

Abstract
Telomeres are composed of repetitive DNA sequences that are replenished by the enzyme telomerase to maintain the self-renewal capacity of stem cells. The RNA component of human telomerase (TERC) is the essential template for repeat addition by the telomerase reverse transcriptase (TERT), and also serves as a scaffold for several factors comprising the telomerase ribonucleoprotein (RNP). Unique features of TERC regulation and function have been informed not only through biochemical studies but also through human genetics. Disease-causing mutations impact TERC biogenesis at several levels including RNA transcription, post-transcriptional processing, folding, RNP assembly, and trafficking. Defects in TERC reduce telomerase activity and impair telomere maintenance, thereby causing a spectrum of degenerative diseases called telomere biology disorders (TBDs). Deciphering mechanisms of TERC dysregulation have led to a broader understanding of noncoding RNA biology, and more recently points to new therapeutic strategies for TBDs. In this review, we summarize over two decades of work revealing mechanisms of human telomerase RNA biogenesis, and how its disruption causes human diseases.
Keywords: dyskeratosis congenita, dyskerin, noncoding RNA, stem cells, telomere, TERC, TERT
1 |. INTRODUCTION
Telomeres are the repetitive DNA sequences that together with protein complexes called shelterin comprise the ends of mammalian chromosomes. Telomeres shorten with each cell division, in part due to incomplete replication of linear chromosome ends by DNA polymerases. Cellular senescence is triggered to protect genome integrity when telomeres reach a critically short length.1,2 To address the need for replicative capacity in stem cells, the tightly regulated enzyme telomerase is activated to elongate telomeres. Telomerase is a ribonucleoprotein (RNP) that at its core is composed of the reverse transcriptase TERT and the long noncoding RNA (lncRNA) TERC, which serves as the template for telomere repeat addition by TERT. It has long been appreciated that TERT expression is the “on/off switch” that determines whether or not a human cell has telomerase activity. However, genetic studies in rare diseases have revealed the critical importance of TERC RNA levels in governing the amount of telomerase activity in the cell, once TERT is expressed. In other words, TERC levels are limiting for replicative capacity in stem cells.3 Here, we summarize 25 years of molecular, biochemical, and genetic studies that have collectively revealed key mechanisms of TERC transcription and post-transcriptional processing, as well as a spectrum of diseases when these go awry. Most recently, these discoveries suggest strategies to manipulate telomerase via TERC for therapeutic benefit. This review focuses on human TERC. Readers are referred to excellent recent reviews on telomerase RNA structure and function in other eukaryotes.4–8
2 |. HUMAN TELOMERASE RNA
Preceding the explosion of ncRNA discovery in the genomics era, telomerase RNA was one of the relatively few lncRNAs of established importance in eukaryotic biology and human diseases. Elegant biochemical studies implicated an RNA component in telomerase, whose in vitro enzymatic activity is capable of synthesizing telomere repeats from an oligonucleotide primer.9 Isolation of the ciliate telomerase RNA demonstrated its template sequence and function.10 The subsequent cloning of various telomerase RNAs from ciliates and yeast demonstrated a large diversity of sizes and sequences, hindering identification of mammalian telomerase RNAs by homology alone. To address this, PCR-based cyclic selection of RNAs carrying the predicted human telomeric template sequence ultimately led to cloning and functional characterization of the 451-nucleotide lncRNA encoding human TERC.11 When TERC was inhibited by anti-sense oligonucleotides or mutated to alter the template, telomerase activity was reduced and telomere attrition ensued in cell lines, confirming its role in telomere biology. Notably, TERC expression alone did not correlate with telomerase activity in human cells,11 consistent with findings that TERT expression is the gatekeeper of cellular immortality.12
In terms of transcriptional regulation, early studies showed that TERC was alpha-amanitin sensitive and thus transcribed by RNA polymerase (pol) II,11 similarly to fungal telomerase RNAs but unlike those in ciliates that use RNA pol III.10 Neither a transcriptional termination site nor a poly-adenylated precursor transcript was identified. Rather, the finding of a predominant 3′ end without poly-adenylation13 suggested that nascent TERC transcripts were processed by other mechanisms, which remained largely undefined for almost two decades.
In early telomerase RNP biogenesis studies, Collins and colleagues noted that TERC had features of another class of ncRNAs. Mature TERC transcripts contained a consensus box H motif (5′-ANANNA-3′), terminated three nucleotides (nt) downstream of an ACA trinucleotide, and were predicted to fold into a hairpin-hinge-hairpin-tail structure,14 all of which are characteristics of box H/ACA small nucleolar RNAs (snoRNAs)15,16 (Figure 1). Although murine telomerase RNA differs substantially from human TERC in size (451 nt vs 397 nt) and sequence (65% identity),11,19 the box H/ACA sequence motifs and predicted secondary structure were found to be conserved,14 as was seen in other vertebrate telomerase RNAs.20 The box H/ACA structure was shown to be required for TERC accumulation in cells and sufficient for transcriptional termination and 3′ end formation, albeit by unknown mechanisms.14,21 Unlike snoRNAs, but as in small nuclear RNAs (snRNAs), TERC 5′ ends are modified with a trimethylguanosine cap,22 which localizes nascent transcripts to subnuclear structures and protects the template sequence from exonucleolytic degradation. These studies demonstrated that TERC, unlike telomerase RNA in ciliates and yeast, was a bona fide snoRNA, but has distinct biogenesis characteristics.21 These observations were also critical for linking TERC and telomere maintenance defects to human diseases.
FIGURE 1.
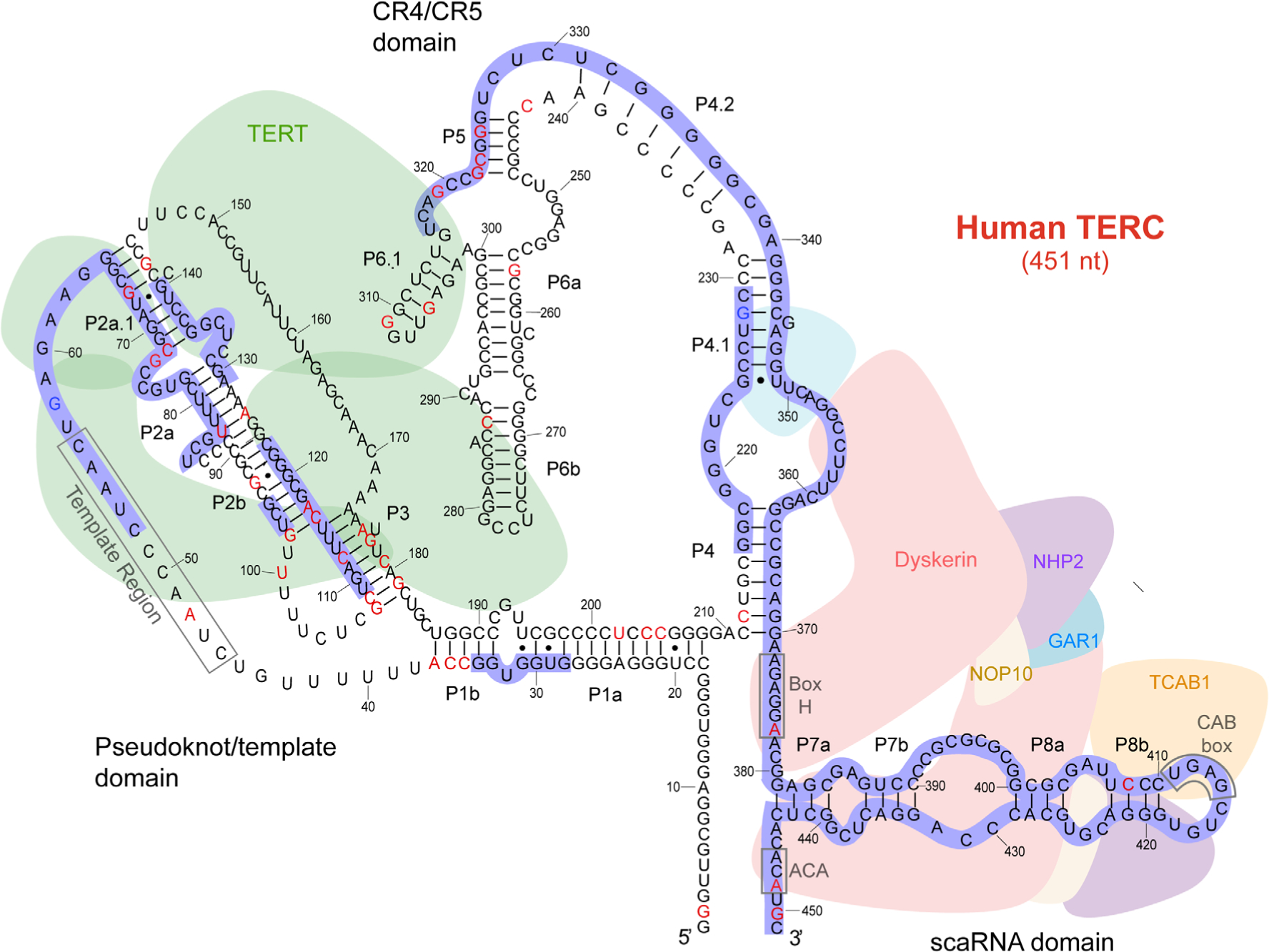
Model of telomerase RNA component (TERC) structure, function, and human dyskeratosis congenita/telomere biology disorder (DC/TBD) mutations. Adapted from References 17 and 18. The primary sequence of the 451 nucleotide TERC is depicted. Secondary structure and functional interactions are based on data from phylogenetic, biochemical, human genetic, and structural studies including cryo-electron microscopy. Conserved regions are indicated as P1a through P8b. The TERT-binding pseudoknot/template and CR4/CR5 domains, and the dyskerin/TCAB1-associated scaRNA domain are shown. Critical sequence motifs are defined: telomere repeat-encoding template region, Box H, ACA motif, and CAB box. Human genetic lesions reported in the literature include point mutations (red nucleotides) and regions where deletions have been described (blue outline). Known polymorphisms are shown as blue nucleotides. Point mutations, deletions, and the resulting DC/TBD phenotypes are detailed in Table 1
3 |. DYSKERATOSIS CONGENITA AND TELOMERE BIOLOGY DISORDERS: A SPECTRUM OF DISEASES UNIFIED BY HUMAN GENETICS
Dyskeratosis congenita (DC) is a rare disease described over 100 years ago, with onset usually occurring in childhood.23 In its classic form, DC is characterized by a diagnostic triad of skin pigmentation abnormalities, oral mucosal lesions, and degenerative changes in the nails. Beyond these features, case reports over the years noted that DC patients frequently had other problems including low blood counts (anemia, thrombocytopenia, and/or leukopenia), microcephaly, learning/developmental disorders, short stature, failure to thrive, difficulty swallowing, dental abnormalities, premature graying of hair, osteopenia, and pathology of the lungs, liver, and other vital organs.24 DC was thus recognized as a multisystem syndrome early on, but for decades the molecular basis and pathophysiology was unknown. A high male to female ratio and pedigree studies indicated X-linked inheritance in some forms of DC.25 Through linkage studies of the UK DC patient registry established in the early 1990s, Dokal and Vulliamy discovered hemizygous mutations in males in a gene they named DKC1, which encoded a protein (dyskerin) of unknown function.26 Dyskerin showed homology to yeast Cbf5p27 and rat NAP57,28 highly conserved pseudouridine synthases known to bind snoRNAs as guides for rRNA modification.16 These findings led to speculation that DC might be driven by defects in ribosome biogenesis.26
The observation of a box H/ACA motif in human TERC led the Collins group to alternate hypotheses, that dyskerin might also bind TERC, and that telomerase might be compromised by DKC1 mutations to result in the degenerative clinical features of DC. Indeed, dyskerin was found to associate directly with telomerase via TERC, and DKC1-mutant patients’ cells had decreased TERC levels and short telomeres.29 Soon thereafter, mutations in TERC itself were identified in autosomal dominant DC.30 Together these findings firmly established DC as a disorder of telomere maintenance, and demonstrated the importance of TERC and telomerase in human health. These studies represent an important early example of the impact of genetics studies on understanding the composition and biogenesis of human telomerase.
The discoveries of DKC1 and TERC mutations in DC patients ultimately revealed a spectrum of telomere biology disorders (TBDs).31,32 Clinical features of patients in the DC registry suggested that another severe disorder of infancy, Hoyeraal-Hreidarsson syndrome (HHS), might also be caused by mutations in DKC1.33 HHS is characterized by prenatal growth retardation, microcephaly, cerebellar hypoplasia, aplastic anemia, and immunodeficiency in infants, early clinical features that can precede the classic DC triad.34 The same DKC1 mutations that cause DC were found in children with HHS, demonstrating that HHS is an early onset, more severe form of DC.33
On the other end of the spectrum, independent genetic discoveries and detailed analyses of pedigrees revealed cryptic presentations of DC in previously healthy adults with seemingly isolated clinical problems. Heterozygous TERC mutations were found in adult patients with a variety of hematologic presentations including aplastic anemia, myelodysplastic syndrome (MDS), and paroxysmal nocturnal hemoglobinuria, in the absence of classic DC manifestations.35–44 In some families, the same TERC mutation presented with hematologic disease in one individual but pulmonary fibrosis (PF) or liver disease in others. In keeping with this, germline TERC mutations were found in families afflicted by PF, as well as cohorts of liver disease patients presenting in late adulthood.45–47 Some patients with acute myeloid leukemia (AML) or squamous cell cancers of the oral and anogenital regions were also found to carry DC-associated mutations,38 reinforcing DC as a cancer predisposition syndrome.48 Taken together, these findings indicated that genetic mutations in TERC present across a spectrum of disease (Table 1), with diverse TBD-associated manifestations presenting anywhere from childhood to mid-life and later.
TABLE 1.
TERC gene mutations implicated in various telomere biology disorders (based on Podlevsky et al, Nucleic Acids Research, 2007, and references therein. http://telomerase.asu.edu)
| Phenotype | TERC gene mutation |
|---|---|
| Myelodysplastic syndrome/acute myeloid leukemia | C35U, del52_55, U83G, del110_113, C212G, C287G, C309U, G319A, G322A, C323U, A377G, del389_390 |
| Dyskeratosis congenita/hoyeraal hreidarsson syndrome | A37G, A48G, del52_55, del54_57, G73U, G93C, del95_96, del96_97, U100A, GC107_108AG, delins129_140GU, G143A, U202G, C205U, C212U, del216_229, C242U, del316_451, del378_451, C408G, A448G |
| Aplastic anemia | G2C, del28_34, C36U, A37G, G67A, C72G, del79, U83G, G107U, del109_123, del110_113, C116U, A117C, A126G, G143A, A176C, G178A, C180U, G182A, C204G, C212G, G257A, G305A, del341_360, A377G |
| Pulmonary fibrosis/lung disease | del52_86, U80A, G98A, C108U, G182C, G325U, del375_377 |
| Liver fibrosis | A37G, del28_34, G319A, del341_360 |
Notes: Nucleotide position based Genbank accession number NR_001566 for the RNA sequence. Mutations in italics denote those verified by in vitro functional testing in the published literature. Literature references can be found at http://telomerase.asu.edu.
Studies in DC/TBD pedigrees with TERC mutations have revealed several interesting genetic features beyond simple Mendelian inheritance, complicating genotype-phenotype correlations. First, in some families, transmission of heterozygous TERC mutations results in earlier onset and more severe disease in subsequent generations.43 This disease anticipation is thought to result from inheritance of not only the genetic mutation compromising telomere maintenance, but also short telomeres from the carrier parent. A similar phenomenon is observed in autosomal dominant DC/TBD due to heterozygous germline loss-of-function mutations in TERT.49,50 A second unusual genetic feature has been revealed in some TERC pedigrees wherein obligate carriers showed underrepresentation or absence of the mutation when genotyping blood DNA. To explain this, single nucleotide polymorphism (SNP) array analysis of non-blood tissue revealed somatic reversion of the TERC mutation in the hematopoietic system.51 The reversion event was evident in both myeloid and lymphoid blood cells, indicating positive selection for restored TERC levels and telomere maintenance at the hematopoietic stem cell (HSC) level. Remarkably, in one patient, multiple independent events of somatic reversion could be documented by SNP arrays, indicating a significant advantage of eliminating the mutant TERC locus in HSCs. A third con-founder of genotype-phenotype correlations is illustrated by a patient found to have a heterozygous TERC mutation but more pronounced disease manifestations compared to family members. Here, it was shown that the patient had also inherited a pathogenic TERT mutation, resulting in oligogenic phenotypic contributions from multiple partial loss-of-function alleles.52 Taken together, these studies illustrate complexities in diagnosis and establishing genotypic-phenotype relationships in patients with germline TERC mutations.
From a clinical standpoint, the detection of TERC mutations and their associated disease burden are likely to be underestimated for several reasons. These include lack of familiarity with DC/TBD amongst clinicians, low suspicion of germline mutations in adult patients with MDS, cirrhosis or PF, and overlooking TERC (as a ncRNA) in clinical and research whole exome sequencing pipelines. Functional telomere length testing is a valuable diagnostic test, useful for increasing suspicion of underlying TBD, but with limitations in sensitivity and specificity in older age groups.53 In summary, germline TERC mutations present across a spectrum of clinical severity and onset, but are likely underdiagnosed in patients without obvious syndromic features or family histories.
4 |. TERC MUTATIONS AND MOLECULAR MECHANISMS OF DISEASE
In addition to DKC1, it is now clear that several other gene mutations result in decreased TERC levels to cause TBDs. These are discussed in detail below. However, it is first instructive to consider the various molecular mechanisms by which mutations in TERC itself have been found to result in loss of function.
4.1 |. TERC mutations
More than 60 disease-associated mutations in TERC have been identified to date17 (Table 1). TERC is 451 nt, and in aggregated databases of whole genome sequencing only two positions show polymorphism (n.58G>A, n.228G>A) at greater than 0.1% frequency (Figure 1). Different impairments caused by TERC mutations have been described, including defects in transcription, template function, secondary structure, assembly into H/ACA-RNPs, and interactions with TERT. With regard to primary transcriptional defects, mutations in the −99 Sp1 site and −58 CCATT box of the TERC promoter have been described in three different families with BMF or MDS.37,42,54 However, there was limited data in these reports showing a direct effect on transcription. In terms of requirements for RNP assembly, point mutations and deletions in the box H/ACA motifs would be expected to impact dyskerin binding in cells (Figure 1). Consistent with this, the accumulation of variants with mutations in the box H or ACA domains is abrogated in cells, despite retaining template function in in vitro reconstitution assays.55 A box H/ACA deletion truncating the last 74 nt of TERC as well as 747 nt downstream was shown to impact not only accumulation of the RNA transcript,21 but chromatin configuration of the allele in cis as well, suggesting transcriptional regulatory function of the sequences immediately 3′ of the gene.56 An extensive examination of multiple pseudoknot/template domain mutations indicated that most of these diminish holoenzyme catalytic activity55 (Figure 1). Not only the template but also the conserved region (CR) CR4/CR5 domains of TERC are required for telomerase assembly and activity in vitro and in vivo.57 Mutations in the CR4/CR5 region have been found which do not affect accumulation or assembly with H/ACA-RNP, but rather impair interactions with TERT and prevent formation of active telomerase holoenzyme.55,57,58 Interestingly, the secondary structure of the CR4/CR5 region has also been shown to be responsive to binding by TCAB1 (telomerase Cajal body protein 1), which is required for subsequent interactions with TERT.59 These studies indicate that certain CR4/CR5 residues mutated in DC may serve as an “activity switch,” which relays TCAB1 binding of the box H/ACA regions and licenses TERT assembly into the holoenzyme. In summary, human disease-associated mutations in TERC disrupt its biogenesis and function, leading to impaired telomerase function. Importantly, until now TERC mutations in patients have been found in heterozygous form, and biallelic or null mutations have not been reported, indicating that complete loss-of-function is unlikely to be tolerated in humans.
5 |. MUTATIONS IN SEVERAL FACTORS IMPACT TERC TO CAUSE DISEASE
To date, mutations in 15 genes have been implicated in DC/TBDs, accounting for ~70% of cases.60,61 Of these, besides TERC itself, loss or alteration of function in seven other genes—DKC1, NOP10, NHP2, NAF1 (nuclear assembly factor 1), TCAB1, PARN (poly[A]-specific ribonuclease), and ZCCHC8 (zinc finger CCHC-type containing 8)—impairs telomere maintenance by compromising TERC processing and biogenesis at different steps.
5.1 |. Mutations impacting the H/ACA-RNP
The nascent H/ACA-RNP complex consists of four proteins: dyskerin, NAF1, NHP2, and NOP10.62 Mutations in the genes encoding any of these four proteins cause DC.26,63–65 NAF1 is exchanged for GAR1 after H/ACA-RNP maturation (Figure 1). Of note, GAR1 mutations have not been reported in DC/TBDs.
Each H/ACA-RNP complex associates with one of hundreds of ncRNAs via the box H/ACA motif,66 and the associated RNA directs the RNP to different substrates and subcellular locations. Dyskerin is a highly conserved enzyme that catalyzes the conversion of uridine to pseudouridine in cellular RNAs.67 Box H/ACA snoRNAs serve as anti-sense guides for the pseudouridylation of ribosomal RNAs (rRNAs) in the nucleolus, and also play a role in rRNA processing.68,69 Small Cajal body (sca)RNAs, including TERC, are a subset of snoRNAs with a binding motif for TCAB1, which re-routes H/ACA-RNPs to the Cajal body instead of nucleolus. There, certain scaRNAs act as guides to target snRNAs involved in RNA splicing for pseudouridylation, although many substrates are unknown.70–72 There is no evidence that TERC serves to target dyskerin to other RNAs for pseudouridylation. However, TERC itself may contain pseudouridine modifications including two residues in the loop of hairpin P6.1 (Figure 1), a domain critical for telomerase catalytic activity.73 Although it is not entirely clear why TERC co-opted a box H/ACA architecture for association with the H/ACA-RNP, possible reasons include exploitation of snoRNA 3′ end processing mechanisms, post-transcriptional fine-tuning of steady state RNA levels, subnuclear compartmentation for efficient telomere elongation.
Despite the fact that dyskerin, NOP10, NHP2, and NAF1 are found in numerous different snoRNPs, mutations in the genes encoding these factors phenocopy DC/TBDs. Although the spectrum of diseases associated with these mutations ranges from classic DC to PF, hemizygous mutations in DKC1 and biallelic mutations in NOP10 and NHP2 tend to associate with earlier onset, multisystem disease. Heterozygous truncation mutations in NAF1 have been found in adult patients with DC, MDS, liver disease, and PF.63 In all cases, steady state levels of TERC are reduced. TERC is limiting for telomerase activity and telomere elongation in DKC1-mutant patient cells.74 Importantly, some of these mutations cause a greater deficiency in TERC than heterozygous loss-of-function TERC mutations. As with TERC itself, mutations in DKC1, NOP10, NHP2, and NAF1 have not been found in null states, which are unlikely to be tolerated. It remains unclear whether other sno/scaRNAs or pseudouridylation are meaningfully impacted by these mutations, with evidence for and against relevant effects in various studies.29,75–82 However, it is clear that TERC is affected by these mutations. Importantly, the range and make-up of disease phenotypes overlap whether caused by mutations in H/ACA-RNP components or in genes regulating telomere maintenance by other mechanisms, such as RTEL1 (regulator of telomere elongation helicase 1) and TINF2 (TERF1 interacting nuclear factor 2). These observations suggest that defects in TERC alone may be sufficient to explain pathophysiology in DC/TBDs driven by mutations in DKC1, NOP10, NHP2, or NAF1.
How does one explain the apparent preferential impact of these mutations on TERC, compared to hundreds of other sno/scaRNAs? TERC is unique amongst this class of RNAs in being encoded by an autonomous RNA pol II-transcribed gene, unlike other sno/scaRNAs that are intron-embedded and processed from host gene transcripts. It has been suggested that nascent TERC RNA is more sensitive to efficient recruitment of pre-assembled dyskerin/NOP10/NHP2/NAF1 complexes due to its lack of 3′ end protection, unlike sno/scaRNAs that are initially contained within a lariat structure during their biogenesis.83 Impairments of dyskerin/NOP10/NHP2/NAF1 assembly due to partial loss-of-function in any of these factors might be expected to compromise TERC more severely than other sno/scaRNAs. Another way to view it is that more severe alterations or loss of function in factors that impact sno/scaRNA biogenesis more globally may not be compatible with early human development.
5.2 |. Mutations impacting TERC secondary structure and Cajal body localization
TCAB1 (also known as WDR79/WRAP53) binds the Cajal body localization sequence (CAB box) in the terminal loop of scaRNAs including TERC84,85 (Figure 1). TCAB1 incorporation into the dyskerin holoenzyme is believed to occur after H/ACA-RNP assembly. TCAB1 serves to localize scaRNPs including telomerase to Cajal bodies instead of the nucleolus. Upon identification of TCAB1’s function in telomere biology, candidate screening of a DC registry identified biallelic TCAB1 mutations in two unrelated patients.86 The mutations were found to compromise TCAB1 protein stability and nuclear localization, and in turn H/ACA-RNP and TERC trafficking to Cajal bodies. Although mislocalization of an otherwise intact telomerase RNP was shown to be a driver of impaired telomere maintenance in the setting of these mutations, subsequent studies showed that telomerase activity was also compromised by loss-of-function TCAB1 mutations.87 Specifically, TCAB1 was found to be required for proper folding of the CR4/CR5 domain of TERC, to enable association with TERT.59 Importantly, unlike core H/ACA-RNP proteins, total TCAB1 loss is tolerated in human cells and results in impaired telomerase activity in the null state.88 These data again indicate a particular sensitivity of TERC biogenesis and post-transcriptional trafficking to disruption of a factor that regulates multiple scaRNPs.
5.3 |. Mutations impairing TERC 3′ end processing
Mechanisms of TERC transcriptional termination and 3′ end processing were unknown until recently. In 2015, whole exome sequencing of DC and HHS patients in the UK DC registry uncovered biallelic mutations in the PARN gene, which encodes a 3′ exoribonuclease.89 At the same time, Garcia and colleagues found heterozygous PARN mutations in familial PF cohorts.90 The majority of patients with PARN mutations in both groups had very short telomeres. These molecular and clinical findings firmly linked PARN to the spectrum of TBDs, with severity and phenotype correlating with gene dosage. However, the role of PARN in telomere biology was unclear. The canonical function of PARN was thought to be catalyzing the turnover of cytoplasmic mRNAs by degrading long poly(A) tails.91 Mutations in PARN were thus proposed to cause telomere disease via effects on mRNA levels, particularly impacting telomere components and p53.89 However, in PARN-mutant patient cells, decreased TERC RNA levels were also found,89,92,93 the molecular basis of which was unclear given that TERC does not have a poly(A) tail. A few key additional observations led to insights on how PARN mutations might cause TERC deficiency. First, although early studies demonstrated a precise 3′ end for TERC, subsequent deep sequencing data indicated a larger diversity, including genomically extended and oligo-adenylated TERC ends.94 Second, beyond a role in mRNA metabolism, PARN had been proposed to regulate ncRNA metabolism, particularly box H/ACA snoRNA 3′ end maturation and stabilization.95 Given that TERC shared the same motif with box H/ACA snoRNAs, it was hypothesized that PARN might also regulate the productive maturation of TERC, which would explain the finding of diminished TERC levels and telomerase deficiency in patients with DC and IPF.93 In cells derived from DC patients with biallelic PARN mutations, increased 3′ extended species of TERC were detected.92,93 Deep sequencing revealed that these species were both genomically extended and oligo-adenylated, yielding short (~ 10 nt) tails on TERC, which would be expected to be degraded by the RNA exosome (Figure 2A). In keeping with this, TERC transcripts were found to be destabilized by PARN deficiency, leading to decreased telomerase activity and telomere length in patient cells, and their accumulation was reversible by restoring expression of PARN.93 Other sno/scaRNAs were found to be altered in 3′ end processing,92,93 but RNA sequencing per-formed on PARN-mutant patient cells did not indicate global effects on mRNAs.93 These observations led to the conclusion that impaired TERC 3′ end maturation, and not mRNA turnover, is the major contributor to diseases caused by PARN mutations.93 In support of this, later studies showed no genome-wide alterations in mRNA representation or poly(A) tail length when PARN was depleted in human cells.96
FIGURE 2.
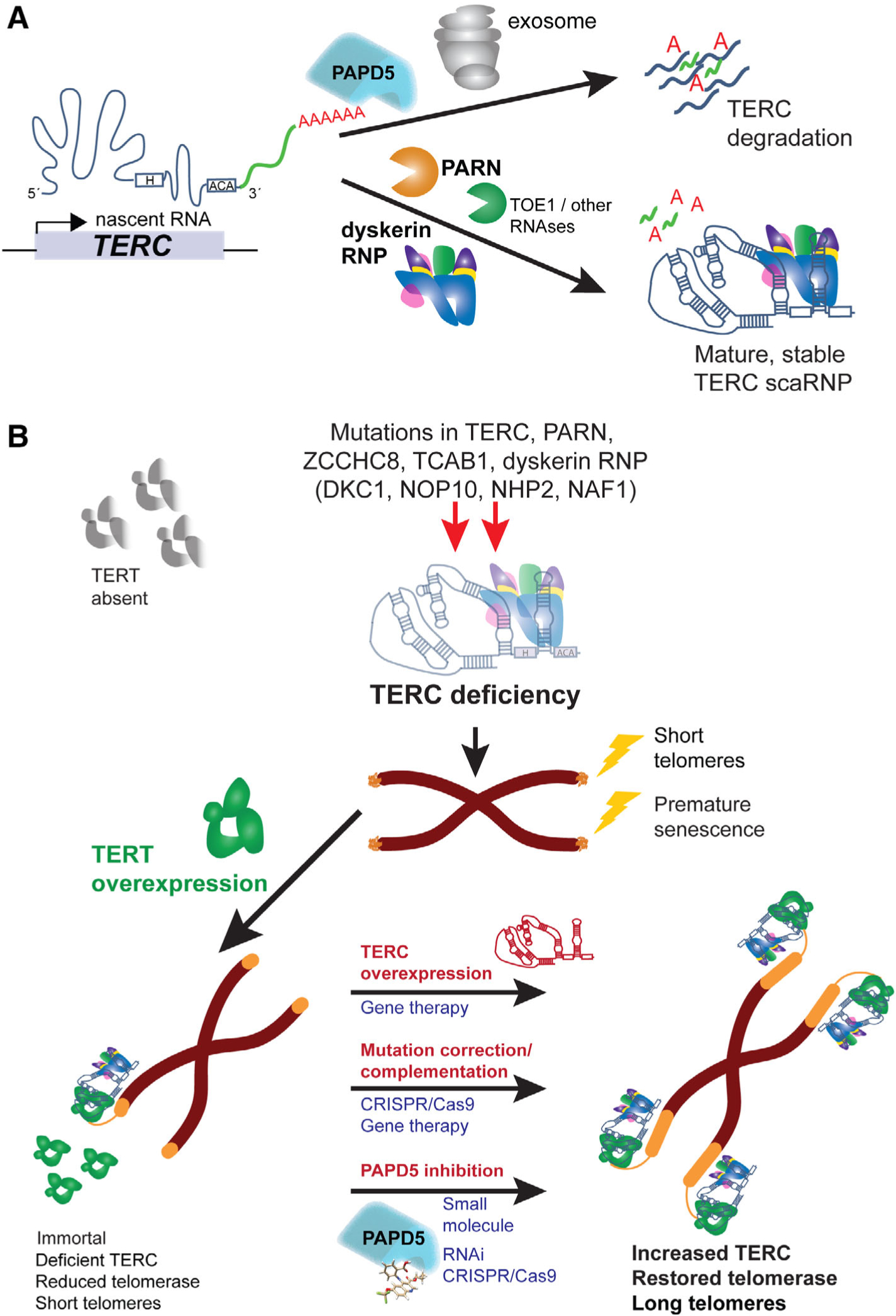
Model for telomerase RNA component (TERC) processing in health and dyskeratosis congenita/telomere biology disorders (DC/TBDs), and potential therapeutic strategies. A, TERC post-transcriptional regulation by RNA-binding proteins, polymerases, and nucleases. Nascent TERC transcripts contain genomically encoded 3′ extensions (green). The extensions can be substrates for adenylation by PAPD5, which stimulates destruction of TERC by the RNA exosome. Conversely, PARN and other exoribonucleases trim 3′ extensions to result in mature TERC. Association with the H/ACA ribonucleoprotein (RNP) is an early event and aids in TERC 3′ maturation/end definition, and scaRNP stability, trafficking, and function. Early processing events including transcriptional termination, processing of long transcripts, and NAF1 H/ACA-RNP exchange are not illustrated. B, TERC deficiency in DC/TBDs and therapeutic prospects. Hypomorphic mutations in TERC or other associated RNA processing and binding factors results in TERC deficiency. A patient somatic cell is depicted, in which TERT is absent, telomerase inactive, and telomeres critically short, resulting in senescence. Overexpression of TERT immortalizes cells carrying these mutations in vitro, but telomerase activity remains reduced and telomeres short because TERC is limiting. Strategies shown to restore TERC and thus telomerase and telomere elongation in patient cells in vitro are shown in red. Potential therapeutic modalities to translate each strategy are shown in blue
Simultaneous investigations of human TERC biogenesis mechanisms in cell lines corroborated and extended these findings. Transient PARN depletion in human cell lines results in accumulation of TERC 3′ extended forms.93,97–99 The non-canonical polymerase PAPD5 is primarily responsible for the addition of non-templated adenosines to extended nascent TERC.97–100 In yeast, the PAPD5 ortholog Trf4 is part of the TRAMP (Trf4, Air2, and Mtr4) complex, which serves a quality control function by oligo-adenylating misfolded RNAs and targeting them for degradation by the nuclear RNA exosome.101 Although it is unclear whether human PAPD5 functions in a TRAMP-like complex, knockdown of PAPD5 decreases oligo-adenylation and increases mature TERC levels in normal cells.99 Furthermore, PAPD5 inhibition by RNA interference in PARN-depleted cell lines or PARN-mutant patient cells reduces TERC oligo-adenylation and increases steady-state TERC levels, telomerase activity and telomere length.97,98,100 A role for the RNA exosome downstream of PAPD5-mediated oligo-adenylation has been indicated by demonstration that knockdown of the core exosome components RRP6/EXOSC10 or RRP40/EXOSC3 increases mature TERC levels in PARN-deficient cells.98,99 These findings collectively yield a model wherein steady state levels of TERC are controlled by competition between productive maturation of nascent TERC transcripts by PARN vs oligo-adenylation by PAPD5, which targets RNAs for degradation by the nuclear exosome97–100 (Figure 2A).
Although these studies have illuminated biogenesis intermediates and critical final steps in TERC maturation, questions remain of precisely how and where TERC transcription terminates, and what factors govern the earliest steps in TERC maturation. Recent studies shed some light. First, 4-thiouridine pulse-chase labeling of nascent TERC transcripts demonstrates definitively that TERC species with short oligo-A tails are converted into mature TERC species, rather than solely representing intermediates for degradation.102 Fusion of the TERC box H/ACA domain to U1 snRNA in an expression construct resulted in the accumulation of a U1-H/ACA hybrid transcript that was oligo-adenylated in a dyskerin- and PARN-sensitive manner. Thus, the TERC H/ACA domain is sufficient to dictate end-formation, as seen in an earlier study,21 and confers regulation of nascent transcripts by dyskerin and PARN. Importantly, complete elimination of both PARN and PAPD5 resulted in normal levels of mature TERC, indicating that PARN/PAPD5-independent pathways for TERC post-transcriptional processing must exist.102 The findings also reinforce and refine the model of competition between PARN and PAPD5 in TERC biogenesis, wherein PARN is specifically required to counteract rapid PAPD5-mediated destabilization of short, extended TERC transcripts.
With regard to the nature and fate of longer nascent TERC transcripts, knocking down the exosome components RRP40/EXOSC3 and RRP6/EXOSC10 results in the accumulation of nascent TERC transcripts extended ≥9 nts beyond the canonical end, as well as increasing mature forms.103 At the same time, there is a reduction in short, extended TERC forms seen with PARN deficiency, suggesting that longer TERC transcripts processed by the RNA exosome ultimately lead to the formation of short ones that are then substrates for PARN.103 Using in vitro transcribed TERC box H/ACA domain-containing RNA and nuclear extracts depleted of factors, it has been shown that processing long forms of TERC requires RRP6/EXOSC10. Surprisingly, dyskerin is also required for RRP-mediated conversion of long TERC forms into short PARN substrates. To explain this, extended TERC primary transcripts were shown to engage in tertiary interactions with nucleotides in the first hairpin structures and box H motifs of the box H/ACA domain. Transcripts folded in this manner are resistant to processing unless tertiary interactions are displaced by dyskerin binding. These results indicate a new role for dyskerin in TERC processing, beyond stabilization and trafficking. Whether this specific role is disrupted by human DKC1 mutations remains to be determined. However, consistent with these results, knockdown of RRP40/EXOSC3 or PAPD5 was shown to increase TERC maturation and telomere length in the context of a pathogenic DKC1 mutation in human embryonic stem cells.104
Recent genetic findings in TBDs support a broader role of exosome components in processing nascent TERC RNA. In a family with PF, heterozygous mutations have been identified in ZCCHC8,105 a component of the nuclear exosome targeting (NEXT) complex.106 Patient cells carrying the mutation showed decreased ZCCHC8 protein but normal levels of two other NEXT components, RBM7 and SKIV2L2/MTR4. ZCCHC8 deficiency was accompanied by increased short and long forms of TERC, and 20% decrease in steady state levels TERC levels in patient fibroblasts. Notably, ZCCHC8 null cancer cell lines could be generated and recapitulated a defect in TERC levels and telomerase activity, although long-term effects on telomere length were not described. Interestingly, homozygous frameshift ZCCHC8 mutations (predicted to be null) have been reported in a patient with neurocognitive defects, but there is no information on telomere biology in this setting.107 It is not clear how ZCCHC8 deficiency results mechanistically in net reduction of TERC levels or function. It is known that ZCCHC8 is required for targeting aberrant 3′ extended ncRNA transcripts to the nuclear exosome. To the extent that the exosome is involved in degradation of TERC after PAPD5-mediated oligo-adenylation of short transcripts, it might be counterintuitive that inhibiting trafficking to the exosome would result in a reduction in transcripts. However, it is possible that ZCCHC8/NEXT and the RNA exosome are also required for productive maturation of long TERC precursors by RRP6.103 Whether there is a role for NEXT/RNA exosome in processing short TERC transcripts to their final form remains unknown.
5.4 |. Other human disease genes potentially involved in TERC maturation
Biallelic germline mutations in TOE1 (target of EGR1), encoding a CAF1 family 3′-exoribonuclease related to PARN, are associated with pontocerebellar hypoplasia (PCH) type VII.108 TOE1-deficient patient cells harbor an increase in pre-snRNAs with non-genomically encoded adenosine residues, reminiscent of aberrant TERC species in PARN-deficient cells.108 Interestingly, other forms of PCH are caused by germline mutations in exosome components, including RRP40/EXOSC3 and EXOSC8. Cerebellar hypoplasia is a feature of severe forms of TBDs such as HHS. These findings raise the question of whether clinical manifestations common to both PCH and TBDs can be explained by defective 3′ end processing of the same ncRNAs. Although TERC levels and telomere length have not been reported in PCH type VII patients, genome-wide ncRNA profiling indicates a potential role for TOE1 in TERC biogenesis.96 TOE1 depletion has minimal effects on TERC steady state levels by itself, but RNA species with very short 3′ extensions are increased in general. Combined TOE1 and PARN knockdown results in additive effects in destabilizing TERC, as well as a global increase in RNA 3′ adenylation. Notably, inhibition of PAPD5 is sufficient to reverse the effects of both TOE1 and PARN depletion. In a detailed evaluation of the effects on telomere biology, TOE1 depletion in cell lines by CRISPR/Cas9 resulted in the accumulation of very short TERC adenylated species109 without changes in overall TERC levels. However, with stable heterozygous TOE1 deletion, telomerase activity was decreased and telomeres shortened over time. These studies show that TOE1 plays a role in TERC biogenesis nonredundant with that of PARN, likely removing the final few 3′ nucleotides and also contributing to TERC maturation in PARN null cells (Figure 2A). These results also lead to the speculation that impaired telomere maintenance contributes to disease manifestations in PCH VII patients, which remains to be studied.
5.5 |. Therapeutic potential of modulating TERC processing
TERC deficiency limits telomere maintenance in cells from DC patients with mutations in TERC, DKC1, or PARN.74,100,110,111 Ectopic TERT expression immortalizes cells carrying these lesions, but is insufficient to efficiently elongate telomeres (Figure 2B). Therefore, although stem cell self-renewal may be conferred by TERT expression, the replicative capacity of TERT-negative daughter cells is likely to remain low because of short telomere reserve. Overexpression of TERC in these settings restores telomerase activity and telomere elongation, as does DKC1 gene correction,112,113 suggesting potential treatment approaches via genome-editing or gene therapy (Figure 2B). However, DC and TBDs affect several organs, and genetic strategies to correct TERC deficiency in stem cells throughout the body are intractable at present.
The discovery that TERC is regulated by multiple components of the ncRNA post-transcriptional machinery has opened up a range of new targets through which TERC levels might be manipulated. Indeed, many of these are enzymes potentially amenable to small molecule inhibitors, which could provide systemic treatments. As described above, several studies have demonstrated that genetic inhibition of PAPD5 can restore TERC levels in PARN- or dyskerin-deficient states.96,98–100,102,104,114 Stable long-term shRNA-mediated PAPD5 depletion is tolerated in PARN-mutant patient iPSCs and restores TERC levels, telomerase activity and telomere length100 (Figure 2B). Along the same lines, long-term PAPD5 knockdown in human embryonic stem cells engineered to express a pathogenic DKC1 mutation (A353V) increases telomere length and improves in vitro hematopoietic differentiation capacity.104 In terms of tolerability, PAPD5 null human cell lines and pluripotent stem cells are able to be isolated and propagated indefinitely, and show decreased TERC oligo-adenylation and increased TERC levels.102,114 These observations provide the basis for considering PAPD5 a “druggable” target to restore TERC and telomere length in certain forms of DC/TBDs100 (Figure 2B).
Recently a high-throughput enzymatic screen identified novel and specific PAPD5 small molecule inhibitors that restored telomerase activity in DC patients’ iPSCs.114 One such PAPD5 inhibitor, a quinoline called BCH001, showed the capacity to increase TERC maturation and RNA levels and elongate telomeres in PARN-mutant patient stem cells (Figure 2B). Importantly, telomere elongation by BCH001 in patient cells was dependent on TERT, expressed either endogenously in iPSCs or ectopically in fibroblasts. These results indicate that small molecule telomerase modulation could be achieved without immortalizing cells or circumventing the gatekeeper function of TERT, important safety considerations for systemic therapy. A second class of small molecule PAPD5 inhibitors, dihydroquinolizinones (DHQ), was identified in a hepatitis B surface antigen suppressor screen, and unexpectedly found to act via PAPD5 inhibition.115,116 DHQ molecules increase TERC and telomere length in DC patient iPSCs in vitro.114 In an in vivo mouse xenotransplant model using CRISPR/Cas9-engineered, PARN-deficient human hematopoietic stem and progenitor cells, oral administration of the DHQ molecule RG7834 restored TERC processing and telomere maintenance. Taken together, these results provide encouraging evidence that pharmacologic manipulation of the TERC biogenesis machinery can be used to modulate telomerase in stem cells throughout the body. Although data have thus far been provided for pathogenic PARN and DKC1 mutations,114 it is possible that other DC/TBD-associated mutations that affect TERC processing and accumulation (eg, NOP10, NHP2, NAF1, TCAB1, ZCCHC8, certain TERC alleles) may also be amenable to a PAPD5 inhibitor strategy. If successful, such an approach may provide a much-needed systemic therapy for patients with DC/TBDs and perhaps other degenerative disorders.
6 |. PERSPECTIVES
Disease-causing mutations have revealed TERC biogenesis mechanisms and illuminated the sensitivity of human stem cells to TERC levels. These insights now allow one to consider how TERC manipulation might be achieved to treat DC/TBDs and other diseases. Fundamental questions remain. How does TERC transcription terminate? What is the precise location, trafficking, timing, and order of various stages of TERC processing? How do elements like chromatin, RNA modifications, and post-translation modifications impact TERC processing? Are there cell-type specific regulatory mechanisms that impact TERC levels? Is DC/TBD pathology in the setting of hypo-morphic mutations in DKC1, NOP10, NHP2, NAF1, PARN, TCAB1, or ZCCHC8 satisfactorily explained by effects on TERC alone? If not, can effects on other ncRNAs and their consequences be defined conclusively? Are the functions and specificities of TERC regulatory factors and pathways sufficiently conserved in animal models to test new therapeutics? Will reversing TERC deficiency pharmacologically be effective in treating or preventing DC/TBDs? As history has shown, staying close to the patients can be expected to yield answers to these and other important questions.
Significance statement.
Telomerase is critical for stem cell homeostasis. It is now clear that in humans, mutations in telomerase cause a range of life-threatening diseases driven at least in part by premature stem cell failure. Over the past 20 years, genetic discoveries have revealed that the long noncoding RNA component of telomerase (TERC) is a critical, limiting factor for telomerase activity in stem cells. Remarkably, recent work on TERC biogenesis and processing has illuminated potential targets for manipulation of telomerase. This article reviews molecular and genetic discoveries in human TERC biogenesis and telomere biology disorders, which have led to a better understanding and potential treatments for stem cell diseases.
ACKNOWLEDGMENTS
This work was funded by NIH grant R01 DK107716, U.S. Department of Defense grant W81XWH-19-1-0572, Harrington Discovery Institute, and Harvard Stem Cell Institute.
Footnotes
CONFLICT OF INTEREST
The authors are named as inventors on patent applications related to manipulating TERC in telomere diseases.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1.Allsopp RC, Harley CB. Evidence for a critical telomere length in senescent human fibroblasts. Exp Cell Res. 1995;219:130–136. [DOI] [PubMed] [Google Scholar]
- 2.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. [DOI] [PubMed] [Google Scholar]
- 3.Greider CW. Telomerase RNA levels limit the telomere length equilibrium. Cold Spring Harb Symp Quant Biol. 2006;71:225–229. [DOI] [PubMed] [Google Scholar]
- 4.Podlevsky JD, Chen JJ. Evolutionary perspectives of telomerase RNA structure and function. RNA Biol. 2016;13:720–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webb CJ, Zakian VA. Telomerase RNA is more than a DNA template. RNA Biol. 2016;13:683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu RA, Upton HE, Vogan JM, Collins K. Telomerase mechanism of telomere synthesis. Annu Rev Biochem. 2017;86:439–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musgrove C, Jansson LI, Stone MD. New perspectives on telomerase RNA structure and function. Wiley Interdiscip Rev RNA. 2018;9:e1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasianovich Y, Wellinger RJ. Life and death of yeast telomerase RNA. J Mol Biol. 2017;429:3242–3254. [DOI] [PubMed] [Google Scholar]
- 9.Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–898. [DOI] [PubMed] [Google Scholar]
- 10.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, Funk WD, Wang SS, et al. The RNA component of human telomerase. Science. 1995;269:1236–1241. [DOI] [PubMed] [Google Scholar]
- 12.Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279:349–352. [DOI] [PubMed] [Google Scholar]
- 13.Zaug AJ, Linger J, Cech TR. Method for determining RNA 3′ ends and application to human telomerase RNA. Nucleic Acids Res. 1996; 24:532–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell JR, Cheng J, Collins K. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol Cell Biol. 1999;19:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganot P, Caizergues-Ferrer M, Kiss T. The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev. 1997;11:941–956. [DOI] [PubMed] [Google Scholar]
- 16.Kiss T, Fayet-Lebaron E, Jady BE. Box H/ACA small ribonucleoproteins. Mol Cell. 2010;37:597–606. [DOI] [PubMed] [Google Scholar]
- 17.Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ. The telomerase database. Nucleic Acids Res. 2008;36:D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen THD, Tam J, Wu RA, et al. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature. 2018;557:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blasco MA, Funk W, Villeponteau B, Greider CW. Functional characterization and developmental regulation of mouse telomerase RNA. Science. 1995;269:1267–1270. [DOI] [PubMed] [Google Scholar]
- 20.Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–514. [DOI] [PubMed] [Google Scholar]
- 21.Fu D, Collins K. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol Cell. 2003;11: 1361–1372. [DOI] [PubMed] [Google Scholar]
- 22.Jady BE, Bertrand E, Kiss T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J Cell Biol. 2004;164:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73:103–112. [DOI] [PubMed] [Google Scholar]
- 24.Dokal I Dyskeratosis congenita in all its forms. Br J Haematol. 2000; 110:768–779. [DOI] [PubMed] [Google Scholar]
- 25.Knight SW, Vulliamy T, Forni GL, Oscier D, Mason PJ, Dokal I. Fine mapping of the dyskeratosis congenita locus in Xq28. J Med Genet. 1996;33:993–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19:32–38. [DOI] [PubMed] [Google Scholar]
- 27.Jiang W, Middleton K, Yoon HJ, Fouquet C, Carbon J. An essential yeast protein, CBF5p, binds in vitro to centromeres and microtubules. Mol Cell Biol. 1993;13:4884–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meier UT, Blobel G. NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J Cell Biol. 1994;127:1505–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402: 551–555. [DOI] [PubMed] [Google Scholar]
- 30.Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. [DOI] [PubMed] [Google Scholar]
- 31.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361: 2353–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knight SW, Heiss NS, Vulliamy TJ, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999;107:335–339. [DOI] [PubMed] [Google Scholar]
- 34.Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170:457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du HY, Pumbo E, Ivanovich J, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fogarty PF, Yamaguchi H, Wiestner A, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–1630. [DOI] [PubMed] [Google Scholar]
- 37.Keith WN, Vulliamy T, Zhao J, et al. A mutation in a functional Sp1 binding site of the telomerase RNA gene (hTERC) promoter in a patient with paroxysmal nocturnal haemoglobinuria. BMC Blood Disord. 2004;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirwan M, Vulliamy T, Marrone A, et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum Mutat. 2009;30:1567–1573. [DOI] [PubMed] [Google Scholar]
- 39.Ly H, Calado RT, Allard P, et al. Functional characterization of telomerase RNA variants found in patients with hematologic disorders. Blood. 2005;105:2332–2339. [DOI] [PubMed] [Google Scholar]
- 40.Marrone A, Sokhal P, Walne A, et al. Functional characterization of novel telomerase RNA (TERC) mutations in patients with diverse clinical and pathological presentations. Haematologica. 2007;92:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood. 2004;104:3936–3942. [DOI] [PubMed] [Google Scholar]
- 42.Ortmann CA, Niemeyer CM, Wawer A, Ebell W, Baumann I, Kratz CP. TERC mutations in children with refractory cytopenia. Haematologica. 2006;91:707–708. [PubMed] [Google Scholar]
- 43.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36:447–449. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi H, Baerlocher GM, Lansdorp PM, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–918. [DOI] [PubMed] [Google Scholar]
- 45.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 46.Calado RT, Brudno J, Mehta P, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology. 2011;53: 1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz de Leon A, Cronkhite JT, Katzenstein AL, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 2010;5:e10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102: 15960–15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jongmans MC, Verwiel ET, Heijdra Y, et al. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collopy LC, Walne AJ, Cardoso S, et al. Triallelic and epigenetic-like inheritance in human disorders of telomerase. Blood. 2015;126: 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alder JK, Hanumanthu VS, Strong MA, et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc Natl Acad Sci USA. 2018;115:E2358–E2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aalbers AM, Kajigaya S, van den Heuvel-Eibrink MM, van der Velden VH, Calado RT, Young NS. Human telomere disease due to disruption of the CCAAT box of the TERC promoter. Blood. 2012; 119:3060–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robart AR, Collins K. Investigation of human telomerase holoenzyme assembly, activity, and processivity using disease-linked sub-unit variants. J Biol Chem. 2010;285:4375–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell JR, Collins K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol Cell. 2000;6:361–371. [DOI] [PubMed] [Google Scholar]
- 58.Boyraz B, Bellomo CM, Fleming MD, Cutler CS, Agarwal S. A novel TERC CR4/CR5 domain mutation causes telomere disease via decreased TERT binding. Blood. 2016;128:2089–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L, Roake CM, Freund A, et al. An activity switch in human telomerase based on RNA conformation and shaped by TCAB1. Cell. 2018;174:218–30.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016;13:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2019;12:1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiss T, Fayet E, Jady BE, Richard P, Weber M. Biogenesis and intra-nuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:407–417. [DOI] [PubMed] [Google Scholar]
- 63.Stanley SE, Gable DL, Wagner CL, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8:351ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105:8073–8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lafontaine DL, Bousquet-Antonelli C, Henry Y, Caizergues-Ferrer M, Tollervey D. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998;12:527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol. 1999;19:7461–7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meier UT. The many facets of H/ACA ribonucleoproteins. Chromosoma. 2005;114:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terns M, Terns R. Noncoding RNAs of the H/ACA family. Cold Spring Harb Symp Quant Biol. 2006;71:395–405. [DOI] [PubMed] [Google Scholar]
- 70.Darzacq X, Jady BE, Verheggen C, Kiss AM, Bertrand E, Kiss T. Cajal body-specific small nuclear RNAs: a novel class of 2′-O-methylation and pseudouridylation guide RNAs. EMBO J. 2002;21:2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kiss AM, Jady BE, Darzacq X, Verheggen C, Bertrand E, Kiss TA. Cajal body-specific pseudouridylation guide RNA is composed of two box H/ACA snoRNA-like domains. Nucleic Acids Res. 2002;30: 4643–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richard P, Darzacq X, Bertrand E, Jady BE, Verheggen C, Kiss T. A common sequence motif determines the Cajal body-specific localization of box H/ACA scaRNAs. EMBO J. 2003;22:4283–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim NK, Theimer CA, Mitchell JR, Collins K, Feigon J. Effect of pseudouridylation on the structure and activity of the catalytically essential P6.1 hairpin in human telomerase RNA. Nucleic Acids Res. 2010; 38:6746–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong JM, Collins K. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 2006;20: 2848–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Batista LF, Pech MF, Zhong FL, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu BW, Apicella M, Mills J, et al. Impaired telomere maintenance and decreased canonical WNT signaling but normal ribosome biogenesis in induced pluripotent stem cells from X-linked dyskeratosis congenita patients. PLoS One. 2015;10:e0127414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thumati NR, Zeng XL, Au HH, Jang CJ, Jan E, Wong JM. Severity of X-linked dyskeratosis congenita (DKCX) cellular defects is not directly related to dyskerin (DKC1) activity in ribosomal RNA biogenesis or mRNA translation. Hum Mutat. 2013;34:1698–1707. [DOI] [PubMed] [Google Scholar]
- 78.Bellodi C, McMahon M, Contreras A, et al. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep. 2013;3: 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Penzo M, Rocchi L, Brugiere S, et al. Human ribosomes from cells with reduced dyskerin levels are intrinsically altered in translation. FASEB J. 2015;29:3472–3482. [DOI] [PubMed] [Google Scholar]
- 80.Schwartz S, Bernstein DA, Mumbach MR, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoon A, Peng G, Brandenburger Y, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. [DOI] [PubMed] [Google Scholar]
- 82.Balogh E, Chandler JC, Varga M, et al. Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc Natl Acad Sci USA. 2020;117:15137–15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Egan ED, Collins K. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol Cell Biol. 2012;32:2428–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Venteicher AS, Abreu EB, Meng Z, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323:644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tycowski KT, Shu MD, Kukoyi A, Steitz JA. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell. 2009;34:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freund A, Zhong FL, Venteicher AS, et al. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell. 2014;159:1389–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vogan JM, Zhang X, Youmans DT, et al. Minimized human telomerase maintains telomeres and resolves endogenous roles of H/ACA proteins, TCAB1, and Cajal bodies. Elife. 2016;5.1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tummala H, Walne A, Collopy L, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125:2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47:512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Virtanen A, Henriksson N, Nilsson P, Nissbeck M. Poly(A)-specific ribonuclease (PARN): an allosterically regulated, processive and mRNA cap-interacting deadenylase. Crit Rev Biochem Mol Biol. 2013; 48:192–209. [DOI] [PubMed] [Google Scholar]
- 92.Dhanraj S, Gunja SM, Deveau AP, et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J Med Genet. 2015;52:738–748. [DOI] [PubMed] [Google Scholar]
- 93.Moon DH, Segal M, Boyraz B, et al. Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat Genet. 2015;47:1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goldfarb KC, Cech TR. 3′ terminal diversity of MRP RNA and other human noncoding RNAs revealed by deep sequencing. BMC Mol Biol. 2013;14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Berndt H, Harnisch C, Rammelt C, et al. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA. 2012;18:958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Son A, Park JE, Kim VN. PARN and TOE1 constitute a 3′ end maturation module for nuclear non-coding RNAs. Cell Rep. 2018;23: 888–898. [DOI] [PubMed] [Google Scholar]
- 97.Nguyen D, Grenier St-Sauveur V, Bergeron D, Dupuis-Sandoval F, Scott MS, Bachand F. A polyadenylation-dependent 3′ end maturation pathway is required for the synthesis of the human telomerase RNA. Cell Rep. 2015;13:2244–2257. [DOI] [PubMed] [Google Scholar]
- 98.Shukla S, Schmidt JC, Goldfarb KC, Cech TR, Parker R. Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat Struct Mol Biol. 2016;23:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tseng CK, Wang HF, Burns AM, Schroeder MR, Gaspari M, Baumann P. Human telomerase RNA processing and quality control. Cell Rep. 2015;13:2232–2243. [DOI] [PubMed] [Google Scholar]
- 100.Boyraz B, Moon DH, Segal M, et al. Post-transcriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J Clin Invest. 2016;126:3377–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vanacova S, Wolf J, Martin G, et al. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005;3:e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roake CM, Chen L, Chakravarthy AL, Ferrell JE Jr, Raffa GD, Artandi SE. Disruption of telomerase RNA maturation kinetics precipitates disease. Mol Cell. 2019;74:688–700. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tseng CK, Wang HF, Schroeder MR, Baumann P. The H/ACA complex disrupts triplex in hTR precursor to permit processing by RRP6 and PARN. Nat Commun. 2018;9:5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fok WC, Shukla S, Vessoni AT, et al. Posttranscriptional modulation of TERC by PAPD5 inhibition rescues hematopoietic development in dyskeratosis congenita. Blood. 2019;133:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gable DL, Gaysinskaya V, Atik CC, et al. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 2019;33:1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lubas M, Christensen MS, Kristiansen MS, et al. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell. 2011;43:624–637. [DOI] [PubMed] [Google Scholar]
- 107.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478: 57–63. [DOI] [PubMed] [Google Scholar]
- 108.Lardelli RM, Schaffer AE, Eggens VR, et al. Biallelic mutations in the 3′ exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing. Nat Genet. 2017;49:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Deng T, Huang Y, Weng K, et al. TOE1 acts as a 3′ exonuclease for telomerase RNA and regulates telomere maintenance. Nucleic Acids Res. 2019;47:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kirwan M, Beswick R, Vulliamy T, et al. Exogenous TERC alone can enhance proliferative potential, telomerase activity and telomere length in lymphocytes from dyskeratosis congenita patients. Br J Haematol. 2009;144:771–781. [DOI] [PubMed] [Google Scholar]
- 111.Westin ER, Chavez E, Lee KM, et al. Telomere restoration and extension of proliferative lifespan in dyskeratosis congenita fibroblasts. Aging Cell. 2007;6:383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paulsen BS, Mandal PK, Frock RL, et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR/Cas9-mediated genome editing. Nat Biomed Eng. 2017;1: 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Woo DH, Chen Q, Yang TL, et al. Enhancing a Wnt-telomere feedback loop restores intestinal stem cell function in a human organotypic model of dyskeratosis congenita. Cell Stem Cell. 2016;19:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nagpal N, Wang J, Zeng J, et al. Small-molecule PAPD5 inhibitors restore telomerase activity in patient stem cells. Cell Stem Cell. 2020; 26:896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mueller H, Lopez A, Tropberger P, et al. PAPD5/7 are host factors that are required for hepatitis B virus RNA stabilization. Hepatology. 2019;69:1398–1411. [DOI] [PubMed] [Google Scholar]
- 116.Mueller H, Wildum S, Luangsay S, et al. A novel orally available small molecule that inhibits hepatitis B virus expression. J Hepatol. 2018; 68:412–420. [DOI] [PubMed] [Google Scholar]