic design using one button to control all of its functions is widely considered innovative and effective (especially now with version X, which does not even have a home button!).2 Unfortunately, one cannot say that this approach has been closely followed by the Quantitative-Structure-Activity Relationship (QSAR) modeling field. Just the opposite - the field has progressed from relatively simple use of linear or multi-linear correlation methods in the original publications by Hansch et al.3–5 to the progressive use of increasingly more sophisticated multivariate machine learning approaches and different descriptor types (reviewed in Cherkasov et al.6).
Several important procedures have been developed to increase the reliability and predictive power of models, such as chemical and biological data curation,7–9 best practices for model development and validation,10,11 and calculation of the modelability index (MODI)12. The addition of these protocols has made the modeling process more complex but also, more robust, leading to the improved reliability and external predictive power of models built with any types of descriptors and model development algorithms. However, in the pursuit of higher statistical accuracy and predictive power of models, many scientists, including our group, have also begun to advocate for consensus (or ensemble/combinatorial) QSAR modeling.13,14 This approach relies on building many parallel QSAR models using different descriptor sets, several machine learning or statistical modeling techniques, and the integration of multiple models for consensus prediction of external compounds.
It has been established that the consensus QSAR approach has, on average, higher reliability than any of the contributing models.13–22 In addition, this approach was shown to be helpful for chemogenomics data curation.8,9 However, needless to say, the consensus approach requires multiple parallel efforts for developing different and diverse QSAR models, often accomplished by multiple modeling groups. For instance, the recent CERAPP project23 led by EPA has combined contributions from 17 research groups, with the respective paper co-authored by 41 scientists who have developed dozens of models for a single endpoint. While this project has demonstrated the power of teamwork and collegial approach to model development, it cannot be regarded as sustainable and applicable to any endpoint of interest. This consideration calls for the development of no less robust but implementation-wise simpler approaches.
In addition to the complexity and expense of execution, statistical QSAR models, and especially, consensus models, have been criticized for being a “black box”, i.e., not interpretable, which led to limited use of statistical QSAR models in regulatory decision support.24,25 At the same time, methods relying on structural alerts and read-across have found much greater acceptance.26 However, despite their perceived benefit, qualitative approaches such as structural alerts, chemical grouping, and read-across have been shown to be extremely over sensitive if used alone to predict biological activity.27 Indeed, it has been argued that to understand the underlying toxicity mechanisms, these qualitative approaches should be used only after proper statistical validation by QSAR.27
As discussed above, both qualitative and quantitative SAR modeling approaches have respective advantages as well as deficiencies. Although ensemble QSAR models represent a great advance in the field, the complexity of this approach, which requires the knowledge and integration of various QSAR approaches, is a serious barrier to their broad use. Conversely, mechanistic and alert-based approaches have been criticized for limited statistical accuracy. Recently, in an effort to establish both transparent and statistically accurate QSAR approach, we have developed a chemical-biological read-across (CBRA) approach for chemical toxicity assessment.28 This approach was later adopted by several groups.29–31
Using a similar methodology, herein we propose a simple approach for developing high quality QSAR models that relies on a single k-Nearest Neighbors (kNN) algorithm and multiple types of chemical descriptors used for consensus chemical similarity calculations. The latter approach - intrinsic to a single model development protocol - is in contrast with the much more complex and laborious consensus QSAR modeling approach discussed above. The proposed method is termed Multi-Descriptors Read Across (MuDRA). In benchmarking studies using several experimental datasets, we have built predictive QSAR models that were fully compliant with the OECD principles of model validation32 and demonstrated that these models offer several advantages over conventional ensemble QSAR models.
Interestingly, mudra is a word in Sanskrit, and one of its meanings is “an energetic seal of authenticity employed… in Indian religions”.33 We expect that MuDRA will become recognized as the most authentic, single universal QSAR modeling approach that can be relied upon for building models for most, if not all, target properties of interest.
Materials and Methods
Datasets
We have employed five different datasets previously used to build QSAR models for aquatic toxicity13, Ames mutagenicity34, hepatotoxicity35, hERG36, skin sensitization37, and endocrine receptor activity23. The datasets were thoroughly curated following the practices developed by our group earlier.7–9 Information regarding dataset size including compound distribution between modeling and external validation sets along with corresponding references is available in Table 1. To make the comparison fair, we have used the same external set both for QSAR and MuDRA. For aquatic toxicity, Ames mutagenicity, and hepatotoxicity we used the same external set available in the original publication. Since hERG and skin sensitization datasets were modeled following a 5-fold external cross validation procedure using the entire datasets in the original publications, we randomly selected 20% of the dataset as external set using stratified sampling to standardize the analysis within this study. In the recent Collaborative Estrogen Receptor Activity Prediction Project (CERAPP)23 more than 40 QSAR models for endocrine receptor activity were reported using either agonist, antagonist or binding activity data. For this endpoint only, we retrieved the external set the authors compiled from the literature containing 6,319 compounds with agonist activity, 6,539 compounds with antagonist activity, and 7,283 compounds with binding activity. We employed MuDRA to predict activity for these compounds and benchmarked our models against those reported in the CERAPP publication.
Table 1.
Summary of datasets employed in this study.
Calculation of Modelability Index (MODI) and model accuracy
The concept of “data set modelability”12 implies an a priori estimate of the feasibility to obtain predictive QSAR models for a given set of chemicals. MODI was calculated as follows (Eq. 1):
| Equation 1 |
where K is the number of classes (in all the studied cases, K = 2), is the number of compounds of i-th activity class that have their first nearest neighbors (NN) belonging to the same activity class i; is the total number of compounds belonging to the class i. Models were evaluated by the Correct Classification Rate (CCR, computed as the average of sensitivity and specificity of the model), sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
Molecular descriptors
We calculated four different types of molecular descriptors: ECFP4-like circular fingerprints (Morgan), PaDEL, Simplex Representation of Molecular Structure (SiRMS)38, and DRAGON. Morgan fingerprints with 2048 bits and atom radius of 2 were calculated using RDKit (http://www.rdkit.org). PaDEL descriptors39 were calculated using the PaDEL-Descriptor software freely available from http://www.yapcwsoft.com/dd/padeldescriptor/. SiRMS descriptors (number of tetratomic fragments with fixed composition and topological structure) were generated by the HiT QSAR software40. Finally, DRAGON descriptors were generated using DRAGON software (v.5.5, Talete SRL, Milan, Italy). All chemical descriptors, with the exception of Morgan fingerprints, were normalized to take values between 0 and 1. Constant and near constant variables were removed; and, if two or more variables were highly correlated (correlation coefficient above 0.9), only one of them was chosen randomly.
Traditional consensus QSAR modeling
Binary QSAR models were developed and rigorously validated according to the best practices of QSAR modeling.11 For each dataset, all four types of descriptors in combination with random forest were used.41 The consensus model was built by averaging the predicted values from each individual model. Consensus model considered majority of votes (at least three out of four) for the final classification. Cases when two models disagreed with the other two models were considered inconclusive. The consensus AD (Consensus with Applicability Domain) model was developed in a similar way; however, only predictions from individual models when the queried compound was inside the applicability domain (z-cutoff method16) were considered. In every case, only the modeling set was used to develop the models, while the external sets were used for evaluation and for comparison of model’s accuracy with the MuDRA method (see next section).
Multi-Descriptors Read-Across (MuDRA)
The MuDRA approach is based on the chemical-biology read-across (CBRA) method developed by us previously28. The CBRA approach relies on both structural similarity and comparisons of biological responses to chemicals in multiple short-term assays (“biological” similarity). Likewise, the MuDRA approach infers the target property of the queried compound from those of their structural analogs identified within each of the multiple chemical spaces defined by the respective descriptor sets (see Figure 1). In each chemical space, a predetermined number of nearest neighbors (NN) is selected based on their similarity to the query compound.
Figure 1.
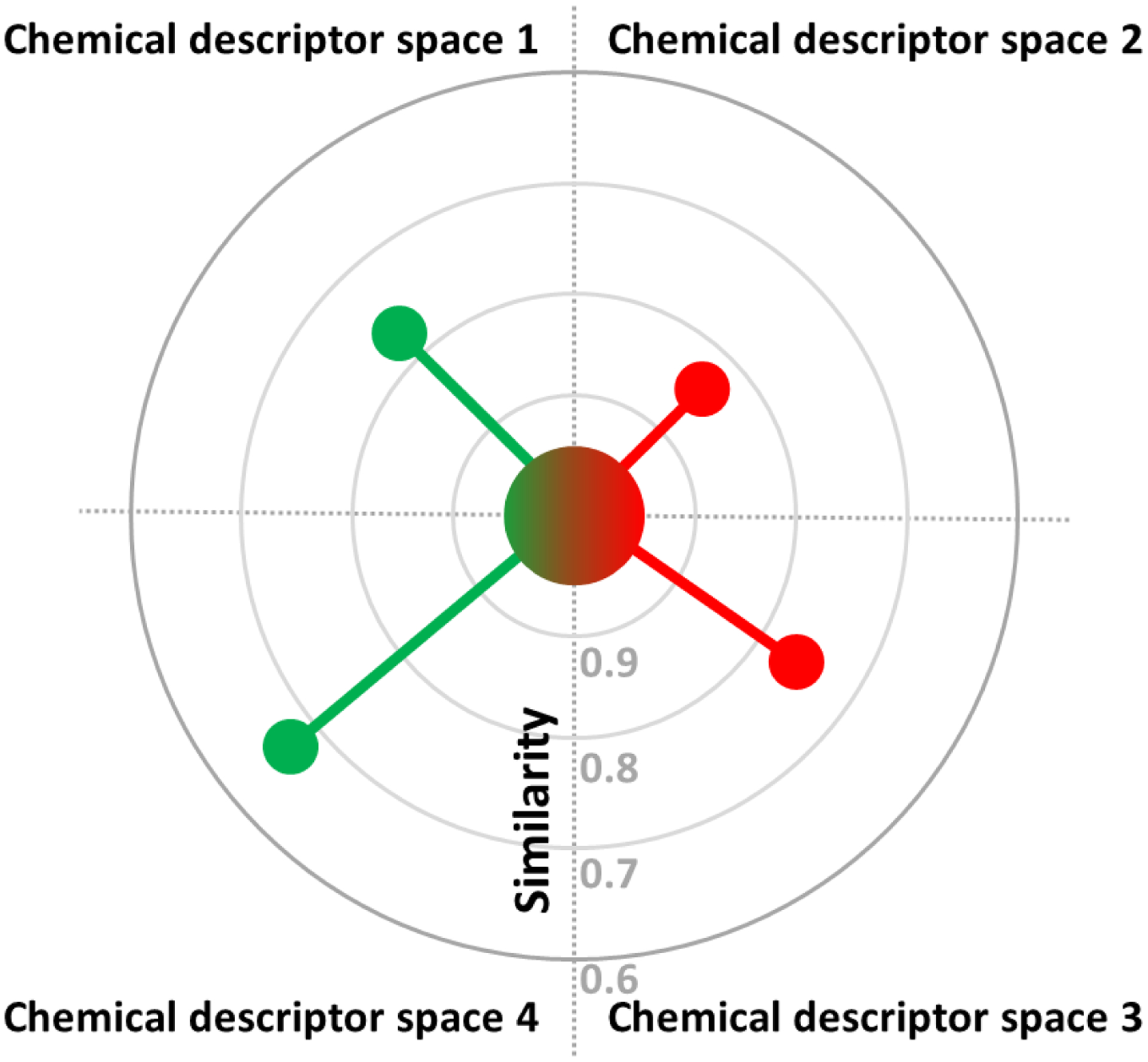
Radial plot representation of the MuDRA method. Activity (toxicity) prediction for a query compound (in the center) is accomplished based on the identification of the nearest neighbor compounds of the query compound in each chemical space (for simplicity of illustration, only one nearest neighbor is shown, but multiple such neighbors are identified typically). Red color circles represent active (or toxic) compounds and green circles represents inactive (or non-toxic) compounds. The activity (or toxicity) of a query compound is predicted as a weighted average of those of the similar compounds where the weight is inversely proportional to the distance from the query (see Eqs. 2 and 3).
The definition of similarity depends on the types of descriptors and similarity metrics that control the outcome of the MuDRA prediction. For quantitative read-across, the predicted activity of a compound (Apred) is calculated using the following equation (Eq. 2) from the similarity (Si) weighted aggregate of the activities Ai of k nearest neighbors.
| Equation 2 |
The pairwise Tanimoto similarity Si between the molecule of interest (A) and its ith neighbor (B) is calculated from the Jaccard distance dJac (see Eq. 3)42, where there are D descriptor spaces with the p1, p2,…,pD descriptors , … , and j=1, … , D. For each ith compound of a dataset, the similarity with compound B in space j is calculated.
| Equation 3 |
For a set of range-scaled continuous descriptors, Tanimoto similarity is normalized between 0 and 1, with 1 corresponding to identical pairs. The similarity-weighted aggregate in Eq. 2 ensures that the activities of more similar neighbors are given higher weights when calculating the predicted activity. This step can be enhanced by considering different descriptor scaling schemes such as range scaling or mean absolute deviation scaling. Compound activity (i.e., the activity predicted by MuDRA) is defined by the similarity with activities of NN in j space, where Bj=1, … , nj (Eq. 4, MuDRA model). In this study, we analyzed 1, 3, 5, and 7 neighbors. Initially, we employed four sets of descriptors (D=4). Activities of nontoxic compounds are assigned “0” while those of toxic compounds are assigned “1”.
| Equation 4 |
Results
MODI
All datasets used in this study are well known and have been previously modeled (cf. Table 1). To ensure that datasets are modelable, we calculated MODI (cf. Eq. 1) for all sets of descriptors. All datasets, except for hepatotoxicity, had MODIs of at least 70% (Table 2).
Table 2.
Calculated MODI for each set of descriptors.
| Datasets | Descriptors | |||
|---|---|---|---|---|
| Morgan | PaDEL | SiRMS | DRAGON | |
| Ames mutagenicity | 0.70 | 0.71 | 0.72 | 0.70 |
| Aquatic toxicity | 0.77 | 0.80 | 0.78 | 0.79 |
| Hepatotoxicity | 0.58 | 0.67 | 0.68 | 0.57 |
| hERG | 0.70 | 0.71 | 0.72 | 0.71 |
| Skin sensitization | 0.75 | 0.72 | 0.72 | 0.72 |
Consensus QSAR vs. MuDRA models for chemical toxicity prediction
The statistical characteristics of consensus QSAR and MuDRA predictions for chosen toxicity endpoints are summarized in Table 3. QSAR models were built using Morgan, PaDEL, SiRMS, and DRAGON descriptors and the random forest algorithm. Statistical characteristics for all individual QSAR models can be found in the Supporting Information. All QSAR models, except for hepatotoxicity, were robust and predictive, with CCR for external sets being in the 66–93% range. The external set for the hepatotoxicity datasets was small (25 compounds), and most of the toxic compounds were mispredicted. For this particular dataset, Low et al.35 were able to produce predictive models by combining both chemical descriptors and toxicogenomics data in a hybrid QSAR model.
Table 3.
Statistical characteristics of external QSAR predictions vs. MuDRA predictions for selected toxicity endpoints.
| Ames mutagenicity (n = 2,181) | ||||||
|---|---|---|---|---|---|---|
| Models | CCR | Sensitivity | PPV | Specificity | NPV | Coverage |
| Consensus | 0.79 | 0.91 | 0.76 | 0.67 | 0.86 | 1.00 |
| Consensus AD | 0.79 | 0.91 | 0.77 | 0.66 | 0.86 | 0.91 |
| 1-NN MuDRA | 0.78 | 0.78 | 0.80 | 0.78 | 0.76 | 1.00 |
| 3-NN MuDRA | 0.80 | 0.80 | 0.82 | 0.80 | 0.77 | 1.00 |
| 5-NN MuDRA | 0.79 | 0.81 | 0.81 | 0.78 | 0.78 | 1.00 |
| 7-NN MuDRA | 0.80 | 0.82 | 0.80 | 0.77 | 0.79 | 1.00 |
| Aquatic toxicity (n = 339) | ||||||
| CCR | Sensitivity | PPV | Specificity | NPV | Coverage | |
| Consensus | 0.86 | 0.93 | 0.85 | 0.80 | 0.89 | 1.00 |
| Consensus AD | 0.93 | 0.97 | 0.92 | 0.89 | 0.96 | 0.81 |
| 1-NN MuDRA | 0.87 | 0.90 | 0.88 | 0.85 | 0.87 | 1.00 |
| 3-NN MuDRA | 0.88 | 0.92 | 0.88 | 0.83 | 0.89 | 1.00 |
| 5-NN MuDRA | 0.86 | 0.88 | 0.88 | 0.84 | 0.85 | 1.00 |
| 7-NN MuDRA | 0.86 | 0.89 | 0.88 | 0.84 | 0.86 | 1.00 |
| Hepatotoxicity (n = 25) | ||||||
| CCR | Sensitivity | PPV | Specificity | NPV | Coverage | |
| Consensus | 0.48 | 0.11 | 0.33 | 0.86 | 0.60 | 1.00 |
| Consensus AD | 0.49 | 0.17 | 0.33 | 0.82 | 0.64 | 0.68 |
| 1-NN MuDRA | 0.50 | 0.20 | 0.40 | 0.80 | 0.60 | 1.00 |
| 3-NN MuDRA | 0.50 | 0.20 | 0.40 | 0.80 | 0.60 | 1.00 |
| 5-NN MuDRA | 0.53 | 0.20 | 0.50 | 0.87 | 0.62 | 1.00 |
| 7-NN MuDRA | 0.50 | 0.20 | 0.40 | 0.80 | 0.60 | 1.00 |
| hERG (n = 1,197) | ||||||
| CCR | Sensitivity | PPV | Specificity | NPV | Coverage | |
| Consensus | 0.80 | 0.87 | 0.82 | 0.72 | 0.80 | 1.00 |
| Consensus AD | 0.80 | 0.87 | 0.82 | 0.73 | 0.80 | 0.93 |
| 1-NN MuDRA | 0.76 | 0.82 | 0.78 | 0.69 | 0.74 | 1.00 |
| 3-NN MuDRA | 0.77 | 0.87 | 0.79 | 0.68 | 0.79 | 1.00 |
| 5-NN MuDRA | 0.78 | 0.87 | 0.78 | 0.68 | 0.80 | 1.00 |
| 7-NN MuDRA | 0.75 | 0.87 | 0.76 | 0.64 | 0.78 | 1.00 |
| Skin sensitization (n = 217) | ||||||
| CCR | Sensitivity | PPV | Specificity | NPV | Coverage | |
| Consensus | 0.66 | 0.64 | 0.90 | 0.68 | 0.28 | 1.00 |
| Consensus AD | 0.78 | 0.82 | 0.94 | 0.75 | 0.46 | 0.42 |
| 1-NN MuDRA | 0.71 | 0.66 | 0.93 | 0.76 | 0.31 | 1.00 |
| 3-NN MuDRA | 0.64 | 0.64 | 0.90 | 0.65 | 0.27 | 1.00 |
| 5-NN MuDRA | 0.62 | 0.62 | 0.89 | 0.62 | 0.25 | 1.00 |
| 7-NN MuDRA | 0.68 | 0.65 | 0.91 | 0.70 | 0.29 | 1.00 |
The MuDRA models showed predictive power similar to that of traditional QSAR models. We also found that the accuracy of MuDRA did not vary considerably with the increase of the number of NNs. This is mainly due the fact that the closest NNs have higher influence, regardless of the chemical space and the number of compounds selected to be included. With the exception of hepatotoxicity, all the MuDRA models were predictive, with CCRs ranging from 62% to 88%. For the Ames mutagenicity data, the MuDRA approach presented a CCR 3–6% higher than the consensus model. Consensus AD showed the highest CCR, but at the expense of somewhat limited coverage (91%). For aquatic toxicity, the Consensus and Consensus AD model presented a CCR of 86% and 93%, respectively. The MuDRA approach produced a CCR as high as 86–88%. QSAR models for hepatotoxicity did not satisfy the standards for external validation. For this endpoint, 10 of the 25 external set compounds were toxic, but only one compound was correctly predicted as toxic by our models. Conversely, the MuDRA approach showed similar behavior, predicting only two compounds correctly. QSAR models for hERG outperformed MuDRA by 2–5% in CCR. Finally, skin sensitization QSAR models showed a CCR of 69% and 71% for consensus and consensus rigor models, respectively. The MuDRA approach for skin sensitization showed the highest variability of predictive accuracy for different numbers of NN used (9%), which may be related to the high structural diversity of skin sensitizers.43–46 In addition, both the QSAR models and the MuDRA approach presented a low NPV (28–31%) due to the low number of non-sensitizers (n = 37) in comparison to the non-sensitizers (n = 180) in the external set.
As a final exercise, we retrieved the literature data compiled for the CERAPP23 project and compared the predictions from MuDRA and those obtained with different models reported in CERAPP publication. This dataset is comprised of 6,319 compounds with agonist activity, 6,539 compounds with antagonist activity, and 7,283 compounds with binding activity compounds. Since not all of the models reported in the CERAPP paper predicted all of the compounds in the external set, we only compared our predictions obtained with MuDRA with those generated by previous models developed in CERAPP for 100% of compounds. The latter subset included five models for agonist activity, four models for antagonist activity, and nine models for binding affinity. In the CERAPP paper, sensitivity, specificity, and CCR (named “balanced accuracy” in the publication) were reported, and thus only these metrics were compared. The comparisons between predictions obtained with QSAR models reported in the CERAPP publication and those generated by MuDRA are summarized in Table 4. As one can see, MuDRA had similar performance as compared to the QSAR models reported in the CERAPP study in terms of CCR, sensitivity, and specificity for all the three endpoints (agonist, antagonist, and binding).
Table 4.
Statistical characteristics of external QSAR predictions reported in CERAPP vs. MuDRA predictions for endocrine receptor activity.
| Agonist (n=6,319) | |||
|---|---|---|---|
| Model | CCR | Sensitivity | Specificity |
| CERAPP (n = 5) | 0.73 (± 0.05) | 0.51 (± 0.13) | 0.95 (± 0.05) |
| MuDRA | 0.74 | 0.65 | 0.83 |
| Antagonist (n=6,532) | |||
| Model | CCR | Sensitivity | Specificity |
| CERAPP (n =) | 0.53 (± 0.02) | 0.11 (± 0.09) | 0.95 (± 0.05) |
| MuDRA | 0.52 | 0.05 | 0.99 |
| Binding (n=7,283) | |||
| Model | CCR | Sensitivity | Specificity |
| CERAPP (n = 9) | 0.57 (± 0.02) | 0.27 (± 0.11) | 0.85 (± 0.08) |
| MuDRA | 0.58 | 0.35 | 0.81 |
Number of descriptors vs. model predictivity
To test how the number of descriptor types affects the model quality, we calculated the average CCR for one, two, three, and four descriptor space components for each studied dataset. As one can see from Figure 2, CCR for Ames, aquatic toxicity, hERG, and skin sensitization increases gradually, stabilizing at 3-NN. The only endpoint the CCR is higher at one and two NN is hepatotoxicity, but as one can see from Table 3 this dataset is not modelable. As Bender47 have shown, the use of different types of descriptors leads to different chemical similarity assessment for the same pairs of compounds. Therefore, a compound may not preserve the same NNs in another chemical space. By using four chemical spaces, the error in predictions given by a particular nearest neighbor is minimized and the predictivity of the method is on par with conventional ensemble QSAR.
Figure 2.
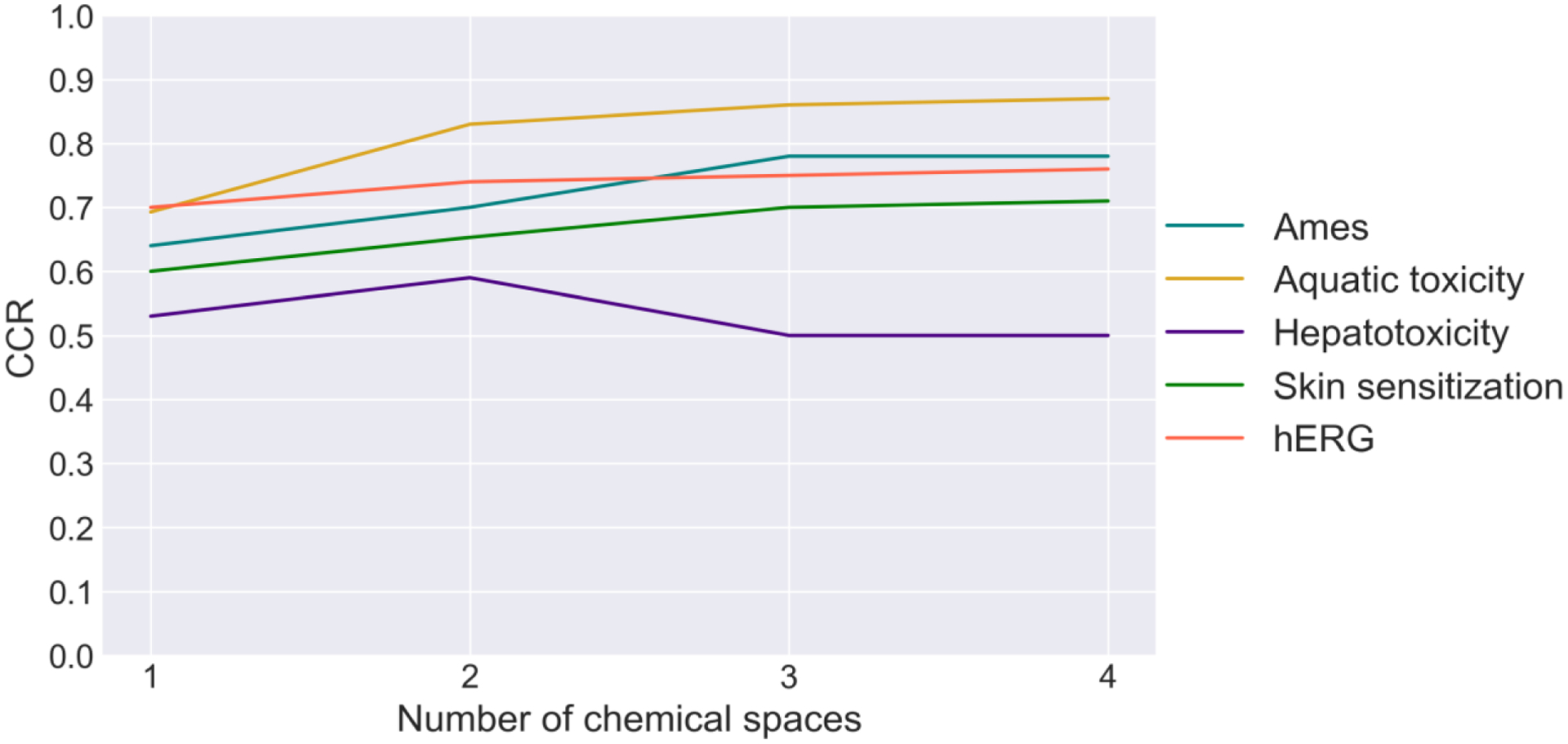
Correct classification rate variation for 1, 2, 3, and 4 chemical spaces.
Discussion
QSAR models vs. MuDRA for chemical toxicity prediction
In chemical toxicity prediction, the expected outcome is a reliable, computationally efficient, and transparent prediction for every queried compound. Developing an ensemble of models requires access to diverse modeling approaches (or software), which is hard to achieve within a single research group, or consolidation of results generated in multiple research groups. For instance, in the CERAPP project23, more than 30 diverse models generated by several research groups using different tools to predict estrogen receptor activity of environmental compounds were integrated. Therefore, we sought to develop a simple, fast, and reliable approach that would produce the same quality predictions of current QSAR models while also providing 100% coverage of the chemical space.
As one can see from Table 3 and Table 4, the use of the MuDRA approach resulted in models with similar accuracy to those built individually with Random Forest. The nearest neighbor aggregation employed by MuDRA ensures that most similar neighbors have higher weights, regardless of the descriptor type. Thus, this feature ensures that the most similar neighbors will drive the outcome of the prediction.
The final prediction of MuDRA differs from qualitative approaches such as structural alerts, chemical grouping, and chemical read-across. Structural alerts are used to flag potential hazards and to group compounds into categories for read-across.48 Chemical read-across is a data gap filling procedure to assess certain endpoint effect of a chemical by using data for the same endpoint from another chemical (or a group of chemicals), which is (are) considered structurally similar.49 These methods have earned acceptance among toxicologists due to their simplicity and ease of interpretation.50 However, as we have shown,27 structural alerts are extremely over-sensitive if used alone to predict biological activity. Moreover, the use of structural alerts may be detrimental in the drug discovery pipeline as well as for the safety assessment due to high false-positive rate.51 If the MuDRA model is built for a high quality and properly curated dataset7–9 following the best practices for model development and validation,6,11 its accuracy is expected to be similar to that of conventional QSAR models. In contrast with the current chemical read-across procedure, the newly-developed MuDRA approach consists of an instance-based learner, i.e., it compares new instances with instances seen in training set, instead of performing explicit generalization.52 We posit that the MuDRA method provides an alternative to the ensemble modeling approach due to the easier implementation and decreased computational cost.
Another important benefit of MuDRA is its transparency, which can aid in the design of new compounds with improved characteristics. We illustrate this approach in Figure 3. As one can see, the predicted compound preserves toxicity if a nitro group is inserted in meta position relative to the bromomethyl group, as observed in Morgan and SiRMS spaces. However, as shown in PADEL space, if bromine is substituted by a bulkier group, as well as the change substitution of a isoquinoline heterocycle by a quinoxaline leads to a non-toxic compound. Moreover, Dragon descriptors demonstrate that, although NN in the chemical space of the integral (whole-molecule) descriptors may be unexpected and substantially different from others, it still may contribute to final correct prediction.
Figure 3.
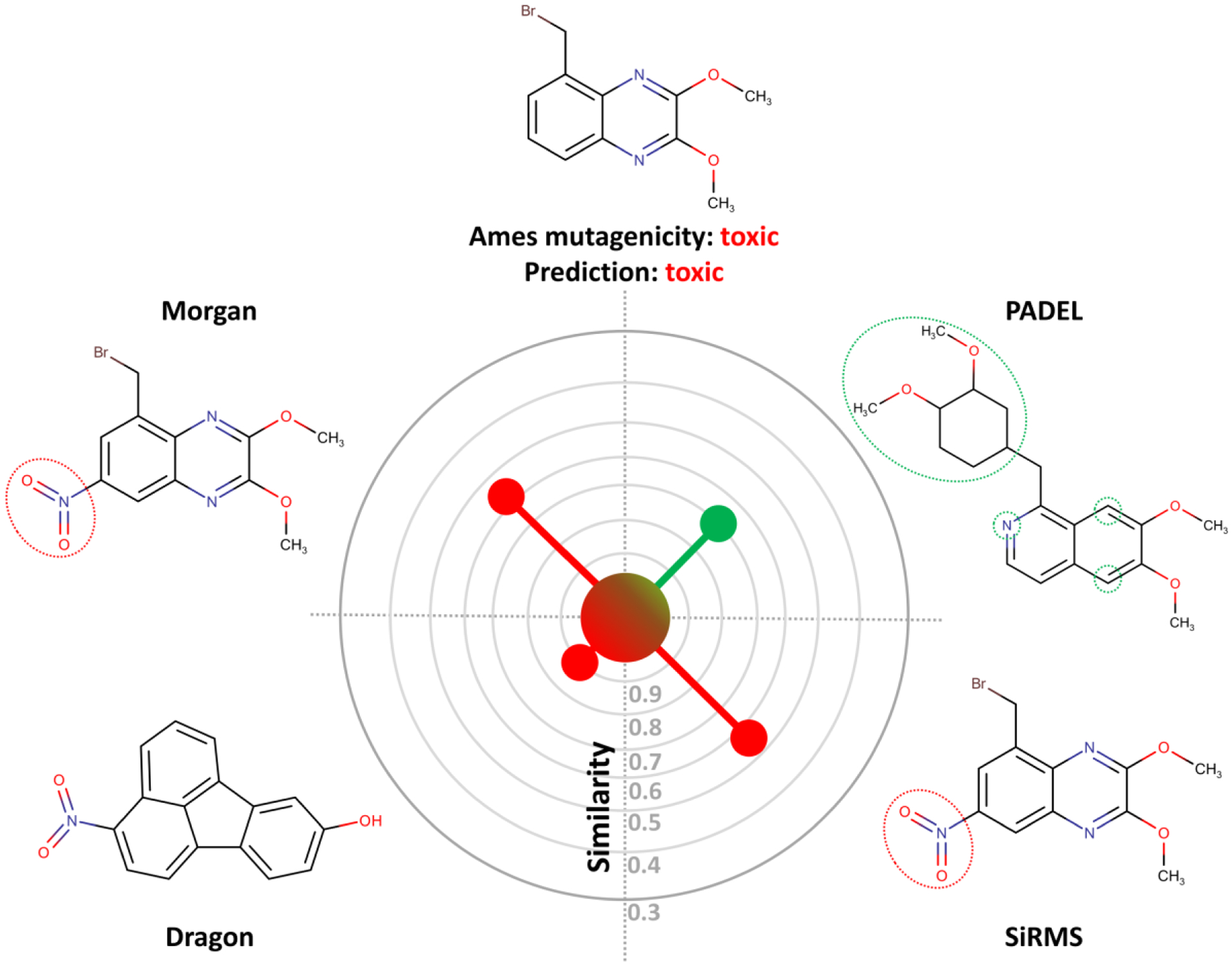
Example of the analysis of NNs in different chemical spaces provided by MuDRA.
We also envision that the MuDRA approach will be useful for the prediction of activity cliffs based on the Matched Molecular Pair (MMP) methodology.53 It should be noted, however, that since MuDRA is based on the activity profiles of structurally similar compounds, the efficiency of this approach would depend entirely on the diversity of MMP network-space and the consistency of the SAR within this network. This observation is also reflected in Figure 3.
In summary, MuDRA was shown to be as reliable as QSAR models, but easier and faster to develop, as well as more transparent while providing 100% coverage. The workflow for MuDRA model generation includes the following steps: (i) calculation of various chemical descriptors for the training set, (ii) calculation of Tanimoto similarity for the query compound against all chemicals in the training set in all descriptor spaces; (iii) activity prediction for the query compound based on the weighted similarity to the nearest neighbors of the query in each chemical space. Since the increase in the number of NNs is not followed by the increase in the accuracy of MuDRA models, we strongly suggest using only one NN per descriptor type. We also emphasize that similar to any QSAR method, the MuDRA model accuracy is estimated for an external set not used to build and/or select the models.
Dissemination
To enable the use of this approach, we have added a MuDRA module to Chembench, our integrated cheminformatics web portal54. More details about Chembench could be found elsewhere54. Similar to other cheminformatics services at Chembench54, MuDRA module is freely accessible at https://chembench.mml.unc.edu/mudra.
To execute the MuDRA approach on Chembench, the user must first upload chemical structures and associated activities (either continuous or binary values) as a modeling dataset in the Datasets module. Then, MODI12 and the descriptors required for MuDRA are calculated. The descriptors available for building MuDRA models include DragonH55, MOE2D56, MACCS keys57, and ISIDA58. These descriptors were selected because they are different in nature and available for QSAR model development on our publicly accessible Chembench portal. It should be noted that several publicly available datasets such as Tox21 stress response and nuclear receptor signaling toxicity assay datasets59 have been included with the MuDRA module.
Once the modeling dataset is uploaded successfully, the user must access the MuDRA module within Chembench and select the dataset. This module facilitates predictions of single compounds, batches of multiple compounds, and virtual chemical libraries. A single compound can be sketched directly using JSME60 or its SMILES can be input. A specific library of interest or a batch of compounds can be uploaded within the Datasets module and used for virtual screening. Chembench has several publicly available chemical libraries, such as the DrugBank61 and the ZINC lead-like library62, that can be also used for virtual screening. Next, the predicted activity of the compound(s) is calculated according to MuDRA equations 2–4.
For a single compound predictions, the outputs are the compound SMILES, the predicted activity, the number of k nearest neighbors (default is to set k = 1), as well as the average Tanimoto similarity, average activity, and k nearest neighbor IDs for all four descriptor types used (Figure 4). For predictions of multiple compounds, the outputs are the ID of the screened compound, the average prediction value, the rounded prediction value (for binary data), the number of k nearest neighbors (default is to set k = 1), as well as the average activity, average similarity, and IDs of the k nearest neighbor IDs for all four descriptor types used (Figure 5). Multiple compound prediction results can be downloaded as a CSV-delimited file.
Figure 4.
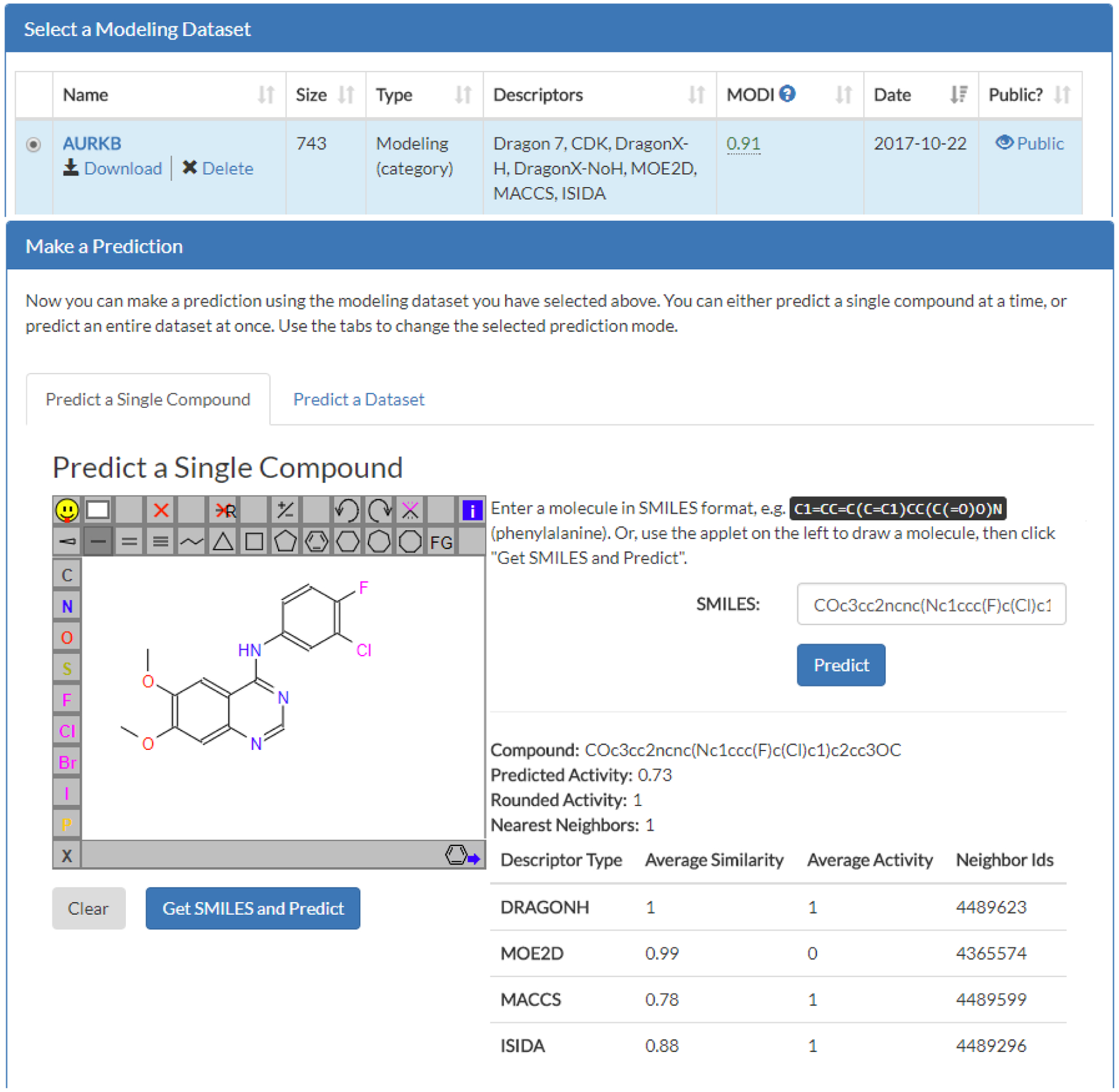
MuDRA prediction for a single compound. In the MuDRA module, a modeling dataset “AURKB” was selected and the single compound to be assessed was sketched. The compound was then predicted and results of MuDRA were output.
Figure 5.
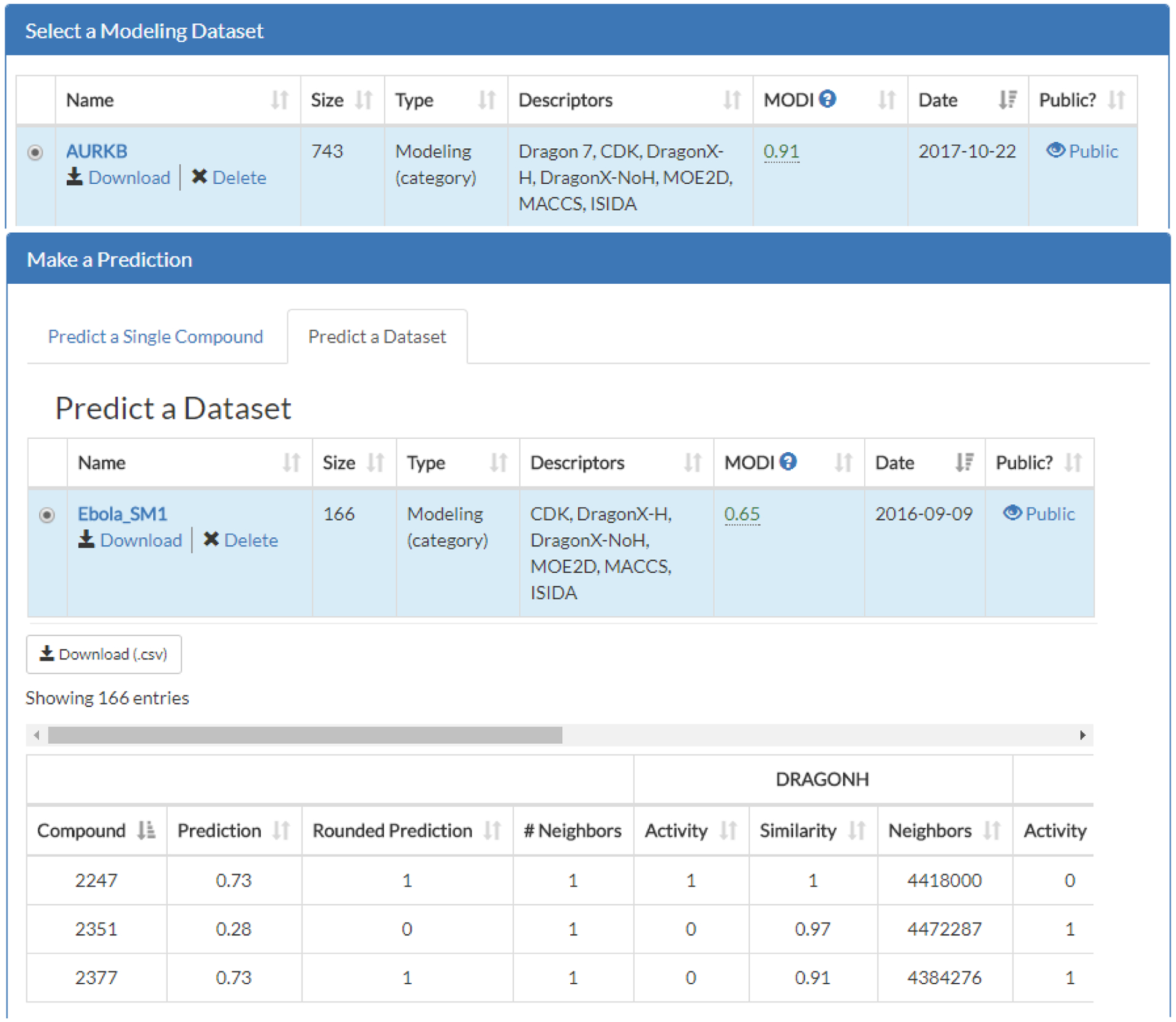
MuDRA predictions for multiple compounds. In the MuDRA module, a modeling dataset “AURKB” was selected and a library of 166 compounds called “Ebola_SM1” was assessed. The compounds were then predicted and results of MuDRA were output. These results are downloadable as a CSV file. It should be noted that the full list of compounds and the associated outputs are not shown here.
Conclusions
We have developed a simple, fast, and reliable approach that yields models of high accuracy with 100% coverage of the prediction set. We posit that because of its simplicity and high accuracy, MuDRA can be successfully used as a computationally efficient but no less reliable, in terms of statistical accuracy and predictive power, alternative to ensemble QSAR. The results obtained on several diverse datasets demonstrated that MuDRA has similar predictive performance to traditional QSAR approaches. At the same time, due to its conceptual simplicity and transparency, MuDRA affords non-experts in the field an alternative to structural alerts and read-across. We have made an implementation of MuDRA freely available on the Chembench web-portal (https://chembench.mml.unc.edu/mudra).
Supplementary Material
Acknowledgements
This study was supported in part by NIH (grant 1U01CA207160). VMA, ENM, and CHA thank CNPq (grant 400760/2014-2). VA also thanks CAPES and FAPEG (201310267001095) for partial support.
Abbreviations
- AD
applicability domain
- CBRA
Chemical-Biology Read-Across
- CCR
correct classification rate
- MuDRA
Multi-Descriptors Read-Across
- MODI
Modelability Index
- NN
nearest neighbor
- NPV
negative predictive value
- PPV
positive predictive value
- QSAR
quantitative structure-activity relationship
- SiRMS
Simplex Representation of Molecular Structure
Footnotes
Supporting information includes curated chemical datasets for all endpoints along with predictions from QSAR models and MuDRA. These materials are available free of charge via the Internet at http://pubs.acs.org.
Conflict of interests
The authors declare no actual or potential conflict of interests.
References
- 1.Occam’s razor https://en.wikipedia.org/wiki/Occam%27s_razor (accessed Mar 1, 2018).
- 2.Apple iPhone https://www.apple.com/iphone/ (accessed Mar 1, 2018).
- 3.Hansch C; Maloney P; Fujita T; Muir R Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar]
- 4.Hansch C; Fujita T P-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc 1963, 86, 1616–1626. [Google Scholar]
- 5.Fujita T; Iwasa J; Hansch C A New Substituent Constant, Π, Derived from Partition Coefficients. J. Am. Chem. Soc 1964, 86, 5175–5180. [Google Scholar]
- 6.Cherkasov A; Muratov EN; Fourches D; Varnek A; Baskin II; Cronin M; Dearden J; Gramatica P; Martin YC; Todeschini R; Consonni V; Kuz’min VE; Cramer R; Benigni R; Yang C; Rathman J; Terfloth L; Gasteiger J; Richard A; Tropsha A QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem 2014, 57, 4977–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fourches D; Muratov E; Tropsha A Trust, but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model 2010, 50, 1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fourches D; Muratov EN; Tropsha A Trust, But Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model 2016, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fourches D; Muratov E; Tropsha A Curation of Chemogenomics Data. Nat. Chem. Biol 2015, 11, 535. [DOI] [PubMed] [Google Scholar]
- 10.Golbraikh A; Tropsha A Beware of q2! J. Mol. Graph. Model 2002, 20, 269–276. [DOI] [PubMed] [Google Scholar]
- 11.Tropsha A Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform 2010, 29, 476–488. [DOI] [PubMed] [Google Scholar]
- 12.Golbraikh A; Muratov E; Fourches D; Tropsha A Data Set Modelability by QSAR. J. Chem. Inf. Model 2014, 54, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu H; Tropsha A; Fourches D; Varnek A; Papa E; Gramatica P; Oberg T; Dao P; Cherkasov A; Tetko IV Combinatorial QSAR Modeling of Chemical Toxicants Tested against Tetrahymena Pyriformis. J. Chem. Inf. Model 2008, 48, 766–784. [DOI] [PubMed] [Google Scholar]
- 14.Svetnik V; Wang T; Tong C; Liaw A; Sheridan RP; Song Q Boosting: An Ensemble Learning Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Model 2005, 45, 786–799. [DOI] [PubMed] [Google Scholar]
- 15.Wang XS; Tang H; Golbraikh A; Tropsha A Combinatorial QSAR Modeling of Specificity and Subtype Selectivity of Ligands Binding to Serotonin Receptors 5HT1E and 5HT1F. J. Chem. Inf. Model 2008, 48, 997–1013. [DOI] [PubMed] [Google Scholar]
- 16.Tropsha A; Golbraikh A Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr. Pharm. Des 2007, 13, 3494–3504. [DOI] [PubMed] [Google Scholar]
- 17.Pissurlenkar RRS; Khedkar VM; Iyer RP; Coutinho EC Ensemble QSAR: A QSAR Method Based on Conformational Ensembles and Metric Descriptors. J. Comput. Chem 2011, 32, 2204–2218. [DOI] [PubMed] [Google Scholar]
- 18.Pradeep P; Povinelli RJ; White S; Merrill SJ An Ensemble Model of QSAR Tools for Regulatory Risk Assessment. J. Cheminform 2016, 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuz’min VE; Muratov EN; Artemenko AG; Varlamova EV; Gorb L; Wang J; Leszczynski J Consensus QSAR Modeling of Phosphor-Containing Chiral AChE Inhibitors. QSAR Comb. Sci 2009, 28, 664–677. [Google Scholar]
- 20.Artemenko AG; Muratov EN; Kuz’min VE; Muratov NN; Varlamova EV; Kuz’mina AV; Gorb LG; Golius A; Hill FC; Leszczynski J; Tropsha A QSAR Analysis of the Toxicity of Nitroaromatics in Tetrahymena Pyriformis: Structural Factors and Possible Modes of Action. SAR QSAR Environ. Res 2011, 22, 575–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neves BJ; Dantas RF; Senger MR; Melo-Filho CC; Valente WCG; de Almeida ACM; Rezende-Neto JM; Lima EFC; Paveley R; Furnham N; Muratov E; Kamentsky L; Carpenter AE; Braga RC; Silva-Junior FP; Andrade CH Discovery of New Anti-Schistosomal Hits by Integration of QSAR-Based Virtual Screening and High Content Screening. J. Med. Chem 2016, 59, 7075–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melo-Filho CC; Dantas RF; Braga RC; Neves BJ; Senger MR; Valente WCG; Rezende-Neto JM; Chaves WT; Muratov EN; Paveley RA; Furnham N; Kamentsky L; Carpenter AE; Silva-Junior FP; Andrade CH QSAR-Driven Discovery of Novel Chemical Scaffolds Active against Schistosoma Mansoni. J. Chem. Inf. Model 2016, 56, 1357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mansouri K; Abdelaziz A; Rybacka A; Roncaglioni A; Tropsha A; Varnek A; Zakharov A; Worth A; Richard AM; Grulke CM; Trisciuzzi D; Fourches D; Horvath D; Benfenati E; Muratov E; Wedebye EB; Grisoni F; Mangiatordi GF; Incisivo GM; Hong H; Ng HW; Tetko IV; Balabin I; Kancherla J; Shen J; Burton J; Nicklaus M; Cassotti M; Nikolov NG; Nicolotti O; Andersson PL; Zang Q; Politi R; Beger RD; Todeschini R; Huang R; Farag S; Rosenberg SA; Slavov S; Hu X; Judson RS CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect 2016, 124, 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casati S; Aschberger K; Barroso J; Casey W; Delgado I; Kim TS; Kleinstreuer N; Kojima H; Lee JK; Lowit A; Park HK; Régimbald-Krnel MJ; Strickland J; Whelan M; Yang Y; Zuang V Standardisation of Defined Approaches for Skin Sensitisation Testing to Support Regulatory Use and International Adoption: Position of the International Cooperation on Alternative Test Methods. Arch. Toxicol 2017, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basketter D; Alépée N; Casati S; Crozier J; Eigler D; Griem P; Hubesch B; de Knecht J; Landsiedel R; Louekari K; Manou I; Maxwell G; Mehling A; Netzeva T; Petry T; Rossi LH Skin Sensitisation--Moving Forward with Non-Animal Testing Strategies for Regulatory Purposes in the EU. Regul. Toxicol. Pharmacol 2013, 67, 531–5. [DOI] [PubMed] [Google Scholar]
- 26.Raunio H In Silico Toxicology - Non-Testing Methods. Front. Pharmacol 2011, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alves VM; Muratov EN; Capuzzi SJ; Politi R; Low Y; Braga RC; Zakharov AV; Sedykh A; Mokshyna E; Farag S; Andrade CH; Kuz’min VE; Fourches D; Tropsha A Alarms about Structural Alerts. Green Chem 2016, 18, 4348–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low Y; Sedykh AY; Fourches D; Golbraikh A; Whelan M; Rusyn I; Tropsha A Integrative Chemical-Biological Read-Across Approach for Chemical Hazard Classification. Chem. Res. Toxicol 2013, 26, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J; Mansouri K; Judson RS; Martin MT; Hong H; Chen M; Xu X; Thomas R; Shah I Predicting Hepatotoxicity Using ToxCast in Vitro Bioactivity and Chemical Structure. Chem. Res. Toxicol 2015, 28, 738–751. [DOI] [PubMed] [Google Scholar]
- 30.Shah I; Liu J; Judson RS; Thomas RS; Patlewicz G Systematically Evaluating Read-across Prediction and Performance Using a Local Validity Approach Characterized by Chemical Structure and Bioactivity Information. Regul. Toxicol. Pharmacol 2016, 79, 12–24. [DOI] [PubMed] [Google Scholar]
- 31.Russo DP; Kim MT; Wang W; Pinolini D; Shende S; Strickland J; Hartung T; Zhu H CIIPro: A New Read-across Portal to Fill Data Gaps Using Public Large-Scale Chemical and Biological Data. Bioinformatics 2017, 33, 464–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.OECD OECD Principles for the Validation, for Regulatory Purposes, of (Quantitative) Structure-Activity Relationship Models http://europa.eu.int/comm/environment/chemicals/reach.htm (accessed Feb 17, 2017).
- 33.Mudra https://en.wikipedia.org/wiki/Mudra (accessed Mar 1, 2018).
- 34.Sushko I; Novotarskyi S; Körner R; Pandey AK; Cherkasov A; Li J; Gramatica P; Hansen K; Schroeter T; Müller K-R; Xi L; Liu H; Yao X; Öberg T; Hormozdiari F; Dao P; Sahinalp C; Todeschini R; Polishchuk P; Artemenko A; Kuz’min V; Martin TM; Young DM; Fourches D; Muratov E; Tropsha A; Baskin I; Horvath D; Marcou G; Muller C; Varnek A; Prokopenko VV; Tetko IV Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model 2010, 50, 2094–3111. [DOI] [PubMed] [Google Scholar]
- 35.Low Y; Uehara T; Minowa Y; Yamada H; Ohno Y; Urushidani T; Sedykh A; Muratov E; Kuz’min V; Fourches D; Zhu H; Rusyn I; Tropsha A Predicting Drug-Induced Hepatotoxicity Using QSAR and Toxicogenomics Approaches. Chem. Res. Toxicol 2011, 24, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braga RC; Alves VM; Silva MFB; Muratov E; Fourches D; Lião LM; Tropsha A; Andrade CH Pred-hERG: A Novel Web-Accessible Computational Tool for Predicting Cardiac Toxicity. Mol. Inform 2015, 34, 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braga RC; Alves VM; Muratov EN; Strickland J; Kleinstreuer N; Tropsha A; Andrade CH Pred-Skin: A Fast and Reliable Web Application to Assess Skin Sensitization Effect of Chemicals. J. Chem. Inf. Model 2017, 57, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 38.Muratov EN; Artemenko AG; Varlamova EV; Polischuk PG; Lozitsky VP; Fedchuk AS; Lozitska RL; Gridina TL; Koroleva LS; Sil’nikov VN; Galabov AS; Makarov V. a; Riabova OB; Wutzler P; Schmidtke M; Kuz’min VE Per Aspera Ad Astra: Application of Simplex QSAR Approach in Antiviral Research. Future Med. Chem 2010, 2, 1205–26. [DOI] [PubMed] [Google Scholar]
- 39.Yap CW PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem 2011, 32, 1466–1474. [DOI] [PubMed] [Google Scholar]
- 40.Kuz’min VE; Artemenko AG; Muratov EN Hierarchical QSAR Technology Based on the Simplex Representation of Molecular Structure. J. Comput. Aided. Mol. Des 2008, 22, 403–421. [DOI] [PubMed] [Google Scholar]
- 41.Breiman LE O. Random Forests. Mach. Learn 2001, 45, 5–32. [Google Scholar]
- 42.Willett P; Barnard JM; Downs GM Chemical Similarity Searching. J. Chem. Inf. Comput. Sci 1998, 38, 983–996. [Google Scholar]
- 43.Alves VMVM; Muratov EN; Fourches D; Strickland J; Kleinstreuer N; Andrade CH; Tropsha A Predicting Chemically-Induced Skin Reactions. Part I: QSAR Models of Skin Sensitization and Their Application to Identify Potentially Hazardous Compounds. Toxicol. Appl. Pharmacol 2015, 284, 262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alves VM; Muratov EN; Fourches D; Strickland J; Kleinstreuer N; Andrade CH; Tropsha A Predicting Chemically-Induced Skin Reactions. Part II: QSAR Models of Skin Permeability and the Relationships between Skin Permeability and Skin Sensitization. Toxicol. Appl. Pharmacol 2015, 284, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alves VM; Capuzzi SJ; Muratov EN; Braga RC; Thornton TE; Fourches D; Strickland J; Kleinstreuer N; Andrade CH; Tropsha A QSAR Models of Human Data Can Enrich or Replace LLNA Testing for Human Skin Sensitization. Green Chem 2016, 18, 6501–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alves VM; Capuzzi SJ; Braga RC; Borba J; Silva A; Luechtefeld T; Hartung T; Andrade CH; Muratov EN; Tropsha A A Perspective and a New Integrated Computational Strategy for Skin Sensitization Assessment. ACS Sustain. Chem. Eng 2018, 6, 2845–2859. [Google Scholar]
- 47.Bender A How Similar Are Those Molecules after All? Use Two Descriptors and You Will Have Three Different Answers. Expert Opin. Drug Discov 2010, 5, 1141–51. [DOI] [PubMed] [Google Scholar]
- 48.Enoch SJ; Roberts DW Approaches for Grouping Chemicals into Categories. In Chemical Toxicity Prediction: Category Formation and Read-Across; Cronin M; Madden J; Enoch S; Roberts D, Eds.; Royal Society of Chemistry, 2013; pp. 30–43. [Google Scholar]
- 49.Cronin MTD An Introduction to Chemical Grouping, Categories and Read-Across to Predict Toxicity. In Chemical Toxicity Prediction: Category Formation and Read-Across; 2013; pp. 1–29. [Google Scholar]
- 50.Cronin MTD Evaluation of Categories and Read-Across for Toxicity Prediction Allowing for Regulatory Acceptance. In Chemical Toxicity Prediction: Category Formation and Read-Across; Cronin M; Madden J; Enoch S; Roberts D, Eds.; Royal Society of Chemistry, 2013; pp. 155–167. [Google Scholar]
- 51.Capuzzi SJ; Muratov EN; Tropsha A Phantom PAINS: Problems with the Utility of Alerts for P an- A Ssay IN Terference Compound S. J. Chem. Inf. Model 2017, 57, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albert MK; Aha DW Analyses of Instance-Based Learning Algorithms. 1991, 553–558.
- 53.Griffen E; Leach AG; Robb GR; Warner DJ Matched Molecular Pairs as a Medicinal Chemistry Tool. J. Med. Chem 2011, 54, 7739–7750. [DOI] [PubMed] [Google Scholar]
- 54.Capuzzi SJ; Kim ISJ; Lam WI; Thornton TE; Muratov EN; Pozefsky D; Tropsha A Chembench: A Publicly Accessible, Integrated Cheminformatics Portal. J. Chem. Inf. Model 2017, 57, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Talete SRL DRAGON 7.0 http://www.talete.mi.it/products/dragon_description.htm (accessed Mar 1, 2018).
- 56.Chemical Computing Group QuaSAR-Descriptor https://www.chemcomp.com/journal/descr.htm (accessed Mar 1, 2018).
- 57.RDKit Module MACCSkeys http://rdkit.org/Python_Docs/rdkit.Chem.MACCSkeys-pysrc.html (accessed Mar 1, 2018).
- 58.Ruggiu F; Marcou G; Varnek A; Horvath D ISIDA Property-Labelled Fragment Descriptors. Mol. Inform 2010, 29, 855–868. [DOI] [PubMed] [Google Scholar]
- 59.Capuzzi SJ; Politi R; Isayev O; Farag S; Tropsha A QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci 2016, 4, 3. [Google Scholar]
- 60.Bienfait B; Ertl P JSME: A Free Molecule Editor in JavaScript. J. Cheminform 2013, 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wishart DS; Knox C; Guo AC; Cheng D; Shrivastava S; Tzur D; Gautam B; Hassanali M DrugBank: A Knowledgebase for Drugs, Drug Actions and Drug Targets. Nucleic Acids Res. 2008, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sterling T; Irwin JJ ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.