

Abstract
Primary sclerosing cholangitis (PSC) is a chronic cholestatic disease with no approved treatments. C‐C chemokine receptor types 2 and 5 (CCR2/CCR5) play an important role in inflammation and fibrosis and are potential therapeutic targets for PSC. We evaluated the efficacy and safety of cenicriviroc (CVC), a dual antagonist of CCR2 and CCR5, for the treatment of PSC. This was a single‐arm, open‐label, exploratory study of CVC in adults with a clinical diagnosis of PSC, serum alkaline phosphatase (ALP) ≥1.5 times the upper limit of normal (ULN), with or without inflammatory bowel disease, across eight sites in the United States and Canada. The primary endpoint was percent change in ALP over 24 weeks; key secondary efficacy endpoints were proportion of participants who achieved ALP normalization and overall response (decrease to <1.5 times the ULN or 50% decrease). Of the 24 participants, 20 completed the study. The mean age was 43 years, 50% were female, and the mean body mass index was 25 kg/m2. From a median ALP baseline of 369 U/L (range: 173, 1,377 U/L), a median absolute reduction of 49.5 U/L (range: −460, 416 U/L) was achieved at week 24, corresponding to a median reduction of 18.0% (range: −46%, 89%). No participant achieved ALP normalization or a 50% decrease; 2 participants (10%) achieved a reduction in ALP to < 1.5 times the ULN, and 4 had ≥25% increase. Twenty participants (83.3%) reported at least one adverse event; most were mild to moderate in severity. The most frequent events were rash, fatigue, and dizziness. Conclusion: After 24 weeks of CVC treatment, adults with PSC achieved a modest reduction (median 18%) in the surrogate endpoint of ALP. CVC was well tolerated, and no new safety signals were observed. ClinicalTrials.gov identifier: NCT02653625.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- APRI
aspartate aminotransferase‐to‐platelet ratio index
- AST
aspartate aminotransferase
- CCR
C‐C chemokine receptor
- CI
confidence interval
- CVC
cenicriviroc
- FIB‐4
Fibrosis‐4
- GGT
gamma‐glutamyl transferase
- IBD
inflammatory bowel disease
- IgG
immunoglobulin G
- ITT
intent‐to‐treat
- MCP‐1
monocyte chemotactic protein‐1
- NASH
nonalcoholic steatohepatitis
- PSC
primary sclerosing cholangitis
- TE
transient elastography
- TEAE
treatment‐emergent adverse event
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary sclerosing cholangitis (PSC) is a chronic cholestatic disease characterized by inflammation and fibrosis of the intrahepatic and extrahepatic bile ducts of the liver.( 1 ) Currently, PSC has an incidence of 1 in 100,000 worldwide and a prevalence of 6‐16 per 100,000 in North America and Europe, and is therefore considered a rare disease.( 2 , 3 , 4 , 5 ) PSC is characterized by multifocal bile duct strictures and dilatations, leading to cholestasis, liver fibrosis, cirrhosis, portal hypertension, liver failure, and cholangiocarcinoma, a major cause of death in patients with PSC.( 6 , 7 , 8 ) The etiology of PSC is unclear; however, inflammatory bowel disease (IBD) is a major risk factor, with current estimates suggesting that 75% of patients with PSC have IBD.( 9 ) The pathogenesis of PSC is purported to be based on a genetic architecture of defined susceptibility genes and activation of the hepatic innate immune response by as yet undefined antigens. Consequently, proinflammatory cytokines and chemokines are secreted, which in turn induce cholangiocyte injury, attracting and activating inflammatory cells, thus perpetuating the inflammatory response.( 10 , 11 ) This results in progressive bile duct damage, cholestasis, and eventually fibrosis with organ dysfunction.
There are no approved effective therapies for PSC, and current pharmacological options, most of which have not demonstrated sufficient clinical benefit, offer only management of complications and symptoms. Disease management is dependent on the stage of PSC and extent of progression, with liver transplantation the only strategy that can alter outcomes and survival. Ursodeoxycholic acid (UDCA) therapy improves the liver function profile in some patients; however, whether UDCA has a beneficial effect on long‐term outcomes and survival remains unclear.( 12 , 13 ) Therefore, there is a need to identify safe treatments that can alter the disease course and offer sustained clinical benefit to patients living with PSC.
Monocyte chemotactic protein‐1 (MCP‐1) is a major chemokine that regulates the migration and infiltration of monocytes and macrophages. The role of monocytes, macrophages, Kupffer cells, and hepatic stellate cells in liver inflammation and fibrogenesis has been demonstrated.( 14 ) C‐C chemokine receptors type 2 (CCR2) and type 5 (CCR5) are the main receptors for MCP‐1 and RANTES (regulated on activation, normal T cell expressed and secreted chemokine), expressed on monocytes, macrophages, Kupffer cells, and hepatic stellate cells, and their role in the activation of these cells has been established.( 15 , 16 ) Moreover, the overexpression of MCP‐1 has been observed in PSC patient cholangiocytes and in livers of PSC preclinical models.( 17 , 18 ) These observations suggest that strategies that block the activation of MCP‐1 may be of therapeutic benefit in PSC.
Cenicriviroc (CVC) is a novel, once‐daily, dual CCR2 and CCR5 antagonist with nanomolar potency.( 19 ) CVC has demonstrated anti‐inflammatory and antifibrotic activity across multiple animal models of liver disease.( 19 , 20 , 21 , 22 , 23 ) Preclinical studies, including a rat bile duct ligation model of cholestasis, demonstrated that CVC (in combination with all‐trans retinoic acid) was associated with reduced liver necrosis, hepatic fibrosis, and bile duct proliferation; these therapeutic effects were also confirmed in Mdr2−/− mice.( 22 ) Mechanistically, CVC blocks the recruitment, migration, and infiltration of proinflammatory monocytes and macrophages into the liver through CCR2 antagonism. Recently, treatment with CVC was found to attenuate macrophage recruitment, liver injury, and fibrosis in a mouse model of PSC.( 24 ) In addition, CVC has shown antifibrotic activity and evidence of target engagement in the form of an increase in MCP‐1 in participants with nonalcoholic steatohepatitis (NASH).( 25 ) Given the key role that CCR2 and CCR5 play at the junction of inflammation and fibrosis,( 16 , 26 , 27 , 28 ) herein we evaluated the effect of CVC in adult participants with PSC by assessing its effects on alkaline phosphatase (ALP), a proposed surrogate marker for PSC disease progression, over 24 weeks of treatment. Findings from this study will expand our understanding of CVC activity in PSC.
Methods
Study Design
This was a single‐arm, open‐label, exploratory, phase 2 study evaluating the effects of CVC 150 mg daily in adult participants with PSC over 24 weeks of treatment. A total of 25 participants were planned for enrollment across four sites in the United States and four sites in Canada. The study was conducted from March 2016 to August 2017. The study consisted of a screening visit, during which participants were assessed for eligibility, which occurred within 6 weeks of the baseline visit. The first dose was administered at the baseline visit (day 1), and participants returned to the clinic for visits at weeks 4, 8, 12, 16, and 24. A follow‐up visit occurred at week 28. CVC was administered orally once daily every morning with food. All authors had access to study data and reviewed and approved the final manuscript.
Participants
Eligible participants were adults (aged 18‐75 years) with a clinical diagnosis of PSC based on cholangiographic evidence of multifocal strictures of the intrahepatic and/or extrahepatic bile ducts or histologic evidence requiring the presence of fibro‐obliterative lesions: evidence of chronic cholestatic liver disease for at least 6 months and a serum ALP ≥ 1.5 times the upper limit of normal (ULN). Participants with or without IBD were allowed. The key exclusion criteria were alternative causes of liver disease, small duct PSC, and cirrhosis. Full eligibility criteria are provided within the study protocol in the Supporting Information. The study protocol was approved by the local institutional review board or independent ethics committee of each center. The study was conducted in accordance with the Declaration of Helsinki, applicable local laws and regulations, and International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use E6 Guideline for Good Clinical Practice. All participants provided written, informed consent before study initiation. Participants could continue their standard‐of‐care medications but had to be on stable therapy for at least 28 days (≥ 3 months for UDCA therapy at a dose of ≤ 20 mg/kg/day) before baseline, whereas concomitant use of fibrates was not allowed during the study. A detailed list of disallowed medications is provided in the study protocol in the Supporting Information.
Study Outcomes and Assessments
Several liver parameters (ALP, alanine aminotransferase [ALT], aspartate aminotransferase [AST], serum albumin, serum bile acids, direct bilirubin, total bilirubin, and gamma‐glutamyl transferase [GGT]) were measured over 24 weeks of treatment and compared with baseline levels. At week 24, changes in transient elastography (TE), immunoglobulin G (IgG) total, IgG1, IgG4, and partial Mayo score for IBD activity were also evaluated.
Safety and tolerability endpoints included assessment of adverse events, fasting clinical laboratory measurements (hematology, chemistry, and urinalysis), vital sign measurements, electrocardiograms, and physical examinations.
Statistical Analyses
The primary endpoint was the percent change in ALP from baseline through week 24. A potential range of −20% for worsening to 80% for improvement in percent ALP change from baseline to 24 weeks was assumed. Based on this supposition, a 25% SD was applied and the study was designed with 80% power using a two‐sided 95% confidence interval (CI) for the mean percent change from baseline to 24 weeks to have its lower limit exceed 15%, if the true mean percent change over the 24 weeks was at least 30%.
The percent change from baseline was defined as 100 × (value at each visit – baseline value) / baseline value. The baseline value was defined as the last nonmissing value on or before the baseline visit (day 1). Percent changes in ALP from baseline were summarized using descriptive statistics. A repeated‐measures mixed model was fitted to the data for all pretreatment and on‐treatment visits. A linear mixed‐effect repeated‐measures model was used with percent change from baseline as the dependent variable, the visit as a fixed effect, the baseline score and baseline score by visit interaction as covariates, and subject as a random effect, to model correlations among the within‐subject repeated measures. Through this model, 95% CIs were calculated for the mean percent change at each visit, with percent change from baseline at week 24 being the primary endpoint estimate. Moreover, a comparison of pretreatment change (screening value – baseline value) per week with the on‐treatment change (value at each visit – baseline value) per week was made through an estimate specification and the corresponding 95% CI. Descriptive comparisons were made for participants with a history of IBD versus no history of IBD at screening, and for participants who received UDCA for at least 3 months versus no UDCA for at least 3 months before screening.
Secondary efficacy endpoints were the proportion of participants who achieved ALP normalization (as defined by the central laboratory reference ranges) at week 24; the proportion of participants who achieved ALP decrease to < 1.5 times the ULN; and the proportion of participants who achieved a 50% decrease in ALP from baseline at each visit. For descriptive responder criteria, such as either 50% ALP decrease from baseline to week 24 or a decrease to < 1.5 times the ULN, the proportion of participants achieving the targets and the two‐sided 95% CIs were provided for each pretreatment and on‐treatment visit using exact binomial CI.
Evaluation of efficacy and participant demographics was based on the intent‐to‐treat (ITT) population, which included all enrolled participants who received at least one dose of study treatment. Evaluation of safety endpoints was conducted using the safety population, which included all enrolled participants who received at least one dose of study drug. The two analysis sets were identical; however, participants who received only one dose of study drug and did not return for any postbaseline visits were included in the ITT analysis set, although they only provided data for safety assessments and no efficacy measurements. Data were analyzed as recorded, and no imputations were made for missing data. Additional details related to statistical analysis are described in the study protocol and statistical analysis plan included in the Supporting Information.
Results
Participant Disposition
A total of 48 patients were screened, of whom 24 met all of the protocol‐defined eligibility criteria and were included in both the ITT and safety population analysis sets. Twenty participants completed the 24‐week study, and 4 participants prematurely discontinued treatment: 1 due to adverse events (asthenia, fatigue, arthralgia, myalgia, and decreased appetite), 1 due to noncompliance with administration of the study drug, 1 due to a protocol violation, and 1 participant due to elevation in liver enzymes that required further evaluation (Supporting Fig. S1).
Baseline characteristics of the enrolled participants are summarized in Table 1. The mean age was 43 years (range: 23, 66 years), 50% were female, and the mean body mass index was 25 kg/m2 (range: 19, 33 kg/m2). Most of the participants had IBD at baseline (17 participants; 71%), mostly with ulcerative colitis (14 participants; 58%). Assessment of IBD activity indicated normal to mild disease in all 19 participants who were evaluated; assessment was not performed for 5 participants.
Table 1.
Participant Demographics and Baseline Characteristics
| Characteristic | CVC 150 mg (n = 24) |
|---|---|
| Demographics | |
| Age (years), mean (SD) | 43.3 (12.9) |
| Female, n (%) | 12 (50.0) |
| White, n (%) | 21 (87.5) |
| Hispanic or Latino, n (%) | 2 (8.3) |
| BMI (kg/m2), mean, (SD) | 25.2 (3.7) |
| IBD, n (%) | |
| Ulcerative colitis | 14 (58.3) |
| Crohn’s disease | 2 (8.3) |
| Indeterminate IBD | 1 (4.2) |
| No IBD | 7 (29.2) |
| IBD activity: Physician’s Global Assessment*, n (%) | |
| Normal | 15 (62.5) |
| Partial Mayo score | 4 (16.7) |
| Mild disease | 4 (16.7) |
| Moderate disease | 0 |
| Severe disease | 0 |
| Assessment not performed | 5 (20.8) |
| Received UDCA for at least 3 months before screening, n (%) | 7 (29.2) |
The partial Mayo score was used only for participants with IBD.
Abbreviation: BMI, body mass index.
Prior or concomitant medication use was reported by all 24 participants. A summary of prior and concomitant medication use is presented in Supporting Table S1. UDCA use for at least 3 months before screening was reported in 7 participants (29%) (Table 1).
Primary Endpoint
The primary efficacy endpoint for this study was the percent change in ALP from baseline to week 24. At baseline, the median ALP was 369.0 U/L (range: 173, 1,377 U/L). The linear mixed‐model analysis of mean (95% CI) change in ALP from baseline to week 24 was −3.111% (−15.47, −9.248). At the end of the 24‐week treatment period, changes in ALP were highly variable among participants, with a median reduction from baseline of −49.5 U/L (range: −460, 416 U/L), which corresponds to a median 18% reduction (range: −46%, 89%) (Table 2). A total of 11 participants (46%) had an increase in ALP of at least 18% during the study. Data from individual participants are depicted in Fig. 1 to show the intersubject variability, with some participants showing decreases in percent ALP throughout the study. ALP levels fluctuated by ±10% between screening and baseline in 13 of 24 participants, indicating that intrasubject variability occurred regardless of treatment initiation.
Table 2.
Summary of Change From Baseline to Week 24 in Serum ALP—Primary Endpoint
| Serum ALP | Baseline Absolute Value (U/L) | Week 24 Absolute Value (U/L) | Week 24 Change From Baseline (U/L) | Week 24 Change From Baseline (%) |
|---|---|---|---|---|
| n | 24 | 20 | 20 | 20 |
| Mean (SD) | 420.7 (262.9) | 382.7 (230.8) | −32.7 (175.1) | −4.5 (34.8) |
| Median | 369.0 | 295.5 | −49.5 | −18.0 |
| Min, Max | 173, 1,377 | 109, 917 | −460, 416 | −46, 89 |
| LS mean | −3.111 | |||
| 95% CI of LS mean | (−15.5; 9.2) |
Baseline is defined as the last available predose value. A linear mixed‐effect repeated‐measures model was used with percent change from baseline as the dependent variable, visit (screening, weeks 4, 8, 12, 16, and 24) as fixed factors, baseline score and baseline score by visit interaction as covariates, and subject included as a random effect to model correlations among the within‐subject repeated measures. ALP (%) on‐treatment change = (value at each visit – baseline value) / baseline value × 100. Denominator degrees of freedom were estimated using the Kenward–Roger method.
Abbreviations: LS, least squares; Max, maximum; Min, minimum.
Fig. 1.
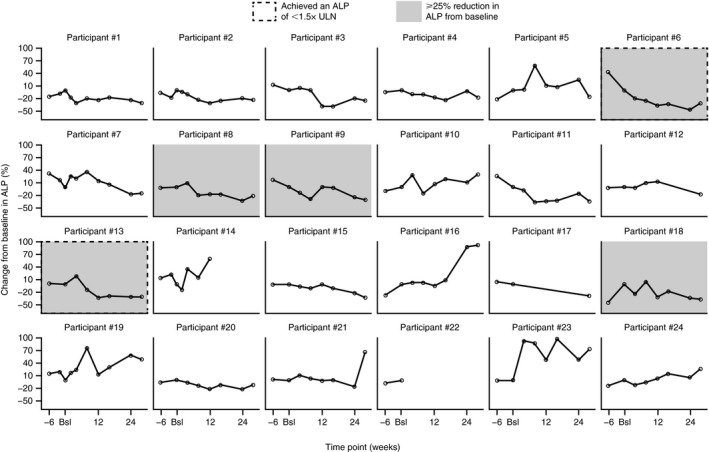
Percentage change in ALP levels from baseline in each individual participant over 24 weeks. Data are based on the intent‐to‐treat population analysis set and were evaluated at the screening visit (week −6), baseline (day 1), weeks 4, 8, 12, 16, and 24, and at the safety follow‐up visit (week 28). Abbreviation: Bsl, baseline.
Analysis of the primary endpoint was performed for four subgroups: (1) participants who did or (2) did not receive UDCA therapy for at least 3 months before screening, and (3) participants with or (4) without history of IBD at screening. At baseline, the median ALP for participants who received UDCA therapy for at least 3 months before screening was 386.0 U/L (range: 212, 680 U/L; n = 7), whereas the mean ALP for participants who did not receive prior UDCA therapy was 354.0 U/L (range: 173, 1,377 U/L; n = 17) (Fig. 2A). Following 24 weeks of CVC treatment, the percent ALP change in the 16 participants who did not receive prior UDCA therapy (median: −20.3% [range: −46%, 89%]) was higher compared with the 4 participants who received UDCA therapy at least 3 months before screening (median: 0.9% [range: −17%, 59%]); however, the higher rate of missing data from participants with prior UDCA therapy may have affected these results. Overall, these results indicate a potential treatment effect of CVC on ALP levels over 24 weeks in participants who did not receive prior UDCA therapy.
Fig. 2.
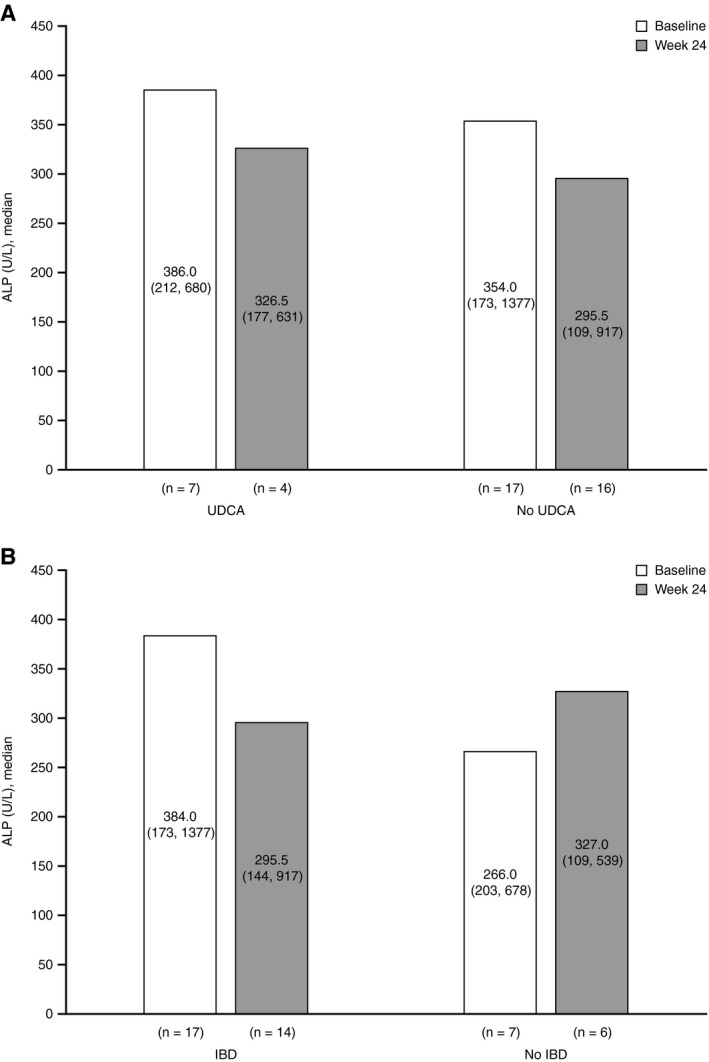
Summary of change from baseline to week 24 in serum ALP by UDCA status (A) and IBD status (B) at screening. Results are displayed as median (minimum, maximum).
At baseline, the median ALP for participants with IBD was 384.0 U/L (range: 173, 1,377 U/L; n = 17), while the median ALP for participants without IBD was 266.0 U/L (range: 203, 678 U/L; n = 7). Following treatment, no differences in percent ALP change were observed between participants with IBD (median: −18% [range: −33%, 89%]) and without IBD (median: −18.5% [range: −46%, 25%]) at screening (Fig. 2B).
Secondary Efficacy Endpoints
The key secondary efficacy endpoints included the proportion of participants who at week 24 achieved ALP normalization, a decrease to < 1.5 times the ULN, or a 50% decrease from baseline at each visit. Of the 20 participants who completed the study, 2 (10%; [0.68; 0.98]95%CI) achieved the secondary endpoint of a decrease in ALP to < 1.5 times the ULN, with the first participant achieving the endpoint at week 8 and the second at week 12 of the study; both participants had baseline ALP values > 1.5 times the ULN (203 U/L [1.9 times the ULN] and 230 U/L [1.8 times the ULN], respectively). No participants achieved ALP normalization or a 50% decrease from baseline at any time during the study. The overall response rate (i.e., the proportion of participants who achieved a 50% decrease in ALP from baseline or a decrease to < 1.5 times the ULN) was 10% (2 participants; [0.68; 0.98]95%CI) at week 24. As no participants achieved the secondary efficacy endpoint of a 50% decrease in ALP from baseline, a post hoc analysis was performed to evaluate the number of participants who achieved a 25% decrease in ALP from baseline. Of the 20 total participants, 5 (25%) achieved this endpoint ([0.50; 0.91]95%CI) at week 24; all 5 of these participants had elevated GGT levels at week 24. A total of 4 (20%) had a ≥ 25% increase in ALP at week 24. A summary of key secondary efficacy endpoints is provided in Table 3.
Table 3.
Summary of Key Secondary Efficacy Endpoints at Week 24
| Secondary Efficacy Endpoints | Overall | ||
|---|---|---|---|
| n | Yes | 95% CI | |
| Proportion of participants who achieved ALP normalization | 20 | 0 (0.0%) | (0.83; 1.00) |
| Proportion of participants who achieved ALP of < 1.5 times the ULN | 20 | 2 (10%) | (0.68; 0.98) |
| Proportion of participants who achieved a 50% decrease in ALP from baseline | 20 | 0 (0.0%) | (0.83; 1.00) |
| Overall responders (i.e., proportion of participants who achieved a 50% decrease in ALP from baseline or a decrease to < 1.5 times the ULN) | 20 | 2 (10%) | (0.68; 0.98) |
| Additional post hoc analysis | |||
| Proportion of participants who achieved a 25% decrease in ALP from baseline | 20 | 5 (25%) | (0.50; 0.91) |
95% CIs were obtained by binomial test for proportions.
Other efficacy outcomes included changes in liver parameters other than ALP (ALT, albumin, AST, direct bilirubin, GGT, total bilirubin, serum bile acids, IgG total, and IgG1). A high variability in all of these parameters was observed, and no clear trends were evident at week 24 compared with baseline levels. A summary of change in liver biomarkers by ALP reduction (≥ 25% vs. < 25%) is provided in Table 4. Median decreases in GGT, ALT, and AST were greater in participants with a ≥ 25% ALP reduction than in those with a < 25% ALP reduction.
Table 4.
Summary of Change from Baseline to Week 24 in Liver Biomarkers by ALP Reduction (≥ 25% vs. < 25%)
| Biomarker | Participants With ≥ 25% ALP Reduction (n = 5) | Participants With < 25% ALP Reduction (n = 15)* | |||
|---|---|---|---|---|---|
| Absolute Value | Change From Baseline (%) | Absolute Value | Change From Baseline (%) | ||
| GGT (U/L) | Mean (SD) | −163.4 (61.7) | −28.1 (14.5) | −5.1 (186.6) | −0.6 (24.5) |
| Median | −135.0 | −33.7 | −39.0 | −7.2 | |
| Min, Max | −235, −109 | −45, −12 | −491, 362 | −32, 44 | |
| ALT (U/L) | Mean (SD) | −73.6 (145.5) | −26.6 (34.3) | −11.9 (65.4) | 4.8 (35.1) |
| Median | −19.0 | −20.8 | −2.0 | −4.3 | |
| Min, Max | −332, 20 | −79, 17 | −233, 50 | −52, 83 | |
| AST (U/L) | Mean (SD) | −72.6 (134.5) | −32.9 (35.5) | −4.6 (48.4) | 6.4 (44.7) |
| Median | −17.0 | −27.4 | 1.0 | 3.0 | |
| Min, Max | −312, 7 | −83, 16 | −152, 58 | −57, 119 | |
| Total bilirubin (mg/dL) | Mean (SD) | −0.1 (0.2) | −11.1 (31.7) | 0.5 (1.6) | 62.3 (178.5) |
| Median | −0.2 | −28.6 | 0.1 | 20.0 | |
| Min, Max | −0.3, 0.2 | −33.3, 40.0 | −0.2, 6.2 | −40.0, 688.9 | |
| Direct bilirubin (mg/dL) | Mean (SD) | −0.1 (0.1) | −28.7 (27.9) | 0.4 (1.3) | 110.0 (319.2) |
| Median | −0.1 | −33.3 | 0.0 | 0.0 | |
| Min, Max | −0.3, 0.0 | −60.0, 0.0 | −0.1, 5.0 | −50.0, 1250.0 | |
| Albumin (g/dL) | Mean (SD) | −0.1 (0.1) | −3.13 (2.6) | 0.01 (0.3) | 0.3 (7.3) |
| Median | −0.1 | −2.3 | 0.1 | 2.3 | |
| Min, Max | −0.3, 0.0 | −6.8, 0.0 | −0.7, 0.3 | −17.5, 7.9 | |
| TE | |||||
| LSM (kPa) | Mean (SD) | 1.2 (0.7) | 16.3 (9.9) | 1.37 (5.9) | 15.6 (57.5) |
| Median | 0.9 | 14.3 | −0.3 | −4.8 | |
| Min, Max | 0.5, 2.0 | 7.8, 32.8 | −5.0, 18.6 | −41.0, 182.4 | |
| Noninvasive biomarkers of liver fibrosis | |||||
| APRI score | Mean (SD) | −1.1 (2.3) | −32.2 (38.5) | −0.1 (0.6) | 5.0 (48.0) |
| Median | −0.1 | −26.2 | −0.01 | −4.8 | |
| Min, Max | −5.2, 0.1 | −85.2, 15.6 | −2.1, 0.6 | −56.5, 116.4 | |
| FIB‐4 Index score | Mean (SD) | −1.0 (1.7) | −24.3 (30.2) | 0.04 (0.4) | 6.3 (40.7) |
| Median | −0.1 | −15.8 | 0.03 | 8.8 | |
| Min, Max | −3.9, 0.04 | −65.3, 9.1 | −0.6, 1.0 | −39.2, 86.2 | |
Baseline was selected as the last available predose value.
n = 13 for noninvasive biomarkers of liver fibrosis.
Abbreviations: LSM, liver stiffness measure; Max, maximum; Min, minimum.
Details on further secondary efficacy outcomes are included in the Supporting Information.
Safety and Tolerability
Throughout the 24‐week study, a total of 20 participants (83.3%) reported at least one treatment‐emergent adverse event (TEAE), most of which were grade 1 (mild) or grade 2 (moderate) in severity (19 participants; 79.1% of the study population) (Table 5). The most common TEAEs were fatigue, rash, and dizziness, occurring in 4 participants (16.7%) each (Table 6). One serious TEAE of gallbladder polyp was reported during the study, which was considered mild in severity and unrelated to study drug; it did not result in a change in CVC dosages and was resolved following a cholecystectomy. No study‐related serious TEAEs were reported and no deaths occurred during the study.
Table 5.
Summary of Adverse Events
| Adverse Events | CVC 150 mg (n = 24), n (%) |
|---|---|
| Participants with ≥ 1 TEAEs | 20 (83.3) |
| Grade 1 | 14 (58.3) |
| Grade 2 | 5 (20.8) |
| Grade 3 | 1 (4.2) |
| Grade 4 or 5 | 0 |
| Deaths | 0 |
| Participants with ≥ 1 SAEs | 1 (4.2) |
| Gallbladder polyp | 1 (4.2) |
| Participants with ≥ 1 study drug‐related SAEs | 0 |
| Participants who discontinued treatment due to a TEAE | 2 (8.3) |
| Asthenia, fatigue, arthralgia, myalgia, decreased appetite | 1 (4.2) |
| Elevation in liver biochemistry requiring further evaluation | 1 (4.2) |
| Participants with ≥ 1 treatment‐related TEAEs | 10 (41.7) |
| Rash | 3 (12.5) |
| Rash pruritic | 1 (4.2) |
| Fatigue | 2 (8.3) |
| Flatulence | 1 (4.2) |
| Nausea | 1 (4.2) |
| Vomiting | 1 (4.2) |
| Dizziness | 1 (4.2) |
| Headache | 1 (4.2) |
| Paresthesia | 1 (4.2) |
| Elevation in liver biochemistry requiring further evaluation | 1 (4.2) |
| Blood cholesterol increased | 1 (4.2) |
| Weight decreased | 1 (4.2) |
A TEAE is any adverse event that has an onset date and time on or after the date and time of first administration of study drug. Adverse events that worsen after administration of study medication are also categorized as TEAEs.
Abbreviation: SAE, serious adverse event.
Table 6.
Summary of TEAEs Affecting > 5% of Participants
| TEAEs | CVC 150 mg (n = 24), n (%) |
|---|---|
| TEAEs affecting > 5% of participants | |
| Fatigue | 4 (16.7) |
| Rash | 4 (16.7) |
| Dizziness | 4 (16.7) |
| Headache | 3 (12.5) |
| Cough | 3 (12.5) |
| Vomiting | 3 (12.5) |
| Pruritus | 2 (8.3) |
| Rash pruritic | 2 (8.3) |
| Nausea | 2 (8.3) |
| Pyrexia | 2 (8.3) |
| Arthralgia | 2 (8.3) |
| Musculoskeletal chest pain | 2 (8.3) |
| Lower respiratory tract infection | 2 (8.3) |
A TEAE is any adverse event that has an onset date and time on or after the date and time of first administration of study drug. Adverse events that worsen after administration of study medication are also categorized as TEAEs.
Further details on safety are included in the Supporting Information.
Discussion
In this open‐label, single‐arm, proof‐of‐concept study, we evaluated the effects of CVC in adults with PSC by assessing changes in the surrogate endpoint of ALP for up to 24 weeks of treatment. Currently, there are no established surrogate endpoints for measuring PSC progression; therefore, regulatory guidance is still limited for clinical development. The International PSC Study Group published a consensus report that provided five candidate surrogate endpoints for clinical trials of PSC: ALP, TE, histology, a combination of ALP and histology, and bilirubin.( 29 , 30 ) Of these, histology, ALP, and TE are considered the most promising endpoints, although the use of histology as a primary surrogate endpoint is limited by the invasive nature of liver biopsy, at least during the early stages of drug development.
PSC is more common in men than women, has a mean patient age of 41 years, and is strongly associated with IBD.( 2 , 31 ) Although the age of our population was close to this mean, 50% were female, which is unusual. This could have skewed our results, although there is no clear evidence that sex affects outcome. Overall, the participant population in this study was reflective of the general PSC population. The primary efficacy endpoint for this study was the percent change in ALP over 24 weeks of treatment. Although percent change in ALP was used herein, the optimal measures of change in ALP, whether absolute value, percent change, or otherwise, are not known. After 24 weeks, from a mean baseline of 420.7 U/L, mean ALP levels decreased to 382.7 U/L, corresponding to a 32.7 U/L reduction. However, variability in both mean absolute values and percent change was observed, with some participants showing decreases that were observed throughout the study (Fig. 1). As the data were not normally distributed, with large intersubject variations and small sample sizes, the results were presented as median percent changes rather than mean percent changes. These showed a modest treatment effect at week 24, with a median absolute reduction in ALP of 49.5 U/L (range: −460, 416 U/L), corresponding to a median reduction of 18% (range: −46%, 89%); however, this decline does not appear to be clinically significant. ALP levels tend to fluctuate over the course of disease and can spontaneously normalize, making it difficult to determine whether observed changes were due to the effects of CVC or normal fluctuation.( 32 , 33 ) Herein, no participant achieved ALP normalization or a 50% decrease from baseline at any time, but 2 participants were overall responders (i.e., achieved a 50% decrease in ALP or a decrease to < 1.5 times the ULN). However, in a post hoc analysis, 25% of participants (5 participants) achieved a 25% decrease in ALP from baseline to week 24, with improvements observed in other liver biochemistry parameters. There have been some conflicting data on the correlation between improvements in ALP and clinical benefits in a setting of UDCA therapy. One study found no significant changes in ALP or clinical improvement with medium‐dose UDCA treatment (17‐23 mg/kg/day) over a 5‐year follow‐up period, while a second study of very‐high‐dose UDCA (28‐30 mg/kg/day) demonstrated ALP improvements yet clinical worsening over the same study period.( 13 , 34 ) Nonetheless, a recent analysis found that both UDCA‐treated and placebo‐treated patients who normalized their ALP had better prognosis than those who did not.( 35 )
Earlier preclinical data suggest that CCR2‐dependent mechanisms help control the balance between proinflammatory and resident macrophages in the colon, indicating a potential role in IBD.( 36 , 37 ) However, we did not observe any difference in change in ALP between participants who did or did not have IBD at screening: the median percent change in ALP at week 24 was −18% and −18.5%, respectively. Conversely, participants who did not receive stable UDCA therapy before screening showed a greater percentage reduction in ALP compared with participants who received prior UDCA therapy (median change: −20.3% vs. 0.9%, respectively). However, given the significant imbalance in the number of participants in each subgroup (with IBD at screening [n = 14] vs. without IBD [n = 6]; with UDCA at screening [n = 4] vs. without UDCA [n = 16]), and given that UDCA can potentially blunt responses to other drugs tested in PSC,( 38 ) no definite conclusions can be drawn from the results on the effect of CVC on these potential predictors of response. Furthermore, we observed highly variable data in other noninvasive liver parameters, including an increase in TE and decreases in median AST‐to‐platelet ratio index (APRI) and Fibrosis‐4 (FIB‐4) Index scores from baseline to week 24 (Table 4). However, given the short duration of this study, it is unlikely that changes in liver fibrosis would have been observed.
The variation in TE data during the study is likely due to changes in the degree of cholestasis and inflammation, whereas the APRI score has not been well studied in cholestatic disease. The modest reduction in FIB‐4 Index score is encouraging for a relatively short‐term study, in which intervention with CVC is unlikely to have a major effect on hepatic fibrosis. Improvements in fibrosis would likely require a longer period of CVC exposure. Greater decreases in APRI and FIB‐4 Index scores were observed in participants who achieved a decrease in ALP of ≥ 25% from baseline (n = 5) compared with those who achieved a decrease of < 25% (n = 15). It should be noted that the median liver stiffness measure in this study was 9.3 kPa, indicative of mild to moderate fibrosis, and the effects of CVC on fibrosis may be different in patients with more advanced fibrosis, as seen in the CENTAUR study in participants with NASH.( 25 )
No new safety signals were observed in this study; TEAEs were predominantly mild to moderate in severity, and the most frequent were rash, fatigue, and dizziness. These findings are in line with the safety profile from the clinical experience of over 1,100 participants treated with CVC, including those with human immunodeficiency virus and NASH.( 25 , 39 )
Some limitations are associated with this study, which necessitate caution in the interpretation of these findings. The main limitations were the small sample size and single‐arm study design, which meant that each participant was his or her own control. The variability of ALP changes among participants with PSC in this study further highlights the challenges of using this biomarker to assess the efficacy of therapeutics in this disease. In addition, unlike UDCA, the mechanism of action of CVC (inhibition of monocyte/macrophage infiltration) may have therapeutic anti‐inflammatory or antifibrotic benefits that are not captured by serum liver biochemistries or even TE during a short, exploratory study such as this one. We did not include liver histology in the study due to its invasive nature. Although histology may have informed the pharmacologic effects of CVC on macrophage liver recruitment, it is unlikely to have demonstrated any improvements associated with clinical outcomes after only 24 weeks of treatment. Recent data associating clinical outcomes with serum markers of fibrosis—including the Enhanced Liver Fibrosis test and PRO‐C3—offer hope that these markers may offer a more dynamic means of measuring treatment effects in similar small proof‐of‐concept studies.( 40 , 41 , 42 , 43 , 44 )
Data from other phase 2 studies in patients with PSC also demonstrated modest changes in ALP. In a study of obeticholic acid, a farnesoid X receptor agonist, the mean ALP decreased by 105 U/L with the 1.5‐3.0 mg dose and 110 U/L with the 5‐10 mg dose, compared with a 27 U/L reduction with placebo after 24 weeks of treatment. Patients receiving obeticholic acid had greater reductions in ALP at week 24 in the absence of UDCA than with UDCA.( 38 ) In a 12‐week study of cilofexor (GS‐9674), another farnesoid X receptor agonist, dose‐dependent reductions in ALP were observed after 12 weeks of treatment (−21 U/L with the 30 mg dose and −73 U/L with the 100 mg dose) compared with an 8 U/L increase with placebo.( 45 ) In a phase 2 study in patients with PSC, 24‐norursodeoxycholic acid (a side chain‐shortened C23 homologue of UDCA) doses of 500, 1,000, and 1,500 mg/day reduced ALP levels by 12.3%, 17.3%, and 26.0%, respectively, after 12 weeks of treatment, with an increase (1.2%) observed with placebo.( 46 ) Furthermore, in a recent phase 2 study of NGM282, an engineered analog of fibroblast growth factor‐19, minimal decreases in mean ALP were observed with the 3 mg dose (−3 U/L), whereas increases were seen with the 1 mg dose (26 U/L) and placebo (5 U/L) after 12 weeks of treatment.( 47 ) Collectively, these recent results highlight not only the challenges of treating PSC but also the need to identify biomarkers beyond ALP.
Given that PSC is an inflammatory liver disease, the role of macrophages has recently been explored, with evidence suggesting that pharmacologic inhibition of macrophage recruitment to the liver through monocyte chemo‐attractants such as the CCR2/CCL2 axis may reduce PSC‐like liver injury.( 24 ) In addition to CCR2 reduction, data from the CENTAUR study indicate that CVC reduces baseline levels of systemic markers of inflammation, including interleukin‐1, interleukin‐6, high‐sensitivity C‐reactive protein, and fibrinogen, highlighting the potential for further treatment opportunities in PSC.( 25 , 48 )
In conclusion, although there are currently no available treatments for PSC, this exploratory study evaluated the preliminary efficacy and safety of CVC in PSC for the first time. Our findings suggest that treatment with CVC 150 mg for 24 weeks resulted in no unexpected safety signals and a modest decrease (median of 18%) in the endpoint of ALP in this PSC population. Although not meeting the primary endpoint, our study provides encouraging exploratory data that CVC may have a role in targeting inflammation in PSC. More work is required to define the optimum CVC dose to inhibit hepatic recruitment of CCR5/CCR2 leukocytes, and given the modest changes seen with CVC, larger, more robust studies are clearly needed. Importantly, novel biomarkers are needed that reflect clinically meaningful changes, to design future studies and the optimal duration to target secondary endpoints, such as fibrosis.
Author Contributions
BE, EL, PV, and LF contributed to the study concept and design. BE, CLB, AJML, EL, PV, EBM, JA, KKY, PP, JJF, GM, and CL were principal investigators, supervised study‐related activities at respective study sites, and contributed to the analysis and interpretation of data. All authors contributed to the drafting, critical revision for intellectual content, and final approval of the manuscript. [Corrections added on February 26, 2021, after online publication: Author Contributions have been added.].
Supporting information
Supplementary Material
Acknowledgments
The sponsor and authors thank the study participants and their families, the study investigators, research coordinators, and study staff. Writing and editorial assistance was provided to the authors by Germaine D. Agollah, PhD, of Allergan plc, and Rob Kite, BSc (Hons), of Complete HealthVizion, Inc., Chicago, IL, USA, funded by Allergan plc, Dublin, Ireland. [Corrections added on February 2, 2021, after first online publication: The preceding sentence has been added.]
Supported by Allergan plc, Dublin, Ireland.
Potential conflict of interest: Dr. Levy consults for and received grants from CymaBay, Genfit, GSK, Pliant, TARGET Pharmasolutions and Cara Therapeutics. She consults for Mirum, Flashlight, and Shire. She received grants from Gilead, Enanta, Zydus, Eli Lilly, Intercept, Alnylam, Durect, High Tide, Genkyotex, Novartis Mitsubishi and NGM. She receives royalities from Up‐to‐Date. Dr. Martins owns stock in and is employed by AbbVie. Dr. Feld consults for and received grants from AbbVie and Gilead. He consults for Abbott, Enanta and Roche. He received grants from Janssen and Wako/Fujifilm. Dr. Fischer is employed by Advernum. He is a former Allergan/AbbVie employee and shareholder. Dr. Lefebvre is a former employee of Tobira/Allergan. Dr. Martins owns stock in and is employed by Allergan. Dr. Montano‐Loza advises Intercept and Pilant. He received grants from Gilead and Novartis. Dr. Pockros advises, is on the speakers’ bureau, and received grants from Gilead, AbbVie and Intercept.
Trial registration: NCT02653625, PERSEUS.
Data Availability Statement
Data reported in this manuscript are available within the article and its Supporting Information.
Allergan will share de‐identified participant‐level data and/or study‐level data, including protocols and clinical study reports, for phase 2‐4 trials completed after 2008 that are registered on ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union, and the primary manuscript from the trial must be published before data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found at http://www.allerganclinicaltrials.com/.
References
Author names in bold designate shared co‐first authorship.
- 1. Maggs JR, Chapman RW. An update on primary sclerosing cholangitis. Curr Opin Gastroenterol 2008;24:377‐383. [DOI] [PubMed] [Google Scholar]
- 2. Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003;125:1364‐1369. [DOI] [PubMed] [Google Scholar]
- 3. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013;58:2045‐2055. [DOI] [PubMed] [Google Scholar]
- 4. Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, et al. Incidence of primary sclerosing cholangitis: a systematic review and meta‐analysis. Hepatology 2011;53:1590‐1599. [DOI] [PubMed] [Google Scholar]
- 5. Kaplan GG, Laupland KB, Butzner D, Urbanski SJ, Lee SS. The burden of large and small duct primary sclerosing cholangitis in adults and children: a population‐based analysis. Am J Gastroenterol 2007;102:1042‐1049. [DOI] [PubMed] [Google Scholar]
- 6. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660‐678. [DOI] [PubMed] [Google Scholar]
- 7. Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology 2013;145:521‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson Å, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36:321‐327. [DOI] [PubMed] [Google Scholar]
- 9. Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013;382:1587‐1599. [DOI] [PubMed] [Google Scholar]
- 10. Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4‐NF‐κB‐mediated inflammatory response in mice. Gastroenterology 2011;141:1498‐1508.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spirlì C, Nathanson MH, Fiorotto R, Duner E, Denson LA, Sanz JM, et al. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 2001;121:156‐169. [DOI] [PubMed] [Google Scholar]
- 12. Lindström L, Hultcrantz R, Boberg KM, Friis‐Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2013;11:841‐846. [DOI] [PubMed] [Google Scholar]
- 13. Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harada K, Nakanuma Y. Innate immunity in the pathogenesis of cholangiopathy: a recent update. Inflamm Allergy Drug Targets 2012;11:478‐483. [DOI] [PubMed] [Google Scholar]
- 15. Marra F, DeFranco R, Grappone C, Parola M, Milani S, Leonarduzzi G, et al. Expression of monocyte chemotactic protein‐1 precedes monocyte recruitment in a rat model of acute liver injury, and is modulated by vitamin E. J Investig Med 1999;47:66‐75. [PubMed] [Google Scholar]
- 16. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310‐G1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N‐ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014;59:2263‐2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakken KE, Nygård S, Haaland T, Berge KE, Arnkværn K, Ødegaard A, et al. Multiple inflammatory‐, tissue remodelling‐ and fibrosis genes are differentially transcribed in the livers of Abcb4 (‐/‐) mice harbouring chronic cholangitis. Scand J Gastroenterol 2007;42:1245‐1255. [DOI] [PubMed] [Google Scholar]
- 19. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 2016;11:e0158156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C‐C motif) receptor 2‐positive monocytes aggravate the early phase of acetaminophen‐induced acute liver injury. Hepatology 2016;64:1667‐1682. [DOI] [PubMed] [Google Scholar]
- 21. Puengel T, Krenkel O, Kohlhepp M, Lefebvre E, Luedde T, Trautwein C, et al. Differential impact of the dual CCR2/CCR5 inhibitor cenicriviroc on migration of monocyte and lymphocyte subsets in acute liver injury. PLoS One 2017;12:e0184694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu D, Cai SY, Mennone A, Vig P, Boyer JL. Cenicriviroc, a cytokine receptor antagonist, potentiates all‐trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int 2018;38:1128‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270‐1283. [DOI] [PubMed] [Google Scholar]
- 24. Guicciardi ME, Trussoni CE, Krishnan A, Bronk SF, Lorenzo Pisarello MJ, O'Hara SP, et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatol 2018;69:676‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo‐controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol 2012;3:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets 2011;10:509‐536. [DOI] [PubMed] [Google Scholar]
- 28. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009;50:185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ponsioen CY, Chapman RW, Chazouillères O, Hirschfield GM, Karlsen TH, Lohse AW, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an International PSC Study Group consensus process. Hepatology 2016;63:1357‐1367. [DOI] [PubMed] [Google Scholar]
- 30. Ponsioen CY, Lindor KD, Mehta R, Dimick‐Santos L. Design and endpoints for clinical trials in primary sclerosing cholangitis. Hepatology 2018;68:1174‐1188. [DOI] [PubMed] [Google Scholar]
- 31. Boonstra K, de Vries EM, van Geloven N, van Erpecum KJ, Spanier M, Poen AC, et al. Risk factors for primary sclerosing cholangitis. Liver Int 2016;36:84‐91. [DOI] [PubMed] [Google Scholar]
- 32. Balasubramaniam K, Wiesner RH, LaRusso NF. Primary sclerosing cholangitis with normal serum alkaline phosphatase activity. Gastroenterology 1988;95:1395‐1398. [DOI] [PubMed] [Google Scholar]
- 33. Olsson R, Broomé U, Danielsson Å, Hägerstrand I, Järnerot G, Lööf L, et al. Spontaneous course of symptoms in primary sclerosing cholangitis: relationships with biochemical and histological features. Hepatogastroenterology 1999;46:136‐141. [PubMed] [Google Scholar]
- 34. Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5‐year multicenter, randomized, controlled study. Gastroenterology 2005;129:1464‐1472. [DOI] [PubMed] [Google Scholar]
- 35. Stanich PP, Björnsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis 2011;43:309‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2‐dependent macrophages promotes colonic inflammation. J Immunol 2010;184:6843‐6854. [DOI] [PubMed] [Google Scholar]
- 37. Ye X, Liu S, Hu M, Song Y, Huang H, Zhong Y. CCR5 expression in inflammatory bowel disease and its correlation with inflammatory cells and β‐arrestin2 expression. Scand J Gastroenterol 2017;52:551‐557. [DOI] [PubMed] [Google Scholar]
- 38. Kowdley KV, Bowlus CL, Levy C, Vuppalanchi R, Floreani A, Andreone P, et al. The AESOP trial: a randomized, double‐blind, placebo‐controlled, phase 2 study of obeticholic acid in patients with primary sclerosing cholangitis. Hepatology 2017;66:1254A. Abstract number: LB‐2. [Google Scholar]
- 39. Thompson M, Saag M, DeJesus E, Gathe J, Lalezari J, Landay AL, et al. A 48‐week randomized phase 2b study evaluating cenicriviroc versus efavirenz in treatment‐naive HIV‐infected adults with C‐C chemokine receptor type 5‐tropic virus. AIDS 2016;30:869‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Vries EMG, Färkkilä M, Milkiewicz P, Hov JR, Eksteen B, Thorburn D, et al. Enhanced liver fibrosis test predicts transplant‐free survival in primary sclerosing cholangitis, a multi‐centre study. Liver Int 2017;37:1554‐1561. [DOI] [PubMed] [Google Scholar]
- 41. Vesterhus M, Holm A, Hov JR, Nygård S, Schrumpf E, Melum E, et al. Novel serum and bile protein markers predict primary sclerosing cholangitis disease severity and prognosis. J Hepatol 2017;66:1214‐1222. [DOI] [PubMed] [Google Scholar]
- 42. Vesterhus M, Hov JR, Holm A, Schrumpf E, Nygård S, Godang K, et al. Enhanced liver fibrosis score predicts transplant‐free survival in primary sclerosing cholangitis. Hepatology 2015;62:188‐197. [DOI] [PubMed] [Google Scholar]
- 43. Nielsen MJ, Veidal SS, Karsdal MA, Orsnes‐Leeming DJ, Vainer B, Gardner SD, et al. Plasma Pro‐C3 (N‐terminal type III collagen propeptide) predicts fibrosis progression in patients with chronic hepatitis C. Liver Int 2015;35:429‐437. [DOI] [PubMed] [Google Scholar]
- 44. Leeming DJ, Birot S, Goodman Z, McKibben A, Seyedkazemi S, Muthian S, et al. Percent change in plasma collagen type III formation (PRO‐C3) is related to change in liver fibrosis stage: CENTAUR study. Poster P2324 presented at the American Association for the Study of Liver Diseases: The Liver Meeting; November 9‐13, 2018; San Francisco, CA. [Google Scholar]
- 45. Trauner M, Gulamhusein A, Hameed B, Caldwell SH, Shiffman ML, Landis CS, et al. The nonsteroidal farnesoid X receptor agonist GS‐9674 improves liver biochemistry and decreases serum bile acids in patients with primary sclerosing cholangitis: a phase 2, randomized, placebo‐controlled trial. Oral presentation 43 presented at the American Association for the Study of Liver Diseases: The Liver Meeting; November 9‐13, 2018; San Francisco, CA. [Google Scholar]
- 46. Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Färkkilä M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol 2017;67:549‐558. [DOI] [PubMed] [Google Scholar]
- 47. Hirschfield GM, Chazouillères O, Drenth JP, Thorburn D, Harrison S, Landis CS, et al. NGM282, an engineered analogue of FGF19, significantly improves markers of bile acid synthesis, hepatic injury and fibrosis in PSC patients: results of a phase 2, multicenter, randomized, double‐blind, placebo‐controlled trial. J Hepatol 2018;68:S101. Abstract number: LBO‐002. [Google Scholar]
- 48. Ratziu V, Sanyal A, Francque S, Wong VW, Loomba R, Goodman Z, et al. Cenicriviroc treatment for adults with non‐alcoholic steatohepatitis: year 2 analysis of the phase 2b CENTAUR study. Oral presentation presented at the European Association for the Study of the Liver: The International Liver Congress; April 11‐15, 2018; Paris, France. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Data reported in this manuscript are available within the article and its Supporting Information.
Allergan will share de‐identified participant‐level data and/or study‐level data, including protocols and clinical study reports, for phase 2‐4 trials completed after 2008 that are registered on ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union, and the primary manuscript from the trial must be published before data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found at http://www.allerganclinicaltrials.com/.