

Abstract
The CENP‐A nucleosome is a key structure for kinetochore assembly. Once the CENP‐A nucleosome is established in the centromere, additional proteins recognize the CENP‐A nucleosome to form a kinetochore. CENP‐C and CENP‐N are CENP‐A binding proteins. We previously demonstrated that vertebrate CENP‐C binding to the CENP‐A nucleosome is regulated by CDK1‐mediated CENP‐C phosphorylation. However, it is still unknown how the phosphorylation of CENP‐C regulates its binding to CENP‐A. It is also not completely understood how and whether CENP‐C and CENP‐N act together on the CENP‐A nucleosome. Here, using cryo‐electron microscopy (cryo‐EM) in combination with biochemical approaches, we reveal a stable CENP‐A nucleosome‐binding mode of CENP‐C through unique regions. The chicken CENP‐C structure bound to the CENP‐A nucleosome is stabilized by an intramolecular link through the phosphorylated CENP‐C residue. The stable CENP‐A‐CENP‐C complex excludes CENP‐N from the CENP‐A nucleosome. These findings provide mechanistic insights into the dynamic kinetochore assembly regulated by CDK1‐mediated CENP‐C phosphorylation.
Keywords: CENP‐A, CENP‐C, Cryo‐EM, kinetochore, phosphorylation
Subject Categories: Cell Cycle; Chromatin, Epigenetics, Genomics & Functional Genomics; Structural Biology
Observation of how an extended CENP‐C peptide binds centromeric nucleosomes explains regulation of kinetochore assembly by CDK1 phosphorylation and the mutual exclusion of CENP‐C/CENP‐N recognition.

Introduction
The centromere is an essential genomic region for accurate chromosome segregation. In most organisms, the centromere is localized to a particular locus on each chromosome by sequence‐independent epigenetic mechanisms. The histone H3 variant centromere protein A (CENP‐A) is a key epigenetic marker for centromere specification (Allshire & Karpen, 2008; Black & Cleveland, 2011; Perpelescu & Fukagawa, 2011; Westhorpe & Straight, 2013; Fukagawa & Earnshaw, 2014). CENP‐A forms a nucleosome with other canonical histones (H2A/B and H4) that share a fundamental architecture with a canonical histone H3‐containing nucleosome (Black & Cleveland, 2011; Tachiwana et al, 2011). However, downstream kinetochore components are specifically associated with CENP‐A nucleosomes, but not with H3 nucleosomes, to form a functional kinetochore structure (Foltz et al, 2006; Izuta et al, 2006; Okada et al, 2006; Hori et al, 2008; Amano et al, 2009; Nishino et al, 2012; Weir et al, 2016; Yan et al, 2019). Therefore, to facilitate recognition by kinetochore proteins, the CENP‐A nucleosome should be structurally different from the H3 nucleosome. The functional sites of CENP‐A have been identified at the N‐terminal, RG loop, and C‐terminal tail regions in the primary sequence (Black & Cleveland, 2011; Tachiwana et al, 2011). Furthermore, structural information on the CENP‐A nucleosome bound to kinetochore proteins is essential for an in‐depth understanding of the mechanism by which kinetochore proteins distinguish the CENP‐A nucleosome from the canonical histone H3 nucleosome.
Among kinetochore proteins, CENP‐C (Kato et al, 2013b; Falk et al, 2015; Falk et al, 2016; Guo et al, 2017; Ali‐Ahmad et al, 2019; Watanabe et al, 2019) and CENP‐N (Carroll et al, 2009; Pentakota et al, 2017; Chittori et al, 2018; Tian et al, 2018; Allu et al, 2019) are direct CENP‐A binding proteins. Binding of these kinetochore proteins to the CENP‐A nucleosome triggers the assembly of other kinetochore proteins. Therefore, it is critical to address how CENP‐C and CENP‐N, individually and/or together, specifically bind to the CENP‐A nucleosome. Various structural studies have been carried out to understand the CENP‐A nucleosome recognition mechanisms of CENP‐C and/or CENP‐N (Carroll et al, 2009; Kato et al, 2013b; Pentakota et al, 2017; Chittori et al, 2018; Tian et al, 2018; Ali‐Ahmad et al, 2019; Allu et al, 2019; Watanabe et al, 2019). These studies have revealed several fundamental structural aspects of the molecular interfaces between the CENP‐A nucleosome and CENP‐C and CENP‐N. First, the central domain and CENP‐C motif in human CENP‐C, which share a sequence motif essential for binding to the CENP‐A nucleosome, recognize the CENP‐A C‐terminal tail and acidic patch of H2A/B in the CENP‐A nucleosome. Second, the CENP‐N N‐terminal domain binds to the RG loop in the CENP‐A target domain (CATD) of CENP‐A. Third, CENP‐C and CENP‐N simultaneously bind to the CENP‐A nucleosome because their CENP‐A binding sites do not overlap. These findings have provided important information concerning the kinetochore architecture in vertebrate cells. However, several questions remain unanswered and need to be addressed. Since relatively short regions of CENP‐C containing either the central domain or the CENP‐C motif have been used for these structural studies (Kato et al, 2013b; Ali‐Ahmad et al, 2019; Allu et al, 2019), it is still unclear whether and how other regions of CENP‐C contribute to its binding to the CENP‐A nucleosome. Moreover, we have demonstrated that CDK1‐mediated CENP‐C phosphorylation outside of the CENP‐C motif facilitates CENP‐C binding to the CENP‐A nucleosome (Watanabe et al, 2019). Thus, a question arises regarding the mechanism by which phosphorylation contributes to CENP‐A‐CENP‐C binding. Furthermore, although previous studies have suggested that CENP‐C and CENP‐N recognize distinct regions on the CENP‐A nucleosome and these proteins might simultaneously bind to the CENP‐A nucleosome (Allu et al, 2019), it is unknown whether the longer CENP‐C fragment and CENP‐N bind to the same surface of the CENP‐A nucleosome without any structural collision.
To address these questions, we performed a structural analysis of the chicken CENP‐A nucleosome in complex with a longer and phosphorylated chicken CENP‐C C‐terminal fragment (amino acid (aa) 601–864, termed CENPC‐CT in this paper), including the CENP‐C motif (aa 655–675, termed CENPC motif in this paper) in the absence or presence of the CENP‐N N‐terminal domain (aa 1–211, termed CENPN‐NT in this paper) using cryo‐electron microscopy (cryo‐EM) single particle image analysis combined with a biochemical approach. The cryo‐EM structure of the CENP‐A nucleosome bound to the short CENPC motif peptide and CENPN‐NT was also determined.
In the present study, we demonstrate a unique and well‐defined structure of CENPC‐CT, featuring a stable CENP‐A‐CENP‐C interaction. Of note, the phosphorylation of T651CENP‐C stabilizes the CENP‐A‐bound structure of CENPC‐CT through an intramolecular interaction with R656CENP‐C in the CENPC motif. At the newly identified CENP‐A‐CENP‐C interface, the C‐terminal folded region extending from the CENPC motif associates with the RG loop of the CENP‐A nucleosome, which is also recognized by CENP‐N. Collectively, our data imply the binding of CENP‐C and CENP‐N to the CENP‐A nucleosome is mutually exclusive and phosphorylation‐dependent. These structural features of the CENP‐A nucleosome complexes provide new insights into kinetochore assembly mechanisms.
Results
The longer C‐terminal fragment of CENP‐C stably associates with the CENP‐A nucleosome
CENP‐C has multiple domains that include the N‐terminal Mis12 complex binding domain, CENP‐L/N and CENP‐H/I/K/M binding domains, CENPC motif, and C‐terminal dimerization domain (Cupin domain) (Fig 1A; Klare et al, 2015; Hara et al, 2018; Watanabe et al, 2019). The entire domain organization of chicken CENP‐C is similar to that of human CENP‐C, except for the absence of the second CENP‐A binding domain (the central domain) in the middle of human CENP‐C (Fig 1A; Kato et al, 2013a). However, as the central domain is not widely conserved (Watanabe et al, 2019), we focused on CENPC‐CT containing the CENPC motif to analyze their CENP‐A binding and purified chicken CENPC‐CT (aa 601–864) as a recombinant protein (Fig 1A). We also prepared a shorter peptide (aa 643–683), including the CENPC motif (CM peptide, Fig 1A). We reconstituted chicken CENP‐A nucleosomes, whereby the CENP‐A N‐terminal region was replaced by the H3 N‐terminal region to stabilize the nucleosome (Watanabe et al, 2019). First, we assessed the binding ability of CENPC‐CT to the CENP‐A nucleosome using an electrophoretic mobility shift assay (EMSA). CENPC‐CT efficiently bound to the CENP‐A nucleosome at a molar ratio of 1:1, but did not bind to the canonical H3 nucleosome, even after adding four times excess of CENPC‐CT than the H3 nucleosome (Fig 1B). We also performed similar assays using the CM peptide. While excess CM peptide (more than 18:1 molar ratio) bound to the CENP‐A nucleosome, it also bound to the H3 nucleosome (Fig 1B), indicating that specific binding to the CENP‐A nucleosome was lost due to excess amounts of the CM peptide. As the CENPC motif binds to the acidic patch of histone H2A/B, excess CM peptide can bind to this region in the H3 nucleosome. However, as CENPC‐CT specifically binds to the CENP‐A nucleosome at a molar ratio of 1:1, other CENP‐C regions in the C‐terminal fragment must facilitate the stable and specific binding of CENP‐C to the CENP‐A nucleosome.
Figure 1. CENP‐A nucleosome binding of the C‐terminal fragment of CENP‐C.
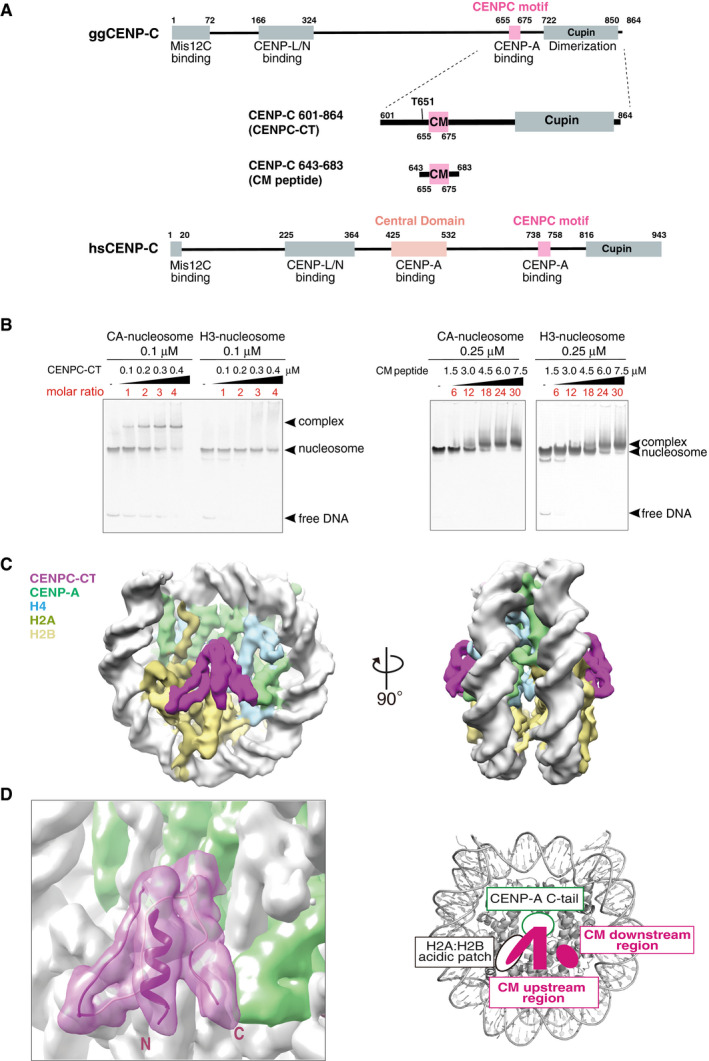
- Schematic diagram of the functional regions of chicken and human CENP‐C molecules. The canonical CENPC motifs for CENP‐A binding (aa 655‐675 in chicken CNEP‐C, aa 738‐758 in human CENP‐C) are colored pink. Another CENP‐A binding region (central domain) in human CENP‐C is colored light pink. The C‐terminal fragment (aa 601‐864: CENPC‐CT) and the CENPC motif‐containing peptide (aa 643‐683; CM peptide) derived from chicken CNEP‐C, which were used for the in vitro CENP‐A nucleosome‐binding assay, are diagrammed.
- Electrophoretic mobility shift assay (EMSA) examination of the binding affinity of CENPC‐CT and CM peptide to a CENP‐A nucleosome. Binding to a canonical H3 nucleosome was also examined using EMSA.
- Cryo‐EM density map of the CENP‐A nucleosome in complex with CENPC‐CT (CA‐CCCT complex) at a 4.5 Å resolution. The side views of the CA‐CCCT complex along the two‐fold axis are shown. The density corresponding to each molecule in the complex is color‐coded, as indicated in the figure.
- Cryo‐EM structure of the CENP‐A nucleosome‐binding region of CENPC‐CT. The left panel shows the cartoon representation of the structure of CENPC‐CT bound to the CENP‐A nucleosome with the cryo‐EM density map. The densities derived from CENPC‐CT and CENP‐A molecules are color‐coded in magenta and pale green, respectively. The functional elements in the CENP‐A nucleosome‐binding region of CENPC‐CT are depicted in the right panel. In addition to the CENPC motif, which recognizes the H2A/H2B acidic patch and the CENP‐A C‐terminal tail (C‐tail), the CM upstream and CM downstream regions are identified in the CENP‐A bound structure of CENPC‐CT. See also Fig EV2C.
To directly examine which regions of CENP‐C are involved in stable and specific binding to the CENP‐A nucleosome, we purified the CENP‐A nucleosome in complex with CENPC‐CT (CA‐CCCT complex) and performed cryo‐EM single particle image analysis of the structure of the complex. Although the CA‐CCCT complexes appeared to be formed in vitro (Fig 1B), most of them were aggregated and/or fell apart, when the sample was concentrated and applied to EM grids. Well‐distributed particles of the CA‐CCCT complex suitable for cryo‐EM single particle image analysis were seldom detected in the micrographs. We have previously demonstrated that phosphorylation of CENP‐C by CDK1 facilitates CENP‐A binding (Watanabe et al, 2019) and hypothesized that phosphorylated CENPC‐CT would form a more stable CA‐CCCT complex suitable for cryo‐EM single particle image analysis. Accordingly, using CENPC‐CT phosphorylated by CDK1, we obtained the appropriate number of particles distributed on the EM grid. The three‐dimensional (3D) reconstitution of the CA‐CCCT complex was determined at a 6.78 Å resolution (Table 1 and Fig EV1A). The EM density of the CA‐CCCT complex revealed the phosphorylated CENPC‐CT fragment bound to each side of the CENP‐A nucleosome (Fig EV2A). Hereafter, we used this phosphorylated CENPC‐CT fragment for further cryo‐EM single particle image analysis. CENPC‐CT contains the dimeric Cupin domain at the extreme C‐terminal end (Fig 1A). The stoichiometry of dimeric CENPC‐CT bound to the single CENP‐A nucleosome was not clear in our cryo‐EM analysis. However, based on size exclusion chromatography analyses of CENP‐A nucleosome complexes, it is likely that a CENPC‐CT dimer associates with each face of the CENP‐A nucleosome (Appendix Fig S1), leading to a stoichiometry of 4 CENPC‐CT fragments per each CENP‐A nucleosome (Appendix Fig S1B‐b). Only one protomer out of the CENPC‐CT dimer clearly binds CENP‐A.
Table 1.
Statistics for Cryo‐EM single particle image analysis and structure refinement.
| Cryo‐EM density map | CA‐CCCT (4.5Å) (EMDB‐30239) (PDB 7BY0) | Asymmetric CA‐CCCT‐CNNT (EMDB‐30241) | CA‐CCpep‐CNNT (EMDB‐30237) (PDB 7BXT) | CA‐CCCT (6.78Å) (EMDB‐30240) |
|---|---|---|---|---|
| Data collection and processing | ||||
| Sample | CA‐CCCT‐CNNT complex | CA‐CCpep‐CNNT complex | CA‐CCCTcomplex | |
| Magnification | 50,000 | 50,000 | 50,000 | 50,000 |
| Voltage (kV) | 200 | 200 | 200 | 200 |
| Electron exposure (e–/Å2 ) | 100 | 100 | 100 | 100 |
| Defocus range (µm) | −0.3 (−7.0) | −0.3 (−7.0) | −0.3 (−7.0) | −0.3 (−7.0) |
| Pixel size (Å) | 1.10 | 1.10 | 1.47 | 1.09 |
| Symmetry | C2 | C1 | C2 | C2 |
| Initial particle images (n) | 3,385,783 | 3,385,783 | 4,192,300 | 3,423,115 |
| Final particle images (n) | 38,542 | 3,959 | 118,294 | 39,542 |
| Map resolution (Å) | 4.5 | 7.8 | 4.2 | 6.78 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 |
| Refinement | ||||
| CCmap_model | 0.87 | 0.84 | ||
| Model compositions | ||||
| Nonhydrogen atoms | 12,693 | 15,715 | ||
| Protein residues | 872 | 1,204 | ||
| Nucleic acids | 285 | 290 | ||
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.007 | 0.010 | ||
| Angles (°) | 0.939 | 1.169 | ||
| Ramachandran plot | ||||
| Favored (%) | 96.79 | 93.36 | ||
| Allowed (%) | 3.21 | 6.64 | ||
| Outliers (%) | 0.00 | 0.00 | ||
| Model validation | ||||
| Rotamer outliers (%) | 0.15 | 0.39 | ||
| Clash score | 11.07 | 9.69 | ||
| Cβ outliers (%) | 0.00 | 0.00 | ||
| CaBLAM outliers (%) | 2.55 | 3.93 | ||
FSC, Fourier shell correlation; R.m.s. deviations, root‐mean‐square deviations.
Figure EV1. Cryo‐EM single particle image analysis of CA‐CCCT, CA‐CCCT‐CNNT, and CA‐CCpep‐CNNT complexes.
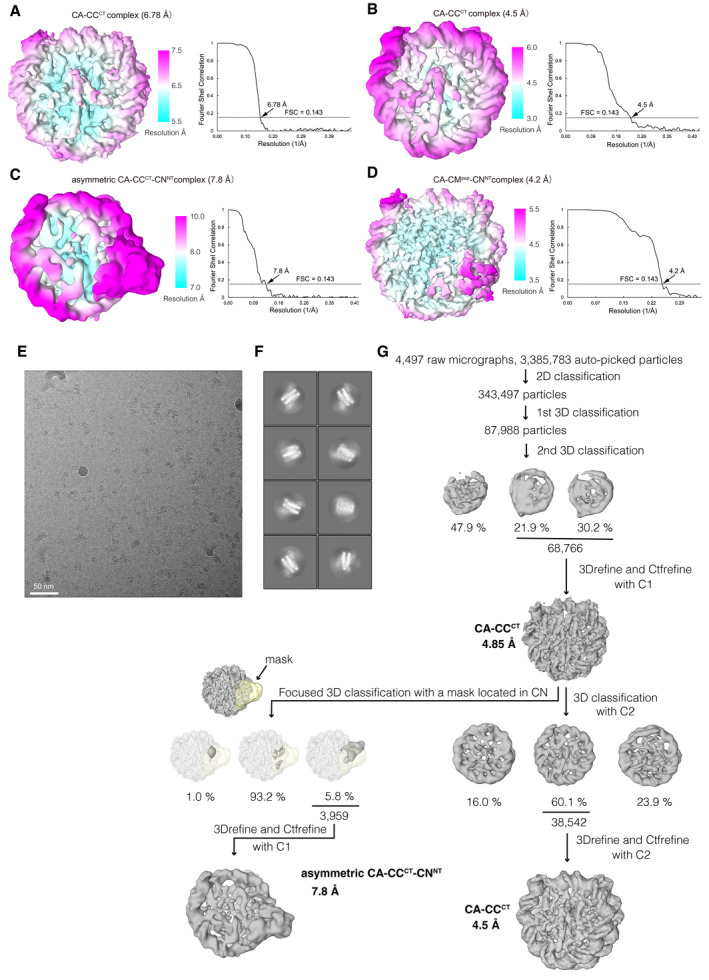
- Cryo‐EM density map of the chicken CA‐CCCT complex at a 6.78 Å resolution, colored according to the local resolution estimated by RELION in the left panel. Gold‐standard Fourier shell correlation (FSC) curve of the Cryo‐EM density map is displayed in the right panel. Reported resolution was based on the FSC = 0.143 criterion.
- Cryo‐EM density map and FSC curve of the chicken CA‐CCCT complex at a 4.5 Å resolution obtained using the CENP‐A nucleosome + CENPC‐CT + CENPN‐NT sample (major component) are shown as in (A).
- Cryo‐EM density map and FSC curve of the asymmetric chicken CA‐CCCT‐CNNT complex at 7.8 Å resolution obtained using CA + CCCT+CNNT sample (minor component) are shown as in (A).
- Cryo‐EM density map and FSC curve of the asymmetric chicken CA‐CCpep‐CNNT complex at a 4.2 Å resolution are shown as in (A).
- A representative micrograph of the CENP‐A complex with CENPC‐CT and CENPN‐NT.
- Representative 2D class averages of the CENP‐A nucleosome + CENPC‐CT + CENPN‐NT sample.
- Flow chart showing the image processing pipeline for the cryo‐EM single particle image analysis of the chicken CA‐CCCT complex and asymmetric chicken CA‐CCCT‐CNNT complex.
Figure EV2. Cryo‐EM structure of the CA‐CCCT complex.
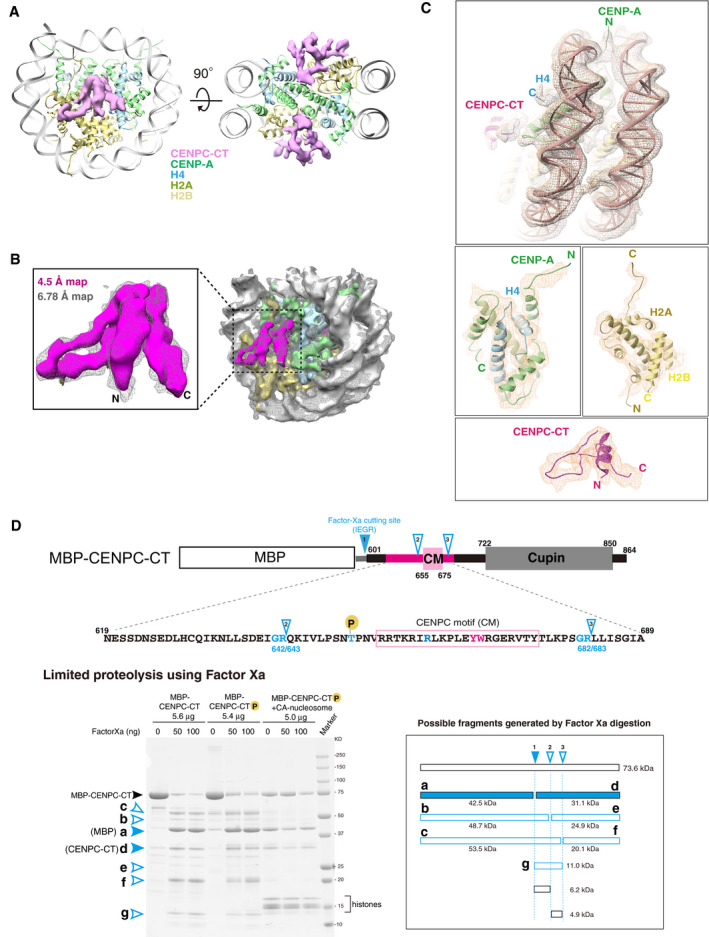
- Cryo‐EM density map of the CENPC‐CT bound to the CENP‐A nucleosome at a 6.78 Å resolution. The EM map for CENPC‐CT is shown in a pink surface representation together with the ribbon representation of the CENP‐A nucleosome. The molecules in the complex are color‐coded as indicated in the figure. The right panel shows a slice view along the two‐fold axis. The two CENPC‐CT fragments symmetrically bind to the CENP‐A nucleosome.
- Comparison of lower (6.78 Å) and higher (4.5 Å) resolution EM densities of the CA‐CCCT complex. The map at 4.5 Å resolution is depicted in a surface representation and superposed on the 6.78 Å resolution map shown as a mesh representation (light gray). The superposed maps corresponding to the CENPC‐CT are enlarged in a left panel.
- Detailed views of the cryo‐EM density map of the CA‐CCCT complex at a 4.5 Å resolution. The map is shown as a mesh representation with the ribbon model of the final cryo‐EM structure.
- Protease sensitivity of CENPC‐CT is altered by the presence of the CENP‐A nucleosome. Schematic diagram showing Factor Xa cleavage sites in MBP‐fused chicken CENPC‐CT with its functional domain organization. The possible minor cleavage sites (Gly‐Arg sequence) are indicated as cutting site 2 (between residues 642 and 643) and 3 (between residues 682 and 683), in addition to a major cleavage site between CENPC‐CT and MBP (site 1). The amino acid sequence of the folded region in CENPC‐CT is indicated below. The lower left panel shows the result of SDS–PAGE analysis of limited proteolysis product of CENPC‐CT in the absence or presence of the CENP‐A nucleosome. Possible fragments generated by Factor Xa digestion are shown in the lower right panel. Bands corresponding to each fragment are indicated in the gel. In the absence of the CENP‐A nucleosome, bands of the limited proteolysis products (b, c, e, f, and g) were observed. These bands were not observed in the presence of CENP‐A nucleosome.
In addition to CENPC‐CT, CENPN‐NT also binds to the CENP‐A nucleosome (Carroll et al, 2009; Pentakota et al, 2017; Chittori et al, 2018; Allu et al, 2019). Therefore, we reconstituted the CENP‐A nucleosome in complex with phosphorylated chicken CENPC‐CT and chicken CENPN‐NT (aa 1–211) (CA‐CCCT‐CNNT complex) and analyzed their structure using cryo‐EM (Table 1). Cryo‐EM single particle image analysis of the CA‐CCCT‐CNNT complex showed a heterogeneous population of particles. We observed the CA‐CCCT complex as the major component (94% of total analyzed particles) even in the presence of CENPN‐NT and the asymmetrical CA‐CCCT‐CNNT complex as a minor component (6% of total analyzed particles) (Fig EV1, EV2, EV3, EV4, EV5). The cryo‐EM density maps of the CA‐CCCT and asymmetrical CA‐CCCT‐CNNT complexes were determined at 4.5 and 7.8 Å resolutions, respectively (Figs EV1B and 1C; see details in later sections). Although CENPC‐CT did not directly interact with CENP‐N, the presence of CENPN‐NT improved the particle distribution on the EM grid, leading to a higher‐resolution cryo‐EM structure of the CA‐CCCT complex. An excess amount of CENPC‐CT tended to nonspecifically bind to DNA and form aggregates. CENPN‐NT may weakly or transiently associate with CENPC‐CT or DNA and prevent the formation of such aggregates.
Figure EV3. Conserved CENP‐A nucleosome‐binding sites in the chicken CENPC‐CT fragment.
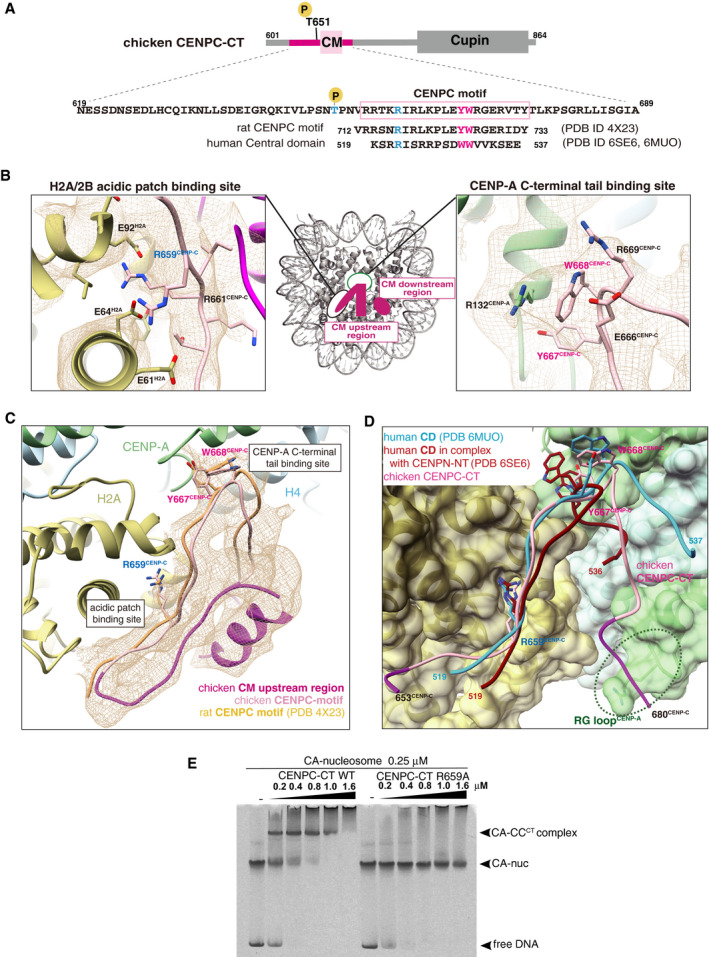
- Schematic diagram showing functional elements in chicken CENPC‐CT. The amino acid sequence of CENPC motif (CM) is enclosed in a pink box. The aligned sequences of rat CENPC motif and human central domain (CD), which were used for previous structural studies, are shown at the bottom. Key residues for CENP‐A nucleosome binding, R659, Y667, and W668 in chicken CENP‐C, are colored in blue (R659) and magenta (Y667 and W668). Corresponding residues in rat and human CENP‐C are also colored.
- Magnified views of the binding sites for the CENP‐A C‐terminal region and the H2A/2B acidic patch are presented in the cryo‐EM map. Side chains of the key residues are indicated as a stick model.
- Cryo‐EM structure of CENPC‐CT bound to the CENP‐A nucleosome. The cryo‐EM density for CENPC‐CT is shown in a mesh representation. The crystal structure of the CENPC motif in complex with the nucleosome (PDB ID: 4X23) is superimposed. The entire backbone structures are well superimposed.
- Structural comparison between the chicken CA‐CCCT complex and the human CD structures (PDB ID: 6MUO and 6SE6) on the CENP‐A nucleosome. The structures of CD bound to the CENP‐A nucleosome superimposed to that of the CENPC motif in the CA‐CCCT complex.
- CENP‐A nucleosome‐binding assays with WT or R659A mutant of CENPC‐CT. The substitution of R659 residue with alanine (R659A) caused a loss of the CENP‐A nucleosome‐binding ability.
Figure EV4. Association of CENPC‐CT with RG loopCENP‐A .
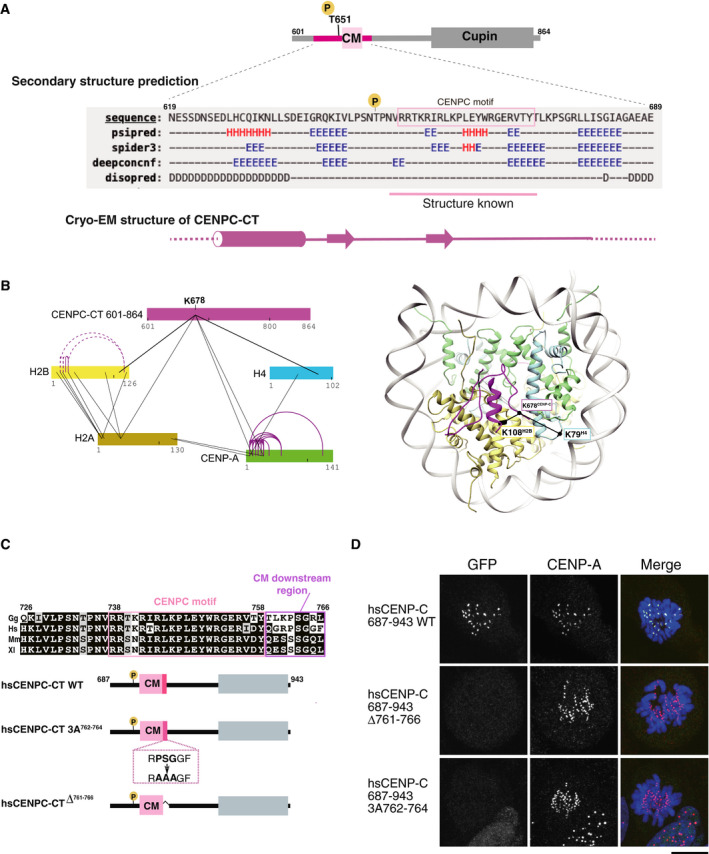
- Secondary structure prediction of CENPC‐CT. A diagram of chicken CENPC‐CT is shown. The secondary structure of the putative CENP‐A binding region of CENPC‐CT was analyzed by six different programs using a HHpred server https://toolkit.tuebingen.mpg.de/tools/hhpred. Predicted secondary structure elements are indicated by H for helix, E for strand, and D for disordered region. The canonical CENPC motif, with previously determined structure in homologues, is highlighted in pink. A schematic diagram of the cryo‐EM structure of CENPC‐CT is shown at the bottom.
- Crosslinking mass spectroscopy (XL‐MS) interactions depicted in relation to CENPC‐CT and histones, including CENP‐A. Color bars represent protein sequences. Black and purple lines show inter‐ and intra‐protein links, respectively. In the right panel, the crosslinked sites between CENP‐C and histones are indicated on the CA‐CCCT complex structure in which K678CENP‐C was linked with K108H2B and K79H4. Detailed XL‐MS data are presented in Appendix Fig S2.
- Alignment of sequences around the CENPC motif region in various species: Gg, chicken; Hs, human; Mm, mouse, and Xl; frog. The CENPC motif and the CM downstream region are depicted by pink and purple boxes, respectively, in the sequence alignment. The residue numbers of human CENP‐C are indicated. Schematic diagram of human CENPC‐CT wild‐type (CENPC‐CT WT: aa 687‐943) corresponding to chicken CNEPC‐CT used for the cryo‐EM analysis is depicted. The conserved PSG residues (aa 762‐764) in the CM downstream region were substituted with AAA (CENPC‐CT 3A762‐764) and CENPC‐CT in which six residues were deleted (CENPC‐CTΔ761‐766) are shown.
- Localization analysis of GFP‐fused human CENPC‐CT WT and mutants shown in (C) on the mitotic chromosomes in CENP‐C knock out human RPE‐1 cells. CENP‐A was used as a centromere marker. Scale bar indicates 10 μm.
Figure EV5. Stabilization of the CENP‐A nucleosome binding of CENPC‐CT via phosphorylation of T651CENP‐C .
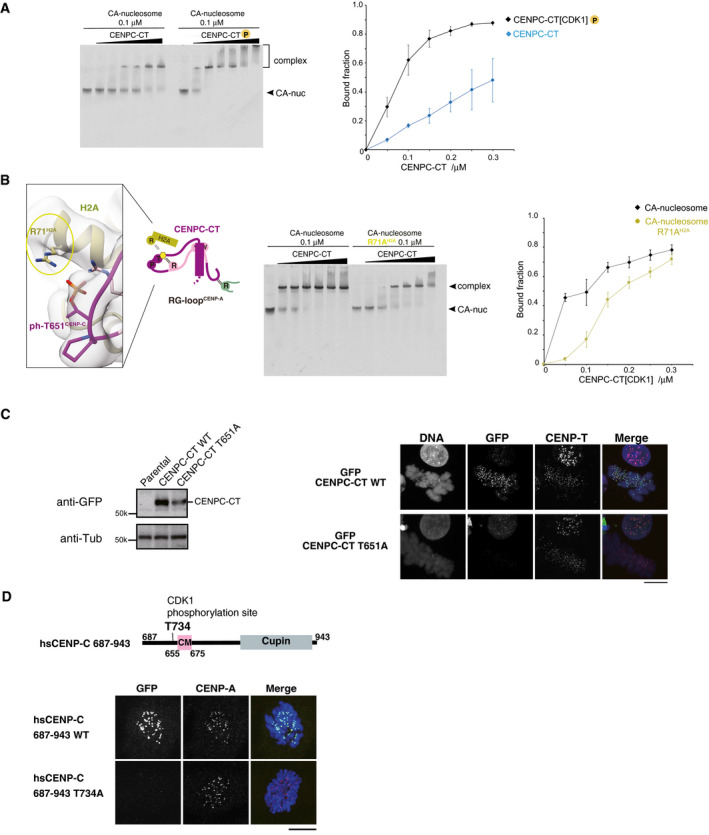
- EMSA was performed to examine the binding affinities of phosphorylated or nonphosphorylated CENPC‐CT (CENPC‐CT[CDK1] or CENPC‐CT). The signal intensities of upper binds (bound fractions) were measured. The graph indicates mean with SD (n = 3).
- Left panel displays a magnified view of the interface between CENPC‐CT and histone H2A in the CENP‐A nucleosome. Arginine 71 of histone H2A (R71H2A) was situated close to the phosphorylated T651CENP‐C in the range of 3.5 to 4.5 Å. Side chains of R71H2A and phosphorylated T651 of CENP‐C (ph‐T651CENP‐C) are indicated in stick model. The middle panel displays results of EMSA performed to examine the significance of the interaction between R71H2A and ph‐T651CENP‐C. Right panel displays quantification of the EMSA results. The graph indicates mean with SD (n = 3).
- Left panel displays the stable expression of GFP‐fused CENPC‐CT or the CDK1 phosphorylation site mutant (CENPC‐CT T651A) in CENP‐C knockout chicken DT40 cells was confirmed by immunoblot analysis. α‐Tubulin (Tub) was probed as a loading control. Parental CENP‐C knockout cells were also analyzed (parental). Right panel displays the results of localization analysis of GFP‐fused CENPC‐CT WT and T651A mutant in CENP‐C knockout chicken DT40 cells. CENP‐T was used as a centromere marker. Scale bar indicates 10 μm.
- Localization analysis of GFP‐fused human CENPC‐CT (687‐943) WT and T734A mutant (equivalent to T651A of chicken CENP‐C) in CENP‐C knockout human RPE1 cells. CENP‐A was used as a centromere marker. Scale bar indicates 10 μm.
The cryo‐EM density map of the CA‐CCCT complex at a 4.5 Å resolution was well fitted on the map at a 6.78 Å resolution and allowed the tracing of the continuous backbone structure of the CENP‐A nucleosome‐binding region in CENPC‐CT (Figs 1C, and EV2B and C). The structure of the CA‐CCCT complex was obtained by docking of the homology model built from the crystal structures of the CENP‐A nucleosome and CENP‐C bound to the nucleosome (Kato et al, 2013b; Arimura et al, 2019) into our cryo‐EM density, further manual model building, and refinement. Cryo‐EM structure analysis revealed a well‐defined structure of CENPC‐CT on the CENP‐A nucleosome and the formation of a stable interface between CENPC‐CT and the CENP‐A nucleosome (Fig 1D). The structured region of CENPC‐CT comprised the conserved CENPC motif and extended regions from both sides of the motif (termed the CM upstream and CM downstream regions) (Fig 1D). The protease sensitivity of CENPC‐CT was altered by the presence of CENP‐A nucleosomes, suggesting that such a stable structure would be induced upon CA‐CCCT complex formation (Fig EV2D).
The model of the CA‐CCCT complex showed interactions of the CENPC motif (aa 655–675) with the C‐terminal tail of CENP‐A and the acidic patch of histone H2A/B (Figs 1D, and EV3A and B), as observed previously in the structure of the complex formed by binding of the CENP‐A nucleosome to human or rat CENP‐C (Fig EV3A, C and D; Kato et al, 2013b; Ali‐Ahmad et al, 2019; Allu et al, 2019). The EM density indicated that the CENP‐A C‐terminal tail was recognized by conserved residues, Y667 and W668, in chicken CENP‐C (Figs 2A and EV3B, right panel). Mutation of the conserved arginine residue in chicken CENP‐C, which was responsible for the acidic patch interaction, to alanine (R659A, Fig EV3B, left panel) led to a loss of CENP‐A binding (Fig EV3E). Furthermore, in the CA‐CCCT complex, the CM downstream region was observed in the vicinity of the RG loop in the CATD of the CENP‐A nucleosome (RG loopCENP‐A), implying an additional CENP‐A binding site in CENP‐C (see details in Fig 2A). The CM upstream region was folded over the CENPC motif and the CM downstream region that provided the extended RG loopCENP‐A binding site (see details in Fig 3). We hypothesized that these newly identified structure regions (Fig 1D) contributed to the stable binding of CENP‐C to the CENP‐A nucleosome. Further analyses were performed to explore this hypothesis.
Figure 2. RG loopCENP‐A recognition through the CM downstream region of CENP‐C.
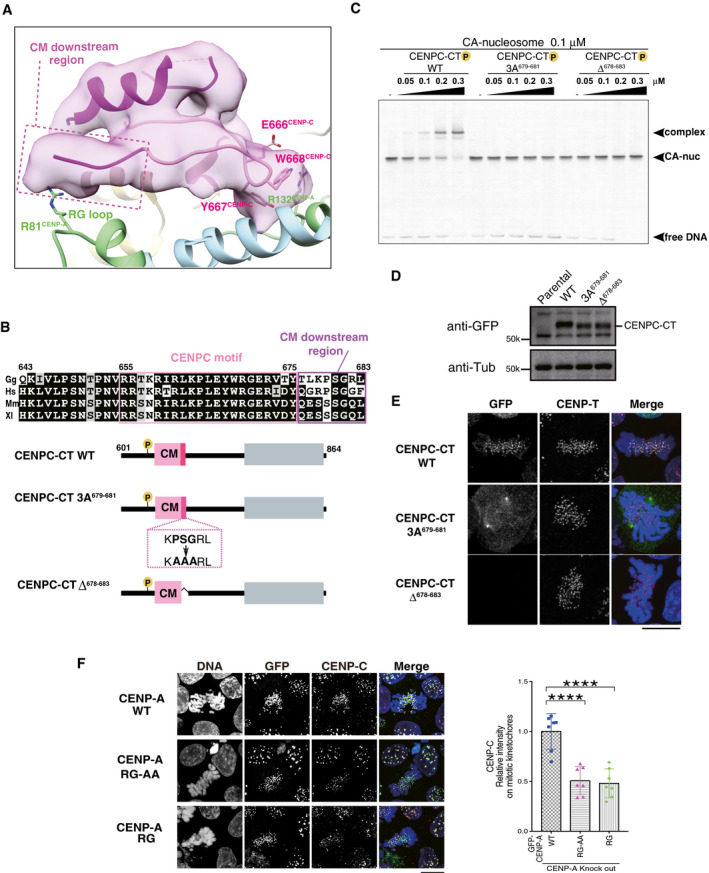
- The RG loopCENP‐A binding site of CENPC‐CT is shown. The backbone structure of the CENPC‐CT is presented in a cartoon model, which is colored in magenta, except for the pink canonical CENPC motif (aa 655‐675). The cryo‐EM density map of CENPC‐CT is overlaid on the cartoon model. The CM downstream region beyond Y675CENP‐C is indicated by a dotted box. Key residues involved in binding to the C‐terminal tail of CENP‐A are indicated as stick models. The cartoon models of CENP‐A and histone H4 are shown in pale green and pale blue, respectively.
- Alignment of sequences around the CENPC motif region of CENP‐C from various species: Gg, chicken; Hs, human; Mm, mouse; and Xl, frog. The CENPC motif and the CM downstream region are depicted by pink and purple boxes, respectively, in the sequence alignment. Schematic diagram of chicken CENPC‐CT wild‐type (CENPC‐CT WT), CENPC‐CT, in which PSG residues in the CM downstream region were substituted with AAA (CENPC‐CT 3A679‐681), and CENPC‐CT, in which six residues were deleted (CENPC‐CT Δ678‐683), are shown.
- EMSA results of the binding affinities of CENPC‐CT WT, CENPC‐CT 3A679‐681, and CENPC‐CT Δ678‐683 to the CENP‐A nucleosome.
- Stable expression of GFP‐fused CENPC‐CT and its mutants (shown in (B)) in CENP‐C knockout chicken DT40 cells. α‐Tubulin (Tub) was probed as a loading control. Parental CENP‐C knockout cells (parental) were also examined.
- Localization analysis of GFP‐fused CENPC‐CT WT, CENPC‐CT 3A679‐681, and CENPC‐CT Δ678‐683 (green) on the mitotic chromosomes in chicken DT40 cells. CENP‐T (red) was used as a centromere marker. DNA was stained using 4',6‐diamidino‐2‐phenylindole (DAPI; blue). Scale bar indicates 10 μm.
- Localization of CENP‐C in CENP‐A knockout DT40 cells stably expressing GFP‐fused chicken CENP‐A WT, RG‐AA, or ∆RG. Scale bar indicates 10 μm. CENP‐C was stained by an anti‐CENP‐C antibody (red), and DNA was stained by DAPI (blue). CENP‐C signals on kinetochores in mitotic cells were quantified in each cell. Bar graph indicates mean ± SD (n = 7; ****, P < 0.0001, unpaired t‐test, two‐tailed).
Figure 3. The CM upstream region of CENP‐C stabilizes the CENP‐A nucleosome binding.
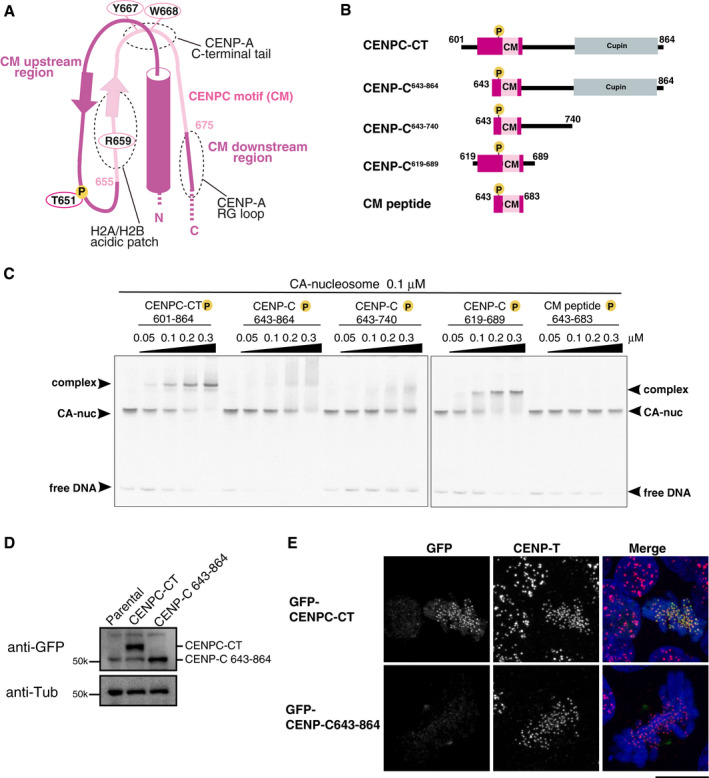
- Schematic representation of the structure of the CENP‐A nucleosome‐binding region in CENPC‐CT. The canonical CENPC motif segment is shown in pink. The CDK1 phosphorylation site (T651) is also indicated. The CM upstream and downstream regions are shown in magenta. Dot circles indicate the recognition sites for H2A/B acidic patch, CENP‐A C‐terminal tail, and RG loopCENP‐A.
- Schematic diagrams of deletion mutants of chicken CENPC‐CT. The fragments contain the canonical CENPC motif and the CM downstream region. CENP‐C643‐864, CENP‐C643‐740, and CM peptide lack the CM upstream region. CENPC‐CT and CENP‐C619‐689 contain the CM upstream region.
- EMSA results of the binding affinities of CENP‐C fragments indicated in (B) to the CENP‐A nucleosome. All fragments were phosphorylated by CDK1.
- Stable expression of GFP‐fused CENPC‐CT and CENP‐C643‐864 (shown in (B)) in CENP‐C knockout chicken DT40 cells. α‐Tubulin (Tub) was probed as a loading control.
- Localization analysis of GFP‐fused CENPC‐CT and CENP‐C643‐864 lacking the CM upstream region (green) on mitotic chromosomes in CENP‐C knockout DT40 cells. CENP‐T (red) was used as a centromere marker. DNA was stained using DAPI (blue). Scale bar indicates 10 μm.
The CM downstream region associates with the RG loop of the CENP‐A nucleosome
The extended density of the C‐terminal side of the CENPC motif, highlighted in Fig 2A as the CM downstream region, was clearly observed in the vicinity of RG loopCENP‐A (Tachiwana et al, 2011; Fang et al, 2015). We could not unambiguously build a model of the RG loop contact site in the cryo‐EM density map at near‐atomic resolution. However, based on the partial backbone model of this region and secondary structure prediction (Fig EV4A), we predicted that the CM downstream region (aa 676–683) would fit in the extended density (Fig 2A and B). In particular, the conserved PSG (aa 679–681) residues were expected to be located close to the RG loopCENP‐A. The association of this region with the CENP‐A nucleosome was also confirmed using crosslinking mass spectroscopy analysis (Appendix Fig S2). Consistent with the model of the CA‐CCCT complex, K678CENP‐C was crosslinked with the histone residues K108H2B and K79H4 surrounding the RG loopCENP‐A (Fig EV4B). To demonstrate the importance of this region experimentally, we generated mutant CENPC‐CT constructs (Fig 2B), in which the conserved PSG sequences in the region were either substituted with AAA (CENPC‐CT 3A679–681) or deleted (CENPC‐CTΔ678–683); we subsequently characterized these mutant proteins. Both mutant proteins were phosphorylated with CDK1, and their binding abilities to the CENP‐A nucleosome were analyzed by EMSA. As shown in Fig 2C, efficient complex formation was observed when phosphorylated CENPC‐CT was added to CENP‐A nucleosomes, whereas none of the mutant proteins showed CENP‐A binding in the same molar ratio range.
Green fluorescent protein (GFP)‐fused mutant fragments were stably expressed in CENP‐C knockout DT40 cells (Fig 2D). As CENP‐C forms a dimer at the extreme C‐terminal region, localization of the C‐terminal fragment must be evaluated in CENP‐C knockout cells (Watanabe et al, 2019). The centromere signals of wild‐type (WT) CENPC‐CT were observed at mitotic chromosomes. However, mutant CENPC‐CTs did not properly localize to centromeres (Fig 2E). Mislocalized CENPC‐CT tended to be associated with spindle poles. We also tested CENP‐C levels in CENP‐A knockout cells expressing the CENP‐A RG loop mutant (GFP‐CENP‐A RG‐AA or GFP‐CENP‐A ΔRG). As shown in Fig 2F, the CENP‐C levels at mitotic centromeres were significantly reduced in these mutant cells. In addition, we characterized the importance of the CM downstream region for centromere localization in human cells (Fig EV4C). Human CENPC‐CT, with mutations in the CM downstream region, did not properly localize to mitotic centromeres, as observed in chicken DT40 mutant cells (Figs 2E and EV4D), suggesting that the association of CENPC‐CT with RG loopCENP‐A for stable CENP‐C‐CENP‐A interaction is conserved in human cells. Based on these results, we propose that the CM downstream region plays an essential role in centromere localization by binding or sensing to the characteristic RG loop of the CENP‐A nucleosome.
The CM upstream region facilitates CENPC‐CT binding to the CENP‐A nucleosome
The continuous cryo‐EM density map demonstrated that the N‐terminal side (CM upstream region) adjacent to the CENPC motif in CENPC‐CT adopted a stable conformation in the CA‐CCCT complex structure. This region contains a single α‐helix at the N‐terminal region followed by a strand that forms a short β‐sheet with the CENPC motif backbone (Fig 3A). The cryo‐EM density map at 4.5 Å resolution was not clear enough for unambiguous side chain assignment of the N‐terminal α‐helix. However, combined with secondary structure prediction (Fig EV4A), we deduced that the extreme N‐terminal segment consisting of residues 601–618 was unstructured and invisible in the cryo‐EM density map. Our cryo‐EM density map suggested that the CM upstream region did not directly bind to the CENP‐A nucleosome. However, the region was associated with the CENPC motif and the CM downstream region, which might be critical for stable CENP‐C‐CENP‐A nucleosome interactions. Notably, the N‐terminal α‐helix was folded over the vicinity of the CENP‐A C‐terminal tail binding site in the CENPC motif and the CM downstream region (Figs 1D, 2A, and 3A). The association of the N‐terminal α‐helix with the CM downstream region appeared to stabilize the RG loopCENP‐A binding interface.
To evaluate the functional significance of the CM upstream region in the CENP‐C‐CENP‐A nucleosome interaction, various truncated CENP‐C constructs were generated (Fig 3B) and tested for their ability to bind to the CENP‐A nucleosome. All the fragments were phosphorylated by CDK1 in vitro. As shown in Fig 3C, while WT CENPC‐CT efficiently bound to the CENP‐A nucleosome at a molar ratio of 1:1 (CENPC‐CT: CENP‐A nucleosome), neither CENP‐C643‐864 nor CENP‐C643–740 fragments lacking the N‐terminal α‐helix and β‐strand bound to the CENP‐A nucleosome. The CENP‐A nucleosome‐binding efficiencies of the CENP‐C643–864 and CENP‐C643–740 fragments were significantly reduced compared with that of WT CENPC‐CT. However, when the N‐terminal segment was added to the CM peptide, binding affinity was recovered (CENP‐C619–689 in Fig 3C). These results suggest that the CM upstream region contributes to efficient CENP‐C binding to the CENP‐A nucleosome. It is worth noting that the Cupin dimerization domain (aa 722–850) in the extreme C‐terminal region was excluded from the folded region of CENPC‐CT and was not required for CENP‐A binding (Fig 3C).
We also examined the role of the CM upstream region in CENP‐C localization in vivo. When the GFP‐fused CENP‐C643–864 construct was stably expressed in CENP‐C knockout cells (Fig 3D), we did not observe its centromeric localization in mitotic chromosomes, whereas GFP‐fused WT CENPC‐CT (aa 601–864) was clearly localized to centromeres (Fig 3E). These data suggest that the CM upstream region of CENPC‐CT contributes to the proper centromere localization of CENP‐C by efficient binding to the CENP‐A nucleosome.
CDK1‐mediated CENP‐C phosphorylation facilitates its binding to the CENP‐A nucleosome
We have previously demonstrated that the chicken CENP‐C T651 residue (T651CENP‐C), which is included in the CM upstream region (Fig 3A), is phosphorylated by CDK1 and that its phosphorylation facilitates binding of the CENPC motif to the CENP‐A nucleosome (Watanabe et al, 2019). As shown in Fig 4A, phosphorylated CENPC‐CT bound more efficiently to the CENP‐A nucleosome than nonphosphorylated CENPC‐CT (Figs 4A, left and EV5A). In contrast, phosphorylation of T651CENP‐C had a negative effect on the CENP‐A nucleosome binding of the CM peptide lacking most of the CM upstream region (Fig 4A, right). These findings indicate that phosphorylated T651CENP‐C enhances CENP‐A nucleosome binding only in the context of the longer C‐terminal fragment. Consistent with this finding, the cryo‐EM structure of the CA‐CCCT complex revealed that the loop conformation containing T651CENP‐C was defined by main chain interactions between the CENPC motif and the K644CENP‐C, V646CENP‐C, and L647CENP‐C residues in the CM upstream region (Fig 4B). The position of phosphorylated T651CENP‐C was appropriately arranged on the CENP‐A nucleosome only in the well‐defined fold of CENPC‐CT. Furthermore, the CA‐CCCT complex structure also suggested that the phosphoryl group attached to T651CENP‐C was involved in an intramolecular interaction with the side chain of R656CENP‐C in the CENPC motif, which was likely to contribute to the stabilization of the loop conformation in the CM upstream region (Fig 4C). Arginine 71 of histone H2A was also situated close to the phosphorylated T651CENP‐C in the range of 3.5–4.5 Å (Fig 4C). EMSA findings indicate that the R71A mutation of H2A in CENP‐A nucleosomes marginally reduced their interaction with phosphorylated CENPC‐CT (Fig EV5B). In addition, the side chain of R655CENP‐C was oriented toward H2B residues, including Q47H2B (Fig 4C). These contacts between CENPC‐CT and histones may further support loop formation around the phosphorylated threonine.
Figure 4. Phosphorylation of T651 stabilizes the loop conformation of CENPC‐CT.
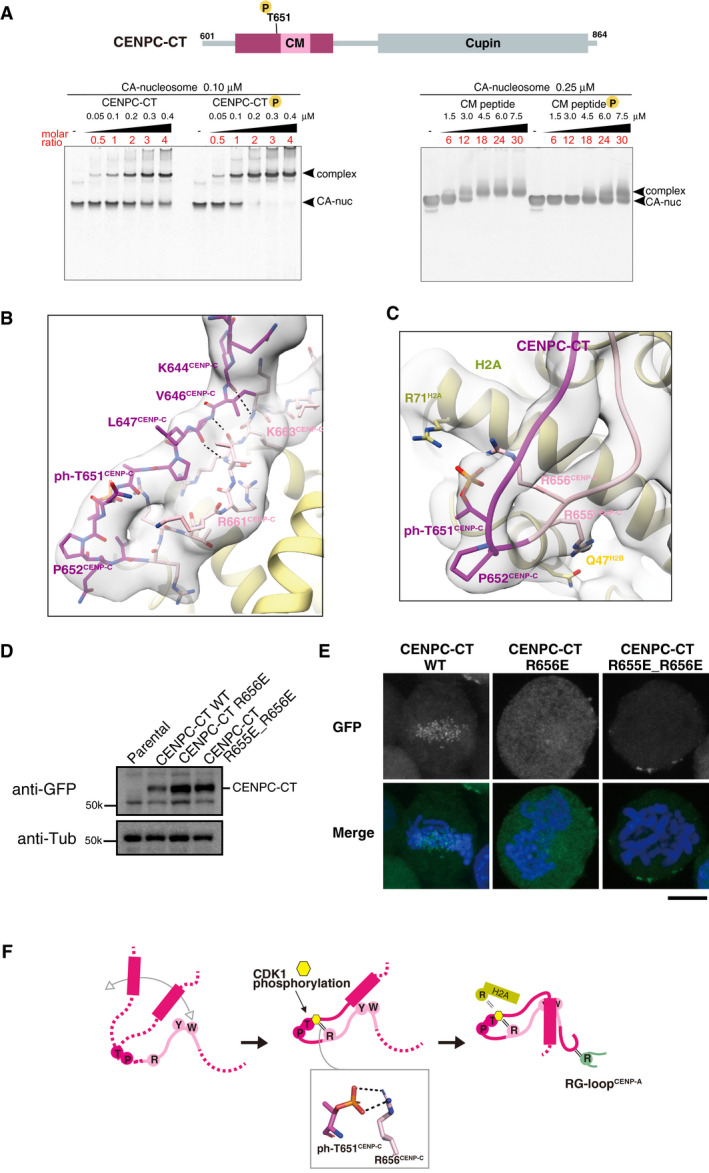
- The position of a CDK1 phosphorylation site, T651CENP‐C, is indicated in the diagram of chicken CENPC‐CT. EMSA was performed to examine the binding affinities of phosphorylated or nonphosphorylated CENPC‐CT (left panel) and CM peptide (right panel) to the CENP‐A nucleosome.
- Structure of the loop region harboring phosphorylated T651CENP‐C. CENPC‐CT is shown in magenta, except for the pink canonical CENPC motif. The cryo‐EM density map is overlaid on the cartoon model. The main chain interactions forming a short β‐sheet are indicated by black dotted lines.
- Magnified view of the structure around phosphorylated T651CENP‐C. Key residues in this region are shown as stick models. The side chains of R656CENP‐C and R71H2A are situated in the vicinity of the phosphoryl group of T651CENP‐C in the distance ranges of 3.5‐4 Å and 3.5‐4.5 Å, respectively.
- All constructs analyzed in (E) were stably expressed in CENP‐C knockout cells.
- Localization analysis of GFP‐fused CENPC‐CT, CENPC‐CTR656E, and CENPC‐CTR655E_R656E (green) on mitotic chromosomes in chicken DT40 cells. DNA was stained using DAPI (blue). Scale bar indicates 10 μm.
- Model indicating the functional role of T651CENP‐C phosphorylation in the CENP‐A‐CENP‐C interaction. Primarily, the CENPC motif associates with CENP‐A nucleosome. Phosphorylation of T651CENP‐C by CDK1 facilitates the loop conformation in the CM upstream region through an intramolecular bridge with the side chain of R656CENP‐C. Subsequently, the N‐terminal α‐helix is folded over to stabilize the CENP‐C‐CENP‐A nucleosome interfaces mediated by the CENPC motif and CM downstream regions.
To test the functional significance of these structural interactions, we expressed GFP‐fused CENPC‐CT with an R656E mutation or a double mutation for R655E and R656E (R655E_R656E) in CENP‐C knockout cells to examine its centromere localization (Fig 4D). While GFP‐fused WT CENPC‐CT clearly localized to mitotic centromeres, CENPC‐CT with R656E or R655E_R656E mutations, which perturbed the loop conformation around T651CENP‐C, did not localize properly to mitotic centromeres (Fig 4E). The CDK1 phosphorylation site mutant, CENPC‐CT T651A, lost the mitosis‐specific centromere localization (Fig EV5C; Watanabe et al, 2019). These findings collectively suggest that CDK1‐mediated T651CENP‐C phosphorylation would stabilize the loop conformation in the CM upstream region through intramolecular interactions with R656CENP‐C, leading to the formation of a stable CA‐CCCT complex by the association between the N‐terminal α‐helix and the CM downstream region (Fig 4F). In our model, although the CM upstream region does not directly bind to the CENP‐A nucleosome, it makes stable contacts with the CENPC motif and the CM downstream region, which enhance CENPC‐CT binding to the CENP‐A nucleosome (Fig 4F).
A majority of the longer CENP‐C C‐terminal fragment excludes CENP‐N from the CENP‐A nucleosome
A previous study demonstrated that CENP‐C and CENP‐N simultaneously bound to the CENP‐A nucleosome (Allu et al, 2019), when a shorter CENP‐C fragment (human CENP‐C519–537) derived from the central domain (Fig 1A) was used. However, in our cryo‐EM single particle image analysis, the simultaneous binding of phosphorylated CENPC‐CT and CENPN‐NT was not clearly identified on the same side of a CENP‐A nucleosome (Fig 5A, left). In a minor component (approximately 6% of the total analyzed particles) of the cryo‐EM single particle image observation of the CA‐CCCT‐CNNT complex, we found an asymmetrical structure in which only one CENPC‐CT was bound to one face of the CENP‐A nucleosome and only one CENPN‐NT was bound to another face (Figs 5A left, and EV1C and G). In the side recognized by CENPN‐NT, a faint electron microscopy density for CENP‐C was observed around the acidic patch of H2A/B (Fig 5A left), suggesting that only a weak CENP‐C binding to the CENP‐A nucleosome through the CENPC motif remained in the presence of CENPN‐NT bound to the RG loopCENP‐A. However, we would like to emphasize that the CENP‐A structure containing both CENPC‐CT and CENPN‐NT was minor and that the phosphorylated CENPC‐CT exclusively bound to the RG loopCENP‐A (CA‐CCCT: 94% of total analyzed particles).
Figure 5. Exclusive CENP‐A nucleosome interaction around the RG loop with CENP‐C and CENP‐N.
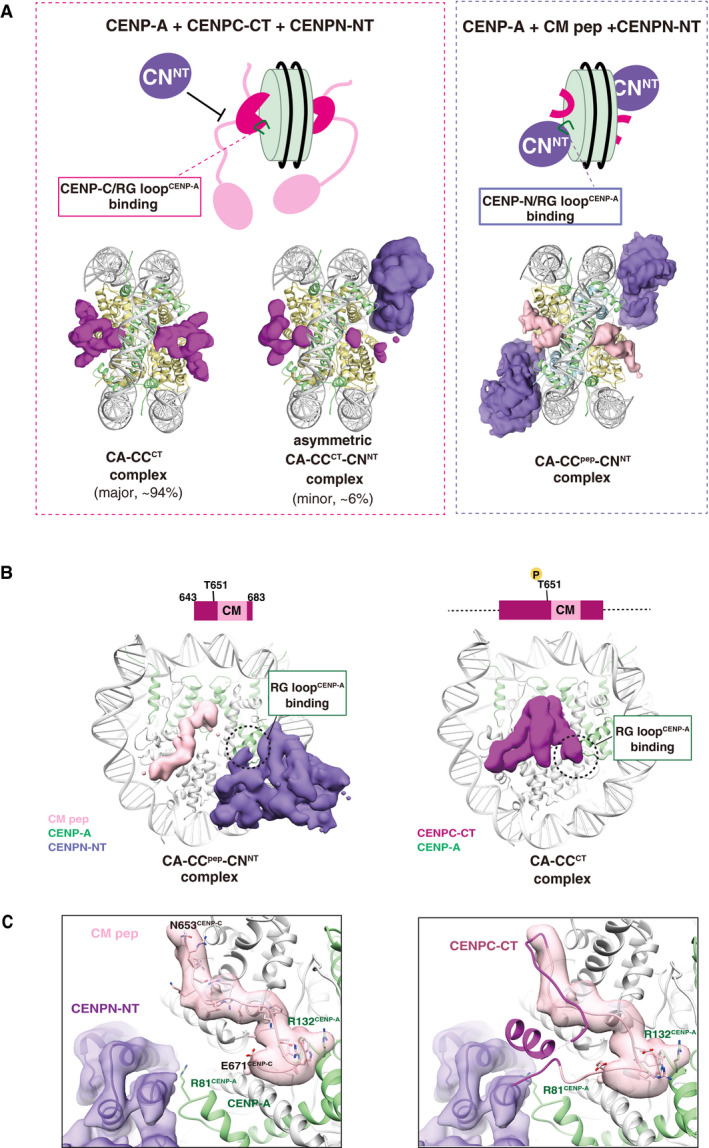
- Structural comparison of the CA‐CCCT with CENPN‐NT (left) and CA‐CCpep with CENPN‐NT (right). The cryo‐EM density maps of the CENPC‐CT fragment, CM peptide, and CENPN‐NT are indicated as a surface representation. The cryo‐EM density maps indicate that the structure of the CENP‐A nucleosomes bound with two CENPC‐CT fragments symmetrically (CA‐CCCT complex) comprised approximately 94% of total analyzed particles. The RG loopCENP‐A is occupied by CENPC‐CT in the CA‐CCCT complex and thereby the CENPN‐NT could not access the same side of the nucleosome. The minor fraction (approximately 6% of total analyzed particles) comprised asymmetric CA‐CCCT‐CNNT complexes. In the structures of the CA‐CCpep‐CNNT (right), CM peptide and CENPN‐NT simultaneously bind to the CENP‐A nucleosome.
- Side views of the CA‐CCpep‐CNNT and CA‐CCCT complexes. The RG loopCENP‐A binding site in each complex is indicated by a dotted circle.
- The left panel contains the structure of the RG loopCENP‐A binding site in the CA‐CCpep‐CNNT complex. R81CENP‐A of the RG loop is recognized by CENPN‐NT. The right panel displays a superposition of the structure of CENPC‐CT in the CA‐CCCT complex on the CA‐CCpep‐CNNT complex shown in the left panel. The model of CM peptide in the CA‐CCpep‐CNNT complex has been removed for clarity. The CENPC motif region in CENPC‐CT is shown in pink. Structural collision between the CM downstream region of CENPC‐CT and CENPN‐NT is shown at RG loopCENP‐A in the CENP‐A nucleosome.
To gain structural insights into the regulation/mode of CENP‐C and CENP‐N binding to the CENP‐A nucleosome, we reconstituted CENP‐A nucleosomes in complex with the CM peptide (aa 643–683) and CENPN‐NT. The cryo‐EM structural model of the CENP‐A nucleosome bound to both a CM peptide and CENPN‐NT (CA‐CCpep‐CNNT complex) was obtained at a 4.2 Å resolution (Table 1, Figs 5A right, B left, and EV1D, Appendix Fig S3). Each side of a CENP‐A nucleosome was bound to one CENPN‐NT molecule and one CENPC motif peptide with no structural collision. Consistent with the results of previous studies (Pentakota et al, 2017; Chittori et al, 2018; Tian et al, 2018; Allu et al, 2019), in the cryo‐EM structure of the CA‐CCpep‐CNNT complex, the N‐terminal domain of CENP‐N bound to the RG loopCENP‐A (Fig 5C, left), while the CM peptide interacted with the CENP‐A C‐terminal tail and the acidic patch of H2A/2B in the CENP‐A nucleosome. The cryo‐EM density map for both the terminal regions of the CM peptide was not observed, indicating that only the canonical CENPC motif region, containing residues 654–671, was used for CENP‐A nucleosome binding.
In the cryo‐EM structure of the CA‐CCCT complex containing the CDK1‐phosphorylated CENPC‐CT fragment (aa 601–864), the CM downstream region extended toward the RG loop of CENP‐A to contact the side chain of R81CENP‐A (Figs 2A and 5C, right panel). Although the CM peptide contains the CM downstream region, the RG loopCENP‐A was occupied by CENPN‐NT in the structure of the CA‐CCpep‐CNNT complex (Fig 5B, left panel). These findings suggest that the CM upstream region of CENP‐C stabilizes the contact of the CM downstream region to the RG loopCENP‐A in the CENP‐A nucleosome. Consequently, the longer CENP‐C C‐terminal fragment, CENPC‐CT, more dominantly associates with the RG loop than CENPN‐NT. The cryo‐EM density map of the CA‐CCCT complex indicated an association between the N‐terminal α‐helix segment and the CM downstream region in CENPC‐CT (Fig 2A). T651CENP‐C phosphorylation appears to facilitate coordinated action of both the CM upstream and downstream regions to stabilize the contact of CENPC‐CT to the RG loopCENP‐A (Fig 4F). Furthermore, superposition of CENPN‐NT with the CA‐CCpep‐CNNT complex to the relative position in the CA‐CCCT complex clearly showed a structural collision between the CM downstream region of CENP‐C and the CENPN‐NT (Fig 5C, right). Once the CENPC‐CT fragment in the well‐defined fold occupies the RG loop in CENP‐A, CENP‐N cannot approach the same site.
Exclusive CENP‐A binding between CENPC‐CT and CENPN‐NT was also examined by a competitive pull‐down assay. Consistent with the cryo‐EM observations, nucleosome histones bound to CENPN‐NT were gradually reduced upon addition of CENPC‐CT (Fig 6A, left). In contrast, tight interaction of phosphorylated CENPC‐CT with the CENP‐A nucleosome was not replaced by the addition of CENPN‐NT (Fig 6A, right).
Figure 6. A model of the dynamic change of binding partners of the CENP‐A nucleosome during the cell cycle progression.
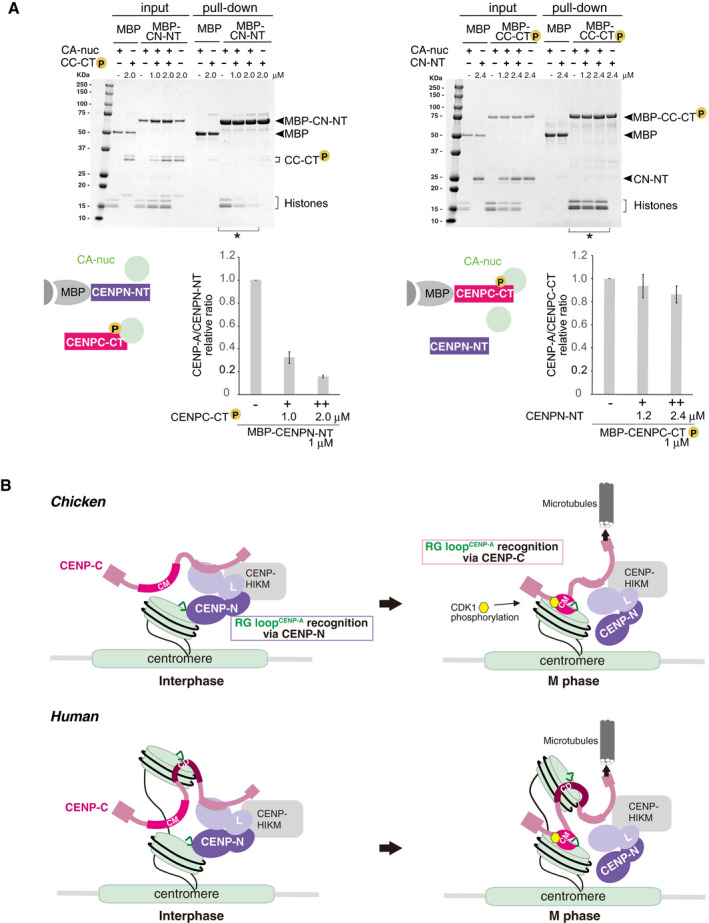
- Competitive pull‐down assays for the CENP‐A nucleosome binding of CENPC‐CT and CENPN‐NT. The left panel displays a complex of MBP‐CENPN‐NT (MBP‐CN‐NT) with the CENP‐A nucleosome (CA‐nuc) incubated in the absence or presence of phosphorylated CENPC‐CT (CC‐CT) and pulled down by MBP affinity. The right panel displays a complex of phosphorylated MBP‐CENPC‐CT (MBP‐CC‐CT) with the CENP‐A nucleosome (CA‐nuc) incubated in the absence or presence of CENPN‐NT (CN‐NT) and pulled down by MBP affinity. The signal intensities of all histones in CENP‐A nucleosomes, which were precipitated with MBP proteins in indicated lanes (*), were quantified. The signal intensities were normalized to MBP‐CN‐NT or MBP‐CC‐CT signals. Bar graph indicates mean with SD (n = 3).
- A model of the dynamic change of binding partners of the CENP‐A nucleosome during the cell cycle progression in chicken (upper) or human (bottom). During the interphase, chicken CENP‐C without phosphorylation at T651 is dissociated from the CENP‐A nucleosome. Then, CENP‐N can bind to the RG loopCENP‐A of the CENP‐A nucleosome in interphase cells. In mitosis, the CENP‐C C‐terminal region is phosphorylated by CDK1, and this phosphorylation facilitates CENP‐A‐CENP‐C interaction. The stable CENP‐A‐CENP‐C interaction excludes CENP‐N from the RG loop. This phospho‐regulation by the CENP‐C C‐terminal region is conserved in chicken and human. In humans, the second CENP‐A binding region (central domain, CD) might associate with the CENP‐A nucleosome throughout the cell cycle. However, it is still unclear how CD is involved in formation of centromeric chromatin structure (bottom).
Based on our cryo‐EM observations and biochemical analysis, we propose that the longer, phosphorylated CENP‐C C‐terminal fragment excludes the CENP‐N N‐terminal domain from the CENP‐A nucleosome.
Discussion
The CENP‐A nucleosome is a key epigenetic marker for centromere specification and kinetochore assembly. To form a functional kinetochore, downstream kinetochore proteins must recognize the CENP‐A nucleosome. To understand kinetochore assembly mechanisms, the structures of the CENP‐A nucleosome bound to its interacting proteins must be determined. CENP‐C and CENP‐N are CENP‐A‐binding proteins, and the structures of complexes containing the CENP‐A nucleosome bound to CENP‐C and/or CENP‐N have been determined (Kato et al, 2013b; Pentakota et al, 2017; Chittori et al, 2018; Tian et al, 2018; Ali‐Ahmad et al, 2019; Allu et al, 2019). However, as relatively short fragments of CENP‐C have been used in previous studies, it is questionable how other CENP‐C regions contribute to CENP‐A binding. In addition, CENP‐C is phosphorylated by CDK1 during mitosis and this phosphorylation facilitates CENP‐A‐CENP‐C interaction (Watanabe et al, 2019). However, the molecular basis of how CENP‐C phosphorylation contributes to efficient CENP‐A binding is still unclear. In the present study, using the phosphorylated chicken CENPC‐CT fragment (aa 601–864), we determined the structure of the CENP‐A nucleosome in complex with CENPC‐CT as well as CENPC‐CT/CENPN‐NT using cryo‐EM single particle image analysis. We identified the unique structure of the CENP‐A nucleosome complexed with phosphorylated CENPC‐CT that involves the extended regions from both sides of the CENPC motif, including the CM upstream and downstream regions (Fig 1D). Both regions contribute to stable and specific CENP‐A binding in coordination with the CENPC motif.
We obtained the cryo‐EM structures of two types of CENP‐A nucleosome‐CENP‐C complexes. The CA‐CCCT complex contains CENPC‐CT on each side of the CENP‐A nucleosome. The asymmetric CA‐CCCT‐CNNT complex contains one CENPC‐CT bound to one face of the CENP‐A nucleosome and one CENPN‐NT bound to another face (Fig 5A). Although we observed the asymmetric CA‐CCCT‐CNNT complex in a minor fraction (6% of total analyzed particles), this structure might exist in vivo. This structure might be similar to the asymmetric structure proposed by Allu et al (2019), which contains one copy of CENP‐N in a single CENP‐A nucleosome. Importantly, in a major fraction (94% of total analyzed particles), the RG loopCENP‐A of the CENP‐A nucleosome was occupied by the CM downstream region in CENPC‐CT on each side of the nucleosome as the CA‐CCCT complex (Figs 2A and EV1B). In addition to cryo‐EM observations, our biochemical analysis demonstrated that phosphorylated CENPC‐CT excludes CENPN‐NT from the CENP‐A nucleosome (Fig 6A). These findings imply that coordinated interactions with three elements of the CENP‐A nucleosome—the C‐terminal tail, RG loop in CENP‐A, and acidic patch in H2A/B—facilitate a well‐defined conformation into CENPC‐CT, thereby strengthening its association with the CENP‐A nucleosome.
Our structural data obtained using CENPC‐CT suggest that CENP‐C and CENP‐N exclusively bind to one surface of CENP‐A nucleosomes. In contrast, previous studies have suggested the simultaneous binding of CENP‐C and CENP‐N to one surface of CENP‐A nucleosomes (Carroll et al, 2010; Pentakota et al, 2017; Ali‐Ahmad et al, 2019; Allu et al, 2019). Previous structural studies used the central domain of human CENP‐C (aa 426–537 or aa 519–537) to examine CENP‐C binding to the CENP‐A nucleosome. These central domain fragments might be too short to reach the RG loopCENP‐A in either the absence or presence of the CENPN‐NT and thus not cause the structural collisions to CENP‐N on the CENP‐A nucleosome (Fig EV3D). Alternatively, it is also possible that such a contrary conclusion might result from the different binding modes between the central domain and the CENPC‐CT. In either case, CENPC‐CT is well conserved between humans and chickens, including the CDK1 phosphorylation site, the CENPC motif, and the CM upstream and downstream regions. Although it is still unclear how the central domain of CENP‐C contributes to its binding to CENP‐A nucleosomes (Fig 6B), we propose that the phospho‐regulation and structure of the CENPC‐CT‐CENP‐A nucleosome complex is conserved in chicken and human cells. The CDK1 phosphorylation site mutant (T734A) of human CENPC‐CT lost mitotic centromere localization, as observed in the chicken CENPC‐CT T651A mutant cells (Fig EV5C and D; Watanabe et al, 2019). Consistent with our model, human CENPC‐CT with mutations in the CM downstream region did not localize to mitotic centromeres (Fig EV4C and D), as observed in chicken cells (Fig 2E).
We have previously proposed that the CENP‐A‐CENP‐C interaction is regulated during cell cycle progression and that CENPC‐CT is not associated with the CENP‐A nucleosome during the interphase stage of both chicken and human cells (Nagpal et al, 2015; Watanabe et al, 2019). Therefore, it is possible that CENP‐N specifically binds to the CENP‐A nucleosome through RG loopCENP‐A, when CENP‐C dissociates from the CENP‐A nucleosome in the interphase (Fig 6B). In fact, unphosphorylated CENPC‐CT does not localize to centromeres (Watanabe et al, 2019; Fig EV5C and D). This may be because CENP‐N and/or other kinetochore components such as CCAN (constitutive centromere‐associated network) (Fukagawa & Earnshaw, 2014) might exclude nonphosphorylated CENP‐C from centromeric chromatin. In human cells, as the central domain of CENP‐C binds to the CENP‐A nucleosome, CENP‐C might bind to the CENP‐A nucleosome in interphase cells (Fig 6B). However, the central domain of CENP‐C is not widely conserved outside mammalian species (Watanabe et al, 2019). CENP‐N also interacts with the CENP‐C middle region (Fig 1A; Klare et al, 2015; McKinley et al, 2015; Nagpal et al, 2015). As the CENP‐N‐binding region of CENP‐C was not used in the present study, it is still unclear how CENP‐N associates with the centromere region. Nevertheless, our structural data clearly demonstrated that phosphorylated CENPC‐CT and CENPN‐NT do not simultaneously bind to the RG loopCENP‐A. CDK1‐mediated CENP‐C phosphorylation occurs during mitosis. Therefore, CENP‐N may directly bind to CENP‐A nucleosomes via its N‐terminal region in the interphase, while it may be associated with the centromere through CENP‐C binding during mitosis (Fig 6B). Future studies are required to determine how CENP‐N dynamically changes its binding partner during cell cycle progression.
The CM upstream region was also observed to facilitate CENP‐A‐CENP‐C binding (Fig 3). Although this region did not directly contact to the CENP‐A nucleosome, it appeared to hold down the CENPC motif and the CM downstream region to stabilize the interaction between CENP‐C and the CENP‐A nucleosome. Interestingly, our cryo‐EM data suggested that the CDK1 phosphorylation site (T651) facilitates the association of the CM upstream region with the CENPC motif and the CM downstream region (Fig 4F). Such a synergistic effect might regulate dynamic changes in kinetochore organization during cell cycle progression. Our model is also consistent with our previous experimental results, in which unphosphorylated CENPC‐CT could bind to the CENP‐A nucleosome in the absence of CENP‐N in vitro, while it did not localize to the centromere in vivo (Watanabe et al, 2019).
In the present study, using the longer and phosphorylated C‐terminal fragment of CENP‐C, we clarified the contribution of multiple CENP‐C regions to its interaction with the CENP‐A nucleosome in coordination with the conserved CENP‐A binding motif (CENPC motif). We also revealed the structural basis of the regulation of the CENP‐A‐CENP‐C interaction by CENP‐C phosphorylation. We believe that our findings provide an important insight for understanding the structural dynamics of kinetochore architecture.
Materials and Methods
Recombinant protein expression and purification
A cDNA fragment encoding the C‐terminal fragment of chicken CENP‐C (aa 601–864, CENPC‐CT) was inserted into a pMAL‐c5X bacterial expression vector (New England BioLabs; pMAL‐c5X‐CENPC‐CT) or a pGEX‐6p bacterial expression vector (GE Healthcare; pGEX‐CENPC‐CT). cDNA fragments encoding shorter C‐terminal fragments of CENP‐C (CENP‐C619–690, CENP‐C643–864, CENP‐C643–740) were each inserted into a pMAL‐c5X‐TEV‐His plasmid (Nishino et al, 2013) (pMAL‐c5X‐TEV‐CC619–690‐His, pMAL‐c5X‐TEV‐CC643–864‐His, pMAL‐c5X‐TEV‐CC643–720‐His). The expression vectors of CENPC‐CT mutants were generated based on the pMAL‐c5X‐CENPC‐CT vector (pMAL‐c5X‐CENPC‐CT 3A679–681 and pMAL‐c5X‐CENPC‐CT Δ678–683). The chicken CENPC‐CT was expressed in Escherichia coli Rosetta™2 (DE3) (Novagen) as a maltose binding protein (MBP)‐fused recombinant protein. Escherichia coli cells were grown in LB‐Lennox media at 37°C until OD600 reached 0.6; protein expression was induced by addition of isopropyl‐β‐D‐thiogalactoside (IPTG) to a final concentration of 0.2 mM, and culture was continued at 17°C overnight. Cells were harvested by centrifugation. The cell pellet was resuspended in buffer A (20 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, and 2 mM tris(2‐carboxyethyl)phosphine (TCEP)) supplemented with protease inhibitor cocktail (Roche) and lysed by sonication on ice. The lysate was clarified by centrifugation and applied to amylose resign (New England Biolabs) pre‐equilibrated with buffer A. After extensive column washing with buffer A containing 1 M NaCl, the immobilized MBP‐fusion protein was eluted from the column with A buffer containing 20 mM maltose and applied to a HiTrap SP HP cation exchange column (GE Healthcare). MBP‐CENPC‐CT was eluted with a linear NaCl gradient from 200 to 750 mM and further purified by size exclusion column chromatography using a HiLoad Superdex 16/60 200 pg column (GE Healthcare) in a buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, and 2 mM TCEP. Peak fractions containing MBP‐CENPC‐CT were combined, concentrated (typically to 3–5 mg/ml), and stored at −80°C until further use for structural and biochemical analyses. The shorter C‐terminal fragments of CENP‐C and mutants were expressed as MBP‐fusion proteins and purified in the similar manner, in which the MBP affinity purification step was replaced by His‐tag affinity column chromatography using Ni‐NTA agarose (Quiagen). GST‐fused CENPC‐CT was expressed in the similar manner to MBP‐CENPC‐CT and purified using glutathione Sepharose FF (GE healthcare) and HiTrap SP (GE healthcare) columns.
For expression of chicken CENP‐N N‐terminal domain (CENPN‐NT, chicken CENP‐N 1–211), a cDNA fragment of encoding CENP‐NT was cloned into pMAL‐5cX‐His vector (pMAL‐c5X‐CNNT‐His). CENPN‐NT was expressed in E. coli Rosetta™2 (DE3) (Novagen) as a recombinant MBP‐fused protein containing a C‐terminal hexa‐histidine tag. Escherichia coli cells were grown in LB‐Lennox media at 37°C, and the expression of MBP‐CENPN‐NT was induced in the same way as MBP‐CENPC‐CT. MBP‐CENPN‐NT was purified by three column chromatography steps using MBP affinity, cation exchange, and size exclusion column chromatography in the similar way to MBP‐CENPC‐CT purification. For competitive pull‐down assay, MBP tag‐free CENPN‐NT was prepared by treatment with TEV protease followed by purification using a cation exchange column chromatography.
Preparation of the phosphorylated CENP‐C C‐terminal fragments
In vitro phosphorylation of MBP‐CENPC‐CT using cyclin B‐CDK1 and purification of cyclin B‐CDK1 were performed as described before (Okumura et al, 1996; Hara et al, 2018; Watanabe et al, 2019). Briefly, MBP‐CENPC‐CT (2 mg/ml) was incubated with the active cyclin B‐CDK1 in reaction buffer (10 mM Tris–HCl, pH 7.5, 2 mM MgCl2, 150 mM NaCl, 100 mM ATP, and 1× complete EDTA‐free proteinase inhibitor) for 1 h at 25°C. Phosphorylated proteins were analyzed by Phos‐tag‐5.0% SDS–PAGE (25 µM Phos‐tag acrylamide and 50 µM MnCl2). After the reaction was terminated by adding EDTA‐NaOH, pH 8.0 (final concentration, 5 mM), CDK‐1‐treated samples were further purified using MBP affinity column chromatography as described before (Watanabe et al, 2019) or using a cation exchange column. For cation exchange column chromatography, the CDK‐1‐treated sample was applied to a Mono S column (GE Healthcare) and eluted with a linear gradient of NaCl from 150 to 1,000 mM. The purified samples were concentrated to 1.25–3 mg/ml and kept at −80°C. Phosphorylation of MBP‐CENPC‐CT mutants was carried out in the same manner. For preparation of phosphorylated CENPC‐CT without a tag, GST‐CENPC‐CT was phosphorylated using cyclenB‐CDK1, and the GST tag was cleaved with HRV3C protease. The phosphorylated tag‐free fragment was purified using a Mono S column (GE Healthcare).
Purification of histones
Chimeric chicken CENP‐A (H31–63‐CAα1‐end*), whose N‐terminal tail (aa 1–54 in chicken CENP‐A) is substituted with that of canonical H3 (aa 1–63 in human H3), was used for chicken type CENP‐A nucleosome reconstitution. For expression of chimeric chicken CENP‐A, H31‐63‐CAα1‐end*, E‐coli strain BL21(DE3) was transformed with a pET15b‐H31‐63‐CAα1‐end* plasmid (Watanabe et al, 2019) and cultured in LB‐Lennox media overnight at 37°C. H31–63‐CAα1‐end* with an N‐terminal His‐tag was produced as inclusion body in bacteria cells. Cells were resuspended in lysis buffer (50 mM Tris pH 7.5, 0.5 M NaCl, 5% glycerol) supplemented with protease inhibitor cocktail (Roche) and were lysed using sonication. After centrifugation (33,000 g for 20 min), pellet was solubilized in unfolding buffer (50 mM Tris pH 7.5, 7 M guanidine‐HCl, and 5% glycerol) with overnight incubation at 37°C and then applied to a Ni‐NTA agarose column (Qiagen) equilibrated with His‐binding buffer (50 mM Tris pH 7.5, 0.5 M NaCl, 6 M urea, 10 mM imidazole). After extensive wash of the column with His‐binding buffer containing 50 mM imidazole, the unmobilized proteins were eluted with His‐elution buffer (50 mM Tris pH 7.5, 0.5 M NaCl, 6 M urea, 300 mM imidazole). The elution was dialyzed against 10 mM Tris buffer (pH 7.5) containing 5 mM DTT and treated with thrombin protease overnight at 4°C to remove the His‐tag. Untagged H31–63‐CAα1‐end* was further purified by ion exchange column chromatography using a HiTrap SP column (GE Healthcare) with a linear gradient of 200–900 mM NaCl in 20 mM sodium acetate buffer (pH 5.2) containing 1 mM EDTA, 6 M urea, and 5 mM DTT. The peak fractions were combined and dialyzed against water. H31–63‐CAα1‐end* was lyophilize using a vacuum centrifugal concentrator (TOMY). Other recombinant histones were prepared as previously described (Watanabe et al, 2019). Briefly, His‐tagged chicken histone H3.2 (gH3.2) or H4 (gH4) was expressed in E. coli BL21(DE3) using pHCE‐AMPFREE‐gH3.2 or pET15‐b‐gH4 plasmid, respectively. His‐tagged gH3.2 or gH4 was extracted as inclusion body and was solubilized in 50 mM Tris buffer (pH8.0) containing 7 M guanidine‐HCl, 5% glycerol, and 5 mM DTT. gH3.2 was purified in the same way as H31–63‐CAα1‐end*. Solubilized inclusion body of gH4 was applied to a HisTrap HP column (GE Healthcare) with a linear gradient of imidazole from 20 to 500 mM in His‐binding buffer. The peak fractions of His‐gH4 were dialyzed against 10 mM Tris buffer (pH 7.5) containing 5 mM DTT and applied to a HiTrap SP column (GE Healthcare). The His‐gH4 fractions were dialyzed against water, and purified His‐gH4 was lyophilize using a vacuum centrifugal concentrator (TOMY).
Histones H2A and H2B were expressed in E. coli Rosetta™2 (DE3) as a homodimer from a pETDuet‐His‐SUMO‐H2A/H2B plasmid which generates N‐terminally 6xHis‐SUMO‐tagged H2A and untagged H2B. Cells expressed H2A/H2B dimer were resuspended in high‐salt buffer (20 mM HEPES pH 7.5, 2 M NaCl, 5% glycerol, and 5 mM TCEP) and disrupted by sonication. The clarified lysate was applied to a Ni‐NTA agarose column (Qiagen). H2A/H2B dimer eluted from the column with suspension buffer containing 300 mM imidazole was incubated with SENP7 protease to remove the His‐SUMO‐tag. The complex was further purified using size exclusion column chromatography (GE Healthcare) in high‐salt buffer. The purified H2A/H2B dimer was concentrated using Amicon Ultra centrifugal filter (Merck).
For preparation of chicken CENP‐A/H4 tetramer, H31–63‐CAα1‐end* and His‐gH4 were mixed at 1:1 molar ratio in refolding buffer (10 mM Tris pH 7.5, 1 M NaCl, 1 mM EDTA, and 5 mM DTT) and assembled as tetramers by three dialysis steps in which NaCl concentration was decreased stepwise to 200 mM. The tetramer was further purified by size exclusion column chromatography using a Superdex 200 column (GE Healthcare) in buffer containing 10 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, and 5 mM DTT and concentrated using Amicon Ultra centrifugal filter (Merck). The gH3.2/H4 tetramer was prepared in the same way.
Reconstitution of chicken CENP‐A nucleosomes
145 bp 601 DNA was expressed as previously described (Tanaka et al, 2004; Arimura et al, 2012). Briefly, E. coli DH5α cells were transformed with the plasmid harboring eight repeats of 601 sequence (pGEM‐8x601) and cultured in 4–6 l of terrific broth media (Invitrogen) at 37°C for 15–16 h. The plasmid was extracted from bacterial cells following an alkaline lysis method and purified by phenol/chloroform extraction, polyethylene glycol (PEG) precipitation, and ethanol precipitation. The 145 bp 601 fragment was generated with EcoRV treatment followed by PEG precipitation and ethanol precipitation. Finally, the 601 DNA fragment was purified by ion exchange column chromatography.
H2A/H2B dimers, chicken (CENP‐A/H4)2 hetero‐tetramers, and 601 DNA were mixed with a molar ratio of 2:1:1.1 at 0.75 mg/ml of final DNA concentration in the presence of 2 M KCl. A gradient dialysis to low salt buffer (2–0.2 M KCl in 20 mM Tris pH 7.5, 1 mM EDTA, 5 mM DTT) was performed at 4°C over 16 h (Dyer et al, 2004). Assembled nucleosomes were then uniquely positioned on the DNA by a thermal shift for 1 h at 55°C. Nucleosome formation was examined using native polyacrylamide gel electrophoresis. Finally, the nucleosome solution was dialysis against 20 mM Tris–HCl buffer (pH 7.5) containing 50 mM NaCl, 1 mM EDTA, and 5 mM DTT. The H3 nucleosome was reconstituted in the same way as chicken CENP‐A nucleosome. The concentration of each nucleosome sample was determined using absorbance at 260 nm.
EMSA analysis
CENPC motif peptides (CM peptide643–683 and CM peptide643–683 containing phosphorylated T651) were commercially synthesized and dissolved in 10 mM Tris–HCl buffer (pH 7.5). Each of CENPC‐CTs or CM peptides was mixed with CENP‐A or H3 nucleosome in binding buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 100 mM NaCl, 2 mM TCEP, 5% glycerol, and 0.5% CHAPS) for 30 min on ice. The mixtures were analyzed by SDS–PAGE using SuperSep 5–20% gels (FUJIFILM WAKO Chemicals). Native PAGE was typically performed at constant 180 V for 120 min at 4°C in 0.5× TAE buffer. The gels were stained with GelRed™ (Biotium, Inc) and visualized by UV illumination at 260 nm. The intensities of bound fractions were measured with Fiji (Schindelin et al, 2012).
Purification of chicken CENP‐A nucleosome complexes
The CENP‐A nucleosome in complex with CENPC‐CT (CA‐CCCT) was prepared by mixing 0.6 nmol of CENP‐A nucleosome and MBP‐CENPC‐CT at a molar ration 1:3.5 in binding buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 2 mM DTT, 1 mM EDTA, 0.1% CHAPS) followed by incubation on ice for 30 min and then purified using a Superpose 6 10/30 column (GE Healthcare) in SEC‐elution buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM EDTA, 5 mM DTT, and 0.1% CHAPS). The fractions were analyzed by native PAGE followed by staining with GelReD™ (Biotium, Inc) and SDS–PAGE followed by staining with SimplyBlue™ SafeStain (Thermo Fisher Scientific). The peak fractions containing stoichiometric complex were combined and concentrated to approximately 0.5 mg/ml (1.6 µM) for EM analysis using Amicon Ultra centrifugal filter (30 kDa molecular weight cut off) and subjected to EM analysis. For preparation of the CA‐CCCT‐CNNT or CA‐CCpep‐CNNT, CENP‐A nucleosome in complex with CENPN‐NT was first purified. 0.8 nmol of CENP‐A nucleosome and 2.2 nmol of MBP‐CENPN‐NT were mixed in binding buffer and incubated on ice for 30 min and then purified using size exclusion column chromatography in the same way as the CA‐CCCT complex. The purified CENP‐A nucleosome—CENPN‐NT (CA‐CNNT) complex (4 µM) was mixed with MBP‐CENPC‐CT at a 1:2 molar ratio, incubated on ice for 30 min, and concentrated to approximately 0.5 mg/ml (1.2 µM) for EM analysis. The CA‐CCpep‐CNNT complex was generated by mixing the CA‐CNNT complex and the CM peptide (chicken CENP‐C, aa 643–683) at a 1:16 molar ratio and concentrated to approximately 1.3 mg/ml (4 µM) using Amicon Ultra centrifugal filter.
Cryo‐EM data collection
A 2.5 µl protein solution of each CENP‐A nucleosome complex was applied to Quantifoil Mo R0.6/1.0 holey carbon grids and frozen in liquid ethan using a Vitrobot IV (FEI, 4°C and 100% humidity). Data collection of each sample was carried out on a CRYO ARM 200 (JEOL, Japan) equipped with a thermal field emission electron gun operated at 200 kV, an Ω‐type energy filter with a 20 eV slit width and a K2 Summit direct electron detector camera (Gatan, USA). An automated data acquisition program, JADAS (JEOL, Japan), was used to collect cryo‐EM image data. Movie frames were recorded using the K2 Summit camera at a calibrated magnification of × 50,000 corresponding to a pixel size of 1.10 Å with a setting defocus range from −1.0 to −3.5 μm. The data were collected with a total exposure of 10 s fractionated into 50 frames, with a total dose of ~ 100 electrons Å−2 in counting mode. A total number of movies were collected; 4,497 for the CA‐CCCT‐CNNT complex, 5,757 for the CA‐CCCT complex, and 5,346 for the CA‐CCpep‐CNNT complex.
Image processing and 3D reconstruction
Motion correction was carried out by MotionCor2 (Zheng et al, 2017) to align all CENP‐A nucleosome complex micrographs, and the CTF parameters were estimated by Gctf (Zhang, 2016). Both programs were performed via the pipeline program Gwatch (https://github.com/FumiakiMakino/Gwatch) (Kato et al, 2019). All CENP‐A complexes were automatically selected by Auto‐picking using the Laplacian and Gaussian in RELION 3.0 (Zivanov et al, 2018), and they were extracted into a box of 192 × 192 pixels. For the CA‐CCCT‐CNNT complex, 343,497 particles were selected after performing 2D classification by RELION 3.0. An initial 3D classification into five classes with C1 symmetry resulted in one class (87,988 particles) that was further used as a reference and second 3D classification. The second 3D classification with C1 symmetry resulted in the two best classes (68,766 particles) that were used as further 3D refinement, CTF refinement procedure, and particles polishing. The map for a well‐defined nucleosome structure and CENPC‐CT, which is the CA‐CCCT complex, at 4.85 Å resolution was obtained after solvent mask post‐processing. However, a faint density was also observed around the CENP‐N binding site on the CENP‐A nucleosome previously reported (Pentakota et al, 2017; Chittori et al, 2018). Therefore, the data set was analyzed by two different ways. The first analysis was performed for isolate CA‐CCCT‐CNNT complexes using the focused 3D classification with a mask located in CENP‐N region with C1 symmetry. It resulted in asymmetric CA‐CCCT‐CNNT (EMD‐30241) at 7.8 Å resolution after 3D refinement, CTF refinement procedure, and solvent masked post‐processing from one class (3,959 particles).The second analysis for further high‐resolution structure of CA‐CCCT (EMD‐30239, PDB‐7BY0) was performed using 3D classification with C2 symmetry and resulted in the best one class (38,542 particles), which achieved 4.48 Å resolution after 3D refinement, CTF refinement with C2 symmetry, and solvent mask post‐processing (Fig EV1G). For the CA‐CCCT complex, 542,992 particles were selected after performing 2D classification. 78,923 particles were selected after repeating 3D classification three times with C1 symmetry. Then, 39,542 particles were selected after 3D classification with C2 symmetry. The map of CA‐CCCT at 6.78 Å resolution (EMD‐30240) was obtained after 3D refinement, particle polishing, CTF refinement, and solvent mask post‐processing. For the CA‐CCpep‐CNNT complex, 421,635 particles were selected after performing 2D classification. Particle extraction was performed again with rescaled size into 1.47 Å/pix. 118,294 particles were selected after 3D classification into five classes with C1 symmetry. The map of the CA‐CCCT‐CNNT complex at 4.2 Å resolution (EMD‐30237, PDB‐7BXT) was obtained after 3D refinement, particle polishing, CTF refinement, and solvent mask post‐processing.
Model building
The homology model of chicken CENP‐A (H31‐63‐CAα1‐end*) was built using SWISS‐MODEL (Waterhouse et al, 2018) based on the crystal structure of CENP‐A nucleosome (PDB ID 5ZBX); the model of chicken CENP‐A containing nucleosome was prepared by substituting chains A and E in the crystal structure with the chicken CENP‐A homology model. The chicken CENP‐A nucleosome model was fit into the cryo‐EM density of the CA‐CCCT complex at 4.5 A resolution using UCSF Chimera (Pettersen et al, 2004). The DNA sequence in the model was manually replaced by 145 base pair 601 DNA sequence in program Coot (Emsley et al, 2010). The model of the CENPC motif peptide taken from the crystal structure of CENP‐C bound to nucleosome (PDB ID 4X23) was fit into the map in UCSF Chimera, manually replaced its sequence by that of chicken CENP‐C and modified in Coot. The model for the further region of CENPC‐CT was manually built. For model building of the CA‐CCpep‐CNNT complex, the homology model of chicken CENPN‐NT (amino acid 1–211) was build using the crystal structure of human CENP‐N (PDB ID 6EQT) as a template. The models of chicken CENP‐A nucleosome bound to CENPC motif and chicken CENP‐N NT fit into the cryo‐EM density using UCSF Chimera. Based on map density, the complex models were iteratively modified and locally refined in Coot. Each of the final models was subjected to real space refinement in PHENIX (Liebschner et al, 2019). The coordinates and maps were deposited in the protein database. Cryo‐EM structures of the asymmetric CA‐CCCT‐CNNT complex were built by fitting the models of CENP‐A nucleosome, CENPC motif peptide, and CENPN‐NT into the density map in UCSF Chimera followed by rigid body refinement using PHENIX.
Cell culture
Chicken DT40 cells were cultured in DMEM medium (Nacalai tesque) supplemented with 10% fetal bovine serum (FBS; Sigma), 1% chicken serum (Gibco), 10 μM 2‐mercaptoethanol, and penicillin/streptomycin (final: 100 unit/ml and 100 μg/ml, respectively) (Thermo Fisher) at 38.5°C with 5% CO2. CENP‐C conditional knockout cells (Kwon et al, 2007) were used for examination of centromere localization GFP‐fused CENPC‐CTs. Various CENPC‐CT mutant DNAs were cloned into a pEGFP‐C3 vector (Clontech), which were transfected into CENP‐C knockout cells with Gene Pulser II electroporator (Bio‐Rad). The transfected cells were selected in the medium containing 2 mg/ml G418 (Santa Cruz Biotechnology).
Human RPE‐1 cells were cultured at 37°C in DMEM medium (Nacalai Tesque) supplemented with 10% fetal bovine serum (FBS; Sigma) and penicillin–streptomycin (Thermo Fisher) (RPE‐1 culture medium). A conditional knockout cells of human CENP‐C (hsCENP‐C) termed as cKO‐hsCENP‐C RPE‐1 cells were used (Watanabe et al, 2019). To establish cKO‐hsCENP‐C RPE‐1 cell lines stably expressing either GFP‐fused hsCENP‐C687–943 WT, hsCENP‐C687–943 T734A, hsCENP‐C687–943 ∆761–766, or hsCENP‐C687–943 3A 762–764, each CENP‐C C‐terminal construct was integrated in the genome in cKO‐hsCENP‐C RPE‐1 cells using the Sleeping Beauty transposon system as previously described (Watanabe et al, 2019). Since the transgene cassette has a neomycin resistance gene, the cell lines were selected in RPE‐1 culture medium containing 500 μg/ml G418.
Immunoblotting
Expression of each protein in culture cells was analyzed by immunoblotting. For whole‐cell samples, DT40 cells were harvested, washed with PBS, and suspended in 1× Laemmli sample buffer (LSB; final 104 cells/μl), followed by sonication and heating for 5 min at 96°C. Proteins were separated on SuperSep Ace, 5–20% (Wako) and transferred to Immobilon‐P (Merck) using HorizeBLOT (ATTO). Primary antibodies used in this study were rabbit anti‐gCENP‐C (Fukagawa et al, 1999), rabbit anti‐GFP (MBL), and mouse anti‐α‐tubulin (Sigma). Secondary antibodies were HRP‐conjugated anti‐rabbit IgG (Jackson ImmunoResearch) and HRP‐conjugated anti‐mouse IgG (Jackson ImmunoResearch). To increase sensitivity and specificity, Signal Enhancer Hikari (Nacalai Tesque) was used for all antibodies. The antibodies were incubated with the blotted membranes for 1 h at room temperature or for overnight at 4°C. Proteins reacting with antibodies were detected with ECL Prime (GE Healthcare) and visualized with ChemiDoc Touch (Bio‐Rad). Acquired images were processed using Image Lab 5.2.1 (Bio‐Rad) and Photoshop CC (Adobe).
Immunofluorescence and image acquisition
DT40 cells or human RPE‐1 cells expressing various GFP‐fused CENPC‐CTs were centrifuged onto slide glasses by the Cytospin3 centrifuge (Shandon) and fixed with 4% paraformaldehyde (PFA; Thermo Fisher) in PBS for 10 min at room temperature. The fixed cells were permeabilized in 0.5% NP‐40 in PBS for 10 min at room temperature and incubated with rabbit anti‐CENP‐T (1:1,000) (Hori et al, 2008) in 0.5% BSA in PBS for 1 h at 37°C as a primary antibody. The reacted cells were washed with 0.5% BSA (Equitech‐Bio Inc) in PBS at three times and were incubated with Cy3‐conjugated mouse anti‐rabbit IgG (1:2,000; Jackson ImmunoResearch) in 0.5% BSA in PBS for 30 min at 37°C as a secondary antibody. DNA was stained with 1 μg/ml DAPI in PBS for 1 min and mounted with VECTASHIELD mounting medium (Vector Laboratories). Immunofluorescence images were acquired every 0.2 μm step of Z‐slice (total 4–8 μm thick) as a Z‐stack image using a Zyla 4.2 sCMOS camera (ANDOR) mounted on a Nikon Eclipse Ti inverted microscope with an objective lens (Plan Apo lambda 100×/1.45 NA; Nikon) with a spinning disk confocal scanner unit (CSU‐W1; YOKOGAWA) controlled by NIS elements (Nikon). The images in figures are the maximum intensity projection of the Z‐stack generated with NIS elements and were processed by Photoshop CC (Adobe).
Quantification and statistical analysis
The fluorescence signal intensities of CENP‐C on kinetochores were quantified using Imaris (Bitplane). The fluorescence signals on ~ 50–100 kinetochores in each of seven cells were quantified and subtracted with mean of background signals in nonkinetochore regions. The mean of the kinetochore signals in each cell is shown. Data are shown as mean ± SD. The unpaired t‐test (two‐tailed) was used, with P < 0.05 considered significant.
Limited proteolysis
MBP‐CENPC‐CT, phosphorylated MBP‐CENPC‐CT (MBP‐CENPC‐CT[CDK‐1]), and purified MBA‐CENPC‐CT[CDK‐1] in complex with CENP‐A nucleosome were treated with approximately 1:100 or 1:50 molar ratio of Factor Xa protease (New England Biolabs) in digestion buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 5% glycerol, 2 mM TCEP) for 60 min at 4°C. The protease was inactivated by boiling at 96°C for 5 min. The products were analyzed by SDS–PAGE followed by staining with SimplyBlueTM SafeStain (Thermo Fisher Scientific).
XL‐MS
The chicken CENP‐A nucleosome bound with the phosphorylated CENP‐C fragment, MBP‐CENPC‐CT601–864[CDK1], or MBP‐CENPC‐CT619–689[CDK1] was crosslinked with BS3 (DOJINDO). The chicken CENP‐A nucleosome at a final concentration of 0.4 µM was mixed with 3.5‐fold molar excess of MBP‐CENPC‐CT601‐864[CDK1] of 7‐fold molar excess of MBP‐CENPC‐CT619–689[CDK1] in XL‐binding buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM TCEP, 0.5% CHAPS) and incubated on ice for 30 min. The BS3 solution was prepared fresh in XL‐binding buffer to obtain a stock concentration of 20 mM and added to the CENP‐A nucleosome complexes at a final concentration of 1.0 or 2.0 mM. The CENP‐A nucleosome complexes were each crosslinked for 40 min at room temperature, and the reaction was quenched by addition of 100 mM Tris–HCl pH 7.5. The crosslinked samples were separated by SDS–PAGE and visualized by staining with SimplyBlue™ SafeStain (Thermo Fisher Scientific). Peptide sample preparation by in‐gel digestion using trypsin (Pierce) was performed (Oya et al, 2019). Crosslinked peptides were enriched by SDB‐SCX stage‐tip (GL science) with a single‐step protocol (Schmidt & Sinz, 2017). Mass spectra were obtained on an LTQ‐Orbitrap Velos pro (Thermo Fisher Scientific) coupled to a nanoflow UHPLC system (ADVANCE UHPLC; AMR Inc.) with Advanced Captive Spray SOURCE (AMR Inc.). The enriched peptides mixtures were loaded onto a C18 0.3 × 20 mm, 5 μm (particle size) trap column (CELI) and then fractionated by C18, 0.075 × 150 mm, 3 μm (particle size) column (CELI) via a linear gradient from B 5%(0 min)‐35%(100 min)‐45%(120 min) followed by washing at 90% B and re‐equilibration at 5% B at a flow rate of 300 nl/min. The total run lengths were 145 min. The mass spectrometer was programmed to carry out 11 successive scans consisting of, first, a full‐scan MS over the range 350–1,600 m/z by orbitrap at 60,000 resolution, and second and 11, automatic data‐dependent MS/MS scans of the ten most intense ion signals in the first precursor scan that were at least triply charged by orbitrap at 7,500 resolution. MS/MS spectra were obtained by setting a normalized collision energy of 35% HCD using a 4 m/z isolation window and exclusion time of 90 s for molecules of the same m/z value range. The raw data files were processed to find the crosslinked peptides using Thermo Scientific Proteome Discoverer 2.2 software (Thermo Fisher Scientific) with the add‐on XlinkX algorithm node (Klykov et al, 2018). The following settings were used in XlinkX: precursor ion mass tolerance, 10 ppm; product ion mass tolerance, 20 ppm; fixed modification, Cys carbamidomethylation; variable modification, Met oxidation; allowed number of mis‐cleavages, 4.
Competitive MBP pull‐down assay
15 µl of amylose resign (New England Biolabs) was equilibrated with pull‐down buffer consisting of 20 mM HEPES NaOH pH 7.5, 200 mM NaCl, 3% glycerol, 5 mM TCEP, 1 mM EDTA, and 0.1% CHAPS. MBP‐CENPC‐CT[CDK1] or MBP‐CENPN‐NT (1–1.5 µM final concentration) as a bait was incubated with CENP‐A nucleosome as a prey at 0.4 µM concentration in the absence or presence of CENPN‐NT or phosphorylated CENPC‐CT as a competitor. The reaction volume was topped up to 50 µl and incubated for 1 h at 4°C. Beads were span down at 1,000 g for 1 min, and the supernatant was removed. The beads were washed with 200 µl of pull‐down assay buffer three times. After completely removing supernatant, the samples were boiled in 15 µl SDS‐loading buffer and analyzed on 10–20% SDS–PAGE gel. Bands were visualized with Coomassie brilliant blue staining. Pull‐down assays were repeated three times. The intensities of MBP‐CENPC‐CT[CDK1] or MBP‐CENPN‐NT were measured with Fiji (Schindelin et al, 2012). The intensities of all histones in the CENP‐A nucleosome were measured as the signals for the CENP‐A nucleosome and normalized by the intensities of MBP proteins. The intensity values were normalized with the value of CENP‐A/MBP‐CENPN‐NT or CENP‐A/MBP‐CENPC‐CT in the absence of CENPC‐CT or CENPN‐NT, respectively. Data are shown as mean ± SD of three independent assays.
Author contributions
MA and FM performed all cryo‐EM experiments. MA also performed all biochemical experiments. RW and MH prepared the phosphorylated CENP‐C and nucleosome samples. RN performed crosslinking mass spectroscopy analyses. TK and KN helped cryo‐EM single particle image analyses. YA, RF, and HK prepared recombinant histones. EO purified CDK1. TF performed immunofluorescence analyses and supervised all experiments. MA and TF wrote the manuscript, discussing with all authors.
Conflict of interest
The authors declare that they have not conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors are very grateful to, K. Oshimo, Y. Fukagawa, and R. Fukuoka for technical assistance. This work was supported by JSPS KAKENHI Grant Number 25221106, 20H05389, 17H06167, 16H06279, and 15H05972 to TF, JSPS KAKENHI Grant Number to 18K06084 to MA, JSPS KAKENHI Grant number 25000013, Platform Project for Supporting Drug Discovery and Life Science Research (BINDS) from AMED under Grant Number JP19am0101117, the Cyclic Innovation for Clinical Empowerment (CiCLE) Grant Number JP17pc0101020 from AMED to KN, and JSPS KAKENHI Grant Numbers 18H05534 and 25116002 and the Platform Project for Supporting Drug Discovery and Life Science Research (BINDS) from AMED under Grant Number JP19am0101076 to HK. KN is supported by JEOL YOKOGUSHI Research Alliance Laboratories of Osaka University.
The EMBO Journal (2021) 40: e105671.
Data availability
The cryo‐EM density maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/): accession number EMD‐30239 for the CA‐CCCT complex at 4.5 Å, EMD‐30241 for the CA‐CCCT‐CNNT complex, EMD‐30237 for the CA‐CCpep‐CNNT complex, and EMD‐30240 for the CA‐CCCT complex at 6.78 Å. Atomic coordinates have been deposited in the Protein Data Bank (https://pdbj.org/): accession number PDB ID 7BY0 for the CA‐CCCT complex at 4.5 Å, and 7BXT for the CA‐CCpep‐CNNT complex. Raw cryo‐EM images were also deposited into EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) with IDs: EMPIAR‐10565 for the CA‐CCCT‐CNNT complex, EMPIAR‐10569 for the CA‐CCCT complex and EMPIAR‐10571 for the CA‐CCpep‐CNNT complex.
References
- Ali‐Ahmad A, Bilokapic S, Schafer IB, Halic M, Sekulic N (2019) CENP‐C unwraps the human CENP‐A nucleosome through the H2A C‐terminal tail. EMBO Rep 20: e48913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, Karpen GH (2008) Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9: 923–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allu PK, Dawicki‐McKenna JM, Van Eeuwen T, Slavin M, Braitbard M, Xu C, Kalisman N, Murakami K, Black BE (2019) Structure of the human core centromeric nucleosome complex. Curr Biol 29: 2625–2639.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Suzuki A, Hori T, Backer C, Okawa K, Cheeseman IM, Fukagawa T (2009) The CENP‐S complex is essential for the stable assembly of outer kinetochore structure. J Cell Biol 186: 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H (2012) Structural analysis of the hexasome, lacking one histone H2A/H2B dimer from the conventional nucleosome. Biochemistry 51: 3302–3309 [DOI] [PubMed] [Google Scholar]
- Arimura Y, Tachiwana H, Takagi H, Hori T, Kimura H, Fukagawa T, Kurumizaka H (2019) The CENP‐A centromere targeting domain facilitates H4K20 monomethylation in the nucleosome by structural polymorphism. Nat Commun 10: 576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BE, Cleveland DW (2011) Epigenetic centromere propagation and the nature of CENP‐a nucleosomes. Cell 144: 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Silva MC, Godek KM, Jansen LE, Straight AF (2009) Centromere assembly requires the direct recognition of CENP‐A nucleosomes by CENP‐N. Nat Cell Biol 11: 896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF (2010) Dual recognition of CENP‐A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittori S, Hong J, Saunders H, Feng H, Ghirlando R, Kelly AE, Bai Y, Subramaniam S (2018) Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP‐N. Science 359: 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol 375: 23–44 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LE, Van Duyne GD, Vinogradov SA et al (2015) Chromosomes. CENP‐C reshapes and stabilizes CENP‐A nucleosomes at the centromere. Science 348: 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Lee J, Sekulic N, Sennett MA, Lee TH, Black BE (2016) CENP‐C directs a structural transition of CENP‐A nucleosomes mainly through sliding of DNA gyres. Nat Struct Mol Biol 23: 204–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Liu Y, Wei Y, Deng W, Yu Z, Huang L, Teng Y, Yao T, You Q, Ruan H et al (2015) Structural transitions of centromeric chromatin regulate the cell cycle‐dependent recruitment of CENP‐N. Genes Dev 29: 1058–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR 3rd, Cleveland DW (2006) The human CENP‐A centromeric nucleosome‐associated complex. Nat Cell Biol 8: 458–469 [DOI] [PubMed] [Google Scholar]
- Fukagawa T, Pendon C, Morris J, Brown W (1999) CENP‐C is necessary but not sufficient to induce formation of a functional centromere. EMBO J 18: 4196–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukagawa T, Earnshaw WC (2014) The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 30: 496–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LY, Allu PK, Zandarashvili L, McKinley KL, Sekulic N, Dawicki‐McKenna JM, Fachinetti D, Logsdon GA, Jamiolkowski RM, Cleveland DW et al (2017) Centromeres are maintained by fastening CENP‐A to DNA and directing an arginine anchor‐dependent nucleosome transition. Nat Commun 8: 15775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Ariyoshi M, Okumura EI, Hori T, Fukagawa T (2018) Multiple phosphorylations control recruitment of the KMN network onto kinetochores. Nat Cell Biol 20: 1378–1388 [DOI] [PubMed] [Google Scholar]
- Hori T, Amano M, Suzuki A, Backer CB, Welburn JP, Dong Y, McEwen BF, Shang WH, Suzuki E, Okawa K et al (2008) CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell 135: 1039–1052 [DOI] [PubMed] [Google Scholar]
- Izuta H, Ikeno M, Suzuki N, Tomonaga T, Nozaki N, Obuse C, Kisu Y, Goshima N, Nomura F, Nomura N et al (2006) Comprehensive analysis of the ICEN (Interphase Centromere Complex) components enriched in the CENP‐A chromatin of human cells. Genes Cells 11: 673–684 [DOI] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y (2013a) A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP‐C. Science 340: 1110–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Zhou BR, Feng H, Bai Y (2013b) An evolving tail of centromere histone variant CENP‐A. Cell Cycle 12: 3133–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Makino F, Miyata T, Horvath P, Namba K (2019) Structure of the native supercoiled flagellar hook as a universal joint. Nat Commun 10: 5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klare K, Weir JR, Basilico F, Zimniak T, Massimiliano L, Ludwigs N, Herzog F, Musacchio A (2015) CENP‐C is a blueprint for constitutive centromere‐associated network assembly within human kinetochores. J Cell Biol 210: 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klykov O, Steigenberger B, Pektas S, Fasci D, Heck AJR, Scheltema RA (2018) Efficient and robust proteome‐wide approaches for cross‐linking mass spectrometry. Nat Protoc 13: 2964–2990 [DOI] [PubMed] [Google Scholar]
- Kwon MS, Hori T, Okada M, Fukagawa T (2007) CENP‐C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol Biol Cell 18: 2155–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ et al (2019) Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75: 861–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Sekulic N, Guo LY, Tsinman T, Black BE, Cheeseman IM (2015) The CENP‐L‐N complex forms a critical node in an integrated meshwork of interactions at the centromere‐kinetochore interface. Mol Cell 60: 886–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal H, Hori T, Furukawa A, Sugase K, Osakabe A, Kurumizaka H, Fukagawa T (2015) Dynamic changes in CCAN organization through CENP‐C during cell‐cycle progression. Mol Biol Cell 26: 3768–3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T, Takeuchi K, Gascoigne KE, Suzuki A, Hori T, Oyama T, Morikawa K, Cheeseman IM, Fukagawa T (2012) CENP‐T‐W‐S‐X forms a unique centromeric chromatin structure with a histone‐like fold. Cell 148: 487–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T, Rago F, Hori T, Tomii K, Cheeseman IM, Fukagawa T (2013) CENP‐T provides a structural platform for outer kinetochore assembly. EMBO J 32: 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Cheeseman IM, Hori T, Okawa K, McLeod IX, Yates JR 3rd, Desai A, Fukagawa T (2006) The CENP‐H‐I complex is required for the efficient incorporation of newly synthesized CENP‐A into centromeres. Nat Cell Biol 8: 446–457 [DOI] [PubMed] [Google Scholar]
- Okumura E, Sekiai T, Hisanaga S, Tachibana K, Kishimoto T (1996) Initial triggering of M‐phase in starfish oocytes: a possible novel component of maturation‐promoting factor besides cdc2 kinase. J Cell Biol 132: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oya E, Nakagawa R, Yoshimura Y, Tanaka M, Nishibuchi G, Machida S, Shirai A, Ekwall K, Kurumizaka H, Tagami H et al (2019) H3K14 ubiquitylation promotes H3K9 methylation for heterochromatin assembly. EMBO Rep 20: e48111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentakota S, Zhou K, Smith C, Maffini S, Petrovic A, Morgan GP, Weir JR, Vetter IR, Musacchio A, Luger K (2017) Decoding the centromeric nucleosome through CENP‐N. Elife 6: e33442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perpelescu M, Fukagawa T (2011) The ABCs of CENPs. Chromosoma 120: 425–446 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R, Sinz A (2017) Improved single‐step enrichment methods of cross‐linked products for protein structure analysis and protein interaction mapping. Anal Bioanal Chem 409: 2393–2400 [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi‐Takanaka Y, Oda T, Sato M, Park SY et al (2011) Crystal structure of the human centromeric nucleosome containing CENP‐A. Nature 476: 232–235 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Tawaramoto‐Sasanuma M, Kawaguchi S, Ohta T, Yoda K, Kurumizaka H, Yokoyama S (2004) Expression and purification of recombinant human histones. Methods 33: 3–11 [DOI] [PubMed] [Google Scholar]
- Tian T, Li X, Liu Y, Wang C, Liu X, Bi G, Zhang X, Yao X, Zhou ZH, Zang J (2018) Molecular basis for CENP‐N recognition of CENP‐A nucleosome on the human kinetochore. Cell Res 28: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Hara M, Okumura EI, Herve S, Fachinetti D, Ariyoshi M, Fukagawa T (2019) CDK1‐mediated CENP‐C phosphorylation modulates CENP‐A binding and mitotic kinetochore localization. J Cell Biol 218: 4042–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JR, Faesen AC, Klare K, Petrovic A, Basilico F, Fischbock J, Pentakota S, Keller J, Pesenti ME, Pan D et al (2016) Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537: 249–253 [DOI] [PubMed] [Google Scholar]
- Westhorpe FG, Straight AF (2013) Functions of the centromere and kinetochore in chromosome segregation. Curr Opin Cell Biol 25: 334–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Yang J, Zhang Z, McLaughlin SH, Chang L, Fasci D, Ehrenhofer‐Murray AE, Heck AJR, Barford D (2019) Structure of the inner kinetochore CCAN complex assembled onto a centromeric nucleosome. Nature 574: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: Real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The cryo‐EM density maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb/): accession number EMD‐30239 for the CA‐CCCT complex at 4.5 Å, EMD‐30241 for the CA‐CCCT‐CNNT complex, EMD‐30237 for the CA‐CCpep‐CNNT complex, and EMD‐30240 for the CA‐CCCT complex at 6.78 Å. Atomic coordinates have been deposited in the Protein Data Bank (https://pdbj.org/): accession number PDB ID 7BY0 for the CA‐CCCT complex at 4.5 Å, and 7BXT for the CA‐CCpep‐CNNT complex. Raw cryo‐EM images were also deposited into EMPIAR (https://www.ebi.ac.uk/pdbe/emdb/empiar/) with IDs: EMPIAR‐10565 for the CA‐CCCT‐CNNT complex, EMPIAR‐10569 for the CA‐CCCT complex and EMPIAR‐10571 for the CA‐CCpep‐CNNT complex.