

Abstract
The evolution of gamma-secretase modulators (GSMs) through the introduction of novel heterocycles with the goal of aligning activity for reducing the levels of Aβ42 and properties consistent with a drug-like molecule are described. The insertion of a methoxypyridine motif within the tetracyclic scaffold provided compounds with improved activity for arresting Aβ42 production as well as improved properties, including solubility. In vivo pharmacokinetic analysis demonstrated that several compounds within the novel series were capable of crossing the BBB and accessing the therapeutic target. Treatment with methoxypyridine-derived compound 64 reduced Aβ42 levels in the plasma of J20 mice, in addition to reducing Aβ42 levels in the plasma and brain of Tg2576 mice.
Keywords: Gamma secretase modulator, Alzheimer’s disease, amyloid beta, Aβ42 reduction, structure activity relationship, methoxypyridines
Graphical Abstract
1. Introduction
The population of Americans affected by Alzheimer’s disease (AD) is currently estimated to be 5.8 million, and as individuals of the large baby boomer generation are progressing beyond the age of 65, the population of those with AD is expected to nearly triple over the next 35 years.1 As a result, AD healthcare spending is projected to cost in excess of $1 trillion by 2050.1 Despite massive research efforts, no disease modifying treatments currently exist for AD, and consequentially, the disease is reaching epidemic proportions and remains a major unmet clinical need.
AD is a devastating neurological condition which insidiously affects regions of the brain critical for cognition. Originally described by Alois Alzheimer, the eponymous disease predominantly affects the elderly and is characterized by senile plaques composed of aggregated amyloid beta (Aβ) peptides and neurofibrillary tangles, paired helical filaments of the hyper-phosphorylated microtubule-associated protein tau.2–4 Although disease symptoms largely appear late in life, a large body of evidence indicates that the disease begins with extracellular deposition of Aβ peptides in regions of the brain that are vital for cognition decades prior. Many years after the initial deposition of Aβ peptides, neurofibrillary tangles appear, which closely correlate with the extent of cognitive decline associated with disease progression.5, 6 The time period between the initial deposition of senile plaques resulting from aberrant biological processes and prior to the manifestation of clinical symptoms, offers the most promising window for therapeutic intervention.
The hallmark neuritic plaques diagnostic for AD form as a result of an imbalance of amyloid peptide variants, which increases the relative levels of Aβ42, a poorly soluble peptide that readily forms aggregates.7, 8 Genetic evidence implicates subtle shifts in Aβ peptide production biased toward the formation of Aβ42 as playing a causative role in the disease.9 The production of amyloid peptides, including Aβ42, is the result of sequential processing of the amyloid precursor protein (APP) by two aspartyl proteases.10–12 The so-called amyloidogenic pathway commences with the cleavage of APP by β-amyloid precursor protein cleaving enzyme 1 (BACE-1), resulting in the APP-carboxy-terminal fragment (CTF) known as C99. Subsequent cleavages of the membrane bound C99 by γ-secretase provide the APP-intracellular domain (AICD) and the extracellular Aβ peptides, which range from 37 to 43 amino acids in length.13, 14 As a result of their role in amyloid production, BACE-1 and γ-secretase have garnered intense interest as potential therapeutic targets for the development of disease modifying AD therapeutics.15–17
Initially, drug discovery and development efforts focused on γ-secretase inhibitors (GSIs), which suppress the formation of all Aβ peptides including Aβ42.18 While GSIs were shown to be capable of reducing Aβ peptides in plasma and to some extent cerebrospinal fluid (CSF) during clinical trials, counter-efficacy and off-target effects were also observed. The adverse effects were attributed to the inhibition of the processing of the numerous other γ-secretase substrates, which include the Notch 1 receptor, causing the accumulation of CTFs, which resulted in the discontinuation of GSI development.19, 20 As an alternative to inhibiting γ-secretase, allosteric modulation alters the substrate cleavage pattern of the enzyme, suppressing the formation of the longer, fibrillogenic Aβ42 peptide in favor of shorter non-fibrillogenic isoforms, Aβ37 and Aβ38.21–23 Importantly, compounds with this mode of action do not preclude the proteolysis of the numerous critical substrates of γ-secretase and consequentially avoid the inhibition-related side effects associated with GSIs.24
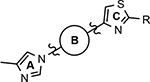
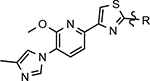
Various classes of γ-secretase modulators (GSMs) have been discovered previously; however, the tetracyclic methyl imidazole-derived GSM scaffold has been intensely pursued by both academic and industry laboratories as a potential disease modifying therapeutic for AD (Figure 1).25–28 γ-Secretase is an intermembrane aspartyl protease complex composed four subunits: presenilin (PS1 or PS2), presenilin enhancer 2 (PEN-2), anterior pharynx defective-1 (Aph-1), and nicastrin.29, 30 Photoaffinity labeling studies of the enzyme complex using a variety of related methyl imidazole-derived GSMs have revealed that these compounds bind to an allosteric site located on the PS1 subunit’s N-terminal fragment (PS1-NTF) distinct from both the binding site of the GSIs as well as the active site of the enzyme and, as a result, do not interfere with the catalytic activity or the processing of the numerous putative substrates.31, 32
Figure 1.

Representative methyl imidazole-derived GSM scaffolds. E2012 was developed by Eisai. PF-06442609 was developed by Pfizer and illustrates that the general GSM tetracyclic system may be fused.25–28
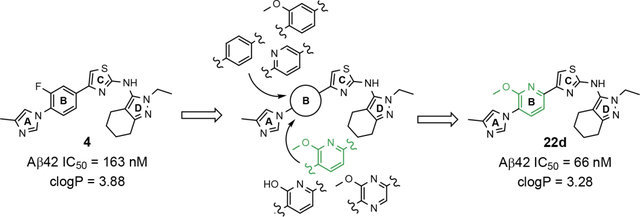
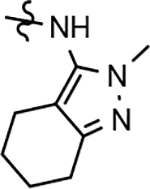
Despite the therapeutic promise that is associated with methyl imidazole-derived GSMs, the discovery of compounds which align potent activity with favorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) has proved challenging. Previously, our lab utilized compound 3 (NGP-555) as a lead in order to discover novel GSMs, which preserve the activity of the parent molecule while simultaneously improving other properties including solubility, through the preparation of novel heterocyclic D-ring analogs (Scheme 1).33 While these analogs exhibited improved properties, the activity of these compounds was greatly diminished as demonstrated by tetrahydroindazole 4. Herein, we describe our ongoing efforts toward the development of a second-generation of GSM compounds through preparation of novel B-ring analogs of our aminothiazole-derived modulators.
Scheme 1.

GSM B-ring optimization. Aminothiazole 3 (NGP-555) represents the initial lead compound for efforts to discover a potent GSM with improved ADMET properties. Tetrahydroindazole D-ring analog 4 was developed with the aim of improving critical properties of compound 3, including solubility. The rings are labeled A-D for clarity.
2. Design and Synthesis
2.1. Design of B-ring GSM analogs
A focused series of aminothiazole B-ring derivatives were designed with the aim of simultaneously improving both the activity for inhibiting the production of Aβ42, as well as refining crucial physicochemical parameters, including aqueous solubility. Previously, the incorporation of heteroatoms within the D-ring of the aminothiazole-based GSMs led to ligands that demonstrated moderately improved chemical properties and good pharmacokinetic behavior.33 However, compounds within this novel series did not maintain the modulator activity associated with the parent aminothiazole 3. Based on literature analysis of methyl imidazole-derived GSMs, various modifications to the B-ring have been reported that favorably influence Aβ activity while also providing ligands with desirable ADMET properties.31, 34, 35 Leveraging our prior efforts toward identifying a potent GSM with improved physical properties, ligand design focused on reducing lipophilicity through the identification of an optimal heterocyclic B-ring alternative within the 2-aminothiazole scaffold.
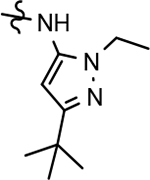
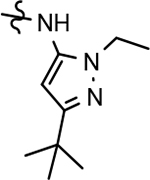
Based on the moderately active tetrahydroindazole D-ring analog 4, a focused series of corresponding B-ring derivatives were conceived in order to assess the functional tolerance of the adjacent chemical environment within the γ-secretase enzyme aimed at simultaneously enhancing modulator activity and improving other problematic properties associated with the ligand through the incorporation of novel bioisosteric functionalities.33 Compound design prioritized scaffolds containing heterocycles for synthesis, since the prior use of this strategy was demonstrated to be generally successful at addressing poor compound solubility as well as other property-related issues. Derivatives containing phenyl, methoxyphenyl, pyridyl, hydroxypyridyl, methoxypyridyl, and methoxypyrazinyl B-rings were subsequently synthesized.
2.2. Synthesis
The preparation of compounds that explore the consequences of modifying the B-ring of the GSM scaffold was based on a convergent synthetic route in which the coupling of two fragments would provide the desired analog (Scheme 2). Synthesis of the western portion of the scaffold containing phenyl and pyridyl B-rings was initiated by the nucleophilic substitution of acetophenones 6a-c using 4-methylimdazole to provide ethanones 7a-c.36 Subsequent acid catalyzed bromination of 7a,c furnished the first desired coupling partners, bromoethanones 8a,c. The construction of the related methoxyphenyl B-ring precursor utilized a similar reaction sequence. Fluoroacetophenone 7b was subjected to a second nucleophilic substitution using sodium methoxide to provide methoxyphenylethanone 9. Bromination furnished the desired methoxyphenyl coupling partner 10 in high yield.
Scheme 2.

Synthesis of phenyl-, methoxyphenyl-, and pyridyl B-ring-containing analogs. Reagents and conditions: a) 4-methylimidazole, K2CO3, DMSO, 55 °C, 16 h, 44–67%; b) Br2, 33% HBr in AcOH, DCM, r.t., 1.5 h, 75–80%; c) NaOMe, DMF, 0 °C to 50 °C, 1 h, 27%; d) Br2, 33% HBr in AcOH, r.t., 0.75 h, 100%.
While the synthesis of the phenyl-, methoxyphenyl-, and pyridyl B-ring-containing analogs was straightforward, the introduction of the methoxypyridine and methoxypyrazine B-rings required longer reaction sequences for the assembly of the desired coupling intermediates (Scheme 3). The preparation of the western portion of the methoxypyridine B-ring analog was initiated through the nucleophilic aromatic substitution of 2,6-dibromo-3-aminopyridine by sodium methoxide providing 6-bromo-2-methoxy-3-aminopyridine 12a.37 N-formylation of intermediate 12a was accomplished through the in situ generation of formic anhydride allowing straightforward access to formamide 13a.38, 39 Conversion to the corresponding ketoformamide was accomplished via nucleophilic substitution of chloropropanone by intermediate 13a. The construction of the A-ring of the tetracyclic was completed through the cyclization of ketoformamide 14a using ammonium acetate.36 Palladium-mediated Heck coupling of bromopyridine 15a and n-butylvinylether followed by acidic hydrolysis allowed the isolation of pyridylethanone 16a. Finally, acid-catalyzed bromination of intermediate 16a provided access to the desired coupling fragment, bromoacetophenone 17a in good yield. While trace amounts of demethylation were observed for the initial transformations from 16 to 17, this was greatly minimized by conducting the reaction at room temperature and under diluted conditions. Similarly, the construction of methoxypyrazine B-ring coupling fragment utilized the same three-step reaction sequence to construct the methyl imidazole A-ring starting from bromomethoxypyrazamine 12b. Stille coupling of bromopyrazine intermediate 15b with tributyl(1-ethoxyvinyl) tin provided pyrazinylethanone 16b, that upon bromination using tailored conditions, provided the desired methoxypyrazine intermediate 17b.
Scheme 3.

Synthesis of methoxypyridine and methoxypyrazine B-ring-containing analogs. Reagents and conditions: a) NaOMe, 1,4-dioxane, reflux, 18 h, 98%; b) Ac2O, formic acid, THF, 0 °C to r.t., 2 h, 95–100%; c) CH3COCH2Cl, K2CO3, KI, DMF, r.t., 4–18 h, 91–93%; d) NH4OAc, acetic acid, 120 °C, 10 h, 85%; e) X=C n-ethylvinylether, Pd(dppf)Cl2, TEA, ethylene glycol, 100 °C, 4 h, 73% or X=N tributyl(1-ethoxyvinyl) tin, Pd(PPh3)2Cl2, dioxane, 100 °C, 18 h, 85%; f) 2N HCl, acetone, r.t., 0.5–18h, 89–96%; g) Br2, 33% HBr in acetic acid, EtOAc, CHCl3, r.t., 0.5–5 h, 85–90%.
The shared eastern portion of the analogs was synthesized from commercially available racemic 2-oxocyclohexane-1-carbonitrile 18 (Scheme 4).33, 36 Condensation of the starting material with ethylhydrazine, furnished 3-aminocyclohexapyrazole 19 in good yield. Benzoyl thiourea 20 was then isolated following the reaction of benzoyl isothiocyanate with intermediate 19. Base-mediated removal of the benzoyl group permitted straightforward isolation of the common coupling partner 21. The desired B-ring analogs were then prepared following Hantzsch condensation of the appropriate bromoketone with thiourea 21, permitting the formation of the thiazole C-ring (Scheme 5).40 In addition, a pyridinol B-ring analog 23, was prepared following demethylation of methoxypyridine 22d using HBr (Scheme 6). The implementation of a convergent synthetic strategy provided access to a variety novel GSM compounds containing focused B-ring alterations.
Scheme 4.

Reagents and conditions: a) ethylhydrazine oxalate, EtOH, reflux, 18 h, 88%; b) benzoyl isothiocyanate, 60 °C, 3.5 h, 91%; c) K2CO3, MeOH, THF, r.t., 18 h, 92%.
Scheme 5.

Reagents and conditions for substrates 8a, 8c, and 10: a) EtOH, DIPEA, 55 °C, 24 h, 18–33%. Reagents and conditions for substrates 17a and 17b: a) EtOH, reflux, 18 h, 40–42%.
Scheme 6.

Reagents and conditions: a) 33% HBr in AcOH, 50 °C, 1.5 h, 92%.
3.0. Results and Discussion
3.1. Activity of novel B-ring GSM analogs
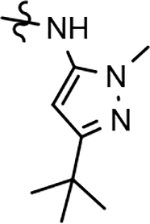
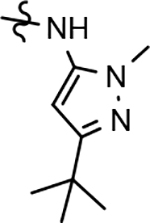
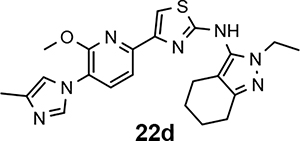
Following preparation, the novel analogs’ ability to suppress the formation of Aβ42 was determined using an enzyme-linked immunosorbent assay (ELISA). The simplification of the B-ring scaffold through the synthesis of desfluoro analog 22a resulted in a greater than 4-fold loss in activity, suggesting that the presence of a substituent may be critical to maintaining and improving potency (Table 1). Replacement of the 3-fluoro substituent within the scaffold was well tolerated as demonstrated by methoxyphenyl analog 22c, which displayed commensurate activity to parent tetrahydroindazole 4. Alternatively, pyridyl derivative 22b revealed that the addition of a heteroatom in place of the substituent within the B-ring was deleterious to γ-secretase activity. 3-Methoxypyridine 22d and 3-hydroxypyridine 23 were then synthesized in order to ascertain if the introduction of substituents within a pyridine derivative was capable of restoring activity. The 3-hydroxypyridine 23 exhibited a modest recovery of potency, whereas the 3-methoxypyridine 22d showed a nearly 3-fold improvement in activity over the parent compound with an IC50 value of 60 nM. Varying the heterocyclic B-ring component of methoxypyridine 22d providing methoxypyrazine 22e was well tolerated and resulted in a minor decrease in ligand activity for attenuating the production of Aβ42 with an IC50 value of 89 nM.
Table 1.
B-ring analogs of GSM 4.
 | ||||
|---|---|---|---|---|
| Cmpd | B-ring | Aβ42 IC50a | clogPb | Kin. Aq. Sol.c |
| 4 |  |
163 ± 10 | 3.88 | ntd |
| 22a |  |
660 ± 131 | 3.74 | ntd |
| 22b | 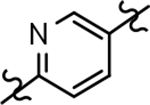 |
>1000 | 3.13 | ntd |
| 22c | 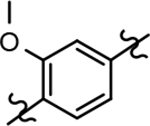 |
171 ± 67 | 3.49 | 4.6 |
| 22d |  |
60 ± 15 | 3.28 | 4.5* |
| 22e | 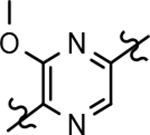 |
89 ± 38 | 2.66 | <1.6 |
| 23 | 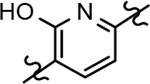 |
468 ± 165 | 3.25 | ntd |
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50%. The IC50 values are the mean ± standard deviation of at least 2 determinations.
Calculated partition coefficient of the simulated ratio of the compound’s concentration in octanol to the compound’s concentration in water using ChemAxon fragment based approach.
Kinetic aqueous solubility measured at pH 7.4 by UV/Vis absorbance in PBS buffer (μM).
nt = not tested - compound did not meet minimum activity threshold.
The preparation of novel B-ring analogs of GSM 4 led to the discovery of methoxypyridine 22d and methoxypyrazine 22e, which demonstrated improved activity for inhibiting the formation of Aβ42. The general reduction in the overall clogP associated with the integration of heteroatoms within the B-ring was anticipated to lead to increased aqueous solubility. Methoxypyridine 22d displayed fair kinetic aqueous solubility, whereas methoxypyrazine 22e remained insoluble despite the observed decrease in clogP (Table 1). The solubility of methoxyphenyl 22c was also evaluated, since GSMs containing this scaffold have been previously developed and demonstrated good activity as well as ideal ADMET properties. Similar to methoxypyridine 22d, methoxyphenyl analog 22c exhibited fair aqueous solubility, which indicates that the character of the aromatic B-ring can influence the solubility of ligands containing a 3-methoxy substituent.
3.2. Expanded SAR of B-ring GSM analogs
Identification of the methoxypyridine 22d, which exhibited robust activity for attenuating the production of Aβ42 in addition to good aqueous solubility, led us to broaden our exploratory set of B-ring analogs to include additional related scaffolds from our modestly active D-ring GSM series in order to foster a better understanding of the role of the B-ring as it pertains to both activity and physical properties. Tetrahydroindazole 24, as well as 3-tert-butylpyrazoles 25 and 26 were selected from two distinct families of GSMs for focused chemical B-ring modification (Figure 2).41 Novel analogs of the selected compounds containing phenyl, pyridyl, methoxyphenyl or methoxypyridyl B-rings were synthesized and evaluated for Aβ42 activity.
Figure 2.

Fluorophenyl-derived GSMs selected for focused B-ring modification in order to broaden SAR.
The synthesis of the initial focused B-ring analog series provided access to the western portion of the desired analogs (Schemes 2 and 3). The preparation of the eastern coupling partner was achieved using previously establish reaction conditions. Briefly, the thiourea intermediate necessary to furnish the additional tetrahydroindazole compounds commenced through the condensation of 2-oxocyclohexane-1-carbonitrile with methylhydrazine and subsequently followed the same reaction sequence as described in Scheme 4.33, 36 Similarly, the thiourea coupling component required to synthesize the substituted tert-butylpyrazoles began with the addition of the appropriate alkylhydrazine to pivaloylacetonitrile (Scheme 7). The 3-aminopyrazole product, 28, was then exposed to benzoyl isothiocyanate, providing intermediate 29, which upon cleavage of the benzoyl group allowed access to the desired thiourea 30. Finally, the additional B-ring modified aminothiazoles were isolated following Hantzsch condensation of the appropriate western and eastern portions of the molecule (Scheme 8).40
Scheme 7.

Reagents and conditions: a) ethylhydrazine oxalate or methylhydrazine, EtOH, reflux, 18 h, 61–66%; b) benzoyl isothiocyanate, acetone, reflux, 3.5 h, 88–96%; c) K2CO3, MeOH, THF, r.t., 18 h, 81–92%.
Scheme 8.

Reagents and conditions: a) EtOH, reflux, 18 h, 33–51%.
The broadened series of compounds were then screened for the ability to suppress the Aβ42 levels. Similar to the initially prepared tetrahydroindazole B-ring analogs, the integration of a phenyl B-ring within the expanded compound series led to a greater than 2-fold loss in potency for tetrahydroindazole 31a and N-ethylpyrazole 33a for suppressing the formation of Aβ42, whereas N-methylpyrazole 32a showed similar activity to the parent (Table 2). The corresponding 3-pyridyl B-ring analogs 31b, 32b, and 33b also displayed substantial losses in activity. The general trend toward significantly reduced affinity within the phenyl and pyridyl analog series reinforced the assertion that the 3-postion B-ring substituent engages in favorable interactions with the target enzyme. Alternatively, 3-methoxyphenyl derivatives 31c and 32c, exhibited a modest improvement in efficacy when compared to the corresponding fluorophenyl compounds. However, the 3-methoxy substituent did not lead to increased affinity for N-ethylpyrazole 33c, which exhibited a slight reduction in potency. This pattern of activity was also observed for the methoxypyridyl B-ring derivatives; a greater than 2-fold improvement in activity was observed for N-methylpyrazole 32d and tetrahydroindazole 31d, whereas N-ethylpyrazole 33d was less active than the corresponding parent ligand. Although the introduction of either the methoxyphenyl or methoxypyridyl B-ring generally led to improved activity for inhibiting the production of Aβ42, a greater magnitude as well as absolute effect upon the peptide was associated with the presence of the methoxypyridyl B-ring. Furthermore, the inclusion of this functionality addressed the limited solubility that was associated with the previously developed compounds, as shown by methoxypyridyl analogs 31d and 33d.
Table 2.
B-ring analogs of tetrahydroindazole-derived GSM 24 and tert-butylpyrazole-derived GSMs 25 and 26.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | B-ring | R | Aβ42 IC50a | clogPb | Kin. Aq. Sol.c | Metabolic Stabilityd | ||
| H | R | M | ||||||
| 31a | 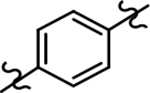 |
 |
453 | 3.40 | 4.7 | 55 | 56 | 84 |
| 31b | 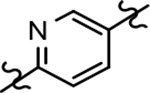 |
 |
>1000 | 2.79 | nte | nte | nte | nte |
| 31c |  |
 |
148 | 3.15 | nte | nte | nte | nte |
| 31d |  |
 |
105 | 2.93 | 15 | 36 | 60 | 34 |
| 32a |  |
 |
275 | 4.27 | nte | nte | nte | nte |
| 32b | 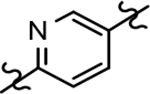 |
 |
590 | 3.66 | nte | nte | nte | nte |
| 32c | 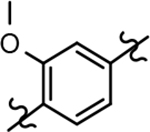 |
 |
253 | 4.02 | 5.5 | 60 | 76 | 92 |
| 32d |  |
 |
136 | 3.80 | ntd | nte | nte | nte |
| 33a |  |
 |
339 | 4.61 | ntd | nte | nte | nte |
| 33b | 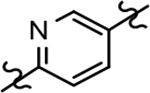 |
 |
292 | 4.00 | <1.6 | 43 | 60 | 87 |
| 33c | 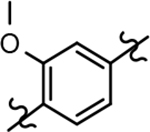 |
 |
96 | 4.36 | ntd | nte | nte | nte |
| 33d |  |
 |
114 | 4.14 | 15 | 38 | 89 | 67 |
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50%. The IC50 values are the mean ± standard deviation of at least 2 determinations.
Calculated partition coefficient of the simulated ratio of the compound’s concentration in octanol to the compound’s concentration in water using ChemAxon fragment based approach.
Kinetic solubility measured at pH 7.4 by UV/Vis absorbance in PBS buffer (μM).
% Remaining after 30 minutes upon incubation with human (H), rat (R) and mouse (M) liver microsomes (1 mg/mL), at 1 μM test compound concentration.
nt = not tested - compound did not meet minimum activity threshold.
3.3. In vitro ADMET and mouse in vivo pharmacokinetics of GSM 22d
The preparation of a focused series of B-ring analogs identified the methoxypyridine-containing scaffold as a promising replacement for the fluorophenyl moiety within the original GSMs based on the criterion of activity as well as solubility. Since the incorporation of this novel B-ring significantly altered architecture of the scaffold, the most active analog, tetrahydroindazole 22d, was selected for comprehensive in vitro ADMET analysis in order to examine the effect of the methoxypyridyl B-ring upon additional critical parameters. These studies showed that tetrahydroindazole 22d displayed an adequate cytochrome P450 inhibition profile (Table 3), as well as acceptable hERG ion channel inhibition potential (all IC50 values > 1 μM). However, the limited metabolic stability with respect to humans is a potential concern for tetrahydroindazole 22d despite the good stability observed in the presence of rodent liver microsomes. In addition, similar to the previously developed fluorophenyl GSM series, analog 22d displayed high plasma protein binding which may limit in vivo exposure due to drug sequestration.
Table 3.
In vitro ADMET properties of pyrazole 22d.
 | |
| IC50 (nM) | 60 |
| Kin. sol. pH 7.4 (μM)a | 4.5 |
| clogP | 3.28 |
| hLM (%)b | 36 |
| rLM (%)b | 71 |
| mLM (%)b | 64 |
| CYP450 IC50 (μM)c | |
| 3A4 | 3.7 |
| 1A2 | >100 |
| 2C9 | 9.0 |
| 2C19 | 5.9 |
| 2D6 | >100 |
| hERG IC50 (μM)d | 8.6 |
| % PPBe | 99.6 |
Kinetic solubility measured at pH 7.4 by UV/Vis absorbance in PBS buffer (μM).
% Remaining after 30 minutes upon incubation with human (hLM), rat (rLM) and mouse (mLM) liver microsomes (1 mg/mL), at 1 μM test compound concentration.
Five recombinant CYP isoforms were tested for inhibition by the test compound using fluorescence-based assays.
Patch-Xpress patch-clamp assay; compounds were tested (n = 3) in a five-point concentration-response on HEK-293 cells stably expressing the hERG channel.
% PPB = plasma protein binding.
Although the ADMET screening of tetrahydroindazole 22d identified potential limitations that may impede compound development, pharmacokinetic evaluation was undertaken in order to assess the impact of the structural changes associated with the novel methoxypyridyl B-ring upon compound behavior in an in vivo setting. Following intravenous administration (iv) of compound 22d (1 mg/kg), high clearance (Cl = 2,159 mL/hr/kg) in male CD-1 mice was observed (Table 4), which represented approximately 40% of mouse hepatic blood flow (90 mL/min/kg) and was associated with a short plasma half-life (0.5 hr). After oral administration (po), compound 22d (5 mg/kg) was rapidly absorbed, reaching Cmax after 0.25 hr. Based on the exposure data for each route of dosing, the bioavailability (F) was estimated to be 53.7%. Given the high clearance of compound 22d, a significant first-pass effect is expected following oral administration. Assuming that the liver is responsible for drug elimination, approximately 40% of the absorbed dose will be metabolized during the first-pass, leaving only 60% of the compound capable of reaching systemic circulation. Since 53.7% of analog 22d is bioavailable, nearly 90% of the oral dose is absorbed from the lumen of the small intestine. The comparison between brain and plasma drug levels of analog 22d shows a ratio of 0.4 following both oral and iv administration, demonstrating that the compound can readily penetrate the blood-brain barrier (BBB) and reach the site of intended action. Although the brain to plasma ratio is indicative of good CNS penetrance, the metric cannot be used as a predictor of in vivo efficacy, since drug action is contingent upon unbound drug rather than absolute levels. Collectively, the pharmacokinetic data demonstrates that despite high clearance in mice, tetrahydroindazole 22d is readily absorbed and is capable of passing the BBB.
Table 4.
In vivo pharmacokinetic profile of tetrahydroindazole 22d.
| Mouse Pharmacokinetics of 22da | ||
| Route | iv | po |
| Dose (mg/kg) | 1 | 5 |
| Cmax (ng/mL) | 824 | 705 |
| Tmax (hrs) | 0.083 | 0.25 |
| t1/2 (hrs) | 0.5 | cndb |
| AUClast (ng·h/mL) | 443 | 1,089 |
| AUCinf (ng·h/mL) | 463 | 1,243 |
| Cl (mL/hr/kg) | 2,159 | |
| Vss (mL/kg) | 1,413 | |
| F (%) | 100 | 53.7 |
| Brain/Plasma Ratio (@ 1 hr post dose) | 0.4 | 0.4 |
Male CD-1 mice; n = 24.
Cannot be determined from data.
3.4. In vitro biomarker assessment of modulator activity of GSM 22d
Since γ-secretase is critical to the processing of numerous substrates, including the Notch 1 receptor, inhibition of the enzyme is known to lead to adverse effects.19, 20 Consequently, compound 22d was subjected to additional scrutiny in order to ensure that the structural modifications did not affect the mode of action. In order to ascertain the preservation function of γ-secretase associated with treatment, vehicle, compound 22d, and a GSI (DAPT) were incubated with H4 human neuroglioma cells over-expressing human APP751 transfected with the Myc-tagged Notch (NΔE) construct.42 Incubation with the GSI led to accumulation of the Notch substrate (NδED) due to inhibition of γ-secretase function (Figure 3), whereas vehicle and compound 22d did not impair enzyme function, as indicated by the presence of the processed Notch intracellular domain (NICD) and the absence of increased substrate. To further assess the modulator activity of compound 22d, the Aβ peptide profile elicited by treatment was determined following incubation with SHSY5Y-APP cells. In concordance with γ-secretase modulation, the levels of Aβ42 and Aβ40 were suppressed while levels of Aβ38 were potentiated (Figure 4). Since the activity of the enzyme was preserved, no change in the total levels of Aβ were observed. In addition, the IC50 value for Aβ42 in the Meso Scale Multiplex assay was consistent with that of the primary activity screen.
Figure 3.

Stable H4 human neuroglioma cells over-expressing human APP751 (H4-APP751 cells) were transfected with the NΔE construct, and then treated with different concentrations GSM compound 22d (lane 3 = 10 μM; lane 4 = 1 μM) or GSI (DAPT, lane 5 = 300 nM, lane 6 = 100 nM) for another 24hrs. Vehicle control treatment shown in lanes 1–2. Cells were harvested 48hrs post transfection and applied to Western blotting analysis. Myc antibody was utilized to assess the NΔED and NICD tagged with Myc on their N-termini. β-Actin was utilized as the loading control. Compound 22d did not inhibit Notching processing; however, DAPT significantly inhibits Notching processing.
Figure 4.

Concentration response curves of compound 22d using orthogonal medium throughput SHSY5Y-APP cell-based screening assays. Aβ38, Aβ40, Aβ42, and total Aβ peptide levels were determined using Meso Scale Sector 6000 Multiplex assays.
3.5. SAR of D-ring GSM analogs
The simultaneous improvement in activity of suppressing Aβ42 formation and solubility that accompanied the incorporation of a methoxypyridine B-ring led us to pursue additional D-ring analogs. The 2-fold or greater increase in potency observed for tetrahydroindazoles 22d and 31d as well as the good solubility exhibited by both compounds supported the examination of previously synthesized and marginally active D-ring tetrahydroindazole compounds in order to examine the broader effect of this B-ring substitution upon ligand activity and physical properties within a more diverse series of analogs aimed at identifying a compound with good drug-like potential.33
Amalgamated tetrahydroindazole analogs 34–52 were prepared through the use of the aforementioned Hantzsch condensation of the appropriate substrates, and the impact of the compounds upon Aβ42 production was determined. The N-alkyl derivatives 34 and 35 displayed a greater than 3-fold increase in efficacy over the corresponding fluorophenyl parent but continued to show the limited aqueous solubility observed within the original series (Table 5). N-tertbutyl tetrahydroindazole 36 similarly exhibited 3-fold increase in activity in addition to ideal solubility despite the added hydrophobic bulk. As observed with the N-alkyl analogs, the N-cycloalkyl tetrahydroindazoles, 37–39, demonstrated improved potency for attenuating the production of Aβ42. Moreover, a direct correlation between the size of the N-cycloalkyl substituent and solubility was observed in which successive ring expansion rapidly diminished aqueous solubility. Heteroatom insertion within the N-cycloalkyl substituent was likewise better tolerated within methoxypyridyl B-ring compounds. N-tetrahydrofuran 40 and N-tetrahydropyran 41 were 2-fold and 7-fold more potent than the analogous fluorophenyl B-ring compounds, respectively, whereas N-methylpiperidine 42 remained inactive. Similar gains in activity and ideal solubility were also observed for tethered N-heterocyclic analogs in which an ethyl or propyl linker was inserted into the substituent. However, despite the significantly improved efficacy, the more flexible N-substituent of analogs 43–45 do not achieve the levels of activity associated with direct N-substitution of the tetrahydroindazole scaffold. Increased efficacy and good solubility were also achieved by both N-fluoroethyl analog 46 and N-trifluoroethyl analog 47, which exhibited a greater than 2-fold improvement in potency when compared to the analogous fluorophenyl with IC50 values of 47 nM and 57 nM, respectively. The same overall trend of augmented activity and ideal solubility associated with the incorporation of the methoxypyridyl B-ring was also observed for 2-methoxyethane 48, 2-hydroxyethane 49, 2-hydroxypropane 50 and 2-hydroxy-2-methylpropane 51; however, analogs 50 and 51 demonstrated superior activity with IC50 values of 50 nM and 38 nM, respectively. Finally, the altered B-ring was even capable of imparting marginal activity to the methylamino-linked tetrahydroindazole 52. Collectively, the introduction of the methoxypyridyl B-ring was largely able to address the solubility of the tetrahydroindazole series while enhancing biochemical activity and supporting the overall rationale of the design.
Table 5.
Methoxypyridine-derived D-ring GSM analogs.
 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | Aβ42 IC50a | Parent Aβ42 IC50b | clogPc | Kin. Aq. Sol.d | Metabolic Stabilitye | CYP450 IC50 (μM)f | hERGg | Papph | Efflux Ratioi | ||||||
| H | R | M | 3A4 | 1A2 | 2C9 | 2C19 | 2D6 | |||||||||
| 34 | 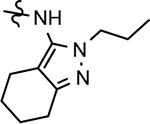 |
48 ± 17 | 148 | 3.75 | <1.6 | |||||||||||
| 35 | 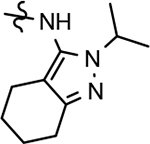 |
46 ± 6 | 195 | 3.69 | 3.3 | 10 | 64 | 45 | ||||||||
| 36 | 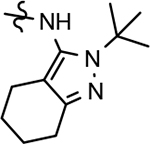 |
71 ± 25 | 216 | 3.77 | 34 | 19 | 63 | 23 | 5.3 | 9.1 | 47 | 12 | >100 | 7.18 | 14.8 | 0.9 |
| 37 | 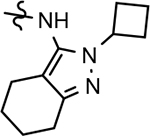 |
64 ± 26 | 99 | 3.73 | 30 | 9.5 | 42 | 44 | 0.59 | >100 | 0.66 | 1.6 | >100 | |||
| 38 | 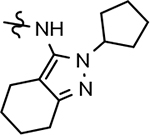 |
83 ± 18 | 225 | 4.12 | 1.8 | |||||||||||
| 39 | 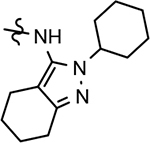 |
63 ± 6 | 280 | 4.52 | <1.6 | |||||||||||
| 40 | 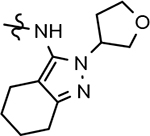 |
102 ± 33 | 289 | 2.71 | 19 | 2.1 | 42 | 70 | ||||||||
| 41 | 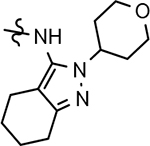 |
102 ± 40 | 766 | 2.77 | 4.7 | |||||||||||
| 42 | 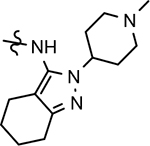 |
>1000 | >1000 | 2.91 | 92 | |||||||||||
| 43 | 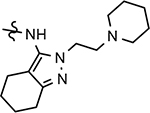 |
208 ± 25 | 475 | 3.64 | 46 | |||||||||||
| 44 | 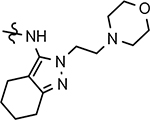 |
177 ± 40 | >1000 | 2.57 | 70 | 0 | 22 | 11 | ||||||||
| 45 | 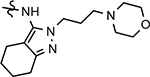 |
126 ± 23 | 270 | 2.62 | 39 | 1.9 | 6.4 | 0.7 | ||||||||
| 46 |  |
47 ± 4 | 110 | 3.09 | 15 | 100 | 100 | 100 | 1.0 | >100 | 6.8 | 4.6 | >100 | 11.4 | 32.6 | 1.9 |
| 47 | 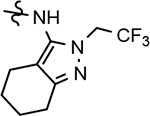 |
57 ± 17 | 179 | 3.91 | 7.4 | 13 | 41 | 39 | ||||||||
| 48 |  |
144 ± 54 | 272 | 2.77 | 20 | 1.8 | 45 | 29 | ||||||||
| 49 | 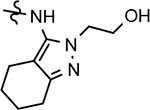 |
144 ± 55 | 362 | 2.19 | 31 | 40 | 78 | 58 | 30 | >100 | >100 | 76 | >100 | 2.63 | 18 | |
| 50 | 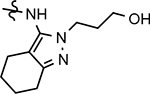 |
50 ± 13 | 229 | 2.24 | 23 | 17 | 52 | 32 | 2.2 | >100 | >100 | 31 | >100 | |||
| 51 |  |
38 ± 14 | 55 | 2.68 | 22 | 12 | 57 | 78 | 1.1 | >100 | 9.0 | 3.9 | >100 | 5.8 | 17.5 | 1.6 |
| 52 |  |
636 ± 44 | >1000 | 3.52 | 3.4 | |||||||||||
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50%. The IC50 values are the mean ± standard deviation of at least 2 determinations.
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50% for the parent 2-fluoro-1,4-phenyl B-ring compound. Connectivity is illustrated by compound 4 in Table 1. The IC50 values are the mean of at least 2 determinations.
Calculated partition coefficient of the simulated ratio of the compound’s concentration in octanol to the compound’s concentration in water using ChemAxon fragment based approach.
Kinetic solubility measured at pH 7.4 by UV/Vis absorbance in PBS buffer (μM).
% Remaining after 30 minutes upon incubation with human (hLM), rat (rLM) and mouse (mLM) liver microsomes (1 mg/mL), at 1 μM test compound concentration.
Five recombinant CYP isoforms were tested for inhibition by the test compound using fluorescence-based assays.
Patch-Xpress patch-clamp assay; compounds were tested (n = 3) in a five-point concentration-response on HEK-293 cells stably expressing the hERG channel.
Apparent permeability (A-B; × 10−6 cm/s) determined in MDR1-MDCK cell monolayers.
Calculated efflux ratio determined using MDR1-MDCK cell monolayers (B-A/A-B).
The novel series of methoxypyridine-derived tetrahydroindazoles also demonstrated distinct structure-activity relationship (SAR) when compared to the parent fluorophenyl series. The methoxypyridine analogs revealed broad tolerance for N-alkyl substitution within the D-ring of the scaffold as demonstrated by the various linear, branched, and cyclic alkyl functionalities that retain good activity for reducing Aβ42 levels as evidenced by analogs 34–39. The heterocyclic substituted analogs, unfortunately, were not as active as their alkyl counterparts and suffered from poor metabolic stability (Table 5). Taken together, these results suggest the presence of a rather large hydrophobic region near the N-substituent of the D-ring. The haloalkyl derivatives, such as N-fluoroethane 46 and N-trifluoroethane 47, also exhibited good modulator activity, indicating that analogs containing smaller N-substituents retain the favorable interactions with γ-secretase of their larger cycloalkyl counterparts. Since variations in the size and connectivity of the N-alkyl substituent result in a modest disparity between the analog activities, the larger N-substituents are likely near a region of open space rather than actively engaging in beneficial interactions with the target enzyme. In addition, 2-hydroxypropane 50 also shows good activity, while 2-hydroxy-2-methylpropane 51 is the most potent analog within the methoxypyridine-derived tetrahydroindazole family with an IC50 value of 38 nM. The tolerance exhibited by these compounds for the introduction of a polar substituent is hypothesized to be the result of an intramolecular hydrogen bond between the hydroxyalkyl moiety and one of the proximal nitrogen atoms within the scaffold which serves to rigidify the D-ring and preserve favorable hydrophobic contacts with the γ-secretase enzyme. Collectively, the significant improvement in activity observed within the tetrahydroindazole series indicates that the methoxypyridyl B-ring engages in beneficial interactions with γ-secretase as well as influences the binding conformation, resulting in access to additional favorable contacts for distal portions of the scaffold.
Although not all of the analogs within this set of derivatives exhibited improved activity for attenuating the production of Aβ42, the broad trend of increased efficacy and solubility demonstrated by the tetrahydroindazole compounds led us to expand the scope of the analogs to include methoxypyridyl B-ring derivatives of other GSMs previously synthesized. Increasing the structural diversity of the methoxypyridine B-ring analogs to include active derivatives of the marginally soluble fluorophenyl 3-tert-butylpyrazole family of compounds as well as the poorly soluble fluorophenyl cyclopentapyrazoles was conducted to explore the impact of this substitution upon physicochemical properties with the overarching aim of identifying a modulator that aligns potent activity with good drug-like behavior.
Through the incorporation of the novel methoxypyridine B-ring, additional D-ring analogs 53–74 were constructed and subsequently evaluated by ELISA for the ability to suppress Aβ42 peptide levels. N-propyl 3-tert-butylpyrazole 53 exhibited a modest loss in activity similar to N-ethylpyrazole 33d from the pilot B-ring screening series, whereas branched N-alkyl derivatives 54 and 55 showed improved potency with IC50 values of 64 nM and 40 nM, respectively (Table 6). However, the hydrophobicity of the N-substituent had a sizable impact on aqueous solubility as evidenced by N-isopropyl analog 54, which displays ideal solubility, while the N-tert-butyl analog 55 remained poorly soluble. The activity of N-trifluoroethyl analog 57 was largely unaffected by the methoxypyridyl B-ring and exhibited only slightly better potency than the corresponding parent compound. Based on these results, the 3-tert-butylpyrazoles support the presence of a hydrophobic pocket near the 3-substituent; however, distinct from the tetrahydroindazole family of compounds, the 3-tert-butylpyrazoles series favors branched N-alkyl substituents. Alterations to the 3-postion substituent of the pyrazole also led to compounds with 2-fold better activity when juxtaposed to the equivalent fluorophenyl ligand as shown by 3-isopropylpyrazole 58 and 3-methylcyclopropylpyrazole 59. However, reducing the hydrophobicity of the 3-substituent was deleterious to the overall activity as demonstrated by 3-isopropylpyrazole 58, whereas the subtle architectural modification of 3-methylcyclopropyl analog 59 led to the most potent derivative of the series with an IC50 value of 32 nM. The trend of improved activity was also observed for derivatives characterized by the presence of a 3-trifluoromethyl substituent as shown by N-ethyl analog 60 and N-trifluoroethyl analog 62 despite the reduction in overall hydrophobicity. In addition, the disparate potency of analogs 60 and 62 belies their remarkable structural similarity and is hypothesized to be the result of electronic effects linked to the presence of multiple electron withdrawing functionalities on the pyrazole ring. The incorporation of the novel methoxypyridyl B-ring generally provided 3-tert-butylpyrazole ligands with improved activity for suppressing the formation of Aβ42; however, increases in aqueous solubility were reserved for compounds with reduced D-ring hydrophobic character.
Table 6.
Methoxypyridine-derived D-ring GSM analogs.
 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R | Aβ42 IC50a | Parent Aβ42 IC50b | clogPc | Kin. Aq. Sol.d | Metabolic Stabilitye | CYP450 IC50 (μM)f | hERGg | Papph | Efflux Ratioi | ||||||
| H | R | M | 3A4 | 1A2 | 2C9 | 2C19 | 2D6 | |||||||||
| 53 |  |
100 ± 4 | 86 | 4.61 | 2.1 | |||||||||||
| 54 |  |
64 ± 3 | 87 | 4.56 | 23 | 21 | 75 | 40 | 2.3 | >100 | 14 | 14 | >100 | |||
| 55 | 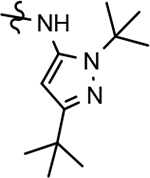 |
40 ± 7 | 85 | 4.64 | <1.6 | |||||||||||
| 56 | 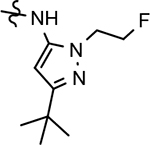 |
73 ± 11 | NA | 3.96 | 3.3 | 49 | 86 | 67 | 0.04 | 64 | 4.1 | 3.3 | >100 | |||
| 57 | 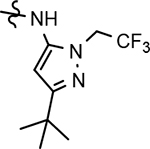 |
73 ± 20 | 87 | 4.78 | 1.7 | 47 | 90 | 93 | 1.0 | 18 | 20 | 9.0 | >100 | |||
| 58 | 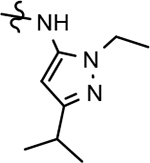 |
184 ± 75 | 394 | 3.48 | ||||||||||||
| 59 | 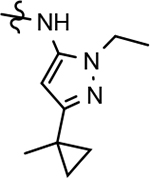 |
32 ± 12 | 67 | 3.64 | 6.7 | 67 | 73 | 81 | 4.1* | >100 | >100 | >100 | >100 | 12.3 | 18.5 | 1.3 |
| 60 | 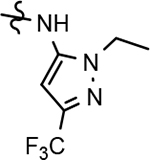 |
58 ± 20 | 98 | 3.40 | 15 | 84 | 96 | 71 | 5.3 | 9.1 | 47 | 12 | >100 | 18.9 | 23.3 | 1.0 |
| 61 | 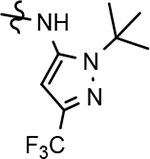 |
126 ± 16 | 121 | 3.89 | <1.6 | |||||||||||
| 62 | 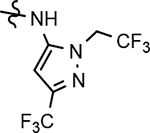 |
199 ± 28 | 771 | 4.03 | 2.0 | |||||||||||
| 63 | 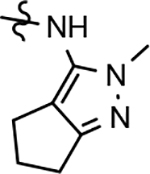 |
80 ± 34 | >1000 | 2.54 | 44 | 76 | 71 | 75* | 4.5 | >100 | >100 | 37 | >100 | 0.92 | 22.3 | 1.9 |
| 64 | 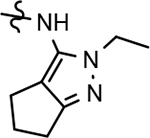 |
33 ± 10 | 41 | 2.88 | 7.8 | 51 | 67 | 54 | 20* | >100 | >100 | >100 | >100 | 1.19 | 29.6 | 0.8 |
| 65 |  |
90 ± 21 | 66 | 3.29 | 5.7 | 23 | 56 | 34 | ||||||||
| 66 |  |
62 ± 1 | 175 | 3.37 | 3.9 | 21 | 55 | 20 | ||||||||
| 67 |  |
21 ± 3 | 92 | 3.33 | 2.7 | 3 | 37 | 26 | ||||||||
| 68 | 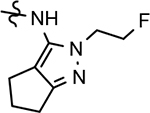 |
64 ± 14 | 2.70 | 23 | 58 | 68 | 60 | 10.5 | >100 | >100 | 35 | >100 | 2.88 | |||
| 69 |  |
36 ± 12 | 94 | 3.51 | 22 | 66 | 72 | 46 | 1.4 | 27 | 11 | 7.1 | >100 | 0.92 | 17.6 | 0.9 |
| 70 |  |
75 ± 11 | >1000 | 1.80 | 29 | 42 | 86 | 73 | <0.05 | >100 | 50 | 9.2 | >100 | |||
| 71 |  |
109 ±13 | 62 | 4.11 | 8.8 | 31 | 61 | 49 | 0.75 | >100 | 48 | 15 | >100 | |||
| 72 | 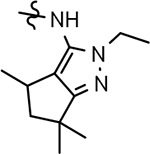 |
72 ± 28 | 49 | 4.44 | 9.2 | 12 | 38 | 21 | 1.5 | >100 | 35 | 33 | >100 | |||
| 73 | 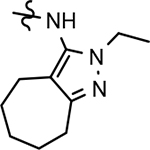 |
32 ± 12 | 114 | 3.67 | 4.3 | 0.1 | 41 | 15 | 11 | >100 | 37 | 85 | >100 | |||
| 74 | 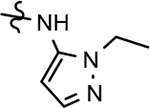 |
>1000 | >1000 | 2.12 | ||||||||||||
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50%. The IC50 values are the mean ± standard deviation of at least 2 determinations.
IC50 represents the concentration in nM of compound required for reducing Aβ42 levels by 50% for the parent 2-fluoro-1,4-phenyl B-ring compound. Connectivity is illustrated by compound 4in Table 1. The IC50 values are the mean of at least 2 determinations.
Calculated partition coefficient of the simulated ratio of the compound’s concentration in octanol to the compound’s concentration in water using ChemAxon fragment based approach.
Kinetic solubility measured at pH 7.4 by UV/Vis absorbance in PBS buffer (μM).
% Remaining after 30 minutes upon incubation with human (hLM), rat (rLM) and mouse (mLM) liver microsomes (1 mg/mL), at 1 μM test compound concentration.
Five recombinant CYP isoforms were tested for inhibition by the test compound using fluorescence-based assays.
Patch-Xpress patch-clamp assay; compounds were tested (n = 3) in a five-point concentration-response on HEK-293 cells stably expressing the hERG channel.
Apparent permeability (A-B; × 10−6 cm/s) determined in MDR1-MDCK cell monolayers.
Calculated efflux ratio determined using MDR1-MDCK cell monolayers (B-A/A-B).
In a similar fashion, methoxypyridyl B-ring derivatives of the cyclopentapyrazole compounds were also synthesized and evaluated for modulator activity. N-methyl analog 63 exhibited remarkable activity considering that the parent fluorophenyl compound was unable to affect Aβ42 levels (Table 6). The potency of N-ethyl compound 64, on the other hand, was not influenced by the methoxypyridyl B-ring; however, the increased size of the N-alkyl substituent led to a greater than 2-fold increase in activity within the series (63 vs. 64) with an IC50 value of 33 nM. Further increase in N-alkyl substituent size in the presence of the methoxypyridyl B-ring was associated with a loss of activity for N-isopropyl analog 65, whereas N-tert-butyl compound 66 exhibited a nearly 3-fold increase in potency. However, neither of the larger N-alkyl branched analogs was able to surpass the activity of N-ethyl derivative 64, indicating that the added bulk is likely perturbing subtle enzyme-ligand interactions by influencing the compound’s binding conformation. The greater than 3-fold improvement in the activity of N-cyclobutyl analog 67 further suggests that the N-substituent of the D-ring plays a significant role in determining access to ligand conformations that facilitate favorable interactions with γ-secretase, since there is no direct correlation between substituent size or hydrophobic content and activity within the alkyl substituted methoxypyridyl B-ring derivatives. In addition to good activity, N-alkyl analogs 63–67 displayed improved solubility when compared to the equivalent fluorophenyl compounds; however, increasing the size of the N-alkyl D-ring substituent led to a localized accumulation of hydrophobicity and a reduction in aqueous solubility. The novel methoxypyridyl B-ring was also responsible for the nearly 3-fold improvement in potency observed for N-trifluoroethyl analog 69, which exhibited an IC50 value of 36 nM as well as imparting good modulator activity to N-hydroxyethyl analog 70. Moreover, these analogs demonstrate that the addition of heteroatoms within the N-substituent is well tolerated and results in compounds that exhibit ideal aqueous solubility. Conversely, analogs 71 and 72, which contain additional methyl substituents upon the fused system, displayed a modest reduction in activity associated with the introduction of the methoxypyridyl moiety, indicating that B-ring is capable of influencing the distal binding conformation, which results in a modest reduction in affinity for the enzyme target. The larger cycloheptapyrazole 73 displayed a greater than 3-fold increase in modulator activity with an IC50 value of 32 nM, thus reinforcing the presence of a broad yet narrow pocket which accommodates the D-ring. In addition, despite the increased alkyl functionalization and increased clogP values, derivatives 71–73 exhibit good aqueous solubility. Finally, reducing the size of the D-ring scaffold was deleterious to activity as evidenced by N-ethyl pyrazole analog 74, indicating the presence of critical hydrophobic interactions between the carbocycle and γ-secretase, which facilitate modulator activity. Overall the previously identified trend of superior activity as well as substantially improved aqueous solubility associated with the methoxypyridyl B-ring was observed for the cyclopentapyrazole series of compounds and paralleled that of the closely related tetrahydroindazole family of analogs. The identification of the methoxypyridyl B-ring proved to be a fortuitous discovery that addressed the primary shortcomings of the initial fluorophenyl aminothiazole series.
3.6. In vitro ADMET of methoxypyridine-derived analogs
The prepared B-ring derivatives significantly broadened our collection of compounds with good activity for suppressing the formation Aβ42 and ideal aqueous solubility. Utilizing these two metrics, compounds were stratified for additional in vitro ADMET studies in order identify other potential property-related liabilities within this novel family of compounds. A tiered ADMET screening paradigm was implemented to allow early triage of molecules with undesirable characteristics. The metabolic stability of methoxypyridyl B-ring analogs was initially evaluated for compounds with fair activity (IC50 values ≤ 100 nM) and good aqueous solubility (solubility ≥ 5 μM). Analogs that exhibited acceptable stability were then evaluated for Cyp inhibition potential and hERG inhibition potential, followed by the evaluation of cell permeability by MDR1-MDCK assay.
ADMET screening of the tetrahydroindazoles showed that N-alkyl and N-cycloalkyl substitution upon the tetrahydroindazole scaffold for the prepared methoxypyridyl B-ring analogs generally show poor metabolic stability as demonstrated by compounds 36 and 37 (Table 5). Since the closely related N-ethyl tetrahydroindazole 22d exhibited better stability, the modestly soluble N-isopropyl analog 35 was also evaluated, but unfortunately also displayed weak stability, suggesting that increasing the size of the alkyl N-substituent facilitates metabolism of the compound. Poor metabolic stability persisted through the N-heterocyclic substituted compounds as demonstrated by N-tetrahydrofuran 40. Methylpiperidine 42 and tethered morpholino analogs 44 and 45 were screened to further explore this trend and demonstrated that poor stability is associated with the N-heterocyclic containing compounds and is exacerbated by the insertion of a linker motif. Alternatively, terminally functionalized N-fluoroethane 46 demonstrated robust metabolic stability, while closely related N-trifluoroethane 47 shared the metabolic instability observed for nearly all the compounds within tetrahydroindazole series. Modestly improved stability was also observed for N-hydroxyethyl analog 49 and N-2-methylpropan-2-ol analog 51. The incongruence with respect to stability across this series of compounds suggests that the novel methoxypyridyl B-ring is not the source of instability.
Guided by the metabolic stability data, N-fluoroethyl analog 46 and N-2-methylpropan-2-ol analog 51 were subjected to further in vitro ADMET analysis. Both analogs exhibited acceptable cytochrome p450 inhibition potential, displayed good brain penetration properties, and lacked potent binding to the hERG ion channel (Table 5). However, based on the totality of the in vitro data for both compounds, N-fluoroethane 46 exhibited superior ADMET characteristics and was selected for further study. Although two compounds with good drug-like properties were identified, the methoxypyridyl-derived tetrahydroindazole family of compounds by and large suffered from impaired metabolic stability, and future design efforts will focus on incorporation of features which aim to address this parameter.
Methoxypyridyl B-ring analogs of 3-tert-butylpyrazole family of compounds that met the development criterion were also subject to ADMET screening to establish drug-like potential. Consistent with the N-alkyl tetrahydroindazoles, N-ethyl analog 33d exhibited fair stability, whereas N-isopropyl derivative 54 was significantly metabolized, indicating that the size of the N-alkyl functionality persistently influenced metabolic stability across related D-ring scaffolds. Conversely, N-fluoroalkyl functionalized analogs, 56 and 57, displayed improved metabolic stability upon incubation with rodent liver microsomes; however, stability in human liver microsomes, while better, remains limited. Altering the connectivity of the 3-tert-butyl moiety providing methylcyclopropyl analog 59 led to a good overall stability profile. Similarly, modifying the scaffold to include a 3-trifluoromethyl substituent (60) resulted in ideal metabolic stability, indicating that both substituents on the pyrazole are capable of moderating metabolism. Collectively, the pyrazole analogs further demonstrate that the introduction of the methoxypyridyl B-ring does impact the physical properties and activities of the ligands; however, it is not the source affecting compound stability. Additional analysis of pyrazoles 59 and 60 established that both compounds possess good Cyp inhibition profiles as well as ideal hERG inhibition potential. These analogs also display moderate permeability and limited efflux, which is consistent with compounds that display high brain penetration. Although most of the 3-tert-butylpyrazoles are readily metabolized, 3-methylcyclopropyl analog 59 and 3-trifluoromethyl analog 60 exhibit acceptable overall ADME profiles, including good stability, which supports the continued investigation of both compounds.
Analysis of the active cyclopentapyrazole compounds reinforced many of the trends observed for the 3-tert-butylpyrazoles and tetrahydroindazoles. Similar to both prior pyrazole-derived D-ring series, good metabolic stability was associated with cyclopentapyrazoles characterized by smaller N-alkyl substituents as demonstrated by analogs 63-66 (Table 6). Moreover, compound 65 shows that N-cycloalkyl substitution serves to exacerbate this trend. The cyclopentapyrazole analogs also illustrate that reducing the size of the fused alkyl functionality of the D-ring is not sufficient to improve metabolic stability. Paralleling the related D-ring analogs, increasing the polarity with the N-ethyl substituent provided compounds with improved metabolic stability. N-trifluoroethane 69 and N-hydroxyethane 70 displayed good microsomal stability in two of the three species tested, whereas N-fluoroethane 68 displayed increased stability across all species evaluated. Conversely, analogs 71 and 72 indicate that reducing the polarity of the D-ring through the introduction of methyl substituents upon the cyclopentane ring is deleterious to metabolic stability. Likewise, poor stability was also associated with increasing the size of the fused ring system as illustrated by cycloheptylpyrazole 73.
The shorter N-alkyl substituted compounds, 63 and 64, as well as polar functionalized cyclopentapyrazoles that showed comparative stability (68, 69, and 70) were subjected to additional screening. The close structural relationship of N-methyl 63 and N-ethyl 64 led to similar overall ADMET profiles; both compounds exhibited weak cytochrome p450 inhibitory potential, good brain penetration potential, and modest hERG inhibition. Alternatively, the profiles of N-fluoroethyl 68, N-trifluoroethyl 69 and N-hydroxyethyl 70 display more variability. Cytochrome p450 inhibition profiles of N-fluoroethane 68 and trifluoroethane 69 were ideal and acceptable, respectively. However, N-hydroxyethane 70 was a potent 3A4 inhibitor, and screening for hERG inhibition potential revealed a potential liability for analog 69. Although ADMET properties precluded the development of many of the cyclopentapyrazoles, N-alkyl substituted compounds 63, 64, and N-fluoroethane 68 generally demonstrated acceptable profiles.
3.7. Pharmacokinetics of GSM analogs
Pharmacokinetic study of selected methoxypyridine-derived analogs was undertaken to assess the effect of the related structural modifications upon in vivo parameters. Following intravenous administration to male CD-1 mice, compound clearance was generally high with the exception of 3-methylcyclopropyl analog 59 and cyclopentapyrazole 63 (Table 7). The clearance observed for compounds 59 and 63 were approximately 13% and 24% of mouse hepatic blood flow (~90 mL/min/kg), whereas the clearance ranged from 30% to 64% of mouse hepatic blood flow for the other dosed analogs. The elevated clearance levels of pyrazole 60 and cyclopentapyrazole 64 were consistent with the measured in vitro metabolic activity; conversely, despite being very metabolically stable, tetrahydroindazole 46 exhibited high clearance (Table 5 and 6). Significant first-pass metabolism is expected for compounds 46, 60, and 64 based on the high rate of clearance. In contrast, less variability was observed with respect to volume of distribution (Vss) of the dosed analogs. A relatively large Vss was measured for all of the compounds; volumes 2 to 5-fold higher than body water in mice (0.65 L/kg), which indicates that the binding of these compounds is biased toward tissue proteins rather than plasma proteins. In addition, compound half-life (t1/2) was generally short and ranged from 0.65 to 3.84 hrs in mice following intravenous administration. The truncated t1/2 is primarily due to the high Cl and, to a lesser extent, the difference in Vss. Brain to plasma ratios for all of the compounds were also measured and, although variable, showed brain penetration that is sufficient for accessing the therapeutic target.
Table 7.
Pharmacokinetic parameters of test article following single iv and po administration to male CD-1 mice.
| Mouse Pharmacokinetics | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Route | Dose (mg/kg) | Cmax (ng/mL) | Tmax (hrs) | t1/2 (hrs) | AUClast (ng·h/mL) | AUCinf (ng·h/mL) | Cl (mL/hr/kg) | Vss (mL/kg) | F (%) | Brain/Plasma Ratio (1 hr post dose) |
| 46 | iv | 1 | 309 | 0.083 | 0.65 | 283 | 288 | 3,468 | 2,826 | 0.81 | |
| po | 5 | 167 | 1.0 | 2.6 | 639 | 669 | 46.4 | 0.68 | |||
| 59 | iv | 1 | 384 | 0.083 | 3.84 | 1,247 | 1,378 | 725 | 3,609 | 0.62 | |
| po | 5 | 592 | 2.0 | cnda | 3,976 | cnda | 63.8 | 0.57 | |||
| 60 | iv | 1 | 359 | 0.083 | 1.6 | 605 | 623 | 1,606 | 3,640 | 0.73 | |
| po | 5 | 470 | 1.0 | cnda | 1,727 | 2,369 | 76.0 | 0.62 | |||
| 63 | iv | 1 | 478 | 0.083 | 1.9 | 723 | 1,308 | 3,180 | 0.21 | ||
| po | 5 | 580 | 1.0 | 1.6 | 2,700 | 74.6 | 0.22 | ||||
| 64 | iv | 1 | 549 | 0.083 | 0.7 | 376 | 383 | 2,608 | 2,234 | 0.37 | |
| po | 5 | 377 | 0.25 | 5.3 | 1,205 | 1,257 | 66.0 | 0.40 | |||
Cannot be determined from data
Following oral administration, all of the selected derivatives showed high oral bioavailability in mice. Tetrahydroindazole 46 exhibited the lowest bioavailability (46.4%) within the series; however, accounting for the approximately 60% of compound that is expected to be metabolized during the first-pass through the liver, the oral absorption is nearly complete in mice. Similarly, the fraction of dose absorbed is greater than 80% for analogs 60, 63, and 64 when accounting for first-pass effects. The period to reach maximum drug concentration (Tmax) following oral administration was relatively short, ranging from 0.25 to 1.0 hr, with the exception of 3-methylcyclopropyl analog 59. The time period to achieve the Tmax is in the range of rodent gastric emptying, which suggests that the rate of intestinal absorption is quite rapid for these compounds. In addition, the longer period to achieving Tmax for compound 59 is likely the result of a slower rate of dissolution resulting from its limited solubility (Table 6).
3.8. Pharmacodynamics of compounds 46 and 64
Although the more active GSMs considered for continued development display similar ADMET parameters and pharmacokinetic profiles, tetrahydroindazole 46 and cyclopentapyrazole 64 were selected for pharmacodynamic study using transgenic J20 mice that over-express human APP.43 Following a 3-day treatment with either vehicle or test article, a dose-dependent reduction in plasma Aβ42 and Aβ40 was observed for compound 64 (Figure 5). Although the dose escalation of tetrahydroindazole 46 led to a greater suppression of Aβ42 and Aβ40 in plasma, the effect seems to plateau between 50 and 100 mg/kg. This effect was also reflected in the exposure data, which shows reduced plasma and brain drug levels at the top dose, suggesting that higher dosing may lead to prolonged absorption while the high clearance identified in the pharmacokinetic study remains unchanged (Table 8). As a result of the shift in the absorption pattern at the high dose, the measured concentration of compound 46 is lower, but likely sustained over a longer period of time resulting the observed pharmacodynamic effect. Consequentially, cyclopentapyrazole 64 exhibited superior activity, suppressing Aβ42 by 76% at a dose of 100 mg/kg. In addition, both compounds also reduced Aβ38 levels. Based on the mode of action, the inhibition of Aβ38 was not expected and was inconsistent with the in vitro data showing potentiation of Aβ38 following GSM treatment (Figure 6).
Figure 5.

Levels of Aβ38, Aβ40, and Aβ42 in plasma following daily oral administration of either vehicle, compound 46 (A), or compound 64 (B) to female 5–6 month-old J20 mice (n = 5/dose) were measured following a 3-day treatment course. Percent reduction in amyloid is indicated for the study. Aβ38, Aβ40, and Aβ42 peptide levels were determined using Meso Scale Sector 6000 Multiplex assays. Statistical analysis was performed using Graphpad Prism software and results are expressed as mean ± SEM, and Anova was used to detect a significant effect. *: p < 0.05. **: p < 0.005. ***: p < 0.0005.
Table 8.
Drug levels in plasma and brain of female J20 mice (n = 5/dose) following a 3-day treatment course of vehicle, tetrahydroindazole 46, or cyclopentapyrazole 64.
| Mouse Brain and Plasma Drug Levelsa | ||||
|---|---|---|---|---|
| Cmpd | Dose (mg/kg) | Plasma Drug Level (ng/mL) | Brain Drug Level (ng/g) | Brain/Plasma Ratio |
| Vehicle | 0 | 0 | 0 | NA |
| 46 | 25 | 72 | 39 | 0.53 |
| 50 | 639 | 282 | 0.42 | |
| 100 | 183 | 105 | 0.57 | |
| 64 | 25 | 86 | 31 | 0.37 |
| 50 | 223 | 83 | 0.38 | |
| 100 | 726 | 337 | 0.46 | |
Tissue samples were collected 4 hrs post final dose administration on Day 3. NA: not applicable.
All samples were analyzed by LC-MS/MS. Compound 46 had a plasma lower limit of quantitation (LLOQ) of 1 ng/mL and brain LLOQ of 5 ng/g. Compound 64 had a plasma lower limit of quantitation (LLOQ) of 1 ng/mL and brain LLOQ of 5 ng/g.
Figure 6.

Concentration response curves of compound 46 (A) and compound 64 (B) using orthogonal medium throughput SHSY5Y-APP cell-based screening assays. Aβ38, Aβ40, Aβ42, and total Aβ peptide levels were determined using Meso Scale Sector 6000 Multiplex assay.
Based on the superior pharmacodynamic effect of cyclopentapyrazole 64 in J20 transgenic mice, an additional efficacy study was conducted using Tg2576 transgenic mice that overexpress human APP containing the Swedish (K,M to N,L) double mutation to further ascertain the impact on the various Aβ peptides.44 Female Tg2576 mice were orally dosed daily with either vehicle or 50 mg/kg of compound 64 for 14 days and the drug levels in plasma and brain were determined in addition to Aβ levels. Consistent with the mode of action, treatment with cyclopentapyrazole 64 decreased the levels of Aβ42 and Aβ40 while potentiating the levels of Aβ38 in plasma (Figure 7). In addition, brain Aβ42 levels were reduced by 23%, demonstrating that the compound is capable of crossing the blood brain barrier and reaching the desired site of pharmacodynamic action in brain. Furthermore, consistent with the in vitro permeability data, the brain to plasma drug levels demonstrated adequate penetration to account for observed effect in brain (Table 9).
Figure 7.

Levels of Aβ38, Aβ40, and Aβ42 in plasma and brain following daily oral administration of either vehicle or compound 64 to female 5–6 month-old Tg2576 mice (n = 12/dose) were measured following a 14-day treatment course. Percent reduction is indicated for the study. Aβ38, Aβ40, and Aβ42 peptide levels were determined using Meso Scale Sector 6000 Multiplex assays. Statistical analysis was performed using Graphpad Prism software and results are expressed as mean ± SEM, and ANOVA was used to detect a significant effect. *: p < 0.05. **: p < 0.005. ***: p < 0.0005.
Table 9.
Drug levels in plasma and brain of female Tg2576 mice (n = 12/dose) following a 14-day treatment course of cyclopentapyrazole 64.
| Mouse Brain and Plasma Drug Levelsa | ||||
|---|---|---|---|---|
| Cmpd | Dose (mg/kg) | Plasma Drug Level (ng/mL) | Brain Drug Level (ng/g) | Brain/Plasma Ratio |
| Vehicle | 0 | 0 | 0 | NA |
| 64 | 50 | 1,131 | 425 | 0.374 |
Tissues samples were collected 4 hrs post final dose administration on Day 14. NA: not applicable.
All samples were analyzed by LC-MS/MS. Compound 64 had a plasma lower limit of quantitation (LLOQ) of 1 ng/mL and brain LLOQ of 5 ng/g.
4.0. Summary and conclusion
A focused effort to improve the activity and ADMET properties of aminothiazole-derived GSMs through the modification of the B-ring led to the identification of methoxypyridine as potential replacement for the fluorophenyl moiety. Integration of this novel structural motif within the B-ring of our scaffolds generally led to both improved activities for reducing Aβ42 and increased solubility across three related aminothiazole series. Subsequent ADMET screening of the highly active GSM analogs led to the selection of subset of structurally diverse compounds for pharmacodynamic evaluation. Based on the aggregate of data, tetrahydroindazole 46 and cyclopentapyrazole 64 were tested in transgenic mice to determine in vivo efficacy for attenuating Aβ42. While compound 64 was capable of reducing Aβ42 in the plasma and brain of Tg2576 mice, the dose required for therapeutic efficacy is likely too high to allow for adequate safety margins with respect to hERG inhibition. Although the discovery of the methoxypyridine led to improved GSM analogs, further optimization of the scaffold with respect to activity is likely necessary.
5.0. Experimental
5.1. Cell-based ELISA assay for measuring extracellular Aβ42 levels
Human SHSY5Y neuroblastoma cells stably overexpressing wild-type human APP751 (SHSY5Y-APP) have been described previously. Briefly, SHSY5Y-APP cells were plated on a 96-well plate at 75K per well. Following overnight incubation at 37 °C, wells were treated with various concentrations of compound in order to generate dose response curves. For treatment, a 25 mM stock solution of compound in DMSO was prepared. The DMSO stock solution was then diluted to 10 μM using cell media. The 10-point dose response curve was generated following treatments that ranged from 0.3 pM – 10 μM for each compound with a 1:3.16 dilution across the plate with a total volume of 125 μL pre well. After being treated for 24 hours, the SHSY5Y-APP cells treated with either vehicle or compound were then inspected for obvious signs of toxicity. The quantity of Aβ42 was then quantified by ELISA.
In order to quantify Aβ42, coating antibody, Aβ35–42 (lot 454.23), was diluted 1:200 in TBS (Tris Buffered Saline, pH 7.4, Sigma T6664). Then, 100 μL of antibody Aβ35–42 was pipetted into each well of the micro plate. The plate was sealed and incubated at 4 °C for 21 hours. The coating antibody was then aspirated, rinsed with 200 μL of TBS, and aspirated again. Then, 200 μL of 1% BSA (bovine serum albumin) in TBS blocking agent was applied to each well followed by a one-hour incubation at room temperature. The wells were then aspirated, and 50 μL of sample from treated cells was added, followed by a two-hour incubation period. Each well of the plate was then washed three times with 200 μL of PBS/0.1%Tween-20. Then, 50 μL of Aβ1–12 mAb 436 (alkaline phosphatase conjugated antibody) diluted to 1:8000 in 1% BSA/TBS/0.1% Tween-20 was added to each well of the plate. The plate was then incubated for another two hours at room temperature and was washed six times with 200 μL of PBS/0.1%Tween-20. Finally, 50 μL of CDP-Star (Sapphire) luminescence substrate was added to each well of the plate and incubated for 15 minutes at room temperature in the dark. Then, the amount of Aβ42 was measured by reading the plate using a GloRunner Pro microplate luminometer by Turner Biosystems. For every plate quantified, a standard Aβ42 curve was generated. The Aβ42 IC50 values were determined from the 10-point concentration response curves (in duplicate at each concentration) by a four-parameter fit nonlinear regression analysis using Graphpad Prism software. The assays were required to demonstrate a Z′ score of ≥ 0.5 and to have a test-retest reliability with an r2 of ≥ 0.75 for the GSM compounds, which represent a broad range of potencies. In addition, upon its discovery, compound 22d was utilized as an internal standard for all subsequently prepared analogs on each assay plate and subjected to the same Z′ score and r2 metrics to ensure the reproducibility and reliability of the data.
5.2. Measurement of extracellular Aβ42, Aβ40, and Aβ38 levels
Samples were tested using the Meso Scale Discovery Aβ triplex immunoassay with 4G8 detection (Catalog # K15141E-1, MSD Rockville, MD) to quantitate levels of Aβ38, Aβ40, and Aβ42. The kit was run according to the manufacturer’s instructions. Briefly, 25 μL of SULFO-TAG conjugated 4G8 detection antibody and 25 μL of sample were added to a blocked MSD multiplex plate pre-coated with capture antibodies that were specific for Aβ38, Aβ40 and Aβ42 and incubated for 2 hr with shaking at room temperature. The plate was read on an MSD SECTOR® Imager 6000 after the addition of read buffer. Concentrations of Aβ38, Aβ40, and Aβ42 in samples were back-fitted from the respective standard curves using MSD Workbench software.
5.3. Pharmacokinetic evaluation
Male CD-1 mice (6–8 week-old) were administered a single dose of the test article via tail vein (iv) or by oral gavage (po) dose route. Formulations were prepared using 3% N-methylpyrrolidione (NMP)/45% polyethylene glycol 300 (PEG300)/12% ethanol/40% sterile water for iv administration and 5% NMP/95% PEG300 for oral administration. Plasma was collected from mice at 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 hr for iv-dosed animals and at 0.25, 0.5, 1, 2, 4, 8, and 24 hr for orally dosed animals (n=3/timepoint). Brains were collected at 1, 2, 8, and 24 hr regardless of the route of administration (n=3/timepoint). Blood was collected from the retro-orbital sinus of mice under isoflurane anesthesia into tubes containing K3EDTA, processed to plasma, and then stored frozen at −70 ± 10°C for subsequent drug levels analysis.
5.4. Drug level bioanalysis
Plasma and brain tissue samples were analyzed for drug levels using a discovery grade HPLC/MS/MS method. Mean plasma drug concentration versus time data were subjected to pharmacokinetic analysis using WinNonlin® (Version 4.1; Pharsight Corp.; Cary, NC). Data were subjected to non-compartmental analysis using WinNonlin® Model 200 (for extravascular administration) or Model 201 (for IV bolus administration); a uniform weighting factor was applied to each data set. Tmax and Cmax values were determined directly from the data. AUClast values were calculated using the log/linear trapezoidal (IV dose) or linear up/log down trapezoidal (PO dose). Half-life values were calculated from the first order rate constant associated with the observed terminal portion of the plasma drug concentration versus time curve, as estimated by regression analysis, using a minimum of three non-zero time points after Tmax. The regression with the largest adjusted R2 (square of the correlation coefficient) was selected as the best fit. Estimated half-life values with an adjusted R2 of less than 0.800 were considered to be unreliable and were excluded from inclusion in the tabulation of kinetic parameters. Bioavailability (F) was calculated as follows:
5.5. Pharmacodynamic study using J20 mice
Female J20 mice (5–6 month-old) were administered vehicle (5% NMP/95% PEG300) or test article at doses of 25, 50, and 100 mg/kg via oral gavage for three consecutive days (n=5/dose). Animals were sacrificed 4 hours post final dose on day 3. Plasma and brain were collected for the analysis of Aβ peptides and drug levels. Plasma and brain Aβ levels were quantified using Meso Scale multiplex kits and MSD SECTOR® Imager 6000. Statistical analysis was performed using Graphpad Prism software, and results were expressed as mean ± SEM. ANOVA was used to detect a significant effect.
5.6. Pharmacodynamic study using Tg2576 mice
Female Tg2576 mice (5–6 month-old) were administered vehicle (5% NMP/95% PEG300) or test article at a dose of 50 mg/kg via oral gavage for 14 consecutive days. Animals were sacrificed 4 hours post final dose on day 14. Plasma and brain were collected for the analysis of Aβ peptides and drug levels. Plasma and brain Aβ levels were quantified using Meso Scale multiplex kits and MSD SECTOR® Imager 6000. Statistical analysis was performed using Graphpad Prism software, and results were expressed as mean ± SEM. ANOVA was used to detect a significant effect.
5.7. Synthetic procedures
All reactions were performed under a dry atmosphere of nitrogen unless otherwise specified. Indicated reaction temperatures refer to the reaction bath, while room temperature (rt) is noted as 25 °C. Commercial grade reagents and anhydrous solvents were used as received from vendors and no attempts were made to purify or dry these components further. Removal of solvents under reduced pressure was accomplished with a Buchi rotary evaporator at approximately 28 mm Hg pressure using a Teflon-linked KNF vacuum pump. Thin layer chromatography was performed using 1” x 3” AnalTech No. 02521 silica gel plates with fluorescent indicator. Visualization of TLC plates was made by observation with either short wave UV light (254 nm lamp), 10% phosphomolybdic acid in ethanol or in iodine vapors. Flash column chromatography was carried out using a Teledyne Isco CombiFlash Companion Unit with RediSep Rf silica gel columns. Proton NMR spectra were obtained either on 300 MHz Bruker Nuclear Magnetic Resonance Spectrometer or 500 MHz Bruker Nuclear Magnetic Resonance Spectrometer and chemical shifts (δ) are reported in parts per million (ppm), and coupling constant (J) values are given in Hz, with the following spectral pattern designations: s, singlet; d, doublet; t, triplet, q, quartet; dd, doublet of doublets; m, multiplet; br, broad; sym, symmetrical. Tetramethylsilane (TMS) was used as an internal reference. Melting points were uncorrected and were obtained using a MEL-TEMP Electrothermal melting point apparatus. Mass spectroscopic analyses were performed either using positive mode electron spray ionization (ESI) on a Varian ProStar LC-MS with a 1200L quadrapole mass spectrometer or using positive mode atmospheric pressure chemical ionization (APCI) on a Shimadzu LC-MS system. High performance liquid chromatography (HPLC) purity analysis was conducted using a Varian Pro Star HPLC system with a binary solvent system A and B using a gradient elution [A, H2O with either 0.05% or 0.1% trifluoroacetic acid (TFA); B, CH3CN with either 0.05% or 0.1% TFA] and flow rate = 1 mL/min, with UV detection at 254 nm. All final compounds were purified to ≥95% purity and these purity levels were measured by a Varian Pro Star HPLC system. The following Varian Pro Star HPLC methods were used to establish compound purity:
Phenomenex Luna C18(2) column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient: 10–95% B (0.0–10 min; hold for 6 min); UV detection at 254 nm.
SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient: 10–100% B (0.0–20 min; hold for 5 min); UV detection at 254 nm.
SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.1% TFA and B = CH3CN with 0.1% TFA; gradient: 0–100% B (0.0–15 min; hold for 5 min)); UV detection at 254 nm.
SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.05% TFA and B = CH3CN with 0.05% TFA; gradient: 5–95% B (0.0–10 min; hold for 8 min)); UV detection at 254 nm.
SunFire C18 column (4.6 × 250 mm); mobile phase, A = H2O with 0.05% TFA and B = CH3CN with 0.05% TFA; gradient: 10–100% B (0.0–20 min; hold for 5 min)); UV detection at 254 nm.
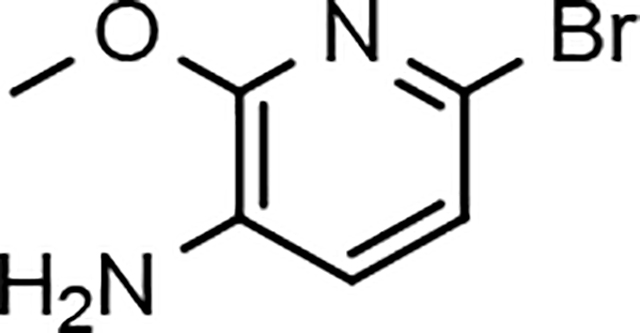
6-bromo-2-methoxypyridin-3-amine (12a)
Sodium methoxide (powder, 600 g, 11.1 mol) was added to a solution of commercially available 2,6-dibromopyridin-3-amine (11a, 400 g, 1.59 mol) in 1,4-dioxane (3500 mL) at room temperature under nitrogen. The resulting mixture was heated to 100 °C for 18 h. The reaction mixture was cooled to 0 °C and quenched by the careful addition of aqueous ammonium chloride (600 g in 1500 mL of water). The mixture was stirred for 15 minutes and concentrated in vacuo to remove as much 1,4-dioxane as possible. The residue was diluted with ethyl acetate (3000 mL), filtered through silica gel (500 g), and eluted with ethyl acetate (3000 mL). The aqueous layer was separated. The combined organic layer was washed with brine (2 × 500 mL), dried over sodium sulfate, filtered, and the solvents were removed under reduced pressure to afford 6-bromo-2-methoxypyridin-3-amine (12a) as a purple solid (314 g, 98%) that was suitable for use without further purification: Multimode MS (M+H) 203; 1H NMR (500 MHz, CDCl3) δ 6.86 (d, J = 7.5 Hz, 1H), 6.76 (d, J = 7.5 Hz, 1H), 3.98 (s, 3H), 3.74 (br s, 2H).
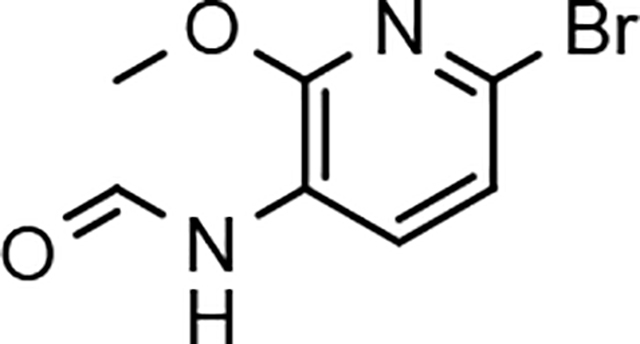
N-(6-bromo-2-methoxypyridin-3-yl)formamide (13a)
Acetic anhydride (522 g, 5.11 mol) was added slowly to formic acid (587 g, 12.8 mol) at 0 °C under nitrogen. The mixture was stirred at 0 °C for 30 min, warmed at room temperature, and stirred for 30 minutes. The mixture was recooled to 0 °C and a solution of 6-bromo-2 methoxypyridin-3-amine (12a, 346 g, 1.70 mol) in THF (2200 mL) was slowly added. Following addition, the ice bath was removed, and the reaction was stirred at room temperature for 2 h. The reaction mixture was poured into 1:1 ice/water (8 L), stirred for 30 minutes, and filtered. The solids were further dried under high vacuum for 18 h to afford N-(6-bromo-2-methoxypyridin-3-yl)formamide (13a) as a red-brown solid (408 g, >99%), which was suitable for use without further purification: Multimode MS (M+H) 231; 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 8.0 Hz, 1H), 8.48 (s, 1H), 7.60 (br s, 1H), 7.07 (d, J = 8.0 Hz, 1H), 4.03 (s, 3H).
N-(6-bromo-2-methoxypyridin-3-yl)-N-(2-oxopropyl)formamide (14a)
Potassium carbonate (powder, 325 mesh, Aldrich catalog #: 347825, 855 g, 6.20 mol) and potassium iodide (29.4 g, 0.177 mmol) were added to a solution of N-(6-bromo-2-methoxypyridin3-yl)formamide (13a, 408 g, 1.77 mol) in N,N-dimethylformamide (4000 mL) at room temperature under nitrogen. To the resulting mixture was slowly added 2-chloroacetone (408 g, 4.43 mol). The mixture was stirred at room temperature overnight. The resulting suspension was carefully poured into 1:1 ice/water (12 L) and stirred vigorously for 25 minutes. The resulting solid was collected by filtration and washed with water. The solid was dried for two days under high vacuum (at 60 °C) to afford N-(6-bromo-2-methoxypyridin-3-yl)-N-(2-oxopropyl)formamide (14a) as a brown solid (474 g, 93%) that was suitable for use without further purification: Multimode MS (M+H) 287; 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.0 Hz, 1H), 4.47 (s, 2H), 3.99 (s, 3H), 2.17 (s, 3H).
6-bromo-2-methoxy-3-(4-methyl-1H-imidazol-1-yl)pyridine (15a)
Ammonium acetate (636 g, 8.26 mol) was added to a stirred solution of N-(6-bromo-2-methoxypyridin-3-yl)-N-(2-oxopropyl)formamide (14a, 474 g, 1.65 mol) in acetic acid (4000 mL) at room temperature. After addition, the mixture was heated to 120 °C for 10 h. After cooling to room temperature, the mixture was concentrated in vacuo (at 60 °C) to remove the majority of the acetic acid. The resulting residue was diluted with ethyl acetate (4000 mL) and water (4000 mL). With fast stirring at room temperature, solid potassium carbonate was slowly added to the reaction mixture (neutralized to pH 8). The layers were separated, and the aqueous phase was extracted with ethyl acetate (2 × 1000 mL). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash column chromatography on silica gel, eluting with heptane/ethyl acetate (gradient of 4:1 to 3:7), to afford 6-bromo-2-methoxy-3-(4-methyl-1H-imidazol-1-yl)pyridine (15a, 380 g, 85%) as a light brown solid: Multimode MS (M+H) 268; 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 1.5 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 6.92 (m, 1H), 4.03 (s, 3H), 2.29 (s, 3H).
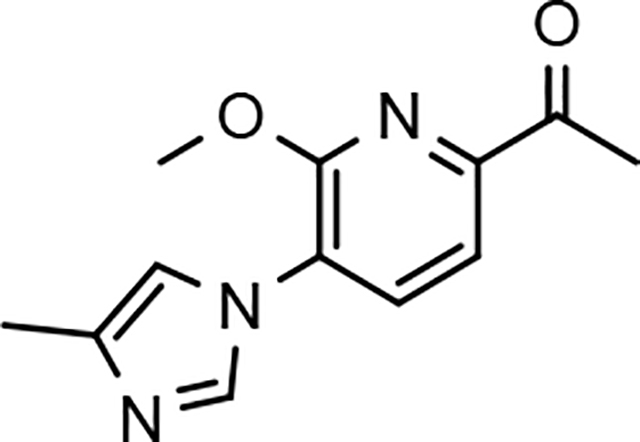
1-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)ethan-1-one (16a)
To a solution of 6-bromo-2-methoxy-3-(4-methyl-1H-imidazol-1-yl)pyridine (15a, 100 g, 0.373 mol), 1-(vinyloxy)butane (74.6 g, 0.746 mmol), and triethylamine (75.3 g, 0.746 mol) in ethylene glycol (1000 mL) at room temperature under nitrogen was added dichloro[1,1’-bis(diphenylphosphino) ferrocene] palladium (II) dichloromethane adduct (8.18 g, 11.2 mmol). The mixture was heated to 100 °C for 4 h (reaction progress was monitored by LCMS; heating the reaction mixture for too long will result in a lower yield) and then cooled to room temperature. The mixture was diluted with 2:1 ethyl acetate/water (3 L) and the resulting layers were separated. The organic layer was washed with brine (2 × 500 mL), dried with sodium sulfate, filtered, and concentrated. The resulting reaction residue was purified by flash column chromatography on silica gel, eluting with heptane/ethyl acetate (gradient of 4:1 to 1:1) to afford 6-(1-butoxyvinyl)-2-methoxy-3-(4-methyl-1H-imidazol-1-yl)pyridine as a yellow solid (78.4 g, 73%) that was suitable for use without further purification. Then aqueous hydrochloric acid (2 N, 550 mL) was added to a solution of 6-(1-butoxyvinyl)-2-methoxy-3-(4-methyl-1H-imidazol-1-yl)pyridine (A6, 78.4 g, 0.273 mol) in acetone (2200 mL) at room temperature. After stirring the reaction mixture for 30 minutes, the organic solvent was removed under reduced pressure, and the mixture was diluted with water (1000 mL) and extracted with ethyl acetate (2000 mL). The organic layer was washed with water (~500 mL), and the ethyl acetate layer was discarded. With fast stirring, potassium carbonate was slowly added (neutralized to pH 8) to the combined aqueous layers. The resulting mixture was extracted with ethyl acetate (2 × 2000 mL). The combined organic layer was washed with brine (2 × 500 mL) and dried over anhydrous sodium sulfate, filtered, and concentrated to afford 1-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)ethanone (16a) as a light yellow solid (60.6 g, 96%) that was suitable for use without further purification: LCMS (M+H) 232; 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 1.5 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.04 (m, 1H), 4.18 (s, 3H), 2.71 (s, 3H), 2.31 (d, J = 0.9 Hz, 3H).
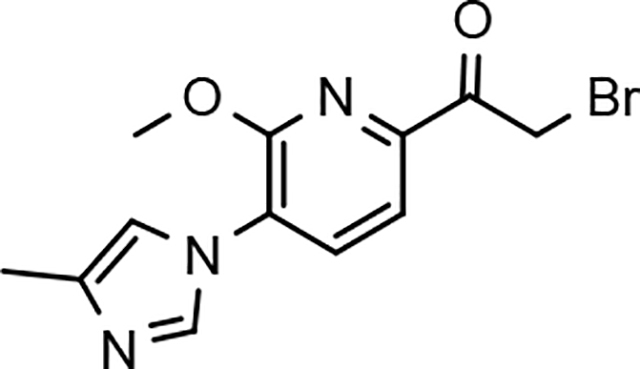
2-bromo-1-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)ethan-1-one (17a)
Hydrogen bromide (2 mL, 33% solution in acetic acid) was added to a solution of 1-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)ethanone (16a, 2.07 g, 8.95 mmol) in chloroform (90 mL) and ethyl acetate (90 mL). To this mixture was added bromine [412 μL, 8.05 mmol, in chloroform (10 mL) and ethyl acetate (10 mL)] drop-wise over ~2 h at room temperature. After complete addition of the bromine, the reaction was stirred a further 2 h at room temperature. The solvents were removed under reduced pressure and the solids taken back up in chloroform (30 mL) and ethyl acetate (30 mL). The solvents were removed again under reduced pressure. Ethyl acetate (30 mL) was then added and the reaction again concentrated under reduced pressure and dried under high vacuum for 18 h to afford 2-bromo-1-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)ethanone hydrobromide (17a) as a tan solid (3.84 g, 92%) that was suitable for use without further purification: APCI MS (M+H) 310.
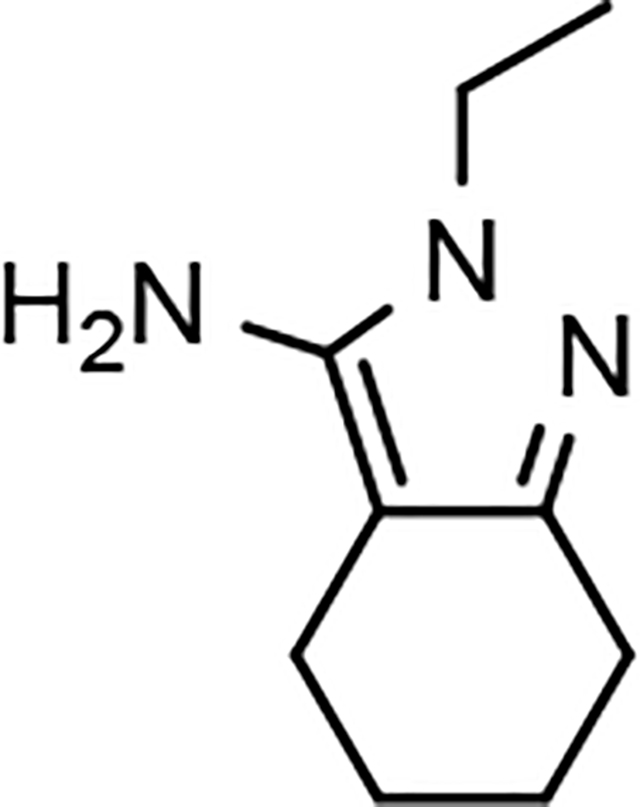
2-Ethyl-4,5,6,7-tetrahydro-2H-indazol-3-amine (19)
A solution of 2-oxocyclohexanecarbonitrile (18, 2.57 g, 20.9 mmol) and ethyl hydrazine oxalate (4.70 g, 31.3 mmol) in ethanol (40 mL) was stirred at reflux for 18 h, cooled to room temperature and the solvent removed under reduced pressure. The residue was diluted with methylene chloride (150 mL) and saturated aqueous sodium bicarbonate (100 mL) was added. The organic layer was separated and the aqueous layer was further extracted with methylene chloride (2 × 100 mL). The combined organic layers were washed with brine (50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The resulting residue was further purified by flash column chromatography on silica gel, eluting with methanol/methylene chloride (gradient of 2:98 to 4:96) to afford 2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-amine (19) as a while solid (2.73 g, 79%): 1H NMR (300 MHz, CDCl3) δ 3.95 (q, J = 7.2 Hz, 2H), 3.24 (br s, 2H), 2.61–2.55 (m, 2H), 2.34–2.28 (m, 2H), 1.83–1.68 (sym m, 4H), 1.38 (t, J = 7.2 Hz, 3H).
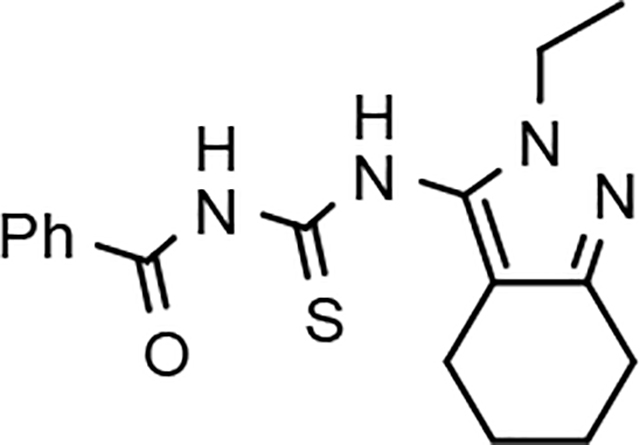
N-((2-Ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)carbamothioyl)benzamide (20)
A solution of 2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-amine (19, 800 mg, 4.84 mmol) and benzoyl isothiocyanate (869 mg, 5.32 mmol) in acetone (35 mL) was stirred at 60 °C for 18 h, cooled to room temperature and the solvent removed under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel, eluting with ethyl acetate/hexanes (gradient of 1:9 to 1:1 over 40 minutes), to afford N-((2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)carbamothioyl)benzamide (20) as an off-white solid (1.41 g, 89%): ESI MS (M+H) 329; 1H NMR (300 MHz, CDCl3) δ 11.81 (s, 1H), 9.28 (s, 1H), 7.93–7.88 (m, 2H), 7.72–7.65 (m, 1H), 7.60–7.53 (m, 2H), 4.06 (q, J = 7.2 Hz, 2H), 2.71–2.65 (m, 2H), 2.49–2.41 (m, 2H), 1.88–1.70 (m, 4H), 1.45 (t, J = 7.2 Hz, 3H).
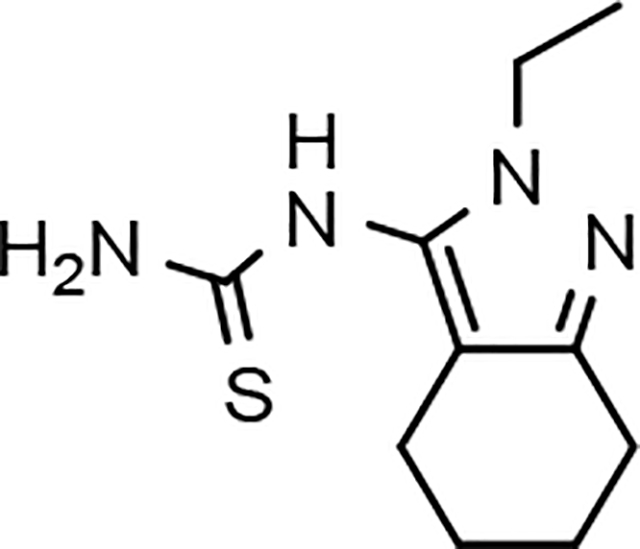
1-(2-Ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiourea (21)
A mixture of N-((2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)carbamothioyl)benzamide (20, 1.41 g, 4.29 mmol) and potassium carbonate (1.13 g, 8.16 mmol) in methanol (43 mL) was stirred at room temperature for 18 h, and concentrated directly onto silica gel, then purified by flash column chromatography on silica gel, eluting with methanol/methylene chloride (gradient of 0:100 to 1:19 over 40 minutes), to afford 1-(2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiourea (21) as an off-white solid (904 mg, 94%): ESI MS (M+H) 225; 1H NMR (300 MHz, DMSO-d6) δ 9.23 (br s, 1H), 8.04 (br s, 1H), 7.06 (br s, 1H), 3.81 (q, J = 7.2 Hz, 2H), 2.54–2.45 (m, 2H), 2.29–2.22 (m, 2H), 1.77–1.56 (m, 4H), 1.24 (t, J = 7.2 Hz, 3H).
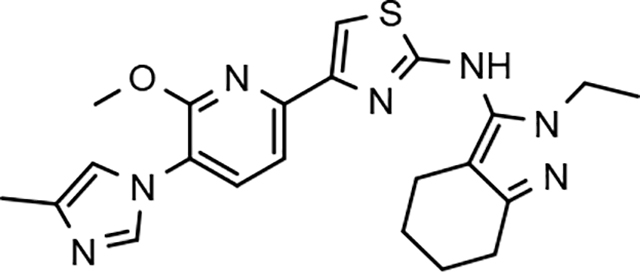
N-(2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (22d)
White solid; mp 198–201 °C; ESI MS (M+H) 436; 1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 7.90 (d, J = 1.0 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 7.51 (s, 1H), 7.50 (d, J = 7.5 Hz, 1H), 7.24 (s, 1H), 4.00 (s, 3H), 3.94 (q, J = 7.0 Hz, 2H), 2.55–2.50 (m, 2H), 2.38–2.32 (m, 2H), 2.16 (s, 3H), 1.77–1.60 (m, 4H), 1.27 (t, J = 7.0 Hz, 3H); HPLC >99% (AUC), (Method C), tR = 9.4 min.
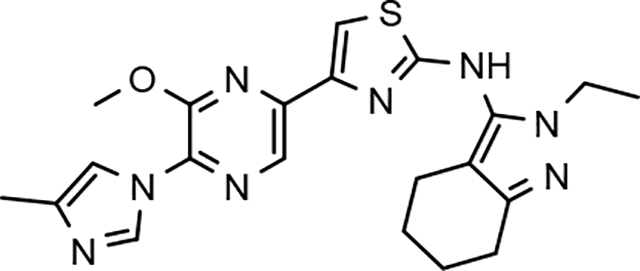
N-(2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyrazin-2-yl)thiazol-2-amine (22e)
Light yellow solid; mp 236–240 °C; APCI MS (M+H) 437; 1H NMR 300 MHz, (DMSO-d6) δ 9.85 (s, 1H), 8.43 (s, 1H), 8.37 (d, J = 1.2 Hz, 1H), 7.63 (s, 1H), 7.56 (s, 1H), 4.12 (s, 3H), 3.94 (q, J = 7.2 Hz, 2H), 2.54 (t, J = 5.7 Hz, 2H), 2.33 (t, J = 5.7 Hz, 2H), 2.18 (s, 3H), 1.80–1.59 (m, 4H), 1.26 (t, J = 7.2 Hz, 3H); HPLC 97.8% (AUC), (Method D), tR = 8.4 min.
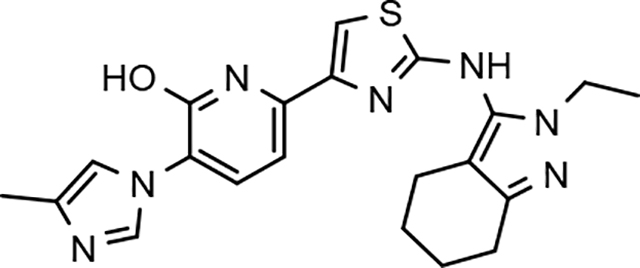
6-(2-((2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)amino)thiazol-4-yl)-3-(4-methyl-1H-imidazol-1-yl)pyridin-2-ol (23)
Light brown solid; mp >300 °C dec; ESI MS (M+H) 422; 1H NMR (500 MHz, DMSO-d6) δ 12.1 (s, 1H), 9.78 (s, 1H), 8.14 (s, 1H), 7.75 (s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.34 (s, 1H), 6.80 (br s, 1H), 3.92 (q, J = 7.0 Hz, 2H), 2.54–2.49 (m, 2H), 2.33–2.29 (m, 2H), 2.14 (s, 3H), 1.76–1.60 (m, 4H), 1.25 (t, J = 7.0 Hz, 3H); HPLC >99% (AUC), (Method B), tR = 8.4 min.
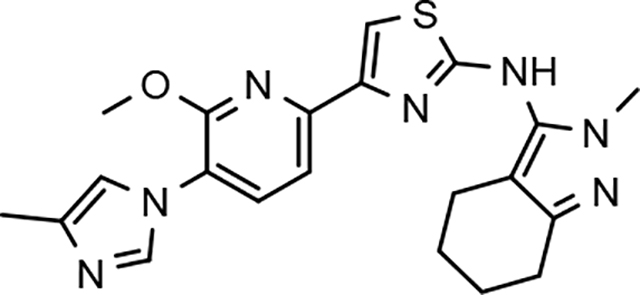
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-methyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (31d)
Light brown solid; mp 177–181 °C; ESI MS (M+H) 422; 1H NMR (500 MHz, DMSO-d6) δ 9.75 (s, 1H), 7.90 (d, J = 1.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.51 (s, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.24 (s, 1H), 4.00 (s, 3H), 3.61 (s, 3H), 2.55–2.50 (m, 2H), 2.38–2.31 (m, 2H), 2.16 (s, 3H), 1.77–1.60 (m, 4H); HPLC 96.9% (AUC), (Method B), tR = 9.1 min.
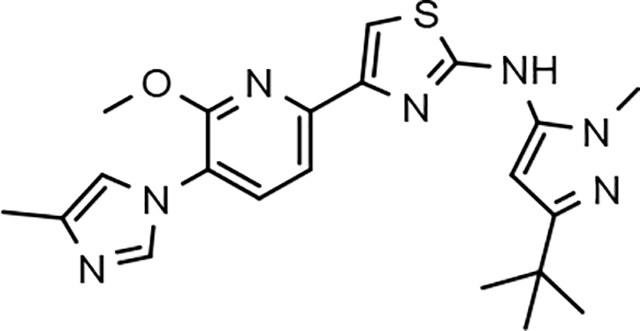
N-(3-(tert-Butyl)-1-methyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (32d)
Off-white solid; APCI MS (M+H) 424; 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 1H), 7.95 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.58 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.26 (s, 1H), 6.33 (s, 1H), 4.02 (s, 3H), 3.67 (s, 3H), 2.16 (s, 3H), 1.24 (s, 9H); HPLC >99% (AUC), (Method B), tR = 9.5 min.
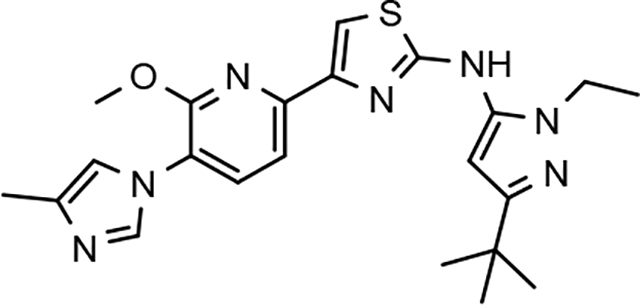
N-(3-(tert-Butyl)-1-ethyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (33d)
Off-white solid; mp 164–169 °C; APCI MS (M+H) 438; 1H NMR (300 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.91 (dd, J = 3.3, 3.6 Hz, 2H), 7.58 (s, 1H), 7.56 (d, J = 3.6 Hz, 1H), 7.25 (s, 1H), 6.33 (s, 1H), 4.03 (q, J = 6.9 Hz, 2H), 4.02 (s, 3H), 2.16 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.25 (s, 9H); HPLC 97.0% (AUC), (Method B), tR = 11.0 min.
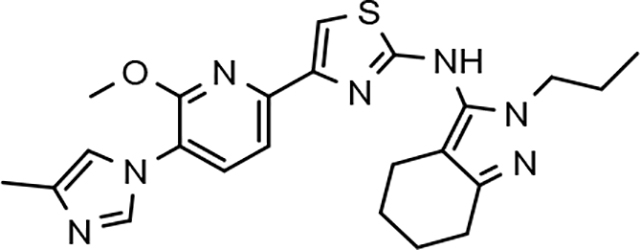
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-propyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (34)
Off-white solid; mp 204–208 °C; ESI MS (M+H) 450; 1H NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H), 7.90 (d, J = 1.0 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 7.50 (d, J = 7.5 Hz, 1H), 7.49 (s, 1H), 7.24 (m, 1H), 4.00 (s, 3H), 3.86 (t, J = 7.0 Hz, 2H), 2.55–2.50 (m, 2H), 2.34–2.30 (m, 2H), 2.16 (s, 3H), 1.77–1.60 (m, 6H), 0.81 (t, J = 7.5Hz, 3H); HPLC 98.8% (AUC), (Method B), tR = 11.0 min.
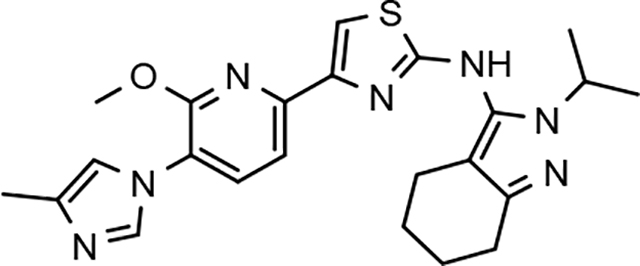
N-(2-isopropyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (35)
White solid; mp 130–134 °C; APCI MS (M+H) 450; 1H NMR (500 MHz, DMSO-d6) δ 9.64 (s, 1H), 7.90 (d, J = 0.6 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 7.5 Hz, 1H), 7.48 (s, 1H), 7.24 (s, 1H), 4.44 (m, J = 6.5 Hz, 1H), 4.00 (s, 3H), 2.55 (m, 2H), 2.32 (m, 2H), 2.16 (s, 3H), 1.73 (m, 2H), 1.65 (m, 2H), 1.32 (d, J = 6.5 Hz, 6H); HPLC 96.1% (AUC), (Method B), tR = 11.2 min.

N-(2-(tert-butyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (36)
Off-white solid; mp 207–210 °C; APCI MS (M+H) 464; 1H NMR (300 MHz, DMSO-d6) δ 9.54 (s, 1H), 7.92 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 8.1 Hz, 1H), 7.47 (s, 1H), 7.25 (s, 1H), 3.99 (s, 3H), 2.50 (m, 2H), 2.27 (m, 2H), 2.16 (s, 3H), 1.78–1.58 (m, 4H), 1.53 (s, 9H); HPLC >99% (AUC), (Method B), tR = 11.8 min.
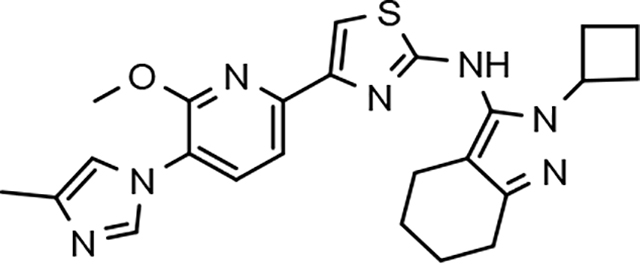
N-(2-cyclobutyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (37)
Off-white solid; mp 141–146 °C; APCI MS (M+H) 462; 1H NMR (300 MHz, DMSO-d6) δ 9.69 (s, 1H), 7.90 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.50 (s, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.24 (s, 1H), 4.71 (m, 1H), 4.00 (s, 3H), 2.57 (m, 2H), 2.46 (m, 2H), 2.37–2.21 (m, 4H), 2.16 (s, 3H), 1.81–1.58 (m, 6H); HPLC >99% (AUC), (Method B), tR = 11.7 min.
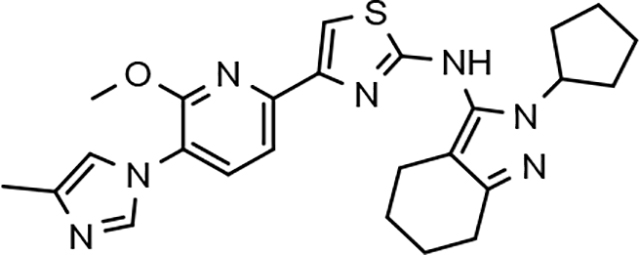
N-(2-cyclopentyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (38)
Brown solid; mp 110–120 °C; ESI MS (M+H) 476; 1H NMR (500 MHz, CDCl3) δ 7.87 (s, 1H), 7.58–7.54 (m, 2H), 7.39 (s, 1H), 6.99 (s, 1H), 6.80 (br s, 1H), 4.69–4.59 (m, 1H), 4.07 (s, 3H), 2.69 (t, J = 7.0 Hz, 2H), 2.42 (t, J = 7.0 Hz, 2H), 2.32 (s, 3H), 2.08–1.55 (m, 12H); HPLC >99% (AUC), (Method A), tR = 9.7 min.
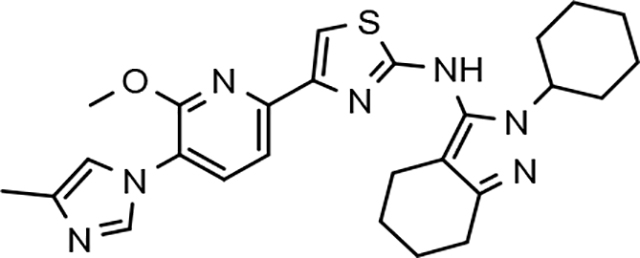
N-(2-cyclohexyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (39)
Orange-brown solid; mp 168–174 °C; APCI MS (M+H) 490; 1H NMR (300 MHz, DMSO-d6) δ 9.68 (s, 1H), 7.93 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.25 (s, 1H), 4.03 (m, 1H), 4.00 (s, 3H), 2.31 (m, 2H), 2.16 (s, 3H), 1.86–1.57 (m, 12H), 1.39–1.08 (m, 4H); HPLC 97.3% (AUC), (Method B), tR = 12.3 min.
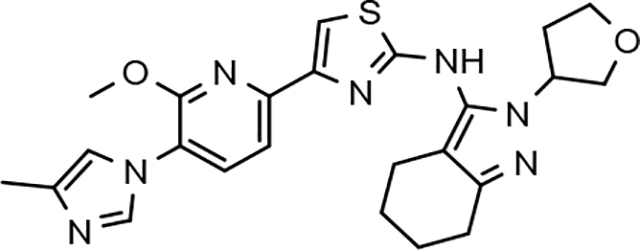
(+/−)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(tetrahydrofuran-3-yl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (40)
Off-white solid; mp 154–158 °C; APCI MS (M+H) 478; 1H NMR (300 MHz, DMSO-d6) δ 9.77 (s, 1H), 7.91 (d, J = 1.5 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.52 (s, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 4.91 (m, 1H), 4.00 (s, 3H), 3.95 (m, 2H), 3.83–3.71 (m, 2H), 2.55 (m, 2H), 2.32 (m, 2H), 2.20 (m, 2H), 2.15 (s, 3H), 1.80–1.59 (m, 4H); HPLC 97.4% (AUC), (Method B), tR = 11.1 min.
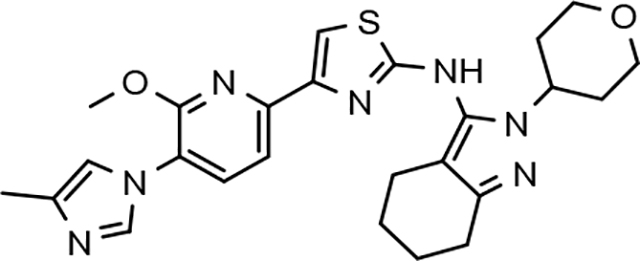
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(tetrahydro-2H-pyran-4-yl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (41)
White solid; mp 186–191 °C; ESI MS (M+H) 492; 1H NMR (300 MHz, DMSO-d6) δ 9.73 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.50 (t, J = 3.9 Hz, 2H), 7.25 (s, 1H), 4.31 (m, 1H), 4.00 (s, 3H), 3.92 (m, 2H), 3.39 (m, 2H), 2.55 (m, 2H), 2.32 (m, 2H), 2.15 (s, 3H), 2.10–1.90 (m, 2H), 1.80–1.59 (m, 6H); HPLC 97.5% (AUC), (Method B), tR = 11.1 min.
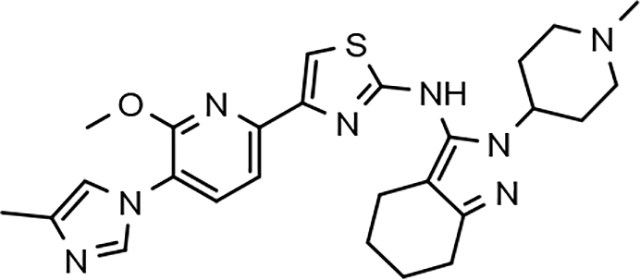
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(1-methylpiperidin-4-yl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (42)
Light brown solid; mp 150–153 °C; APCI MS (M+H) 505; 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 1.2 Hz, 1H), 7.61–7.45 (m, 3H), 7.36 (s, 1H), 6.99 (s, 1H), 4.11 (m, 1H), 4.05 (s, 3H), 2.94 (d, J = 11.4 Hz, 2H), 2.67 (t, J = 6.0 Hz, 2H), 2.40 (t, J = 6.0 Hz, 2H), 2.30 (s, 3H), 2.27 (s, 3H), 2.24 (m, 1H), 2.02 (t, J = 11.4 Hz, 3H), 1.89–1.65 (m, 6H); HPLC 96.4% (AUC), (Method B), tR = 9.3 min.
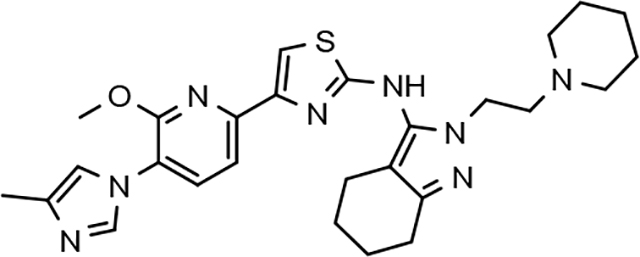
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(2-(piperidin-1-yl)ethyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (43)
Off-white solid; mp 80–90 °C; ESI MS (M+H) 519; 1H NMR (500 MHz, CDCl3) δ 10.90 (br s, 1H), 7.79 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.39 (s, 1H), 6.99 (s, 1H), 4.29–4.12 (m, 2H), 4.07 (s, 3H), 2.85–2.49 (m, 8H), 2.45 (t, J = 6.0 Hz, 2H), 2.30 (s, 3H), 1.95–1.70 (m, 4H), 1.68–1.50 (m, 6H); HPLC >99% (AUC), (Method A), tR = 8.4 min.
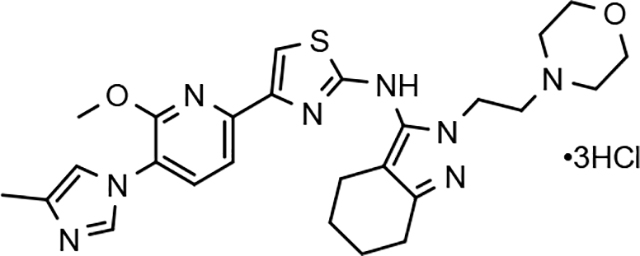
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(2-morpholinoethyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine trihydrochloride (44)
Light brown solid; mp 160–170 °C; ESI MS (M+H) 521; 1H NMR (500 MHz, DMSO-d6) δ 11.31 (br s, 1H), 10.28 (br s, 1H), 9.45 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H), 7.66 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 6.3–5.5 (br s, 2H), 4.47–4.44 (m, 2H), 4.04 (s, 3H), 4.00–3.70 (br s, 4H), 3.59–3.55 (m, 2H), 3.50–3.00 (br s, 4H), 2.57–2.50 (m, 2H), 2.36 (s, 3H), 2.37–2.30 (m, 2H), 1.80–1.60 (m, 4H); HPLC 93.5% (AUC), (Method C), tR = 8.6 min.
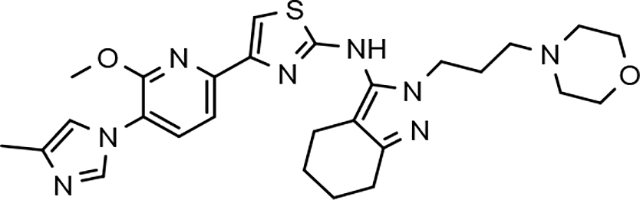
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(3-morpholinopropyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (45)
Yellow solid; mp 80–90 °C; ESI MS (M+H) 535; 1H NMR (500 MHz, CDCl3) δ 10.55 (br s, 1H), 7.81 (d, J = 1.0 Hz, 1H), 7.63–7.59 (m, 2H), 7.39 (s, 1H), 6.99 (s, 1H), 4.15–4.05 (m, 6H), 4.07 (s, 3H), 2.68 (t, J = 6.0 Hz, 2H), 2.59–2.49 (m, 4H), 2.44 (t, J = 6.0 Hz, 2H), 2.31 (s, 3H), 2.24–2.23 (m, 2H), 2.08–2.06 (m, 2H), 1.87–1.82 (m, 2H), 1.77–1.65 (m, 2H); HPLC >99% (AUC), (Method A), tR = 8.5 min.
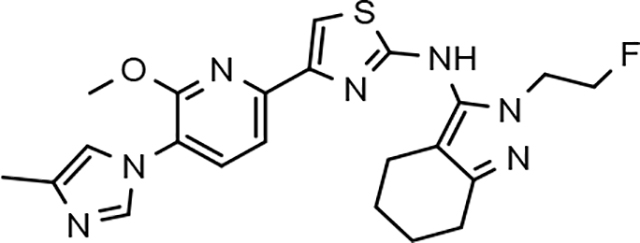
N-(2-(2-fluoroethyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (46)
White solid; mp 190–192 °C; APCI MS (M+H) 454; 1H NMR (300 MHz, DMSO-d6) δ 9.75 (s, 1H), 7.90 (d, J = 1.1 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.52 (s, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.24 (s, 1H), 4.70 (dt, J = 47.1, 4.9 Hz, 2H), 4.23 (dt, J = 26.1, 4.9 Hz, 2H), 4.00 (s, 3H), 2.53 (m, 2H), 2.34 (m, 2H), 2.16 (s, 3H), 1.73 (m, 4H); HPLC >99% (AUC), (Method D), tR = 8.6 min.
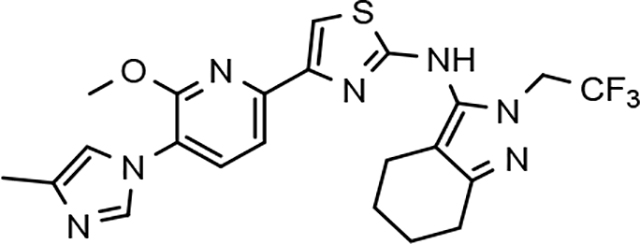
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(2,2,2-trifluoroethyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (47)
Light yellow solid; mp 122–126 °C; ESI MS (M+H) 490; 1H NMR (300 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.55 (s, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.24 (m, 1H), 4.88 (q, J = 8.7 Hz, 2H), 4.00 (s, 3H), 2.60–2.53 (m, 2H), 2.39–2.31 (m, 2H), 2.16 (s, 3H), 1.80–1.60 (m, 4H); HPLC 98.1% (AUC), (Method A), tR = 9.6 min.

4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(2-methoxyethyl)-4,5,6,7-tetrahydro-2H-indazol-3-yl)thiazol-2-amine (48)
White solid; mp 78–82 °C; ESI MS (M+H) 466; 1H NMR (500 MHz, CDCl3) δ 7.98 (br s, 1H), 7.69 (br s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.42 (s, 1H), 7.02 (s, 1H), 4.24 (t, J = 5.0 Hz, 2H), 4.08 (s, 3H), 3.68 (t, J = 5.0 Hz, 2H), 3.40 (s, 3H), 2.68 (t, J = 6.0 Hz, 2H), 2.43 (t, J = 6.0 Hz, 2H), 2.36 (s, 3H), 1.87–1.81 (m, 2H), 1.77–1.72 (m, 2H); HPLC 98.5% (AUC), (Method B), tR = 9.6 min.
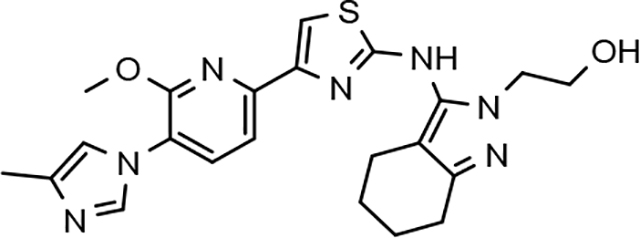
2-(3-((4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-yl)amino)-4,5,6,7-tetrahydro-2H-indazol-2-yl)ethanol (49)
White solid; mp 132–136 °C; ESI MS (M+H) 452; 1H NMR (300 MHz, DMSO-d6) δ 9.66 (s, 1H), 7.93 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.26 (m, 1H), 4.85 (t, J = 5.6 Hz, 1H), 4.00 (s, 3H), 4.00–3.94 (m, 2H), 3.70–3.60 (m, 2H), 2.55–2.50 (m, 2H), 2.36–2.30 (m, 2H), 2.16 (s, 3H), 1.78–1.60 (m, 4H); HPLC >99% (AUC), (Method B), tR = 9.8 min.

3-(3-((4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-yl)amino)-4,5,6,7-tetrahydro-2H-indazol-2-yl)propan-1-ol (50)
Light orange solid; mp 178–182 °C; ESI MS (M+H) 466; 1H NMR (300 MHz, DMSO-d6) δ 9.71 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.50 (s, 1H), 7.24 (m, 1H), 4.55 (dd, J = 5.1, 5.1 Hz, 1H), 4.03–3.90 (s, 2H), 4.00 (s, 3H), 3.42–3.30 (m, 2H), 2.60– 2.47 (m, 2H), 2.37–2.30 (m, 2H), 2.15 (s, 3H), 2.40–2.10 (m, 6H); HPLC >99% (AUC), (Method A), tR = 8.1 min.
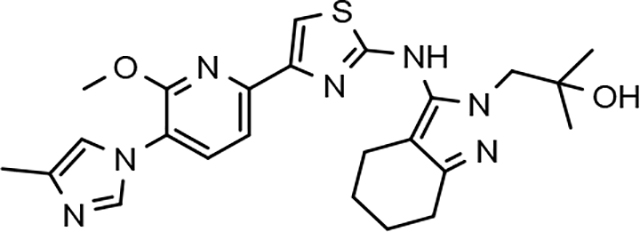
1-(3-((4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-yl)amino)-4,5,6,7-tetrahydro-2H-indazol-2-yl)-2-methylpropan-2-ol (51)
Yellow solid; mp 90–100 °C; ESI MS (M+H) 480; 1H NMR (300 MHz, DMSO-d6) δ 9.60 (s, 1H), 7.91 (s, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.52 (s, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.25 (s, 1H), 4.92 (s, 1H), 4.00 (s, 3H), 3.88 (s, 2H), 2.60–2.51 (m, 2H), 2.38–2.29 (m, 2H), 2.16 (s, 3H), 1.80–1.56 (m, 4H), 1.06 (s, 6H); HPLC 96.8% (AUC), (Method A), tR = 8.4 min.
N-(2-ethyl-4,5,6,7-tetrahydro-2H-indazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-methylthiazol-2-amine (52)
Off-white solid; mp 74–79 °C; APCI MS (M+H) 450; 1H NMR (300 MHz, DMSO-d6) δ 7.92 (d, J = 1.2 Hz, 1H), 7.90 (d, J = 8.1 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.52 (s, 1H), 7.25 (t, J = 1.2 Hz, 1H), 4.00 (s, 3H), 3.90 (q, J = 7.2 Hz, 2H), 3.43 (s, 3H), 2.56 (m, 2H), 2.33 (m, 2H), 2.16 (d, J = 0.9 Hz, 3H), 1.81–1.60 (m, 4H), 1.29 (t, J = 7.2 Hz, 3H); HPLC >99% (AUC), (Method B), tR = 12.5 min.

N-(3-(tert-butyl)-1-propyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine dihydrochloride (53)
Brown solid; mp 200–205 °C; APCI MS (M+H) 452; 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H), 9.39 (s, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.81 (s, 1H), 7.69 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 6.36 (s, 1H), 4.40–3.80 (br s, 2H), 4.05 (s, 3H), 3.96 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H), 1.71 (m, 2H), 1.25 (s, 9H), 0.83 (t, J = 7.2 Hz, 3H); HPLC 95.7% (AUC), (Method B), tR = 10.1 min.
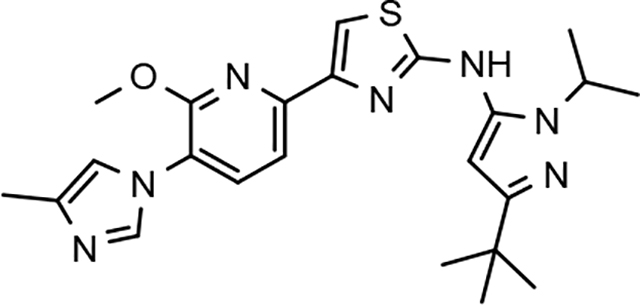
N-(3-(tert-butyl)-1-isopropyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridine-2-yl)thiazol-2-amine (54)
White solid; mp 170–174 °C; ESI MS (M–H) 450; 1H NMR (500 MHz, DMSO-d6) δ 9.98 (s, 1H), 7.91–7.89 (m, 2H), 7.56–7.54 (m, 2H), 7.25 (s, 1H), 6.27 (s, 1H), 4.55–4.50 (m, 1H), 4.02 (s, 3H), 2.16 (s, 3H), 1.34 (d, J = 6.5 Hz, 6H), 1.25 (s, 9H); HPLC >99% (AUC), (Method B), tR = 10.6 min.

N-(1,3-di-tert-butyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (55)
Light yellow solid; mp 206–210 °C; ESI MS (M+H) 466; 1H NMR (300 MHz, DMSO-d6) δ 9.55 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.47 (s, 1H), 7.24 (m, 1H), 6.23 (s, 1H), 4.00 (s, 3H), 2.16 (s, 3H), 1.55 (s, 9H), 1.23 (s, 9H); HPLC >99% (AUC), (Method A), tR = 12.1 min.
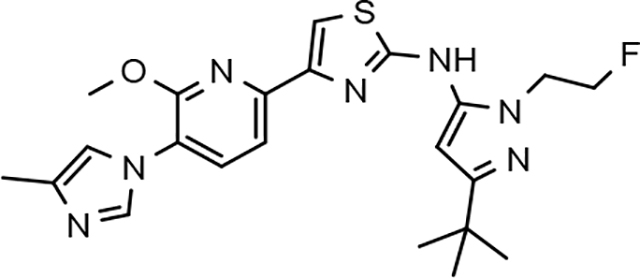
N-(3-(tert-butyl)-1-(2-fluoroethyl)-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (56)
White solid; mp 210–214 °C; ESI MS (M+H) 456; 1H NMR (300 MHz, DMSO-d6) δ 10.2 (s, 1H), 7.94 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.59–7.57 (m, 2H), 7.26 (s, 1H), 6.42 (s, 1H), 4.79 (t, J = 5 Hz, 1H), 4.64 (t, J = 5 Hz, 1H), 4.39 (t, J = 5 Hz, 1H), 4.31 (t, J = 5 Hz, 1H),), 4.29 (s, 3H), 2.16 (s, 3H), 1.26 (s, 9H); HPLC >99% (AUC), (Method A), tR = 9.1 min.
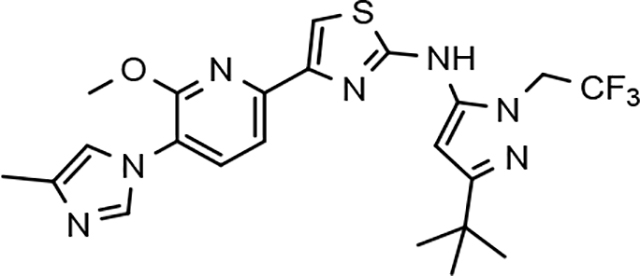
N-(3-(tert-butyl)-1-(2,2,2-trifluoroethyl)-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (57)
Off-white solid; mp 220–225 °C; ESI MS (M+H) 492; 1H NMR (300 MHz, CDCl3) δ 7.94 (s, 1H), 7.61–7.50 (m, 2H), 7.33 (s, 1H), 7.00 (s, 1H), 6.24 (s, 1H), 4.76–4.66 (m, 2H), 4.07 (s, 3H), 2.33 (s, 3H), 2.2–1.7 (br s, 1H), 1.27 (s, 9H); HPLC >99% (AUC), (Method A), tR = 10.1 min.
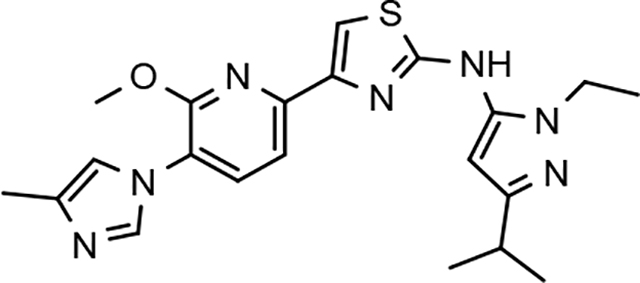
N-(1-ethyl-3-isopropyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (58)
Light yellow solid; mp 170–174 °C; ESI MS (M+H) 424; 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.91 (d, J = 1.2 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.58 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 6.30 (s, 1H), 4.04 (q, J = 7.2 Hz, 2H), 4.02 (s, 3H), 2.84 (septet, J = 6.9 Hz, 1H), 2.16 (s, 3H), 1.27 (t, J = 7.2 Hz, 3H), 1.20 (d, J = 6.9 Hz, 6H); HPLC >99% (AUC), (Method B), tR = 10.5 min.
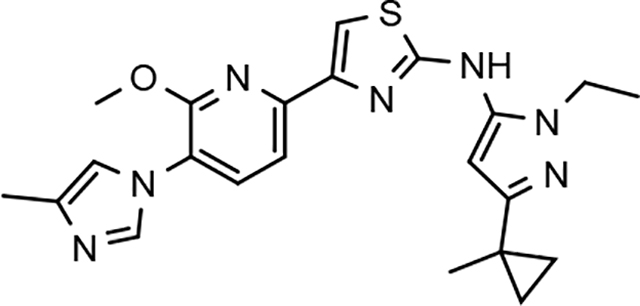
N-(1-ethyl-3-(1-methylcyclopropyl)-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (59)
Off-white solid; mp 178–182 °C; ESI MS (M+H) 436; 1H NMR (300 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.92 (d, J = 1.2 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.58 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.25 (m, 1H), 6.21 (s, 1H), 4.01 (s, 3H), 3.99 (q, J = 7.2 Hz, 2H), 2.16 (s, 3H), 1.38 (s, 3H), 1.25 (t, J = 7.2 Hz, 3H), 0.86 (dd, J = 6.0, 3.6 Hz, 2H), 0.68 (dd, J = 6.0, 3.6 Hz, 2H); HPLC >99% (AUC), (Method A), tR = 8.6 min.
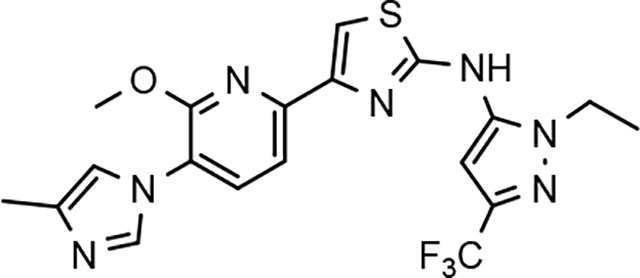
N-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (60)
Light brown solid; mp 222–225 °C; APCI MS (M+H) 450; 1H NMR (500 MHz, DMSO-d6) δ 10.60 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 1.0 Hz, 1H), 7.67 (br s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.24 (s, 1H), 7.00 (br s, 1H), 4.21 (q, J = 7.5 Hz, 2H), 4.03 (s, 3H), 2.16 (d, J = 1.0 Hz, 3H), 1.36 (t, J = 7.5 Hz, 3H); HPLC >99% (AUC), (Method D), tR = 9.1 min.
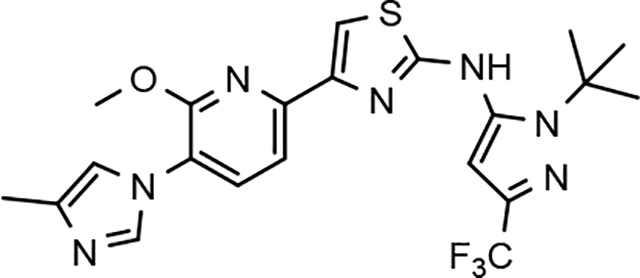
N-(1-(tert-butyl)-3-(trifluoromethyl)-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (61)
White solid; mp 197–200 °C; APCI MS (M+H) 478; 1H NMR (300 MHz, DMSO-d6) δ 9.89 (s, 1H), 7.90 (s, 1H), 7.87 (d, J = 8.7 Hz, 1H), 7.57 (s, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 6.98 (s, 1H), 4.01 (s, 3H), 2.16 (s, 3H), 1.62 (s, 9H); HPLC >99% (AUC), (Method B), tR = 14.0 min.
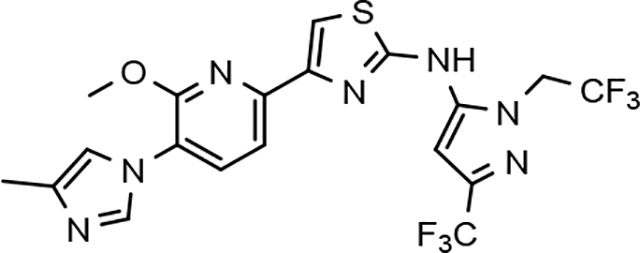
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(1-(2,2,2-trifluoroethyl)-3-(trifluoromethyl)-1H-pyrazol-5-yl)thiazol-2-amine (62)
Off-white solid; mp 230–234 °C; APCI MS (M+H) 504; 1H NMR (300 MHz, DMSO-d6) δ 10.97 (br s, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 1.2 Hz, 1H), 7.70 (br s, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.26 (s, 1H), 7.08 (br s, 1H), 5.24 (m, 2H), 4.04 (s, 3H), 2.16 (s, 3H); HPLC >99% (AUC), (Method B), tR = 13.3 min.
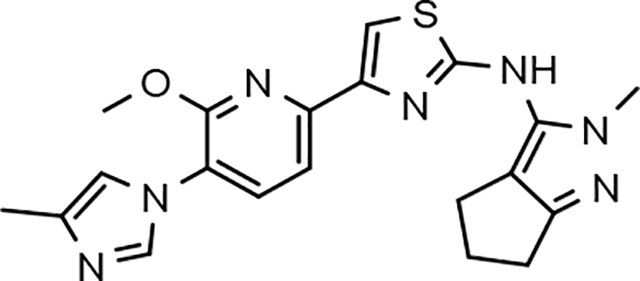
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-methyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)thiazol-2-amine (63)
White solid; mp 196–199 °C; ESI MS (M+H) 408; 1H NMR (300 MHz, DMSO-d6) δ 10.04 (s, 1H), 7.90 (d, J = 1.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.55 (s, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 4.01 (s, 3H), 3.64 (s, 3H), 2.63–2.53 (m, 4H), 2.37–2.23 (m, 2H), 2.16 (m, 3H); HPLC >99% (AUC), (Method D), tR = 8.1 min.
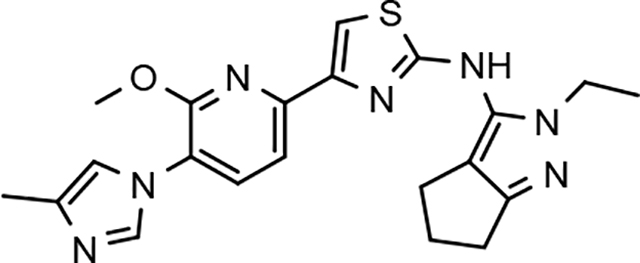
N-(2-ethyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (64)
White solid; mp 228–232 °C dec; ESI MS (M+H) 422; 1H NMR (500 MHz, DMSO-d6) δ 9.95 (s, 1H), 7.90 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.54 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.24 (s, 1H), 4.01 (s, 3H), 3.97 (q, J = 7.0 Hz, 2H), 2.60–2.55 (m, 4H), 2.34–2.28 (m, 2H), 2.16 (s, 3H), 1.27 (t, J = 7.0 Hz, 3H); HPLC >99% (AUC), (Method C), tR = 9.1 min.
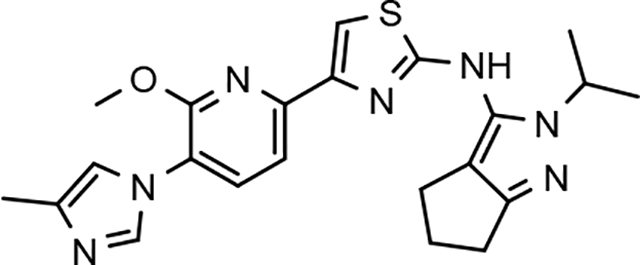
N-(2-isopropyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (65)
Light yellow solid; mp 212–216 °C; ESI MS (M+H) 436; 1H NMR (300 MHz, DMSO-d6) δ 9.93 (s, 1H), 7.91 (d, J = 1.5 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.53 (s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.25 (m, 1H), 4.46 (septet, J = 6.6 Hz, 1H), 4.01 (s, 3H), 2.63–2.50 (m, 4H), 2.38–2.25 (m, 2H), 2.16 (s, 3H), 1.33 (d, J = 6.6 Hz, 6H); HPLC >99% (AUC), (Method B), tR = 10.7 min.
N-(2-(tert-butyl)-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (66)
Brown solid; mp 95–105 °C; ESI MS (M+H) 450; 1H NMR (300 MHz, CDCl3) δ 7.90 (br s, 1H), 7.59–7.53 (m, 2H), 7.41 (s, 1H), 7.01 (s, 1H), 4.07 (s, 3H), 2.75 (t, J = 6.9 Hz, 2H), 2.56 (t, J = 6.9 Hz, 2H), 2.44–2.37 (m, 3H), 2.33 (s, 3H), 1.65 (s, 9H); HPLC >99% (AUC), (Method A), tR = 8.9 min.
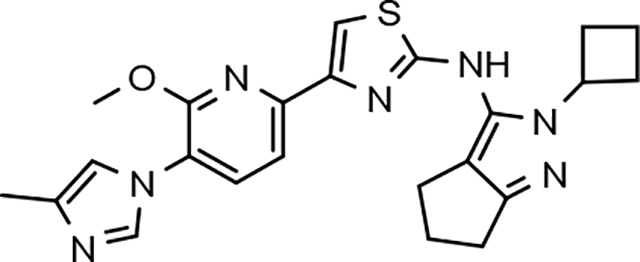
N-(2-cyclobutyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (67)
White solid; mp 180–183 °C; APCI MS (M+H) 448; 1H NMR (300 MHz, DMSO-d6) δ 9.93 (s, 1H), 7.94 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.54 (s, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.26 (s, 1H), 4.73 (m, 1H), 4.00 (s, 3H), 2.59 (m, 6H), 2.29 (m, 4H), 2.16 (s, 3H), 1.73 (m, 2H); HPLC >99% (AUC), (Method B), tR = 11.4 min.
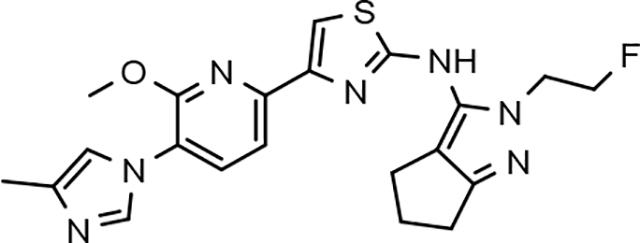
N-(2-(2-fluoroethyl)-2, 4, 5, 6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl) pyridin-2-yl) thiazol-2-amine (68)
White solid; mp 160–163 °C; ESI MS (M+H) 440; 1H NMR (300 MHz, CDCl3) δ 7.91 (s, 1H), 7.64–7.58 (m, 2H), 7.44 (s, 1H), 7.01 (s, 1H), 4.84 (t, J = 5 Hz, 1H), 4.69 (t, J = 5 Hz, 1H), 4.41 (t, J = 5 Hz, 1H), 4.33 (t, J = 5 Hz, 1H), 4.08 (s, 3H), 2.77–2.72 (m, 2H), 2.66–2.61 (m, 2H), 2.60–2.30 (br s, 1H), 2.45–2.40 (m, 2H), 2.34 (s, 3H); HPLC >99% (AUC), (Method A), tR = 8.6 min.
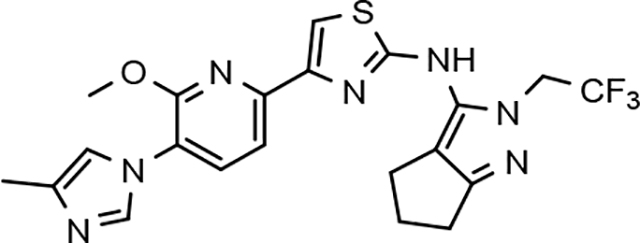
4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)-N-(2-(2,2,2-trifluoroethyl)-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)thiazol-2-amine (69)
Brown solid; mp 80–85 °C; ESI MS (M+H) 476; 1H NMR (300 MHz, DMSO-d6) δ 10.17 (s, 1H), 7.91 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.25 (s, 1H), 5.09–4.85 (m, 2H), 4.02 (s, 3H), 2.67–2.54 (m, 4H), 2.38–2.25 (m, 2H), 2.16 (s, 3H); HPLC 96.9% (AUC), (Method A), tR = 9.3 min.
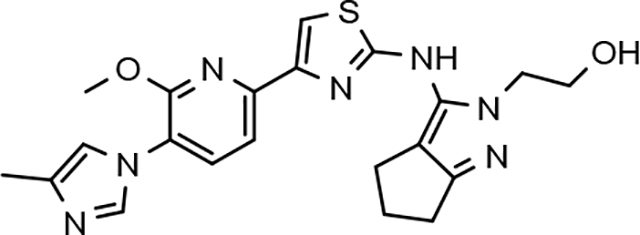
2-(3-((4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-yl)amino)-5,6-dihydrocyclopenta[c]pyrazol-2(4H)-yl)ethanol (70)
Brown solid; mp 140–145 °C; ESI MS (M+H) 438; 1H NMR (300 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.21 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.58 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.37 (s, 1H), 4.93 (br s, 1H), 4.10–3.95 (m, 5H), 3.71–3.60 (m, 2H), 2.63–2.53 (m, 4H), 2.38–2.29 (m, 2H), 2.19 (s, 3H); HPLC >99% (AUC), (Method A), tR = 7.8 min.
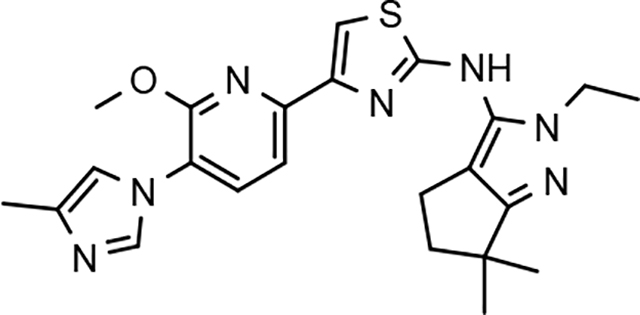
N-(2-ethyl-6,6-dimethyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (71)
White solid; mp 176–180 °C; ESI MS (M+H) 450; 1H NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 7.90 (d, J = 1.0 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.53 (s, 1H), 7.24 (m, 1H), 4.01 (s, 3H), 3.97 (q, J = 7.2 Hz, 2H), 2.55 (t, J = 6.7 Hz, 2H), 2.16 (s, 3H), 2.12 (t, J = 6.7 Hz, 2H), 1.27 (t, J = 7.2 Hz, 3H), 1.23 (s, 6H); HPLC >99% (AUC), (Method A), tR = 8.7 min.
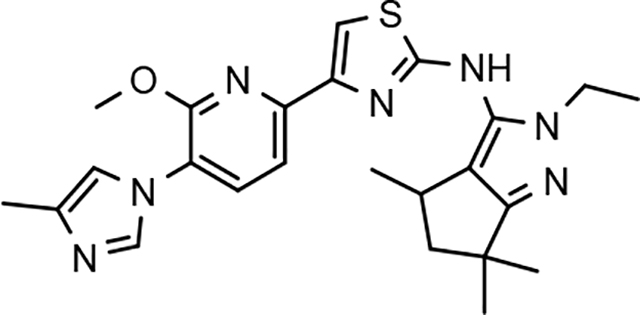
(+/−)-N-(2-ethyl-4,6,6-trimethyl-2,4,5,6-tetrahydrocyclopenta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (72)
Light yellow solid; mp 160–164 °C; ESI MS (M+H) 464; 1H NMR (300 MHz, DMSO-d6) δ 9.90 (s, 1H), 7.91 (d, J = 1.5 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.54 (s, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.25 (m, 1H), 4.02 (s, 3H), 4.03–3.85 (sym m, 2H), 3.10 (q, J = 6.9 Hz, 1H), 2.32 (dd, J = 12.4, 7.0 Hz, 1H), 2.16 (s, 3H), 1.73 (dd, J = 12.4, 8.1 Hz, 1H), 1.32–1.22 (s, 6H), 1.19 (s, 3H), 0.95 (d, J = 6.9 Hz, 3H); HPLC >99% (AUC), (Method A), tR = 9.2 min.
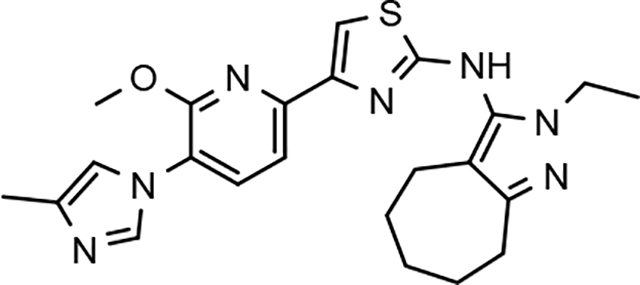
N-(2-ethyl-2,4,5,6,7,8-hexahydrocyclohepta[c]pyrazol-3-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (73)
Yellow solid; mp 217–221 °C; APCI MS (M+H) 450; 1H NMR (300 MHz, DMSO-d6) δ 9.66 (s, 1H), 7.91 (s, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.48 (s, 1H), 7.24 (s, 1H), 3.99 (s, 3H), 3.89 (q, J = 7.2 Hz, 2H), 2.64 (m, 2H), 2.31 (m, 2H), 2.16 (s, 3H), 1.78 (m, 2H), 1.56 (m, 4H), 1.24 (t, J = 7.2 Hz, 3H); HPLC >99% (AUC), (Method E), tR = 11.6 min.
N-(1-ethyl-1H-pyrazol-5-yl)-4-(6-methoxy-5-(4-methyl-1H-imidazol-1-yl)pyridin-2-yl)thiazol-2-amine (74)
Yellow solid; mp 180–187 °C; ESI MS (M+H) 382; 1H NMR (300 MHz, CDCl3) δ 7.90 (s, 1H), 7.59 (s, 2H), 7.55 (s, 1H), 7.41 (s, 1H), 7.00 (s, 1H), 6.31 (s, 1H), 4.20–4.15 (m, 2H), 4.08 (s, 3H), 2.32 (s, 3), 1.90 (br s, 1H), 1.45 (t, J = 7.0 Hz, 3H); HPLC >99% (AUC), (Method A), tR = 8.1 min.
Supplementary Material
Acknowledgements
We acknowledge SRI International for their support in conducting selected pharmacokinetic studies. We also acknowledge the contributions of Charles Cywin of the National Institute of Neurologic Disorders and Stroke (NINDS) and Lorenzo Refolo of the National Institute on Aging (NIA) for guidance and insights throughout the course of this research program. The research presented was funded by the Cure Alzheimer’s Fund (CAF) and by NIH, NIA and NINDS through a Blueprint Neurotherapeutics Award (U01 NS074501).
Footnotes
Declaration of interests
☒The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Drs. S. L. Wagner and R. E. Tanzi are shareholders and cofounders of a privately held company (Neurogenetic Pharmaceuticals, Inc.) that holds rights to a gamma-secretase modulator previously in clinical development.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Association, A. s., Alzheimer’s Disease Facts and Figures. Alzheimers Dement 2019, 15 (3), 321–387. [Google Scholar]
- 2.Glenner GG; Wong CW, Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984, 120 (3), 885–90. [DOI] [PubMed] [Google Scholar]
- 3.Iwatsubo T; Saido TC; Mann DM; Lee VM; Trojanowski JQ, Full-length amyloid-beta (1–42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am J Pathol 1996, 149 (6), 1823–30. [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson DW, The Pathogenesis of Senile Plaques. Journal of Neuropathology & Experimental Neurology 1997, 56 (4), 321–339. [DOI] [PubMed] [Google Scholar]
- 5.Rowe CC; Ellis KA; Rimajova M; Bourgeat P; Pike KE; Jones G; Fripp J; Tochon-Danguy H; Morandeau L; O’Keefe G; Price R; Raniga P; Robins P; Acosta O; Lenzo N; Szoeke C; Salvado O; Head R; Martins R; Masters CL; Ames D; Villemagne VL, Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010, 31 (8), 1275–83. [DOI] [PubMed] [Google Scholar]
- 6.Sperling RA; Aisen PS; Beckett LA; Bennett DA; Craft S; Fagan AM; Iwatsubo T; Jack CR Jr.; Kaye J; Montine TJ; Park DC; Reiman EM; Rowe CC; Siemers E; Stern Y; Yaffe K; Carrillo MC; Thies B; Morrison-Bogorad M; Wagster MV; Phelps CH, Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011, 7 (3), 280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy JA; Higgins GA, Alzheimer’s disease: the amyloid cascade hypothesis. Science 1992, 256 (5054), 184–5. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J; Selkoe DJ, The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 297 (5580), 353–6. [DOI] [PubMed] [Google Scholar]
- 9.Tanzi RE; Bertram L, Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 2005, 120 (4), 545–55. [DOI] [PubMed] [Google Scholar]
- 10.Thinakaran G; Koo EH, Amyloid precursor protein trafficking, processing, and function. J Biol Chem 2008, 283 (44), 29615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Strooper B; Vassar R; Golde T, The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol 2010, 6 (2), 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karran E; Mercken M; De Strooper B, The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 2011, 10 (9), 698–712. [DOI] [PubMed] [Google Scholar]
- 13.Oehlrich D; Berthelot DJ; Gijsen HJ, gamma-Secretase modulators as potential disease modifying anti-Alzheimer’s drugs. J Med Chem 2011, 54 (3), 669–98. [DOI] [PubMed] [Google Scholar]
- 14.Evin G; Barakat A; Masters CL, BACE: Therapeutic target and potential biomarker for Alzheimer’s disease. Int J Biochem Cell Biol 2010, 42 (12), 1923–6. [DOI] [PubMed] [Google Scholar]
- 15.Durham TB; Shepherd TA, Progress toward the discovery and development of efficacious BACE inhibitors. Curr Opin Drug Discov Devel 2006, 9 (6), 776–91. [PubMed] [Google Scholar]
- 16.Stachel SJ, Progress toward the development of a viable BACE-1 inhibitor. Drug Development Research 2009, 70 (2), 101–110. [Google Scholar]
- 17.Vassar R; Kandalepas PC, The beta-secretase enzyme BACE1 as a therapeutic target for Alzheimer’s disease. Alzheimers Res Ther 2011, 3 (3), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bateman RJ; Siemers ER; Mawuenyega KG; Wen G; Browning KR; Sigurdson WC; Yarasheski KE; Friedrich SW; Demattos RB; May PC; Paul SM; Holtzman DM, A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann Neurol 2009, 66 (1), 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomita T, Secretase inhibitors and modulators for Alzheimer’s disease treatment. Expert Rev Neurother 2009, 9 (5), 661–79. [DOI] [PubMed] [Google Scholar]
- 20.Imbimbo BP; Panza F; Frisardi V; Solfrizzi V; D’Onofrio G; Logroscino G; Seripa D; Pilotto A, Therapeutic intervention for Alzheimer’s disease with gamma-secretase inhibitors: still a viable option? Expert Opin Investig Drugs 2011, 20 (3), 325–41. [DOI] [PubMed] [Google Scholar]
- 21.Iwatsubo T; Odaka A; Suzuki N; Mizusawa H; Nukina N; Ihara Y, Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13 (1), 45–53. [DOI] [PubMed] [Google Scholar]
- 22.Weggen S; Eriksen JL; Das P; Sagi SA; Wang R; Pietrzik CU; Findlay KA; Smith TE; Murphy MP; Bulter T; Kang DE; Marquez-Sterling N; Golde TE; Koo EH, A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 2001, 414 (6860), 212–6. [DOI] [PubMed] [Google Scholar]
- 23.Kumar-Singh S; Theuns J; Van Broeck B; Pirici D; Vennekens K; Corsmit E; Cruts M; Dermaut B; Wang R; Van Broeckhoven C, Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 2006, 27 (7), 686–95. [DOI] [PubMed] [Google Scholar]
- 24.Rynearson KD; Tanzi RE; Wagner SL, Discovery of Potent Gamma-Secretase Modulators for the Treatment of Alzheimer’s Disease. In Translational Neuroscience: Fundamental Approaches for Neurological Disorders, 1st ed.; Tuszynski MH, Ed. Springer Science: New York, 2016; pp 359–368. [Google Scholar]
- 25.Portelius E; Van Broeck B; Andreasson U; Gustavsson MK; Mercken M; Zetterberg H; Borghys H; Blennow K, Acute effect on the Abeta isoform pattern in CSF in response to gamma-secretase modulator and inhibitor treatment in dogs. J Alzheimers Dis 2010, 21 (3), 1005–12. [DOI] [PubMed] [Google Scholar]
- 26.Borgegard T; Jureus A; Olsson F; Rosqvist S; Sabirsh A; Rotticci D; Paulsen K; Klintenberg R; Yan H; Waldman M; Stromberg K; Nord J; Johansson J; Regner A; Parpal S; Malinowsky D; Radesater AC; Li T; Singh R; Eriksson H; Lundkvist J, First and second generation gamma-secretase modulators (GSMs) modulate amyloid-beta (Abeta) peptide production through different mechanisms. J Biol Chem 2012, 287 (15), 11810–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettersson M; Johnson DS; Humphrey JM; Am Ende CW; Evrard E; Efremov I; Kauffman GW; Stepan AF; Stiff CM; Xie L; Bales KR; Hajos-Korcsok E; Murrey HE; Pustilnik LR; Steyn SJ; Wood KM; Verhoest PR, Discovery of indole-derived pyridopyrazine-1,6-dione gamma-secretase modulators that target presenilin. Bioorg Med Chem Lett 2015, 25 (4), 908–13. [DOI] [PubMed] [Google Scholar]
- 28.Pettersson M; Johnson DS; Humphrey JM; Butler TW; Am Ende CW; Fish BA; Green ME; Kauffman GW; Mullins PB; O’Donnell CJ; Stepan AF; Stiff CM; Subramanyam C; Tran TP; Vetelino BC; Yang E; Xie L; Bales KR; Pustilnik LR; Steyn SJ; Wood KM; Verhoest PR, Design of Pyridopyrazine-1,6-dione gamma-Secretase Modulators that Align Potency, MDR Efflux Ratio, and Metabolic Stability. ACS Med Chem Lett 2015, 6 (5), 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takasugi N; Tomita T; Hayashi I; Tsuruoka M; Niimura M; Takahashi Y; Thinakaran G; Iwatsubo T, The role of presenilin cofactors in the gamma-secretase complex. Nature 2003, 422 (6930), 438–41. [DOI] [PubMed] [Google Scholar]
- 30.Edbauer D; Winkler E; Regula JT; Pesold B; Steiner H; Haass C, Reconstitution of gamma-secretase activity. Nat Cell Biol 2003, 5 (5), 486–8. [DOI] [PubMed] [Google Scholar]
- 31.Crump CJ; Johnson DS; Li YM, Development and mechanism of gamma-secretase modulators for Alzheimer’s disease. Biochemistry 2013, 52 (19), 3197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pozdnyakov N; Murrey HE; Crump CJ; Pettersson M; Ballard TE; Am Ende CW; Ahn K; Li YM; Bales KR; Johnson DS, gamma-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J Biol Chem 2013, 288 (14), 9710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rynearson KD; Buckle RN; Barnes KD; Herr RJ; Mayhew NJ; Paquette WD; Sakwa SA; Nguyen PD; Johnson G; Tanzi RE; Wagner SL, Design and synthesis of aminothiazole modulators of the gamma-secretase enzyme. Bioorg Med Chem Lett 2016, 26 (16), 3928–37. [DOI] [PubMed] [Google Scholar]
- 34.Pettersson M; Stepan AF; Kauffman GW; Johnson DS, Novel γ-secretase modulators for the treatment of Alzheimer’s disease: a review focusing on patents from 2010 to 2012. Expert Opinion on Therapeutic Patents 2013, 23 (10), 1349–1366. [DOI] [PubMed] [Google Scholar]
- 35.Bursavich MG; Harrison BA; Acharya R; Costa DE; Freeman EA; Hodgdon HE; Hrdlicka LA; Jin H; Kapadnis S; Moffit JS; Murphy DA; Nolan S; Patzke H; Tang C; Wen M; Koenig G; Blain JF; Burnett DA, Design, Synthesis, and Evaluation of a Novel Series of Oxadiazine Gamma Secretase Modulators for Familial Alzheimer’s Disease. J Med Chem 2017, 60 (6), 2383–2400. [DOI] [PubMed] [Google Scholar]
- 36.Wagner SL; Cheng S; Mobley WC; Tanzi RE Compounds and Uses thereof in Modulating Levels of Varoius Amyloid Beta Peptide Alloforms. US 2011/041905, 2011. [Google Scholar]
- 37.Parlow JJ; Case BL; Dice TA; Fenton RL; Hayes MJ; Jones DE; Neumann WL; Wood RS; Lachance RM; Girard TJ; Nicholson NS; Clare M; Stegeman RA; Stevens AM; Stallings WC; Kurumbail RG; South MS, Design, parallel synthesis, and crystal structures of pyrazinone antithrombotics as selective inhibitors of the tissue factor VIIa complex. J Med Chem 2003, 46 (19), 4050–62. [DOI] [PubMed] [Google Scholar]
- 38.Huffman CW, Formylation of Amines. The Journal of Organic Chemistry 1958, 23 (5), 727–729. [Google Scholar]
- 39.Gerack CJ; McElwee-White L, Formylation of amines. Molecules 2014, 19 (6), 7689–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hantzsch A, Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen. Berichte der deutschen chemischen Gesellschaft 1881, 14 (2), 1637–1638. [Google Scholar]
- 41.Raven F; Ward JF; Zoltowska KM; Wan Y; Bylykbashi E; Miller SJ; Shen X; Choi SH; Rynearson KD; Berezovska O; Wagner SL; Tanzi RE; Zhang C, Soluble Gamma-secretase Modulators Attenuate Alzheimer’s beta-amyloid Pathology and Induce Conformational Changes in Presenilin 1. EBioMedicine 2017, 24, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kounnas MZ; Danks AM; Cheng S; Tyree C; Ackerman E; Zhang X; Ahn K; Nguyen P; Comer D; Mao L; Yu C; Pleynet D; Digregorio PJ; Velicelebi G; Stauderman KA; Comer WT; Mobley WC; Li YM; Sisodia SS; Tanzi RE; Wagner SL, Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 2010, 67 (5), 769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mucke L; Masliah E; Yu GQ; Mallory M; Rockenstein EM; Tatsuno G; Hu K; Kholodenko D; Johnson-Wood K; McConlogue L, High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000, 20 (11), 4050–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsiao K; Chapman P; Nilsen S; Eckman C; Harigaya Y; Younkin S; Yang F; Cole G, Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274 (5284), 99–102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.