

Abstract
Various forms of cell death have been identified over the last decades with each relying on a different subset of proteins for the activation and execution of their respective pathway(s). In addition to the three best characterized pathways—apoptosis, necroptosis, and pyroptosis—other forms of regulated cell death including autophagy‐dependent cell death (ADCD), mitochondrial permeability transition pore (MPTP)‐mediated necrosis, parthanatos, NETosis and ferroptosis, and their relevance for organismal homeostasis are becoming better understood. Importantly, it is increasingly clear that none of these pathways operate alone. Instead, a more complex picture is emerging with many pathways sharing components and signaling principles. Finally, a number of cell death regulators are implicated in human diseases and represent attractive therapeutic targets. Therefore, better understanding of physiological and mechanistic aspects of cell death signaling should yield improved reagents for addressing unmet medical needs.
Keywords: apoptosis, caspase, necroptosis, pyroptosis, RIPK1
Subject Categories: Autophagy & Cell Death, Signal Transduction
This review provides an overview of the different forms of cell death and how they are related to each other, as well as the role of cell death regulators in several pathophysiological processes.

Introduction
Cell death plays a central role in all aspects of life. It is involved in the development of multicellular organisms and tissue homeostasis where cell death depletes dispensable cells. Moreover, it is critical for fighting off infections and is associated with multiple diseases that are caused by deregulated or dysfunctional cell death signaling. Consequentially, there is a growing interest in modulating cell death to treat diseases. Various forms of cell death have been described so far, with apoptosis, necroptosis, and pyroptosis being the best understood. In recent years, a more complex picture of cell death modalities has been established as crosstalk and backup mechanisms between different pathways were identified. This review will focus on different forms of cell death, their interconnectivity, and validated targets for treating diseases.
Apoptosis
Apoptosis is the first described form of programmed cell death (Kerr et al, 1972), and it plays a critical role in tissue homeostasis. It contributes to cell turnover, the proper functioning of the immune system, and embryonic development (Voss & Strasser, 2020). There are several key characteristics of apoptosis. Cells undergo morphological changes which lead to cellular, organelle, and DNA fragmentation as well as the formation of apoptotic bodies (Kerr et al, 1972; Zakeri et al, 1993). This is an active, energy consuming process executed by a subset of cellular proteins. Even though, in general, this process is immunological silent, apoptosis has been shown to be involved in inflammatory pathologies as well (Rickard et al, 2014; Yang et al, 2015; Singh et al, 2019).
There are two major pathways that mediate apoptosis: intrinsic and extrinsic pathways. Intrinsic apoptosis is controlled by the equilibrium of the different Bcl‐2 (B‐cell lymphoma 2) family members which can be disrupted by various stimuli leading to cell death. During extrinsic apoptosis, members of the TNF (tumor necrosis factor) superfamily (TNFSF) can induce cell death by binding to their cell surface receptors and activating a deathly signaling cascade causing extrinsic apoptosis. The third modality of apoptosis induction is cell‐based. Cytotoxic T cells can engage cells that present non‐self‐antigens leading to cell death induction by proteases called granzymes. All apoptotic pathways converge on the central proteases of this pathway: caspases, which are either playing a role in transmitting cell death stimulus (initiator caspases) or in the execution (effector caspases).
Intrinsic apoptosis
Intrinsic apoptosis is engaged by cells that are obsolete, deprived from growth factors or damaged (e.g., UV) (Fig 1). These diverse stimuli can tip the balance between different groups of the Bcl‐2 (B‐cell lymphoma 2) proteins leading to the activation of cell death (Kale et al, 2018) The Bcl‐2 superfamily can be divided into three subfamilies: the anti‐apoptotic Bcl‐2 proteins, the pro‐apoptotic BH3‐only (BH: Bcl‐2 homology) proteins, and the death effectors Bax (Bcl‐2‐associated X protein), Bak (Bcl‐2 homologous antagonist/killer), and Bok (Bcl‐2‐related ovarian killer). Normally, the cells keep Bax and Bak in check by the expression of anti‐apoptotic Bcl‐2 family members (Bcl‐2, Bcl‐XL, Mcl‐1, A1, Bcl‐w) which inhibit Bax and Bak pore forming ability (Kale et al, 2018). The BH3‐only family members can inhibit the anti‐apoptotic Bcl‐2 proteins or in some cases also directly engage Bax and Bak (for example Bim) (Kim et al, 2006; Czabotar et al, 2014). Some BH3‐only proteins are regulated by transcriptional regulation (PUMA regulated by p53, DNA damage) (Nakano & Vousden, 2001) or by post‐translational modifications (BIM, BID) (Li et al, 1998; Lei & Davis, 2003). Tipping the equilibrium in favor of pro‐apoptotic Bcl‐2 proteins leads to activation of Bax and Bak and results in the MOMP (mitochondrial outer membrane permeabilization) (Wei et al, 2001). Bok, which can induce MOMP in a constitutively fashion, is regulated differently—by proteasomal degradation pathways (Llambi et al, 2016; Ke et al, 2018). MOMP causes the release of the key mediators of intrinsic apoptosis, cytochrome c (Stein & Hansen, 1999), and endogenous IAP (inhibitor of apoptosis) antagonist, SMAC/ Diablo (second mitochondria derived activator of caspases/direct IAP binding protein with low pI) (Du et al, 2000; Verhagen et al, 2000). Cytochrome c‐bound Apaf1 (apoptotic protease activating factor 1) (Zou et al, 1997) recruits initiator caspase caspase‐9 to form the apoptosome, a platform for the activation of the executioner caspases caspase‐3 and ‐7 (Li et al, 1997). Caspases‐3, ‐7, and ‐9 can be blocked by the major endogenous caspase inhibitor, XIAP (X chromosome‐linked IAP). SMAC can antagonize XIAP and other IAPs thus allowing full caspase activation and apoptosis execution (Du et al, 2000). Caspases cleave a wide variety of cellular proteins to induce characteristic changes of apoptotic death (cellular and nuclear fragmentation, DNA laddering, etc.). For example, ICAD (inhibitor of caspase‐activated DNase) cleavage leads to the activation of CAD (caspase‐activated DNase) that induces genome fragmentation (Enari et al, 1998; Sakahira et al, 1998), whereas cleavage of ROCK‐I (Rho associated protein kinase) induces the contraction and blebbing of cells (Coleman et al, 2001; Sebbagh et al, 2001). A specific form of intrinsic apoptosis is anoikis, which is induced by the loss of pro‐survival signals via integrin binding to the ECM (extracellular matrix) (Frisch & Francis, 1994). Integrins can signal via different pathways (PI3K or FAK), which modulate the Bcl‐2 or BH3‐only family members (Gilmore, 2005). In this fashion, anoikis ensures that cells can only survive in the appropriate compartments/organs within the body. Overcoming this checkpoint plays a major role in metastasis/ invasiveness of cancer (Paoli et al, 2013).
Figure 1. Intrinsic and extrinsic apoptosis.
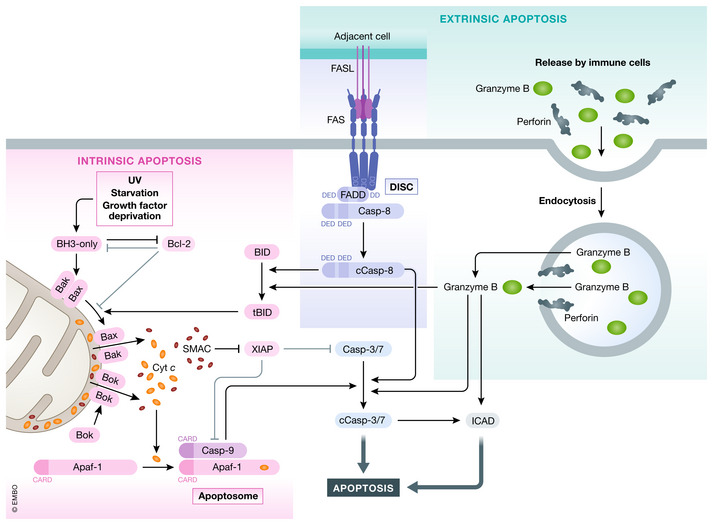
Intrinsic apoptosis can be induced by various stimuli (e.g., GF (growth factor) withdrawal) by shifting the equilibrium of pro‐survival Bcl‐2 and BH3‐only proteins. Sequestering of pro‐survival Bcl‐2 proteins or directed binding of BH3‐only proteins to Bax and Bak induces oligomerization of Bax and Bak leading to MOMP and cytochrome c and Smac release. Bok can lead to MOMP independent of Bcl‐2 family members. Released cytochrome c binds Apaf‐1 and induces apoptosome formation which recruits caspase‐9. Activated caspase‐9 induces caspase‐3/7 cleavage and activation (cCasp3/7) leading to apoptosis. Extrinsic apoptosis can be induced by binding of select group of TNF family ligands to, their receptors leading to DISC formation by recruitment of adapter FADD/TRADD and caspase‐8. Caspase‐8 autoprocesses itself (cCasp8—cleaved/activated caspase‐8) and can directly activate caspase‐3 or cleave Bid to generated tBid and triggers intrinsic apoptosis. Caspase‐3/7/9 activity can be inhibited by XIAP, which itself can be antagonized by Smac. Lastly, apoptosis can also be induced by granzyme B and Perforin released form immune cells. Once taken up by the target cell, granzyme B can induces cell death by caspase‐3 or by directly activating apoptosis effectors (e.g., CAD).
Extrinsic apoptosis
Extrinsic apoptosis is triggered by TNF family ligand‐receptor interactions, most prominently by TNF family ligands: TNF, FasL, TRAIL, and TL1A. The receptor complexes either recruit FADD (Fas‐associated protein with death domain) or TRADD (TNFRSF1A‐associated via death domain) to the oligomerized complex (Wilson et al, 2009). FasL‐mediated signaling will be used to describe extrinsic apoptotic signaling (Fig 1), and TNF signaling will be described for necroptotic signaling. FasL binds to its transmembrane receptor Fas, which recruits FADD (Fas‐associated death domain protein) via death domain (DD) interactions (Chinnaiyan et al, 1995; Boldin et al, 1996). FADD contains a DD and also a death effector domain (DED), which allows the recruitment of caspase‐8 via homotopic domain interaction, forming the death inducing signaling complex—DISC (Boldin et al, 1996; Muzio et al, 1996; Medema et al, 1997). The proximity of multiple caspase‐8 molecules induces the transactivation by proteolytic cleavage (Muzio et al, 1998; Yang et al, 1998). Cleavage results in the p18 and p10 fragments which activate caspase‐3 and caspase‐7 (type I apoptosis) (Stennicke et al, 1998). Insufficient activation of caspase‐3 leads to type II apoptosis in which caspase‐8 cleaves the BH3‐only protein BID (BH3 interacting domain death agonist) to generate its activated form: truncated BID (tBID) (Li et al, 1998). tBID stimulates intrinsic apoptotic pathway by binding directly binding to Bax/Bak inducing MOMP (type II apoptosis) (Desagher et al, 1999; Wei et al, 2000). The two pathways are cell line dependent, and their activation is differentially regulated by XIAP expression (Jost et al, 2009; Varfolomeev et al, 2009).
Granzyme‐mediated apoptosis
Cytotoxic lymphoid cells (predominantly NK cells and cytotoxic T cells) can induce cell death via death receptor ligands (see above) or the granzyme/perforin system (Pardo et al, 2008; Martinez‐Lostao et al, 2015). After recognition of transformed or infected cells, cytotoxic cells release secretory granules that contain perforin and granzyme B (Fig 1). These secreted factors are taken up by endocytosis and released to the cytosol by the perforin‐dependent or ‐independent pathways (Voskoboinik et al, 2015). Once released to the cytosol, granzyme B cleaves caspases and Bid activating apoptotic pathways described above (Boivin et al, 2009). However, human granzyme B can also directly cleave ICAD, a known caspase‐3 target, to induce DNA fragmentation, thereby circumventing the need of caspases (Thomas et al, 2000).
Necroptosis
Necroptosis is a regulated, pro‐inflammatory, and caspase‐independent form of necrotic cell death. Death receptors (DRs), toll‐like receptors (TLRs), and nucleic acid sensing protein ZBP1 (Z‐DNA binding protein 1) are prototypic inducer of necroptosis whose stimulation converges on the activation of RIP3 (receptor‐interacting protein 3) (Newton & Manning, 2016). Following autophosphorylation, RIP3 engages the pseudokinase MLKL (mixed lineage kinase like) and phosphorylates it (Sun et al, 2012; Murphy et al, 2013). Activated MLKL oligomerizes and is transported to the cytoplasmic membrane, where it induces membrane permeabilization and cell death (Cai et al, 2014; Chen et al, 2014; Dondelinger et al, 2014; Samson et al, 2020). It is important to note that activation of MLKL is not the point of no return as several mechanisms have been found that can modulate necroptosis after MLKL activation, which are reviewed by Murphy (Murphy, 2020).
The key mediators for necroptosis contain a signature RHIM domain (RIP homotypic interaction motif): RIP1 is key for death receptor signaling (Sun et al, 2002), TRIF (TIR‐domain‐containing adapter inducing interferon‐β) for toll‐like receptors (Kaiser & Offermann, 2005), ZBP1 for virally encoded nucleic acid induced necroptosis (Kaiser et al, 2008), and RIP3 (Sun et al, 2002) as activator of MLKL.
Necroptosis has been studied extensively based on the TNF receptor‐mediated signaling (Fig 2). TNF binding to TNFR1 triggers the recruitment of RIP1, TRADD, TRAF2 (TNF receptor‐associated factor 2), and c‐IAP1/2 to the receptor‐associated complex (complex I) (Newton, 2020). c‐IAPs then ubiquitinate several proteins, including RIP1 and themselves, in complex I with K11‐ and K63‐linked ubiquitin chains (Bertrand et al, 2008; Mahoney et al, 2008; Varfolomeev et al, 2008; Dynek et al, 2010; Moulin et al, 2012), resulting in LUBAC (linear ubiquitin chain assembly complex) recruitment, and addition of linear ubiquitin chains (Haas et al, 2009). The different ubiquitin chains serve as a platform for recruitment of the kinase complexes IKK and TAK1/TAB2/3 leading to the NF‐κB (nuclear factor κ‐light‐chain‐enhancer of activated B cells) and MAPK (mitogen‐activated protein kinase) pathway activation (Hayden & Ghosh, 2014). These signaling events induce the expression of pro‐survival and pro‐inflammatory genes. Countering these effects are ubiquitin hydrolases, deubiquitinases, which restrict signaling by removing the different ubiquitin chains from the protein (Lork et al, 2017).
Figure 2. Necroptosis.
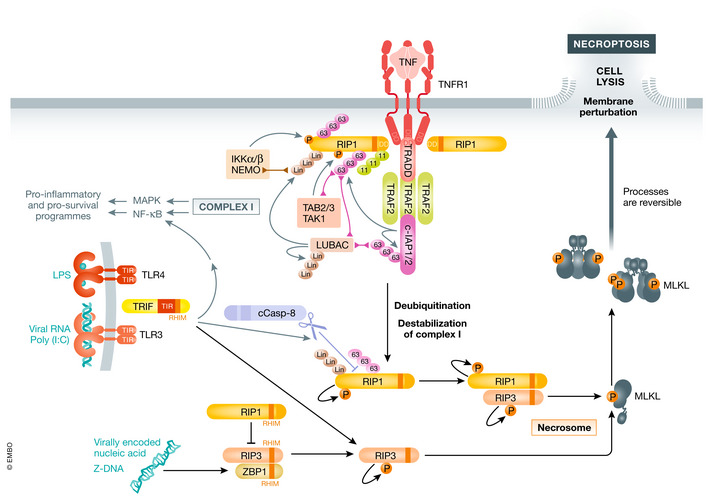
Necroptosis can be induced by different stimuli. Upon binding to its receptor, TNF induces complex formation leading to NF‐κB and MAPK activation. Prolonged signaling and inhibition of caspases leads to RIP1 translocation to the cytosol forming complex II. RIP1 autophosphorylates recruiting RIP3. RIP3 phosphorylates itself as well as MLKL leading to MLKL oligomerization which induces membrane perturbation and cell lysis. LPS or Poly(I:C)‐induced TLR3/4 signaling also can stimulate necroptosis through adaptor TRIF, which engages RIP1 or RIP3. Sensing of Z‐DNA by ZBP1 leads to binding to RIP3 and cell death, which can be inhibited by RIP1.
Once deubiquitinated, RIP1 is released into the cytosol, where it can be auto‐activated by autophosphorylation. Interestingly, in mouse cells TNF‐mediated activation of necroptosis can stimulate RIP1 autophosphorylation already in the TNFR1 complex (Newton et al, 2016b). Nevertheless, a protein complex composed of FADD, caspase‐8, and c‐FLIP (cellular FLICE‐inhibitory protein) can bind RIP1 and regulate RIP1 necroptotic and apoptotic potential by caspase‐8‐mediated cleavage of RIP1 at D324 (Lin et al, 1999; Zhang et al, 2019; Newton et al, 2019a; Lalaoui et al, 2020). Stoichiometry of caspase‐8 and c‐FLIP isoforms determine if cells will survive or undergo apoptosis or necroptosis (discussed in more detail in another section). Only when caspase‐8 is inhibited, depleted, or insufficiently activated, necroptotic signaling can proceed, leading to RIP1 binding to RIP3 and subsequent RIP3 autophosphorylation (Cho et al, 2009; He et al, 2009). RIP1 auto‐activation and cell death induction can be restricted by inhibitory phosphorylation (e.g., on S321 of RIP1) (reviewed in (Delanghe et al, 2020).
The second key mediator for necroptosis induction is TRIF (Fig 2). Upon sensing of viral RNA (TLR3) or LPS (TLR4), TRIF can be recruited to activated TLR3/4 complexes via its TIR (Toll/interleukin‐1 receptor) domain. TRIF can then induce NF‐κB signaling via TRAF2/6‐RIP1 axis, which leads to TNF expression (Kawasaki & Kawai, 2014). However, TRIF can also trigger apoptosis by engaging RIP1 and caspase‐8 (McAllister et al, 2013) or necroptosis by directly activating RIP3 (Kaiser et al, 2013).
The fourth RHIM domain containing protein is ZBP1 or DAI (Fig 2). ZBP1 is activated by binding to viral Z‐DNA or Z‐RNA, a left‐handed fold of DNA/RNA, to mediate immune response against certain viruses (reviewed by (Kuriakose & Kanneganti, 2018)). ZBP1 can directly bind RIP3 to induce necroptosis, while RIP1 inhibits necroptotic signaling by RHIM‐RHIM domain interactions (Lin et al, 2016; Newton et al, 2016b). Most recently, ZBP1 activation has been linked to sensing endogenous Z‐double‐strand RNA and induction of necroptosis in the context of RIP1 RHIM mutation, epithelial cell‐specific knockout of RIP1 (Ripk1E‐KO), or intestinal epithelial cell knockout of FADD (FaddIEC‐KO) deficiency (Jiao et al, 2020).
Pyroptosis
Pyroptosis is a Gasdermin‐dependent form of pro‐inflammatory necrotic cell death. Stimulation of caspase‐1/4/5/11 (caspase‐4/5 are the human homologs to murine caspase‐11) by different inflammasome pathways lead to their activation by autoprocessing. Active caspases can then cleave Gasdermin‐D (GSDMD) into its N‐terminal and C‐terminal domains, GSDMD‐N and GSDMD‐C (He et al, 2015; Kayagaki et al, 2015; Shi et al, 2015). GSDMD‐C is the auto‐inhibitory domain that inhibits the cell lytic properties of GSDMD‐N (Kayagaki et al, 2015; Shi et al, 2015; Kuang et al, 2017). Once the two domains are separated by cleavage, GSDMD‐N translocates to the membrane by binding to phosphatidylinositol phosphates and phosphatidylserine and oligomerizes inducing a lytic cell death by forming multi subunit pores (Aglietti et al, 2016; Ding et al, 2016; Liu et al, 2016; Sborgi et al, 2016). Besides targeting GSDMD, caspase‐1 also processes the pro‐inflammatory cytokines pro‐IL‐1β (interleukin 1β) and pro‐IL‐18 (interleukin 18) to their mature forms (Thornberry et al, 1992; Ghayur et al, 1997; Gu et al, 1997). Neither IL‐1β nor IL‐18 have a secretion sequence, so they are leaking through GSDMD pores (Evavold et al, 2018; Heilig et al, 2018).
Signal for GSDMD cleavage by caspases 1/4/5/11 is propagated from different inflammasomes and mediated by the canonical or the non‐canonical inflammasome pathways (Fig 3). Generally, canonical inflammasomes consist of a sensor for DAMP (damage‐associated molecular pattern) or PAMP (pathogen‐associated molecular pattern) detection (AIM2, NLRP1, NLRP3, NLRC4, and Pyrin), which interact via Pyrin domains (PYD) with the bridging molecule ASC (apoptosis‐associated speck‐like protein containing a CARD). ASC then interacts with caspase‐1 via CARD (caspase activation and recruitment domains) and oligomerizes caspase‐1 to induce its activation (Martinon et al, 2002; Srinivasula et al, 2002; Chauhan et al, 2020).
Figure 3. Pyroptosis.
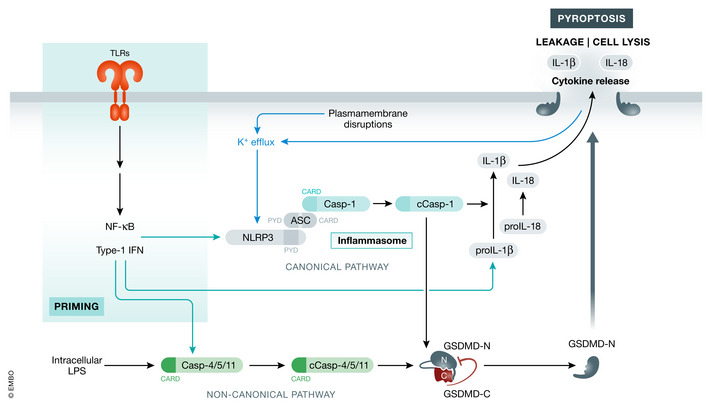
Pyroptotic cell death can be induced by various stimuli that activate inflammasome. The activation of NLRP3 prompts its binding to ASC and caspase‐1 forming the inflammasome. Caspase‐1 processes pro‐IL‐1β and pro‐IL‐18 to their active forms. In parallel, caspase‐1 cleaves GSDMD separating the inhibitory C‐ and active N‐terminal domains. GSDMD‐N then translocates to the membrane inducing cell lysis and cytokine release. Non‐canonical inflammasome activation consists of priming which induces expression of several pathway genes (caspase‐11). Intracellular LPS can be sensed by caspase‐11 leading to its activation and processing of GSDMD and caspase‐1. Cell lysis can then also activate canonical inflammasome signaling. Green arrows indicate upregulation by gene expression, while black arrows indicate interactions/cleavage events or inhibition.
Inflammasome activation is a two‐step process. In a priming step, TLRs recognize PAMPs and induce a NF‐κB and IFN type I (interferon type I)‐dependent gene expression (Rathinam et al, 2012). This leads to upregulation of NLRP3 and caspase‐11. The second step in the activation of non‐canonical inflammasome signaling is triggered by intracellular LPS which binds murine caspase‐11 or human caspase‐4/5 leading to their activation (Kayagaki et al, 2011; Shi et al, 2014). Activated caspase‐11 cleaves GSDMD to trigger pyroptosis (Kayagaki et al, 2015). The process of inflammasome activation is reviewed in more detail elsewhere (Kelley et al, 2019).
Besides GSDMD, other Gasdermins also have reported roles in cell death. For example, Gasdermin E (GSDME) has been shown to be cleaved by caspase‐3 as well as granzyme B (Rogers et al, 2017; Wang et al, 2017; Lee et al, 2018). The interconnectivity of various cell death modalities will be discussed in more detail in following sections.
Other forms of regulated cell death
Other forms of cell death have been identified in the recent years. While some signaling components and pathways are already well understood, others still need further research. The key characteristics of each cell death pathway described below are summarized in Fig 4.
Figure 4. Key features of different forms of regulated cell death.
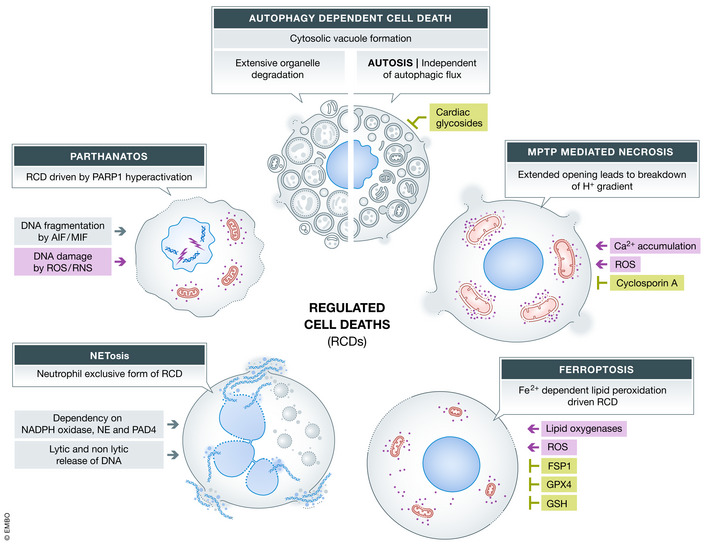
Overview of regulated cell death pathways highlighting the stimuli, key features as well as positive and negative regulators of the pathways. Gray boxes indicate cell death pathway, green boxes show negative regulators/inhibitors, while red boxes indicate activators.
Autophagy‐dependent cell death (ADCD)
Autophagy defines the degradation of cellular components by the lysosomal pathway. This can include organelles, like mitochondria and ER, as well as other cytoplasmic content. Autophagy is a critical process for cellular homeostasis and knockout of autophagy mediators often results in perinatal lethality (Kuma et al, 2017). The process of autophagy can be divided into several steps which are regulated by individual protein complexes. The most common way for autophagy activation is starvation, which initiates autophagy by a kinase complex consisting of Ulk1‐ATG13/101 and FIP200 (Zachari & Ganley, 2017). After initiation of autophagy, nucleation, elongation, and phagophore formation are regulated by different protein complexes. The phagophore is labeled with LC3 to eventually mature to the autophagosome. Upon fusion of those vesicles with lysosomes, the autolysosome is formed and the content is degraded. A detailed description of the process can be found in the following review (Dikic & Elazar, 2018). Interestingly, most of the pathway components seem to have additional non‐autophagic functions (reviewed in (Galluzzi & Green, 2019)). Autophagy‐dependent cell death (ADCD) has been defined as a form of cell death that relies exclusively on the autophagic pathway components, which is an important distinction given that autophagy can also coincide with other forms of cell death (Galluzzi et al, 2018). ADCD can proceed by two different pathways. The first pathway is induced by extensive degradation of organelles which is dependent on the autophagic flux (Dasari et al, 2017). The second form, referred to as Autosis, does not depend on the fusion of autophagosomes and lysosomes (Liu et al, 2013). In both cases, vacuole formation in the cytoplasm can be detected (Bialik et al, 2018). Treatment of cancer cells with resveratrol triggers the autophagic flux‐dependent ADCD, without activating apoptosis or necroptosis (Dasari et al, 2017). The massive degradation by lysosome fusion leads to a breakdown of the cytoplasmic organization with loss of organelles such as endoplasmic reticulum or mitochondria. Autosis can be induced by treatment with TAT‐Beclin‐1 peptides, starvation or hypoxia, which leads to cell swelling and eventually rupture of the plasma membrane. These conditions result in cell death mediated by Na+/K+‐ATPase and can be inhibited by cardiac glycosides (Liu et al, 2013). Autotic cells were also identified in samples of patients with severe anorexia nervosa (Kheloufi et al, 2015). In general, ADCD has been shown in association with physiological process as well as various pathologies including reperfusion injuries and various forms of cancer (Bialik et al, 2018; Denton & Kumar, 2019).
Mitochondrial permeability transition pore (MPTP)‐mediated necrosis
The mitochondrial permeability transition pore can mediate necrosis based on changes in the intracellular microenvironment. Two factors that can induce opening of the pores are oxidative stress and cytosolic/ mitochondrial Ca2+ accumulation. The pores allow the flux of molecules up to 1.5 kDa in size leading to breakdown of the H+ gradient and subsequently halting the ATP synthesis (Lemasters et al, 2009; Izzo et al, 2016). The pore opening has been shown to be reversible and meant to regulate mitochondrial Ca2+ levels while prolonged opening induces cell death (Baines et al, 2005; Korge et al, 2011). Cyclophilin D (CypD) so far is the only protein that has been shown to be critical for MPTP in vivo and in vitro. Accordingly, Ppif −/− mice (gene coding for CypD) showed reduced infarct size after ischemia/reperfusion injury of heart or brain (Baines et al, 2005; Nakagawa et al, 2005; Schinzel et al, 2005). In addition, mitochondria isolated from Ppif −/− showed a reduced swelling upon treatment with Ca2+. Cyclosporin A, a MPTP inhibitor, did not show an additional effect in knockout mitochondria indicating CypD as the target (Basso et al, 2005; Nakagawa et al, 2005; Schinzel et al, 2005). Yet, another study detected pore opening in mitochondria lacking CypD with increasing Ca2+ concentration, which indicates that blocking CypD might not be sufficient (Basso et al, 2005). So far, the structure and components of the pore are still not completely known (Nesci, 2020), but the F1F0 ATP synthase has been shown to be part of it (Bonora et al, 2013; Giorgio et al, 2013). These findings also suggest the F1F0 ATP synthase may be a potential drug target for various pathologies, such as myocardial infarct, reperfusion injuries, and neurodegenerative diseases (Sileikyte & Forte, 2019).
Parthanatos
Parthanatos is a form of regulated cell death dependent on poly(ADP) ribose polymerase 1 (PARP1) (Andrabi et al, 2008). PARP1 is part of the DNA repair machinery which binds DNA single breaks and PARylates itself and other proteins to recruit other components of the machinery (reviewed in (Ray Chaudhuri & Nussenzweig, 2017)). Severe DNA damage by prolonged generation of reactive oxygen species or reactive nitrogen species (RNS) induces recruitment and activation of PARP1 to the DNA (Zhang et al, 1994) leading to the formation of PAR polymers and depletion of NAD+ and ATP, which might be fatal for the cell (Robinson et al, 2019). However, extensive generation of PAR polymers can promote AIF (apoptosis inducing factor mitochondria associated 1) ‐MIF (macrophage migration inhibitory factor) interaction to facilitate MIF catalyzed DNA fragmentation (Yu et al, 2002; Wang et al, 2016). Nevertheless, a PARP‐dependent cell death driving retinal degradation in vivo can be AIF‐independent (Jang et al, 2017). RNS associated with parthanatos have been show to play a role in neural pathologies (Virag & Szabo, 2002; Fatokun et al, 2014).
NETosis
Neutrophils are part of the innate immune system, and their main task is to neutralize pathogens by phagocytosis or degranulation (Segal, 2005). Another form of host defense is the formation of NET (neutrophil extracellular traps). NETosis describes the process of neutrophil DNA release into the extracellular space (Brinkmann et al, 2004). The release of neutrophil DNA containing different proteins with anti‐pathogenic activity can be associated with cell death, but can be independent of it as well (Yipp & Kubes, 2013; Yousefi et al, 2019). For both processes, NADPH oxidase and ROS, including mitochondrial ROS, have been reported to be critical for actin depolarization and NET release (Papayannopoulos et al, 2010; Douda et al, 2015; Stojkov et al, 2017). Elevated ROS leads to myeloperoxidase activation, which leads to activation of neutrophil elastase (NE) (Papayannopoulos et al, 2010). NE associates with actin and processes it thus leading to depolarization and loss of actin dynamics (Metzler et al, 2014). Subsequently, NE can translocate to the nucleus, cleaves histones, and nuclear envelope proteins (Papayannopoulos et al, 2010). In combination with histone processing, histones are also citrullinated by PAD4 (protein arginine deiminase 4), which leads to chromatin decondensation and eventually NET release (Wang et al, 2009; Thiam et al, 2020). A detailed discussion of the pathway can be found in this review (Papayannopoulos, 2018). Further research is needed to understand the molecular details of non‐lytic and lytic forms of NET formation. The role of other forms of cell death and autophagy in the context of NETosis will be discussed below.
Ferroptosis
Ferroptosis is a form of regulated cell death that depends on iron (Fe2+)‐mediated lipid peroxidation induced by ROS (Yang & Stockwell, 2008; Dixon et al, 2012). Reactive oxygen species are constantly generated by different physiological processes. In combination with cellular labile iron, this can lead to ferroptosis (Snezhkina et al, 2019). Fe2+ can act as a catalyst to convert H2O2 to OH• radicals (Fenton reaction) which react with polyunsaturated fatty acids. Further, Fe2+ is a cofactor for lipoxygenases which catalyze the generation of lipid hydroperoxides (Yang et al, 2016; Stoyanovsky et al, 2019). To protect cells from ROS, hydroperoxides are neutralized stepwise by different enzyme families, namely superoxide dismutases, glutathione peroxidases, catalases, and peroxiredoxins. Ferroptotic cell death can be induced by either increased ROS generation or dysregulation of ROS neutralization (Li et al, 2020). Ferroptosis can be negatively regulated by glutathione and GPX4 (glutathione peroxidase 4). Cystine uptake is critical for glutathione synthesis, and interference with its transporter (Xc − Cys/Glu anti‐porter) induces ferroptosis by indirectly reducing GPX4 activity (Dixon et al, 2012). Loss of GPX4 resulted in sensitization to ferroptosis in vitro and in vivo (Friedmann Angeli et al, 2014; Yang et al, 2014). Besides a glutathione dependent system, ferroptosis suppressor protein 1 (FSP1) was identified to protect cells from ferroptosis by reducing coenzyme Q10, which acts as a radical scavenger (Bersuker et al, 2019; Doll et al, 2019). Ferroptosis has been associated with several pathologies as reviewed elsewhere (Bebber et al, 2020; Belavgeni et al, 2020; Li et al, 2020).
The interplay of cell death pathways
Caspase‐8: Between cell survival, apoptosis, and necroptosis
Caspase‐8 was initially identified as a component of extrinsic apoptotic signaling platform DISC (death inducing signaling complex) (Boldin, 1996; Muzio et al, 1996) and later as part of the cytosolic TNF‐induced complex II (Micheau & Tschopp, 2003). Soon, it became obvious that caspase‐8 has a more complex role, especially in the regulation of other cell death pathways (Degterev et al, 2005) (Fig 5). Caspase‐8 lethality can be rescued by deletion of RIP3 or MLKL (Kaiser et al, 2011; Oberst et al, 2011), which indicated that caspase‐8 restricts necroptotic signaling. The enzymatic activity of caspase‐8 determines if cells survive or die via apoptosis or necroptosis. One way of regulating catalytic activity of caspase‐8 in complex II is achieved by its catalytically inactive paralog c‐FLIP (Goltsev et al, 1997; Han et al, 1997; Hu et al, 1997; Inohara et al, 1997; Irmler et al, 1997; Shu et al, 1997; Srinivasula et al, 1997; Rasper et al, 1998). Low levels of c‐FLIPL lead to the formation of caspase‐8 homodimers resulting in self‐processing and apoptosis (Hughes et al, 2016). Pharmacological inhibition of caspase‐8 (e.g., by zVAD‐FMK or Emricasan) or inhibition by FLIP(S/R) leads to necroptosis and RIP1 activation (Fricker et al, 2010). Besides the cellular isoforms of FLIP, some viruses carry their own versions of FLIP (viral FLIP—vFLIP), which can inhibit caspase‐8 (Thome et al, 1997). Overexpression of vFLIP MC159 in combination with IAP antagonist induces necroptosis (Feoktistova et al, 2012). Heterodimers of c‐FLIP and caspase‐8 partially activate caspase‐8 and restrict necroptosis by cleaving RIP1 (at Asp324 human or at Asp325 mouse RIP1) (Oberst et al, 2011; Pop et al, 2011). Cleavage of RIP1 is critical for cell homeostasis, and several patients have been identified with heterozygous mutation at the caspase‐8 cleavage site resulting in a disease called cleavage‐resistant RIP1‐induced auto‐inflammatory (CRIA) syndrome (Lalaoui et al, 2020; Tao et al, 2020). CRIA patients suffer periodic fever with elevated cytokine and chemokine levels (Lalaoui et al, 2020; Tao et al, 2020). Analogous mutation in mice drew a similar picture, with Ripk1D325A/D325A animals being embryonic lethal (Zhang et al, 2019; Newton et al, 2019a; Lalaoui et al, 2020) and heterozygous animals being sensitive in TNF‐induced systemic inflammatory response syndrome (SIRS) (Newton et al, 2019a).
Figure 5. Crosstalk between different forms of cell death.
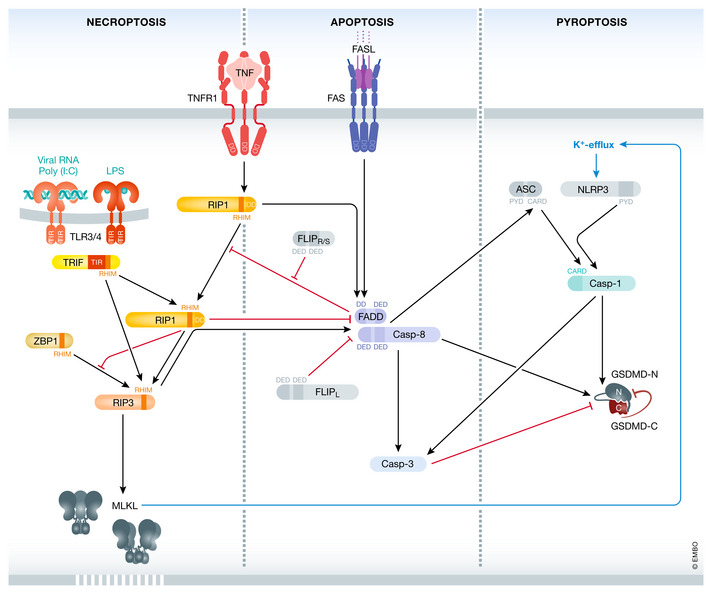
A more complex network between the different cell death pathways has been established over the years. Arrows indicate established interconnectivity between apoptosis, necroptosis, and pyroptosis. For details, please refer to the main text. Black arrows indicate crosstalk events, while red arrows indicate inhibition.
Interestingly, RIP1 also restricts caspase‐8‐mediated cell death as loss of RIP1 sensitized to TNF‐induced apoptosis (Dillon et al, 2014; Rickard et al, 2014). In addition, RIP1 restricts ZBP1‐mediated necroptosis by interfering with RHIM‐mediated interaction of ZBP1 and RIP3 (Lin et al, 2016; Newton et al, 2016b). The interplay of apoptosis and necroptosis is apparent in many inflammatory in vivo models where they are frequently concomitantly activated (Webster & Vucic, 2020). This is not surprising given that majority of signaling proteins are common to both pathways, and the balance of expression or activation of critical factors (RIP3, caspase‐8) can tip the balance in favor of apoptosis or necroptosis.
The addition of pyroptosis to the crosstalk
Caspases are involved in apoptotic, necroptotic, and pyroptotic signaling, and in recent years, a more complex interconnectivity has been revealed. As mentioned before, caspase‐3 can cleave GSDME, and there are conflicting reports of GSDME playing a role during secondary necrosis of apoptotic macrophages (Rogers et al, 2017; Lee et al, 2018). GSDME has also been implicated in mitochondrial pore formation and enhancement of apoptotic signaling (Rogers et al, 2019). GSDMD forms pores at the mitochondrial membranes preceding plasma membrane rupture (de Vasconcelos et al, 2019). Caspase‐3 can inactivate GSDMD by cleavage at Asp84 though the physiological meaning remains unknown (Rogers et al, 2017; Taabazuing et al, 2017). On the other hand, caspase‐1 has been reported to induce apoptosis by cleaving caspase‐3 in Gsdmd −/− cells (Tsuchiya et al, 2019).
Apart from mediating the activation of apoptotic cell death and regulation of necroptosis, caspase‐8 was found to be important for pyroptotic signaling. Several studies provided evidence that caspase‐8 plays a central role inducing cell death, inflammasome activation, and IL‐1β/ IL‐18 processing in Yersinia infection models (Weng et al, 2014; Orning et al, 2018; Sarhan et al, 2018). Caspase‐8 has been shown to activate GSDMD by cleavage, which resulted in K+‐efflux inducing NLRP3 inflammasome activation (Orning et al, 2018; Sarhan et al, 2018). In this setting, Asc −/− or Casp1 −/− Casp11 −/− cells showed reduced IL‐1β release without altering cell death. On the other hand, caspase‐8 can be recruited to the inflammasome. Cytosolic DNA and nigericin were able to induce caspase‐8 activation and apoptosis in an ASC‐dependent fashion (Sagulenko et al, 2013). Further studies from Sagulenko et al showed that caspase‐1/11 can process caspase‐3 during DNA transfection when caspase‐8 is deleted (Sagulenko et al, 2018). IAP antagonism or deletion of XIAP, cIAP1, and cIAP2 led to release of IL‐1β in LPS primed cells in a process that was also dependent on caspase‐1, caspase‐8, and RIP3 (Vince et al, 2012). The interaction of caspase‐8 and ASC is mediated by DED1 and DED2 of caspase‐8 and PYD of ASC, as was shown with recombinant proteins as well as overexpression studies (Vajjhala et al, 2015). This heterotypic interaction also has been shown to be critical for NLRC4‐induced apoptotic signaling when cells do not express caspase‐1 (Lee et al, 2018).
Besides caspase‐8, RIP1 inactivation (Ripk1KD/KD—RIP1 kinase‐dead bone marrow derived macrophages (BMDM)) can reduce cell death induced by Yersinia infection or LPS + TAK1 inhibitor (Peterson et al, 2017; Sarhan et al, 2018). Reduced GSDMD cleavage comparable to Casp8 −/− Ripk3 −/− BMDMs was also detected by Orning et al in Ripk1KD/KD BMDMs (Orning et al, 2018). This indicates that not only caspase‐8 but probably RIP1 also plays an important role in mediating pyroptosis. This is underlined by another publication that implicated FADD in the regulation of inflammasome activation. C. rodentium infection of Fadd −/− Ripk3 −/− BMDMs lead to reduced processing of caspase‐1/8, IL‐1β secretion, and cell death compared to WT or Ripk3 −/− BMDMs (Gurung et al, 2014). Necroptotic cell death also leads to NLRP3 activation by K+ efflux mediated by MLKL in cell intrinsic manner (Conos et al, 2017). In vivo, reduced IL‐1β was detected in Fadd −/− Ripk3 −/− mice compared to WT or Ripk3 −/− animals when treated with LPS, but increased bacterial load when infected with C. rodentium (Gurung et al, 2014). Similar findings were made for Fadd −/− Mlkl −/− BMDMs and mice. Treatment with LPS + ATP resulted in reduced caspase‐1 cleavage and IL‐1β secretion. In vivo challenge with LPS resulted in decreased IL‐1β serum levels in Fadd −/− Mlkl −/− mice (Zhang et al, 2016). However, bacterial infection of Casp8 −/− Ripk3 −/− mice resulted in higher morbidity compared to WT mice. These DKO mice showed reduced cytokine levels, but increased bacterial load indicating that a proper clearance was not achieved (Weng et al, 2014). The same finding was made in Ripk1KD/KD mice when they were infected with Yersinia by oral gavage (Peterson et al, 2017). During LPS‐induced shock, Casp11 −/− and Casp8 −/− Ripk3 −/− mice showed reduced morbidity compared to WT mice, due to the involvement of TNF‐mediated signaling in LPS‐mediated shock (Mandal et al, 2018). The described findings could be explained by the fact that bacterial infection is a long‐term challenge, which becomes worse with a defective clearance. For the LPS challenge, reduced cytokines result in healthier animals as they do not have to fight an infection.
Further understanding of caspase‐8 in the context of pyroptosis was gained by analysis of various transgenic mouse models (Fritsch et al, 2019; Newton et al, 2019b; Schwarzer et al, 2020; Tummers et al, 2020) (Fig 5, Table 1). Mice expressing catalytic‐dead caspase‐8, Casp8C362A/C362A (Newton et al, 2019a) and Casp8C362S/C362S (Fritsch et al, 2019), are embryonic lethal at E11.5, just like Casp8 −/− mice (Varfolomeev et al, 1998). While Casp8 −/− mice can be rescued by loss of RIP3 or MLKL, Mlkl −/− only delayed lethality of Casp8C362A/C362A to birth and Ripk3 −/− in some of the animals to after weaning. Interestingly, processing of caspase‐3, caspase‐7, and cleavage of RIP1 and RIP3 was detectable in caspase‐1‐dependent manner in intestines of Casp8C362A/C362A Mlkl −/− E18.5 embryos (Newton et al, 2019b). This might draw a link to papers reporting caspase‐1‐mediated apoptosis in Gsdmd −/− (Tsuchiya et al, 2019). Fritsch et al and Newton et al showed ASC specks in the intestine of E18.5 Casp8C362A/C362A Mlkl −/− Casp1 −/− Casp11 −/− embryos or 5‐week‐old Casp8C362S/C362S Mlkl −/− Casp1 −/− mice suggesting that caspase‐8 can induce ASC‐dependent inflammasome activation (Fritsch et al, 2019; Newton et al, 2019b). Deletion of ASC in Casp8C362A/C362A Mlkl −/− /Casp8C362S/C362S Mlkl −/− mice lead to survival beyond weaning comparable to Casp8C362A/C362A Mlkl −/− Casp1 −/− /Casp8C362S/C362S Mlkl −/− Casp1 −/− (Fritsch et al, 2019; Newton et al, 2019b). Combined loss of caspase‐1/11 in Casp8C362A/C362A Mlkl −/− mice had an additional protective effect suggesting an interplay of the different cell death signaling pathways. Most mice survived when RIP3 and caspase‐1/11 were deleted in Casp8C362A/C362A (Newton et al, 2019b).
Table 1.
Genotypes and phenotypes of cell death mediator mutations
| Genotype | Phenotype |
|---|---|
| Casp8C362A/C362A | Embryonic lethal (E12.5) |
| Casp8C362A/C362A Mlkl −/− | Perinatal lethal |
| Casp8C362A/C362A Ripk3 −/− | 75% of animals survive past weaning |
| Casp8C362A/C362A Mlkl −/− Ripk1 −/− | Lethal before weaning |
| Casp8C362A/C362A Mlkl −/− Asc −/− | 65% of animals survive past weaning |
| Casp8C362A/C362A Mlkl −/− Casp1 −/− | 40% of animals survive past weaning |
| Casp8C362A/C362A Mlkl −/− Casp1 −/− Casp11 −/− | 75% of animals survive past weaning |
| Casp8C362A/C362A Ripk3 −/− Casp1 −/− Casp11 −/− | Survival past weaning, best survival |
| Casp8C362S/C362S | Embryonic lethal (E12.5) |
| Casp8C362S/C362S Mlkl −/− | Perinatal lethal |
| Casp8C362S/C362S Mlkl −/− Casp1 −/− | Survival beyond parturition |
| Casp8C362S/C362S Mlkl −/− Asc −/− | Survival beyond parturition |
| Casp8D387A/D387A | No overt phenotype |
| Casp8D387A/D387A Mlkl −/− | Inflammatory phenotype |
| Casp8D387A/D387A Mlkl −/− Fasl+/ − | Rescues phenotype |
| Casp8D387A/D387A Mlkl −/− Ripk1+/ − | Rescues phenotype |
| Casp8D387A/D387A Mlkl −/− Fadd+/ − | Rescues phenotype |
| Casp8D387A/D387A Mlkl −/− Fadd −/− | Lethal before weaning |
| Casp8D387A/D387A Mlkl −/− Fadd −/− Ripk1 −/− | Survival past weaning |
| Casp8D387A/D387A Mlkl −/− Fadd −/− Casp1 −/− | Survival past weaning |
| Casp8IEC‐KO | Ileitis |
| Casp8IEC‐KO Mlkl −/− | Rescues phenotype |
| FaddIEC‐KO | Colitis and ileitis |
| FaddIEC‐KO Ripk3 −/− | Rescues phenotype |
| FaddIEC‐KO Ripk3IEC‐KO | Rescues phenotype |
| FaddIEC‐KO Mlkl −/− | Casp8‐driven pathology |
| FaddIEC‐KO Mlkl −/− ASC −/− | Casp8‐driven pathology |
| FaddIEC‐KO Mlkl −/− Gsdmd −/− | Rescues phenotype |
Summary of genotypes and main associated phenotypes discussed in the context of crosstalk between different cell death pathways. Please refer to the original publications for a complete list of crosses and their respective phenotypes. References for listed genotypes are indicated throughout the text.
Caspase‐8 autoprocessing is key for it is full activation but mutagenesis of the cleavage site D387 to alanine, which separates the small and large catalytic subunit, did not cause a developmental phenotype (Philip et al, 2016; Newton et al, 2019a; Tummers et al, 2020). However, a reduced morbidity was observed in in vivo challenge with CD95. Crossing of Casp8D378A/D378A (from now on Casp8DA/DA) to Mlkl −/− mice resulted in inflammation and splenomegaly as well as hypersensitivity to LPS injection (Tummers et al, 2020). The inflammatory phenotype of Casp8DA/DA Mlkl −/− mice was rescued by deletion of one allele of FADD, RIP1, or FASL (Tummers et al, 2020). Interestingly, Casp8DA/DA Mlkl −/− Fadd −/− died within 14 days after birth indicating that FADD has a bivalent role in this model. Lethality of Casp8DA/DA Mlkl −/− Fadd −/− mice was rescued by crossing to Casp1 −/− or to Ripk1 −/− mice but ASC specks were again revealed in ileal tissue (Tummers et al, 2020). Another study focused on ileitis and colitis driven by deletion of either FADD or caspase‐8 in the intestinal epithelial cells (IEC) (Schwarzer et al, 2020). Caspase‐8‐induced pathologies were rescued by deletion of Mlkl −/−; however, MLKL ablation was not significantly protective in FaddIEC‐KO mice (Schwarzer et al, 2020). Interestingly, FaddIEC‐KO ileitis was milder in Ripk3IEC‐KO or in RIP1 kinase‐dead knock‐in mice (Schwarzer et al, 2020) and completely inhibited by whole body RIP3 ablation (Welz et al, 2011). But only the deletion of GSDMD, not of ASC, resulted in loss of the inflammatory phenotype in FaddIEC‐KO Mlkl −/− (Schwarzer et al, 2020).
In all caspase‐8 knock‐in models or FaddIEC‐KO, RIP1 deletion or kinase inhibition resulted in attenuated phenotypes arguing that RIP1 plays important role in caspase‐8‐associated pathologies. Nevertheless, a large body of published work shows a complex role of caspase‐8 in regulation of apoptosis, necroptosis, and pyroptosis. Thus, incompletely activated (either enzymatic dead or cleavage‐resistant) caspase‐8 in the context of Mlkl −/− can provide a scaffold for ASC binding and induce ASC‐dependent inflammasome formation. In FaddIEC‐KO Mlkl −/− mice, it seems more likely that caspase‐8 cleaves GSDMD to induce pyroptosis. These findings exemplify the complexity in the signaling crosstalk between different cell death pathways and raise additional questions concerning other pathway components and their role. For example, what is the role of apoptosis induced by caspase‐3 in those pathologies? What are the proteins or pathways regulated by RIP3? Clearly, future studies are needed to answer these questions.
Autophagy during other forms of cell death
ADCD is defined as a form of cell death that does not share features with other forms of cell death. However, autophagy has been shown to coincide various forms of cell death. In the following, the role of autophagy‐related genes in the context of other forms of cell death will be discussed.
Apoptotic and autophagic pathways can intercross at various levels. Most Bax −/− Bak −/−mice die perinatally and show interdigital skin and increased white blood cells besides other phenotypes (Lindsten et al, 2000). Similarly, genetic deletion of autophagy regulator ATG5 also leads to perinatal lethality (Kuma et al, 2004). However, Atg5 ablation in a Bax −/− Bak −/− mice causes a more severe phenotype with enhanced brain exencephaly and even more delayed reduction of interdigital webbing (Arakawa et al, 2017), compared to Atg5 −/− (Kuma et al, 2004) or Bax −/− Bak −/−. This suggests that autophagy serves as a partial backup mechanism for apoptosis during development.
In addition to Atg5, deletion of other critical mediators of autophagy Atg16l1 or Atg12 also results in perinatal lethality (Kuma et al, 2004; Saitoh et al, 2008; Malhotra et al, 2015). Several autophagy regulators are implicated in human pathologies, such as Atg16l1 polymorphism, which is associated with Crohn’s disease (Hampe et al, 2007). The most common ATG16L1 variant, T300A, is linked with a loss of Paneth cells in humans as well as mice (Cadwell et al, 2008). ATG16L1 T316A mutation (in mouse) introduces a new caspase‐3 cleavage site, and mice harboring this mutation show reduced autophagy and pathogen clearance (Lassen et al, 2014; Murthy et al, 2014). Bacterial infections of these mice lead to increased cytokines levels, especially IL‐1β, indicating a defective clearance of bacteria that results in more severe inflammation (Lassen et al, 2014; Murthy et al, 2014). Similar results have been reported for ATG16L1 conditional knockout mice. Mice with myeloid‐specific deletion of ATG16L1 (Atgl16l1ΔLyz2) succumbed faster than WT mice after LPS injection (Samie et al, 2018; Lim et al, 2019). Similarly, chimeric mice with transplanted Atg16l1 −/− fetal liver cells treated with DSS (dextran sodium sulfate) and infected with MNV (murine norovirus) develop a lethal colitis while WT mice survived, indicating that ATG16L1 is necessary to restrict inflammatory signaling (Saitoh et al, 2008). Interestingly, mice with an intestinal‐specific ATG16L1 knockout (Atgl16l1IEC‐KO) were also more sensitive in a DSS‐induced colitis model (Matsuzawa‐Ishimoto et al, 2017). Comparison of conditional knockout of ATG16L1 in intestinal epithelial cells or mononuclear cells showed that both have increased cytokine secretion; however, Atgl16l1IEC‐KO show a more drastic increase compared to myeloid‐specific KO (Conway et al, 2013). In DSS/MNV‐induced colitis, TNF‐blocking antibodies or RIP1 inhibitors had protective effects, which is in line with their finding that TNF‐treated Atg16l1 −/− organoids are more sensitive to TNF‐induced and RIP1‐mediated death compared to WT organoids (Matsuzawa‐Ishimoto et al, 2017; Matsuzawa‐Ishimoto et al, 2020). Similar findings were made in a different infection model. Atg16l1IEC‐KO mice were more susceptible to Helicobacter hepaticus + αIL‐10R‐induced colitis compared to WT or myeloid‐specific Atg16l1 −/− mice. Epithelial cell‐specific KO mice disease was preventable by administration of TNF (Pott et al, 2018), confirming a TNF‐mediated pathology. Furthermore, mice with intestinal deletion of ATG16L1 were exquisitely sensitive to TNF‐induced hypothermia and lethality in RIP1‐dependent fashion (Patel et al, 2020). Similar results were obtained when Atg5, Atg16l1, Fip200, or Becn1 were deleted in the myeloid cell compartment by LysM‐Cre (Orvedahl et al, 2019). All 4 genotypes were more sensitive to TNF injections compared to their WT littermates. In the case of Atg5ΔLysM, the RIP1 kinase inhibitor Nec‐1 protected the mice from death (Orvedahl et al, 2019). In Atg16l1 −/− BMDMs, TRIF degradation by autophagy was shown to be critical for regulation of inflammatory signaling (Samie et al, 2018). In addition, ATG16L1 can restrict necroptotic signaling by regulating the turnover of RHIM‐containing proteins, especially ZBP1 (Lim et al, 2019). ATG16L1 has been shown to play a critical role in the modulation of TNF‐mediated cell death signal in various in vitro and in vivo systems. Several models proved that loss of ATG16L1 either in intestinal epithelial cells or myeloid cells leads contributes to a more severe phenotype. For this reason, further investigation of the pathways is critical to understand the differences and similarities of interplay between autophagy and TNF‐mediated cell death signaling in different cell types.
Besides ATG16L1, other autophagy pathway components were also implicated in TNF and TRAIL‐mediated apoptosis and necroptosis. Several studies suggested a scaffolding function of autophagy genes and autophagic structures, which will be discussed in more detail below. In Tak1‐deficient cells, TRAIL‐induced necroptotic cell death is dependent on p62, indicating that the autophagosome formation plays a critical role in complex formation (Goodall et al, 2016). The interplay of autophagy and cell death also has been implicated in HIV‐infected T cells (HIV‐TCM) treated with IAP antagonists (also referred to as SMAC mimetics). IAP antagonists induce cell death of HIV‐TCMs in a caspase, RIP1, and autophagosome formation‐dependent manner, while p62 knockdown protected from induced cell death (Campbell et al, 2018). Similarly, necroptosis in L929 cells could be inhibited by autophagy inhibitors wortmannin and pepstatin A or by the knockdown of RIP1 (Yu et al, 2006). In all studies described above, inhibition of autophagosome formation by wortmannin reduced cell death suggesting that autophagosome can directly affect cellular viability. The importance of autophagy as inducer of other forms of cell death was also shown for the clinically tested Bcl‐2 antagonist GX15‐070/obatoclax, which was shown to induce necroptotic cell death in different cancer cell lines depending on ATG5, ATG7, RIP1, and RIP3 (Bonapace et al, 2010; Basit et al, 2013). These studies provide a link between regulated cell death and autophagy and implicate dysfunctional autophagy in cell death activation in physiological settings linked to diseases.
NETosis in the context of inflammatory cell death
NET formation can also be associated with the activation different cell death pathways. Necroptosis has been shown in several studies to by critical for NETosis. Loss of the necroptotic effectors RIP3 or MLKL or the pharmacological inhibition of RIP1, RIP3, or MLKL let to reduced cell death and NET formation when cells were treated with LPS, PMA (Desai et al, 2016), RSV (respiratory syncytial virus) (Muraro et al, 2018), or various crystalline substances (Desai et al, 2017) for an extended period. NET formation was independent of RIP3 or MLKL when neutrophils were treated for shorter periods (Amini et al, 2016). These findings might be linked to the differences of lytic versus non‐lytic NET formation. Injection of monosodium urate (MSU) crystals in air pouches has been shown to promote NET formation and sterile inflammation in vivo (Schauer et al, 2014). In this model, Ripk3 −/−, Mlkl −/− mice, and Necrostatin‐1‐treated mice showed reduced tophus like NET aggregations (Desai et al, 2016; Desai et al, 2017). Similar findings were made using a murine AAV (anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitis) disease model. Ripk3 −/− and Mlkl −/− mice were protected against necrotizing crescentic glomerulonephritis detected in WT mice (Schreiber et al, 2017). Besides necroptosis, several studies showed that GSDMD plays a critical role during neutrophil cell death and NET formation (Chen et al, 2018; Kambara et al, 2018; Sollberger et al, 2018). Chen et al showed that non‐canonical inflammasome activation can lead to GSDMD processing by caspase‐11 leading to cell death resembling NETosis, which was independent of MPO, NE, and PAD4 (Chen et al, 2018). The other two publication that identified GSDMD as a target of the neutrophil elastase during PMA induced NET formation (Sollberger et al, 2018) or E. coli infection (Kambara et al, 2018). Infection with a cytosolic Salmonella strain (ΔsifA) lead to a more severe infection in Gsdmd −/− or Casp11 −/− mice compared to WT mice. Interestingly, DNAse I treatment resulted in aggravation of the bacterial load only in WT mice (Chen et al, 2018), indicating potential defects of NET formation in Gsdmd −/− and Casp11 −/− mice. A second study, however, reported that Gsdmd −/− lead to better bacterial clearance due to increased neutrophil numbers because of an increased lifespan (Kambara et al, 2018).
For a better understanding of necrotic and pyroptotic cell death in the context of NET formation, it would be interesting to perform infection or disease models in tissue specific knockouts. Additionally, it would be interesting to understand and clarify in which physiological contexts distinct cell death forms play a dominant role.
Ferroptosis and necroptosis
Ferroptosis and necroptosis have been implicated in kidney pathologies (Belavgeni et al, 2020), and inhibitors of ferroptosis and necroptosis showed protection in various disease models in mice. However, it is still not completely understood how both different mechanisms work together. Deletion of Ripk3 or Mlkl has been shown to be protective in kidney reperfusion injury models (Linkermann et al, 2013; Newton et al, 2016a; Muller et al, 2017; von Massenhausen et al, 2018). Ripk1KD/KD mice (Newton et al, 2016a) or pharmacological inhibition of RIP1 showed protective effects in IRI (ischemia–reperfusion injury) as well (Linkermann et al, 2012). However, in AKI (acute kidney injury) induced by folic acid ferroptosis has been shown to be the driving form of inflammatory cell death (Martin‐Sanchez et al, 2017). Further analysis of this model leads to better understanding of the interplay of ferroptosis and necroptosis. During later stages of disease, deletion of TWEAK receptor (Fn14) as well as treatment of Necrostatin‐1 reduced the severity of the disease (Martin‐Sanchez et al, 2018). As discussed by Martin‐Sanchez et al, this leads to a model in which ferroptosis in critical for during the initial phase of AKI and necroptosis is taking over at the later stages leading to amplification of tubular cell death. Besides ferroptosis and necroptosis, MPTP and necroptosis have been shown to be critical mediators during IRI, as either genetic ablation of RIP3 and CypD showed complete rescue of lethality (Linkermann et al, 2013).
Clearly, ferroptosis and necroptosis can be triggered by shared stimuli (e.g., ROS) and are both involved in ischemia–reperfusion‐driven pathologies. Consequently, further experimental validation of inhibitors or biomarkers selective for these pathways is needed for better understanding of ferroptosis/necroptosis biology and physiological importance.
Cell death pathways components as therapeutic targets
Cell death pathways play a pivotal role in homeostasis of the body, and their dysregulation can lead to many diseases ranging from auto‐immune disease to neurodegeneration and cancer. As such, these pathways are attractive targets for therapeutic intervention. The initial validation of the relevance of cell death for human diseases came from identification of Bcl‐2 from the genomic region with frequent chromosomal translocations t(14;18) in follicular lymphomas (Tsujimoto et al, 1984). Furthermore, expression of Bcl‐2 protected cells from death and enabled lymphocyte accumulation often leading to cancer (Vaux et al, 1988; McDonnell et al, 1989; Strasser et al, 1991). Soon afterward, the importance of Bcl‐xL, Mcl‐1, and other Bcl‐2 family members was recognized and development of antagonists of anti‐apoptotic Bcl‐2 proteins was started (Czabotar et al, 2014). These compounds are referred to as BH3 mimetics given that they emulate cell death promoting function of BH3 domains.
The first successful BH3 mimetic was ABT‐737, a compound with partial selectivity for Bcl‐2, Bcl‐xL, and Bcl‐w over Mcl‐1 and A1 that efficiently inhibited tumor growth in numerous animal cancer models and validated this targeting approach (Oltersdorf et al, 2005). ABT‐737 was followed by a related orally available BH3 mimetic ABT‐263 or navitoclax (Tse et al, 2008). Navitoclax showed great promise in patients with chronic lymphocytic leukemia. In addition to its single‐agent activity, navitoclax had a great potential for combination therapies, especially those that downregulated Mcl‐1, Bcl‐2 protein not affected by ABT‐263 (Cragg et al, 2009). However, navitoclax clinical path was hindered because it stimulated precipitous loss of platelets (Roberts et al, 2012). Drop in platelet number was caused by Bcl‐xL inhibition, which steered ongoing drug discovery efforts away from this important pro‐survival protein (Czabotar et al, 2014). Thus, emerged Bcl‐2 selective inhibitor ABT‐199 or venetoclax, the first BH3 mimetic and the first cell death regulating small molecule to be approved by the FDA for the treatment of chronic lymphocytic leukemia or small lymphocytic lymphoma (Souers et al, 2013). Venetoclax is undergoing investigation in a number of clinical trials that aim to expand the number of malignancies where it can be beneficial (Strasser & Vaux, 2020). However, there is also increasing awareness that Mcl‐1 is a major resistance factor for Bcl‐2 targeting (Gong et al, 2016). To address this therapeutic need, several Mcl‐1 selective inhibitors have been generated and some have been enrolled in clinical trials (Kotschy et al, 2016; Caenepeel et al, 2018; Tron et al, 2018), (ClinicalTrials.gov). Having selective antagonist to various pro‐survival Bcl‐2 protein would give oncologists a great set of tools to treat patients’ tumors with potent and tolerable anti‐cancer agents.
The favorite strategy for targeting IAP proteins involves SMAC‐mimicking small‐molecule IAP antagonists (Fulda & Vucic, 2012). IAP antagonists were meant to block caspase inhibition by XIAP (Sun et al, 2007). However, the key to single‐agent pro‐apoptotic activity of IAP antagonists results from c‐IAP1/2 antagonism and TNF‐dependent cell death (Gaither et al, 2007; Petersen et al, 2007; Varfolomeev et al, 2007; Vince et al, 2007). The monovalent IAP antagonists emulate one SMAC AVPI motif, while the bivalent antagonists comprise two AVPI‐like motif mimetics connected by a chemical linker (Sun et al, 2007; Varfolomeev et al, 2007). Binding of IAP antagonists triggers a conformational change that opens the c‐IAP1 structure and enables c‐IAP RING domain dimerization, a prerequisite feature of their E3 activation (Dueber et al, 2011; Feltham et al, 2011). This prompt activation of c‐IAP1/2 E3 activity causes their K48‐linked auto‐ubiquitination, subsequent proteasomal degradation (Dueber et al, 2011; Feltham et al, 2011) and activation of canonical NF‐κB signaling (Varfolomeev et al, 2007; Vince et al, 2007). Proteasomal degradation of c‐IAPs leads to NIK stabilization and activation of the non‐canonical NF‐κB pathway (Varfolomeev et al, 2007; Vince et al, 2007). The stimulation of NF‐κB as well as MAPK pathways induces TNF production and activation of TNFR1 signaling (Varfolomeev et al, 2007; Vince et al, 2007). However, with c‐IAP1/2 degraded, RIP1 cannot be ubiquitinated during TNF‐induced signaling and the canonical NF‐κB pathway is poorly activated. Instead, RIP1 will complex with FADD/caspase‐8 and provoke apoptosis, and if caspase‐8 is inhibited or insufficiently activated, RIP3 and MLKL‐dependent necroptotic cell death (Bertrand et al, 2008; He et al, 2009). TNF‐blocking reagents efficiently inhibit IAP antagonist stimulated cell death further demonstrating its’ TNF dependence (Petersen et al, 2007; Varfolomeev et al, 2007; Vince et al, 2007).
Several IAP antagonists such as GDC‐0152, TL3271, and SM‐164 have demonstrated tumor‐inhibiting activity in in vivo cancer models without showing any significant toxicity or weight loss in mice (Lu et al, 2008; Flygare & Fairbrother, 2010; Flygare et al, 2012; Fulda & Vucic, 2012; Morrish et al, 2020a). Based on the positive results from preclinical studies, a number of IAP antagonists have entered phase I/II clinical trials for people with a variety of malignancies (Fulda & Vucic, 2012; Jensen et al, 2020b). Clinical trials with GDC‐0152, LCL161, HGS1029, and TL32711 reported target antagonism, dose proportional pharmacokinetics, and no dose‐limiting toxicity (Morrish et al, 2020a). However, none of these trials reported significant anti‐tumor activity of IAP antagonists and were not pursued further (Morrish et al, 2020a). Nevertheless, the ability of IAP antagonists to activate non‐canonical NF‐κB signaling is prompting an interest in combining them with checkpoint inhibitors (e.g., anti‐PD1 antibody) and anti‐retroviral therapy (Chesi et al, 2016; Nixon et al, 2020; Morrish et al, 2020a). IAP antagonists have also shown a great potential in treating liver pathologies caused by HBV and Plasmodium infections (Ebert et al, 2020; Morrish et al, 2020b). These and other ongoing and future clinical trials will examine the safety and the efficacy of IAP antagonists for the treatment of human malignancies and infections in hopes of bringing new therapies to patients who need them.
Bcl‐2 and IAP antagonists were developed to promote cell death in hematological and solid tumors. However, excessive cell death can be detrimental for healthy organism and cause tissue damage and neurodegeneration. For this reason, RIP1 has been proposed as a safe modality to treat inflammatory and neurodegenerative diseases with no known risk of immunosuppression (Yuan et al, 2019; Mifflin et al, 2020). While RIP3 kinase could also be considered an attractive target, genetic studies and RIP3 targeting efforts have demonstrated toxicity which precludes safe inhibition of RIP3 (Mandal et al, 2014; Newton et al, 2014). Contrarily, genetic inactivation or chemical inhibition of RIP1 kinase activity is well tolerated and pose no known risks (Berger et al, 2014; Newton et al, 2014; Polykratis et al, 2014; Patel et al, 2020; Webster et al, 2020).
Inhibiting RIP1 kinase activity is beneficial in joint and skin inflammation, ileocolitis as well as in the TNF‐induced systemic inflammatory response syndrome (SIRS) model (Berger et al, 2014; Vlantis et al, 2016; Newton et al, 2016a; Patel et al, 2020; Webster et al, 2020). Similarly, the role of RIP1 kinase activity is also evident in the number of neurodegenerative and neuroinflammatory diseases (Yuan et al, 2019; Mifflin et al, 2020). In pancreatic cancers, lung metastases, pancreatitis, and certain viral infections however, the therapeutic effects of RIP1 inhibition have been disputed recently (Newton et al, 2016a; Patel et al, 2020; Webster et al, 2020). Clearly, more studies are needed to delineate the suitable diseases for RIP1 inhibition, with confirmation coming from testing RIP1 inhibitors in clinical trials.
So far, RIP1 inhibitors developed by GlaxoSmithKline (GSK) and Denali have been tested in clinical settings with initial reports indicating that GSK2982772 and DNL104 were generally well tolerated in people (Harris et al, 2017; Weisel et al, 2017b; Grievink et al, 2020; Jensen et al, 2020; Mifflin et al, 2020). Phase I trials with GSK2982772 showed no serious adverse events (AEs) and no suggestion of a safety concern (Weisel et al, 2017), which enabled GSK to initiate several small phase 2 clinical trials for psoriasis, rheumatoid arthritis, and ulcerative colitis. To date, GSK2982772 has not shown significant therapeutic benefit in psoriasis or rheumatoid arthritis, while the data from the ulcerative colitis trial are not yet available (ClinicaTrials.gov). In addition to inflammatory diseases, GSK tested RIP1 inhibitor, GSK3145095, in clinical trial intended to test RIP1 inhibition in pancreatic and other solid tumors (Harris et al, 2019), but was terminated during patient recruitment. DNL104, Denali’s brain‐penetrant RIP1 inhibitor, did not trigger any adverse effects in central nervous system, although they observed abnormal liver function in some healthy subjects (Grievink et al, 2020). Denali has abandoned clinical trials of DNL104 and entered into collaboration with Sanofi to examine another RIP1 inhibitor, DNL747, in clinical trials for Alzheimer’s disease, amylotrophic lateral sclerosis, and multiple sclerosis (Martens et al, 2020). Overall, targeting RIP1 represents an attractive opportunity for therapeutic intervention in inflammatory diseases, and future preclinical and especially clinical studies should further define the optimal indication and patient populations,
An alternative way of targeting inflammatory diseases is to block inflammasome assembly and inflammatory cell death mediated by NLRP3 and GSDMD. GSDMD is a key component of pyroptotic cell death and targeting GSDMD cleavage, membrane association, and/or the ability to form membrane pores is attractive strategy for several devastating diseases such as sepsis or ARDS (acute respiratory distress syndrome) (Chauhan et al, 2020). As GSDMD is a relatively novel target with no known enzymatic activity, GSDMD targeting efforts are still nascent (Shi et al, 2017). Consequently, none of the GSDMD blocking reagents have advanced to clinical trials yet. However, several more established inflammasome regulators have been a focus of drug discovery for a long time. The best example of NLRP3 inflammasome has been implicated in a number of inflammatory, neurodegenerative, and metabolic diseases (Voet et al, 2019). Early verification of the feasibility of NLRP3 therapeutic targeting came from demonstration that chemical compound called glyburide effectively blocked IL‐1b secretion and pyroptotic cell death (Lamkanfi et al, 2009). Another agent, MCC950/CRID3, targets the NACHT domain of NLRP3 and has a higher potency and selectivity for NLRP3 (Coll et al, 2019; Tapia‐Abellan et al, 2019). MCC950/CRID3 has shown efficacy in a number of animal disease models including myocardial infarction, atherosclerosis, dermal and pulmonary inflammation, and multiple sclerosis (Primiano et al, 2016; van der Heijden et al, 2017; van Hout et al, 2017; Perera et al, 2018; Voet et al, 2019). Currently, two MCC950/CRID3‐related compounds (IZD334 and Inzomelid) are undergoing clinical trials to evaluate their safety and tolerability (Clinicaltrials.gov). These and future trials could pave the way for efficacious and safe NLRP3 targeting in patients with CAPS (cryopyrin‐associated periodic syndromes) and other inflammatory diseases.
Caspases play a central role in various cell death pathways and therefore were/are an interesting target for drug development. Several pan‐caspase inhibitors mimicking peptide substrates have been developed with few reaching clinical trials. Emricasan is an irreversible caspase inhibitor which accumulates in the liver, likely because of the first pass effect (Hoglen et al, 2004). In several mouse models (α‐Fas models, D‐Gln/LPS), Emricasan showed protective capacity and was also used in intervention animal studies (Hoglen et al, 2004). Combination therapy of Emricasan and IAP antagonist Birinapant promoted necroptosis in AML cells in vivo and prolonged survival in mouse models (Brumatti et al, 2016). Clinical trials of Emricasan did not raise any safety concerns and showed reduction of serum level transaminases in patients with prior diagnosis of mild hepatic impairment (Valentino et al, 2003). In several other liver pathologies, Emricasan showed promising results. During liver transplantation, Emricasan was tested in two different patients with non‐alcoholic fatty liver acid disease and it showed reduced levels of ALT (alanine aminotransferase) compared to the placebo group (Shiffman et al, 2019). Similar findings were made for patients with hepatitis C virus infection where Emricasan reduced transaminase blood levels without affecting virus titers (Shiffman et al, 2010). However, Emricasan did not have a beneficial effect on portal hypertension in liver cirrhosis patients (Garcia‐Tsao et al, 2020). Lastly, the inhibitor was tested during liver transplantation as cell death driven by reperfusion is a major concern. Treatment showed some therapeutic effect by reducing apoptosis (Baskin‐Bey et al, 2007). As mentioned by the authors, more studies would need to be conducted to confirm these observations. Another caspase inhibitor, GS‐9450, reduced ALT levels in patients suffering from non‐alcoholic steatohepatitis (Ratziu et al, 2012). Therefore, although caspase inhibitors showed some potential in the treatment of various hepatic disease, more studies are needed to evaluate their therapeutic potential.
As described earlier, PARP1 plays a critical role in DNA damage repair pathways. PARP1 inhibitors induce cytotoxicity, especially in BRCA1/2 mutated (homologous recombination‐deficient (HDR)) cancers cells (Bryant et al, 2005; Farmer et al, 2005). This is explained by synthetic lethal interaction between BRCA1/2 and PARP1 during DNA repair (Lord & Ashworth, 2017). PARP inhibitors “trap” PARP1 at the DNA inducing blockage of replication forks leading to cell death in HDR tumors (Murai et al, 2012). Several PARP inhibitors are approved in BRCA1/2 mutated ovarian cancer as well as BRCA mutated HER negative breast cancer with ongoing trials for other forms of cancer (Hoy, 2018; Jiang et al, 2019; Murthy & Muggia, 2019).
Ferroptosis has been implicated in several experimental models of reperfusion injury. In addition, chelation of iron by M30 or α‐lipoic acid showed improvement in neurodegenerative disease models (Kupershmidt et al, 2012; Zhang et al, 2018; Han et al, 2020) suggesting a possible benefit in the clinical setting. In a clinical trials, iron chelator DFO (desferrioxamine mesylate) slowed the progression of dementia in Alzheimer’s patients over the placebo group (McLachlan & Dalton, 1991). While inhibition of ferroptosis may be a promising strategy for neurodegenerative diseases, ferroptosis inducers showed a potential benefit for cancer. In mouse models, induction of ferroptosis reduced the tumor growth/size significantly compared to the controls (Kim et al, 2016). This process was reversible by cotreatment of DFO (Kim et al, 2016). There are several potential strategies targeting ferroptosis in cancer reviewed by (Dixon & Stockwell, 2019). Several agents which can potentially induce ferroptosis by interference with the GSH synthesis or GPX4 have been investigated in clinical trials. The GSH synthesis inhibitor (Buthionine sulfoximine‐BSO) reduced GSH levels in tumor cells and was tolerated in patients in phase I clinical trials (Bailey et al, 1997). The Xc − inhibitor Sulfasalazine (SAS) reduced targeted cancer stem cell populations in a phase I trial (Shitara et al, 2017). Interestingly, the approved cancer drug altretamine, which was originally classified as a alkylating agent, was identified as a potential GPX4 inhibitor (Woo et al, 2015). These examples show the perspective of targeting ferroptosis for various diseases but, clearly, further research is needed to understand its full potential.
In summary, various cell death pathways are implicated in numerous seminal physiological processes. Therefore, they represent attractive targets for therapeutic intervention with the hope of addressing unmet medical needs and helping patients suffering from cancers, neurodegenerative, inflammatory, and other diseases.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Kim Newton, Nobuhiko Kayagaki, and Eugene Varfolomeev for valuable comments. Both authors are Genentech employees.
The EMBO Journal (2021) 40: e106700.
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW, Simon HU, Yousefi S (2016) NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol 46: 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL (2008) Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci 1147: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa S, Tsujioka M, Yoshida T, Tajima‐Sakurai H, Nishida Y, Matsuoka Y, Yoshino I, Tsujimoto Y, Shimizu S (2017) Role of Atg5‐dependent cell death in the embryonic development of Bax/Bak double‐knockout mice. Cell Death Differ 24: 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Mahvi D, Schink J, Pomplun M, Mulcahy RT et al (1997) Phase I study of continuous‐infusion L‐S, R‐buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst 89: 1789–1796 [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW et al (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662 [DOI] [PubMed] [Google Scholar]
- Basit F, Cristofanon S, Fulda S (2013) Obatoclax (GX15‐070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 20: 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin‐Bey ES, Washburn K, Feng S, Oltersdorf T, Shapiro D, Huyghe M, Burgart L, Garrity‐Park M, van Vilsteren FG, Oliver LK et al (2007) Clinical trial of the pan‐caspase inhibitor, IDN‐6556, in human liver preservation injury. Am J Transplant 7: 218–225 [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P (2005) Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 280: 18558–18561 [DOI] [PubMed] [Google Scholar]
- Bebber CM, Müller F, Prieto Clemente L, Weber J, von Karstedt S (2020) Ferroptosis in cancer cell biology. Cancers 12: 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belavgeni A, Meyer C, Stumpf J, Hugo C, Linkermann A (2020) Ferroptosis and necroptosis in the kidney. Cell Chem Biol 27: 448–462 [DOI] [PubMed] [Google Scholar]
- Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes‐Earle A et al (2014) Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN‐deficient mice. J Immunol 192: 5476–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30: 689–700 [DOI] [PubMed] [Google Scholar]
- Bialik S, Dasari SK, Kimchi A (2018) Autophagy‐dependent cell death ‐ where, how and why a cell eats itself to death. J Cell Sci 131: jcs215152 [DOI] [PubMed] [Google Scholar]
- Boivin WA, Cooper DM, Hiebert PR, Granville DJ (2009) Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest 89: 1195–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D (1996) Involvement of MACH, a novel MORT1/FADD‐interacting protease, in Fas/APO‐1‐ and TNF receptor‐induced cell death. Cell 85: 803–815 [DOI] [PubMed] [Google Scholar]
- Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schafer BW, Schrappe M, Stanulla M, Bourquin JP (2010) Induction of autophagy‐dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest 120: 1310–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A et al (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535 [DOI] [PubMed] [Google Scholar]
- Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, Richmond J, Ghisi M, Salmon JM, Silke N et al (2016) The caspase‐8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med 8: 339ra369 [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2‐deficient tumours with inhibitors ofpoly(ADP‐ribose) polymerase. Nature 434: 913–917 [DOI] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S et al (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456: 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, Whittington DA, Huang X, Poppe L, Cheng AC et al (2018) AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov 8: 1582–1597 [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Bruckman RS, Chu YL, Trout RN, Spector SA (2018) SMAC mimetics induce autophagy‐dependent apoptosis of HIV‐1‐infected resting memory CD4+ T cells. Cell Host Microbe 24: 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Vande Walle L, Lamkanfi M (2020) Therapeutic modulation of inflammasome pathways. Immunol Rev 297: 123–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K (2018) Noncanonical inflammasome signaling elicits gasdermin D‐dependent neutrophil extracellular traps. Sci Immunol 3: 1–11 [DOI] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P et al (2014) Translocation of mixed lineage kinase domain‐like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL et al (2016) IAP antagonists induce anti‐tumor immunity in multiple myeloma. Nat Med 22: 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM (1995) FADD, a novel death domain‐containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505–512 [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell 137: 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF (2001) Membrane blebbing during apoptosis results from caspase‐mediated activation of ROCK I. Nat Cell Biol 3: 339–345 [DOI] [PubMed] [Google Scholar]
- Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB et al (2019) MCC950 directly targets the NLRP3 ATP‐hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15: 556–559 [DOI] [PubMed] [Google Scholar]
- Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, Masters SL, Murphy JM, Schroder K, Vaux DL et al (2017) Active MLKL triggers the NLRP3 inflammasome in a cell‐intrinsic manner. Proc Natl Acad Sci USA 114: E961–E969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ et al (2013) Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145: 1347–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Harris C, Strasser A, Scott CL (2009) Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 9: 321–326 [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63 [DOI] [PubMed] [Google Scholar]
- Dasari SK, Bialik S, Levin‐Zaidman S, Levin‐Salomon V, Merrill AH Jr, Futerman AH, Kimchi A (2017) Signalome‐wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ 24: 1288–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119 [DOI] [PubMed] [Google Scholar]
- Delanghe T, Dondelinger Y, Bertrand MJM (2020) RIPK1 kinase‐dependent death: a symphony of phosphorylation events. Trends Cell Biol 30: 189–200 [DOI] [PubMed] [Google Scholar]
- Denton D, Kumar S (2019) Autophagy‐dependent cell death. Cell Death Differ 26: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S, Osen‐Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou J‐C (1999) Bid‐induced conformational change of bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 144: 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai J, Foresto‐Neto O, Honarpisheh M, Steiger S, Nakazawa D, Popper B, Buhl EM, Boor P, Mulay SR, Anders HJ (2017) Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap‐like chromatin. Sci Rep 7: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, Herrmann M, Bilyy R, Muller S, Popper B et al (2016) PMA and crystal‐induced neutrophil extracellular trap formation involves RIPK1‐RIPK3‐MLKL signaling. Eur J Immunol 46: 223–229 [DOI] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN et al (2014) RIPK1 blocks early postnatal lethality mediated by caspase‐8 and RIPK3. Cell 157: 1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell 149: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR (2019) The hallmarks of ferroptosis. Annu Rev Cancer Biol 3: 35–54 [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH et al (2019) FSP1 is a glutathione‐independent ferroptosis suppressor. Nature 575: 693–698 [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW et al (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971–981 [DOI] [PubMed] [Google Scholar]
- Douda DN, Khan MA, Grasemann H, Palaniyar N (2015) SK3 channel and mitochondrial ROS mediate NADPH oxidase‐independent NETosis induced by calcium influx. Proc Natl Acad Sci USA 112: 2817–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a Mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42 [DOI] [PubMed] [Google Scholar]
- Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, Zobel K, Maurer B, Varfolomeev E, Wu P et al (2011) Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 334: 376–380 [DOI] [PubMed] [Google Scholar]
- Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael‐Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS et al (2010) c‐IAP1 and UbcH5 promote K11‐linked polyubiquitination of RIP1 in TNF signalling. EMBO J 29: 4198–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert G, Lopaticki S, O'Neill MT, Steel RWJ, Doerflinger M, Rajasekaran P, Yang ASP, Erickson S, Ioannidis L, Arandjelovic P et al (2020) Targeting the extrinsic pathway of hepatocyte apoptosis promotes clearance of plasmodium liver infection. Cell Rep 30: 4343–4354.e4 [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S (1998) A caspase‐activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391: 43–50 [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC (2018) The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 48: 35–44.e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921 [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM (2014) Parthanatos: mitochondrial‐linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM, Chunduru SK, McKinlay MA, Vaux DL, Silke J et al (2011) Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J Biol Chem 286: 17015–17028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Panayotova‐Dimitrova D, Leverkus M (2012) Pick your poison: the Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11: 460–467 [DOI] [PubMed] [Google Scholar]
- Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, Cohen F, Deshayes K, Doerner K, Eckhardt SG et al (2012) Discovery of a potent small‐molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC‐0152). J Med Chem 55: 4101–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flygare JA, Fairbrother WJ (2010) Small‐molecule pan‐IAP antagonists: a patent review. Expert Opin Ther Pat 20: 251–267 [DOI] [PubMed] [Google Scholar]
- Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN (2010) Model‐based dissection of CD95 signaling dynamics reveals both a pro‐ and antiapoptotic role of c‐FLIPL. J Cell Biol 190: 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16: 1180–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Francis H (1994) Disruption of epithelial cell‐matrix interactions induces apoptosis. J Cell Biol 124: 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM et al (2019) Caspase‐8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575: 683–687 [DOI] [PubMed] [Google Scholar]
- Fulda S, Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 11: 109–124 [DOI] [PubMed] [Google Scholar]
- Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J, Sage D, Slisz J, Tran M, Straub C et al (2007) A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor‐alpha signaling. Cancer Res 67: 11493–11498 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Green DR (2019) Autophagy‐independent functions of the autophagy machinery. Cell 177: 1682–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, Lawitz E, Satapathy SK, Ghabril M, Shiffman ML, Younes ZH et al (2020) Randomized placebo‐controlled trial of emricasan for non‐alcoholic steatohepatitis‐related cirrhosis with severe portal hypertension. J Hepatol 72: 885–895 [DOI] [PubMed] [Google Scholar]
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M et al (1997) Caspase‐1 processes IFN‐gamma‐inducing factor and regulates LPS‐induced IFN‐gamma production. Nature 386: 619–623 [DOI] [PubMed] [Google Scholar]
- Gilmore AP (2005) Anoikis. Cell Death Differ 12(Suppl 2): 1473–1477 [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I et al (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110: 5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D (1997) CASH, a novel caspase homologue with death effector domains. J Biol Chem 272: 19641–19644 [DOI] [PubMed] [Google Scholar]
- Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, Khaw SL, Lessene G, Spencer A, Herold MJ et al (2016) Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood 128: 1834–1844 [DOI] [PubMed] [Google Scholar]
- Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy‐Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A (2016) The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell 37: 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grievink HW, Heuberger J, Huang F, Chaudhary R, Birkhoff WAJ, Tonn GR, Mosesova S, Erickson R, Moerland M, Haddick PCG et al (2020) DNL104, a centrally penetrant RIPK1 inhibitor, inhibits rip1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin Pharmacol Ther 107: 406–414 [DOI] [PubMed] [Google Scholar]
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K et al (1997) Activation of interferon‐gamma inducing factor mediated by interleukin‐1beta converting enzyme. Science 275: 206–209 [DOI] [PubMed] [Google Scholar]
- Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD (2014) FADD and caspase‐8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192: 1835–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T et al (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF‐R1 signaling complex and is required for TNF‐mediated gene induction. Mol Cell 36: 831–844 [DOI] [PubMed] [Google Scholar]
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J et al (2007) A genome‐wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39: 207–211 [DOI] [PubMed] [Google Scholar]
- Han C, Liu Y, Dai R, Ismail N, Su W, Li B (2020) Ferroptosis and its potential role in human diseases. Front Pharmacol 11: 239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DKM, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel RT, Baskin DG, Schwartz SM, Hood L (1997) MRIT, a novel death‐effector domain‐containing protein, interacts with caspases and BclXL and initiates cell death [In Process Citation]. Proc Natl Acad Sci USA 94: 11333–11338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong X et al (2017) Discovery of a first‐in‐class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem 60: 1247–1261 [DOI] [PubMed] [Google Scholar]
- Harris PA, Faucher N, George N, Eidam PM, King BW, White GV, Anderson NA, Bandyopadhyay D, Beal AM, Beneton V et al (2019) Discovery and lead‐optimization of 4,5‐dihydropyrazoles as mono‐kinase selective, orally bioavailable and efficacious inhibitors of receptor interacting protein 1 (RIP1) kinase. J Med Chem 62: 5096–5110 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2014) Regulation of NF‐kappaB by TNF family cytokines. Semin Immunol 26: 253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J (2015) Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res 25: 1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J (2017) NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E‐deficient mice‐brief report. Arterioscler Thromb Vasc Biol 37: 1457–1461 [DOI] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P (2018) The Gasdermin‐D pore acts as a conduit for IL‐1beta secretion in mice. Eur J Immunol 48: 584–592 [DOI] [PubMed] [Google Scholar]
- Hoglen NC, Chen LS, Fisher CD, Hirakawa BP, Groessl T, Contreras PC (2004) Characterization of IDN‐6556 (3‐[2‐(2‐tert‐butyl‐phenylaminooxalyl)‐amino]‐propionylamino]‐4‐oxo‐5‐(2,3,5,6‐te trafluoro‐phenoxy)‐pentanoic acid): a liver‐targeted caspase inhibitor. J Pharmacol Exp Ther 309: 634–640 [DOI] [PubMed] [Google Scholar]
- van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pasterkamp G et al (2017) The selective NLRP3‐inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J 38: 828–836 [DOI] [PubMed] [Google Scholar]
- Hoy SM (2018) Talazoparib: first global approval. Drugs 78: 1939–1946 [DOI] [PubMed] [Google Scholar]
- Hu S, Vincenz C, Ni J, Gentz R, Dixit VM (1997) I‐FLICE, a novel inhibitor of tumor necrosis factor receptor‐1‐ and CD‐ 95‐induced apoptosis. J Biol Chem 272: 17255–17257 [DOI] [PubMed] [Google Scholar]
- Hughes MA, Powley IR, Jukes‐Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M (2016) Co‐operative and hierarchical binding of c‐FLIP and caspase‐8: a unified model defines how c‐FLIP isoforms differentially control cell fate. Mol Cell 61: 834–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Hu Y, Chen S, Nunez G (1997) CLARP, a death effector domain‐containing protein interacts with caspase‐8 and regulates apoptosis. Proc Natl Acad Sci USA 94: 10717–10722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C et al (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388: 190–195 [DOI] [PubMed] [Google Scholar]
- Izzo V, Bravo‐San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26: 655–667 [DOI] [PubMed] [Google Scholar]
- Jang KH, Do YJ, Son D, Son E, Choi JS, Kim E (2017) AIF‐independent parthanatos in the pathogenesis of dry age‐related macular degeneration. Cell Death Dis 8: e2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S, Seidelin JB, LaCasse EC, Nielsen OH (2020) SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci Signal 13: eaax8295 [DOI] [PubMed] [Google Scholar]
- Jiang X, Li W, Li X, Bai H, Zhang Z (2019) Current status and future prospects of PARP inhibitor clinical trials in ovarian cancer. Cancer Manag Res 11: 4371–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, Fisher A, Lane R, Young GR, Kassiotis G et al (2020) Z‐nucleic‐acid sensing triggers ZBP1‐dependent necroptosis and inflammation. Nature 580: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J et al (2009) XIAP discriminates between type I and type II FAS‐induced apoptosis. Nature 460: 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Offermann MK (2005) Apoptosis induced by the toll‐like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174: 4942–4952 [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES (2013) Toll‐like receptor 3‐mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288: 31268–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES (2008) Receptor‐interacting protein homotypic interaction motif‐dependent control of NF‐kappa B activation via the DNA‐dependent activator of IFN regulatory factors. J Immunol 181: 6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston‐Rosanoff D, Daley‐Bauer LP, Hakem R, Caspary T, Mocarski ES (2011) RIP3 mediates the embryonic lethality of caspase‐8‐deficient mice. Nature 471: 368–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, Andrews DW (2018) BCL‐2 family proteins: changing partners in the dance towards death. Cell Death Differ 25: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W et al (2018) Gasdermin D exerts anti‐inflammatory effects by promoting neutrophil death. Cell Rep 22: 2924–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Kawai T (2014) Toll‐like receptor signaling pathways. Front Immunol 5: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, Czabotar PE, Voss AK, Strasser A (2018) Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173: 1217–1230 e1217 [DOI] [PubMed] [Google Scholar]
- Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer 26: 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheloufi M, Boulanger CM, Codogno P, Rautou PE (2015) Autosis occurs in the liver of patients with severe anorexia nervosa. Hepatology 62: 657–658 [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin‐Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2006) Hierarchical regulation of mitochondrion‐dependent apoptosis by BCL‐2 subfamilies. Nat Cell Biol 8: 1348–1358 [DOI] [PubMed] [Google Scholar]
- Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X et al (2016) Ultrasmall nanoparticles induce ferroptosis in nutrient‐deprived cancer cells and suppress tumour growth. Nat Nanotechnol 11: 977–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN (2011) Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem 286: 34851–34857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin‐Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM et al (2016) The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538: 477–482 [DOI] [PubMed] [Google Scholar]
- Kuang S, Zheng J, Yang H, Li S, Duan S, Shen Y, Ji C, Gan J, Xu XW, Li J (2017) Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci USA 114: 10642–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Kuma A, Komatsu M, Mizushima N (2017) Autophagy‐monitoring and autophagy‐deficient mice. Autophagy 13: 1619–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupershmidt L, Amit T, Bar‐Am O, Weinreb O, Youdim MB (2012) Multi‐target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer's‐like neuropathology and age‐related alterations in mice. Mol Neurobiol 46: 217–220 [DOI] [PubMed] [Google Scholar]
- Kuriakose T, Kanneganti TD (2018) ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol 39: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, Liu L, Stoffels M, Kratina T, Lawlor KE et al (2020) Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577: 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ et al (2014) Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci USA 111: 7741–7746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, Cuellar TL, Reichelt M, Kayagaki N (2018) ASC‐ and caspase‐8‐dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci Rep 8: 3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Davis RJ (2003) JNK phosphorylation of Bim‐related members of the Bcl2 family induces Bax‐dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL (2009) Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 1787: 1395–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu C‐j, Yuan J (1998) Cleavage of BID by caspase 8 mediatesthe mitochondrial damage in the faspathway of apoptosis. Cell 94: 491–501 [DOI] [PubMed] [Google Scholar]
- Li J, Cao F, Yin H‐l, Huang Z‐J, Lin Z‐T, Mao N, Sun B, Wang G (2020) Ferroptosis: past, present and future. Cell Death Dis 11: 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 91: 479–489 [DOI] [PubMed] [Google Scholar]
- Lim J, Park H, Heisler J, Maculins T, Roose‐Girma M, Xu M, McKenzie B, van Lookeren Campagne M, Newton K, Murthy A (2019) Autophagy regulates inflammatory programmed cell death via turnover of RHIM‐domain proteins. Elife 8: e44452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M (2016) RIPK1 counteracts ZBP1‐mediated necroptosis to inhibit inflammation. Nature 540: 124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase‐8 prompts TNF‐induced apoptosis. Genes Dev 13: 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K et al (2000) The combined functions of proapoptotic Bcl‐2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S (2012) Rip1 (receptor‐interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81: 751–761 [DOI] [PubMed] [Google Scholar]
- Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H et al (2013) Two independent pathways of regulated necrosis mediate ischemia‐reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shoji‐Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ et al (2013) Autosis is a Na+, K+‐ATPase‐regulated form of cell death triggered by autophagy‐inducing peptides, starvation, and hypoxia‐ischemia. Proc Natl Acad Sci USA 110: 20364–20371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S et al (2016) BOK is a non‐canonical BCL‐2 family effector of apoptosis regulated by ER‐associated degradation. Cell 165: 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lork M, Verhelst K, Beyaert R (2017) CYLD, A20 and OTULIN deubiquitinases in NF‐kappaB signaling and cell death: so similar, yet so different. Cell Death Differ 24: 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bai L, Sun H, Nikolovska‐Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y et al (2008) SM‐164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP‐1/2 and XIAP. Cancer Res 68: 9384–9393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J et al (2008) Both cIAP1 and cIAP2 regulate TNFalpha‐mediated NF‐kappaB activation. Proc Natl Acad Sci USA 105: 11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Warne JP, Salas E, Xu AW, Debnath J (2015) Loss of Atg12, but not Atg5, in pro‐opiomelanocortin neurons exacerbates diet‐induced obesity. Autophagy 11: 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B et al (2014) RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56: 481–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, Tharp GK, Maner‐Smith K, Burd EM, Schaeffer M et al (2018) Caspase‐8 collaborates with caspase‐11 to drive tissue damage and execution of endotoxic shock. Immunity 49(1): 42–55.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P (2020) Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci 41: 209–224 [DOI] [PubMed] [Google Scholar]
- Martinez‐Lostao L, Anel A, Pardo J (2015) How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res 21: 5047–5056 [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 10: 417–426 [DOI] [PubMed] [Google Scholar]
- Martin‐Sanchez D, Fontecha‐Barriuso M, Carrasco S, Sanchez‐Nino MD, Massenhausen AV, Linkermann A, Cannata‐Ortiz P, Ruiz‐Ortega M, Egido J, Ortiz A et al (2018) TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc Natl Acad Sci USA 115: 4182–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Sanchez D, Ruiz‐Andres O, Poveda J, Carrasco S, Cannata‐Ortiz P, Sanchez‐Nino MD, Ruiz Ortega M, Egido J, Linkermann A, Ortiz A et al (2017) Ferroptosis, but Not necroptosis, is important in nephrotoxic folic acid‐induced AKI. J Am Soc Nephrol 28: 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Massenhausen A, Tonnus W, Himmerkus N, Parmentier S, Saleh D, Rodriguez D, Ousingsawat J, Ang RL, Weinberg JM, Sanz AB et al (2018) Phenytoin inhibits necroptosis. Cell Death Dis 9: 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa‐Ishimoto Y, Hine A, Shono Y, Rudensky E, Lazrak A, Yeung F, Neil JA, Yao X, Chen YH, Heaney T et al (2020) An intestinal organoid‐based platform that recreates susceptibility to T‐cell‐mediated tissue injury. Blood 135: 2388–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa‐Ishimoto Y, Shono Y, Gomez LE, Hubbard‐Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR, Lazrak A, Marinis JM et al (2017) Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med 214: 3687–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister CS, Lakhdari O, Pineton de Chambrun G, Gareau MG, Broquet A, Lee GH, Shenouda S, Eckmann L, Kagnoff MF (2013) TLR3, TRIF, and caspase 8 determine double‐stranded RNA‐induced epithelial cell death and survival in vivo. J Immunol 190: 418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ (1989) bcl‐2‐Immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57: 79–88 [DOI] [PubMed] [Google Scholar]
- McLachlan DRC, Dalton AJ (1991) Intramuscular desferrioxamine in patients with Alzheimer's disease. The Lancet 337: 1304–1308 [DOI] [PubMed] [Google Scholar]
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME (1997) FLICE is activated by association with the CD95 death‐inducing signaling complex (DISC). Embo J 16: 2794–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V (2014) A myeloperoxidase‐containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep 8: 883–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I‐mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190 [DOI] [PubMed] [Google Scholar]
- Mifflin L, Ofengeim D, Yuan J (2020) Receptor‐interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov 19: 553–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish E, Brumatti G, Silke J (2020a) Future therapeutic directions for Smac‐mimetics. Cells 9: 406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish E, Mackiewicz L, Silke N, Pellegrini M, Silke J, Brumatti G, Ebert G (2020b) Combinatorial treatment of birinapant and zosuquidar enhances effective control of HBV replication in vivo. Viruses 12: 901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin M, Anderton H, Voss AK, Thomas T, Wong WW‐L, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J et al (2012) IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 31: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S (2017) Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci 74: 3631–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y (2012) Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72: 5588–5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, Saraiva EM, Porto BN (2018) Respiratory syncytial virus induces the classical ROS‐dependent NETosis through PAD‐4 and necroptosis pathways activation. Sci Rep 8: 14166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM (2020) The killer pseudokinase mixed lineage kinase domain‐like protein (MLKL). Cold Spring Harbor Perspect Biol 12: a036376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez‐Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453 [DOI] [PubMed] [Google Scholar]
- Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose‐Girma M, DeVoss J, Diehl L, Graham RR et al (2014) A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506: 456–462 [DOI] [PubMed] [Google Scholar]
- Murthy P, Muggia F (2019) PARP inhibitors: clinical development, emerging differences, and the current therapeutic issues. Cancer Drug Resist 2: 665–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R et al (1996) FLICE, a novel FADD‐Homologous ICE/CED‐3–like protease, is recruited to the CD95 (Fas/APO‐1) death‐inducing signaling complex. Cell 85: 817–827 [DOI] [PubMed] [Google Scholar]
- Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM (1998) An induced proximity model for caspase‐8 activation. J Biol Chem 273: 2926–2930 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658 [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Nesci S (2020) The mitochondrial permeability transition pore in cell death: a promising drug binding bioarchitecture. Med Res Rev 40: 811–817 [DOI] [PubMed] [Google Scholar]
- Newton K (2020) Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harbor Perspect Biol 12: a036368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J et al (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343: 1357–1360 [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin‐McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L et al (2016a) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Manning G (2016) Necroptosis and Inflammation. Annu Rev Biochem 85: 743–763 [DOI] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose‐Girma M, Warming S, Solon M et al (2016b) RIPK1 inhibits ZBP1‐driven necroptosis during development. Nature 540: 129–133 [DOI] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose‐Girma M, Dohse M, Komuves L, Webster JD, Dixit VM (2019a) Cleavage of RIPK1 by caspase‐8 is crucial for limiting apoptosis and necroptosis. Nature 574: 428–431 [DOI] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, Roose‐Girma M, Modrusan Z, Sagolla MS, Webster JD et al (2019b) Activity of caspase‐8 determines plasticity between cell death pathways. Nature 575: 679–682 [DOI] [PubMed] [Google Scholar]
- Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, Mattingly C, Ho PT, Schoof N, Cammon CG et al (2020) Systemic HIV and SIV latency reversal via non‐canonical NF‐kappaB signalling in vivo. Nature 578: 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR (2011) Catalytic activity of the caspase‐8‐FLIP(L) complex inhibits RIPK3‐dependent necrosis. Nature 471: 363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ et al (2005) An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 435: 677–681 [DOI] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA et al (2018) Pathogen blockade of TAK1 triggers caspase‐8‐dependent cleavage of gasdermin D and cell death. Science 362: 1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, McAllaster MR, Sansone A, Dunlap BF, Desai C, Wang YT, Balce DR, Luke CJ, Lee S, Orchard RC et al (2019) Autophagy genes in myeloid cells counteract IFNgamma‐induced TNF‐mediated cell death and fatal TNF‐induced shock. Proc Natl Acad Sci USA 116: 16497–16506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli P, Giannoni E, Chiarugi P (2013) Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta 1833: 3481–3498 [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18: 134–147 [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191: 677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo J, Wallich R, Martin P, Urban C, Rongvaux A, Flavell RA, Mullbacher A, Borner C, Simon MM (2008) Granzyme B‐induced cell death exerted by ex vivo CTL: discriminating requirements for cell death and some of its signs. Cell Death Differ 15: 567–579 [DOI] [PubMed] [Google Scholar]
- Patel S, Webster JD, Varfolomeev E, Kwon YC, Cheng JH, Zhang J, Dugger DL, Wickliffe KE, Maltzman A, Sujatha‐Bhaskar S et al (2020) RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ 27: 161–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R (2018) MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep 8: 8618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SL, Wang L, Yalcin‐Chin A, Li L, Peyton M, Minna J, Harran P, Wang X (2007) Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac‐mimetic‐induced apoptosis. Cancer Cell 12: 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Philip NH, DeLaney A, Wynosky‐Dolfi MA, Asklof K, Gray F, Choa R, Bjanes E, Buza EL, Hu B et al (2017) RIPK1‐dependent apoptosis bypasses pathogen blockade of innate signaling to promote immune defense. J Exp Med 214: 3171–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos‐Marrero M, Grier JT, Sun Y, Wynosky‐Dolfi MA, Zwack EE, Hu B, Olsen TM et al (2016) Activity of uncleaved caspase‐8 controls anti‐bacterial immune defense and TLR‐induced cytokine production independent of cell death. PLoS Pathog 12: e1005910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FKM, Pasparakis M, Kelliher MA (2014) Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF‐induced necroptosis in vivo. J Immunol 193: 1539–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS (2011) FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J 433: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J, Kabat AM, Maloy KJ (2018) Intestinal epithelial cell autophagy is required to protect against TNF‐induced apoptosis during chronic colitis in mice. Cell Host Microbe 23(2): 191–202.e4 [DOI] [PubMed] [Google Scholar]
- Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD, Mangan M, Dower K, Monks BG, Cushing L et al (2016) Efficacy and pharmacology of the NLRP3 Inflammasome inhibitor CP‐456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol 197: 2421–2433 [DOI] [PubMed] [Google Scholar]
- Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SLC, Tawa P, Xanthoudakis S, Nasir J, Martindale D et al (1998) Cell death attenuation by `Usurpin', a mammalian DEDcaspase homologue that precludes caspase‐8 recruitment and activation by the CD‐95 (Fas, APO‐1) receptor complex. Cell Death Differ 5: 271–288 [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA (2012) TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell 150: 606–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Sheikh MY, Sanyal AJ, Lim JK, Conjeevaram H, Chalasani N, Abdelmalek M, Bakken A, Renou C, Palmer M et al (2012) A phase 2, randomized, double‐blind, placebo‐controlled study of GS‐9450 in subjects with nonalcoholic steatohepatitis. Hepatology 55: 419–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18: 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H et al (2014) RIPK1 regulates RIPK3‐MLKL‐driven systemic inflammation and emergency hematopoiesis. Cell 157: 1175–1188 [DOI] [PubMed] [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H et al (2012) Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 30: 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N, Ganesan R, Hegedus C, Kovacs K, Kufer TA, Virag L (2019) Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol 26: 101239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes‐Alnemri T, Alnemri ES (2019) Gasdermin pores permeabilize mitochondria to augment caspase‐3 activation during apoptosis and inflammasome activation. Nat Commun 10: 1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes‐Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES (2017) Cleavage of DFNA5 by caspase‐3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8: 14128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM et al (2013) AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20: 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V, Vitak N, Vajjhala PR, Vince JE, Stacey KJ (2018) Caspase‐1 is an apical caspase leading to caspase‐3 cleavage in the AIM2 inflammasome response, independent of caspase‐8. J Mol Biol 430: 238–247 [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 456: 264–268 [DOI] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391: 96–99 [DOI] [PubMed] [Google Scholar]
- Samie M, Lim J, Verschueren E, Baughman JM, Peng I, Wong A, Kwon Y, Senbabaoglu Y, Hackney JA, Keir M et al (2018) Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol 19: 246–254 [DOI] [PubMed] [Google Scholar]
- Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, Whitehead LW, Frank D, Garnish SE, Fitzgibbon C et al (2020) MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun 11: 3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR et al (2018) Caspase‐8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA 115: E10888–E10897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S (2016) GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35: 1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, Lell M, Manger B, Rech J, Naschberger E et al (2014) Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med 20: 511–517 [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber A, Rousselle A, Becker JU, von Massenhausen A, Linkermann A, Kettritz R (2017) Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci U S A 114: E9618–E9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M (2020) FADD and Caspase‐8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL‐ and GSDMD‐Mediated Death of Intestinal Epithelial Cells. Immunity 52(978–993): e976 [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J (2001) Caspase‐3‐mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol 3: 346–352 [DOI] [PubMed] [Google Scholar]
- Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23: 197–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Gao W, Shao F (2017) Pyroptosis: Gasdermin‐Mediated Programmed Necrotic Cell Death. Trends Biochem Sci 42: 245–254 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Shiffman M, Freilich B, Vuppalanchi R, Watt K, Chan JL, Spada A, Hagerty DT, Schiff E (2019) Randomised clinical trial: emricasan versus placebo significantly decreases ALT and caspase 3/7 activation in subjects with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther 49: 64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G (2010) Clinical trial: the efficacy and safety of oral PF‐03491390, a pancaspase inhibitor ‐ a randomized placebo‐controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther 31: 969–978 [DOI] [PubMed] [Google Scholar]
- Shitara K, Doi T, Nagano O, Imamura CK, Ozeki T, Ishii Y, Tsuchihashi K, Takahashi S, Nakajima TE, Hironaka S et al (2017) Dose‐escalation study for the targeting of CD44v(+) cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric Cancer 20: 341–349 [DOI] [PubMed] [Google Scholar]
- Shu HB, Halpin DR, Goeddel DV (1997) Casper is a FADD‐ and caspase‐related inducer of apoptosis. Immunity 6: 751–763 [DOI] [PubMed] [Google Scholar]
- Sileikyte J, Forte M (2019) The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid Med Cell Longev 2019: 3403075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Letai A, Sarosiek K (2019) Regulation of apoptosis in health and disease: the balancing act of BCL‐2 family proteins. Nat Rev Mol Cell Biol 20: 175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snezhkina AV, Kudryavtseva AV, Kardymon OL, Savvateeva MV, Melnikova NV, Krasnov GS, Dmitriev AA (2019) ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Medicine and Cellular Longevity 2019: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B et al (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3: eaar6689 [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ et al (2013) ABT‐199, a potent and selective BCL‐2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19: 202–208 [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes‐Alnemri T, Croce CM, Litwack G, Tomaselli KJ et al (1997) FLAME‐1, a novel FADD‐like anti‐apoptotic molecule that regulates Fas/TNFR1‐induced apoptosis. J Biol Chem 272: 18542–18545 [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES (2002) The PYRIN‐CARD protein ASC is an activating adaptor for caspase‐1. J Biol Chem 19: 19 [DOI] [PubMed] [Google Scholar]
- Stein JC, Hansen G (1999) Mannose induces an endonuclease responsible for DNA laddering in plant cells. Plant Physiol 121: 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D et al (1998) Pro‐caspase‐3 is a major physiologic target of caspase‐8. J Biol Chem 273: 27084–27090 [DOI] [PubMed] [Google Scholar]
- Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, Yousefi S (2017) ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol 216: 4073–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, Jadhav S, Bolevich SB, Kozlov AV, Vladimirov YA et al (2019) Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol Med 133: 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Vaux DL (2020) Cell Death in the Origin and Treatment of Cancer. Mol Cell 78: 1045–1054 [DOI] [PubMed] [Google Scholar]
- Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW (1991) Enforced BCL2 expression in B‐lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A 88: 8661–8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Nikolovska‐Coleska Z, Lu J, Meagher JL, Yang CY, Qiu S, Tomita Y, Ueda Y, Jiang S, Krajewski K et al (2007) Design, synthesis, and characterization of a potent, nonpeptide, cell‐permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc 129: 15279–15294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain‐like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227 [DOI] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM (2002) Identification of a novel homotypic interaction motif required for the phosphorylation of receptor‐interacting protein (RIP) by RIP3. J Biol Chem 277: 9505–9511 [DOI] [PubMed] [Google Scholar]
- Taabazuing CY, Okondo MC, Bachovchin DA (2017) Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell chemical biology 24(507–514): e504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, Pan H, Bai R, Zhang J, Wang Y et al (2020) A dominant autoinflammatory disease caused by non‐cleavable variants of RIPK1. Nature 577: 109–114 [DOI] [PubMed] [Google Scholar]
- Tapia‐Abellan A, Angosto‐Bazarra D, Martinez‐Banaclocha H, de Torre‐Minguela C, Ceron‐Carrasco JP, Perez‐Sanchez H, Arostegui JI, Pelegrin P (2019) MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol 15: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, Goldman RD, Wagner DD, Waterman CM (2020) NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4‐mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A 117: 7326–7337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DA, Du C, Xu M, Wang X, Ley TJ (2000) DFF45/ICAD Can Be Directly Processed by Granzyme B during the Induction of Apoptosis. Immunity 12: 621–632 [DOI] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M et al (1997) Viral FLICE‐inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386: 517–521 [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J et al (1992) A Novel Heterodimeric Cysteine Protease Is Required for Interleukin‐1‐Beta Processing in Monocytes. Nature 356: 768–774 [DOI] [PubMed] [Google Scholar]
- Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, Cidado J, Embrey KJ, Gangl E, Gibbons FD et al (2018) Discovery of Mcl‐1‐specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 9: 5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P et al (2008) ABT‐263: a potent and orally bioavailable Bcl‐2 family inhibitor. Cancer Res 68: 3421–3428 [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Le Manh T, Hori O, Mahib MR, Yamaguchi Y, Miura M et al (2019) Caspase‐1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10: 2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM (1984) Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226: 1097–1099 [DOI] [PubMed] [Google Scholar]
- Tummers B, Mari L, Guy CS, Heckmann BL, Rodriguez DA, Ruhl S, Moretti J, Crawford JC, Fitzgerald P, Kanneganti TD et al (2020) Caspase‐8‐Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 52: 994–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajjhala PR, Lu A, Brown DL, Pang SW, Sagulenko V, Sester DP, Cridland SO, Hill JM, Schroder K, Stow JL et al (2015) The Inflammasome Adaptor ASC Induces Procaspase‐8 Death Effector Domain Filaments. J Biol Chem 290: 29217–29230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino KL, Gutierrez M, Sanchez R, Winship MJ, Shapiro DA (2003) First clinical trial of a novel caspase inhibitor: anti‐apoptotic caspase inhibitor, IDN‐6556, improves liver enzymes. Int J Clin Pharmacol Ther 41: 441–449 [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Alicke B, Elliott JM, Zobel K, West K, Wong H, Scheer JM, Ashkenazi A, Gould SE, Fairbrother WJ et al (2009) X chromosome‐linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J Biol Chem 284: 34553–34560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ et al (2007) IAP antagonists induce autoubiquitination of c‐IAPs, NF‐kappaB activation, and TNFalpha‐dependent apoptosis. Cell 131: 669–681 [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D (2008) c‐IAP1 and c‐IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)‐induced NF‐kappaB activation. J Biol Chem 283: 24295–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O et al (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276 [DOI] [PubMed] [Google Scholar]
- de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M (2019) Single‐cell analysis of pyroptosis dynamics reveals conserved GSDMD‐mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 26: 146–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM (1988) Bcl‐2 gene promotes haemopoietic cell survival and cooperates with c‐myc to immortalize pre‐B cells. Nature 335: 440–442 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identification of DIABLO, a Mammalian Protein that Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell 102: 43–53 [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M et al (2007) IAP antagonists target cIAP1 to induce TNFalpha‐dependent apoptosis. Cell 131: 682–693 [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G et al (2012) Inhibitor of apoptosis proteins limit RIP3 kinase‐dependent interleukin‐1 activation. Immunity 36: 215–227 [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C (2002) The Therapeutic Potential of Poly(ADP‐Ribose) Polymerase Inhibitors. Pharmacol Rev 54: 375–429 [DOI] [PubMed] [Google Scholar]
- Vlantis K, Wullaert A, Polykratis A, Kondylis V, Dannappel M, Schwarzer R, Welz P, Corona T, Walczak H, Weih F et al (2016) NEMO Prevents RIP Kinase 1‐Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF‐kappaB‐Dependent and ‐Independent Functions. Immunity 44: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet S, Srinivasan S, Lamkanfi M, van Loo G (2019) Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11: e10248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboinik I, Whisstock JC, Trapani JA (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 15: 388–400 [DOI] [PubMed] [Google Scholar]
- Voss AK, Strasser A (2020) The essentials of developmental apoptosis. F1000Res 9: 148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C et al (2016) A nuclease that mediates cell death induced by DNA damage and poly(ADP‐ribose) polymerase‐1. Science 354: aad6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F (2017) Chemotherapy drugs induce pyroptosis through caspase‐3 cleavage of a gasdermin. Nature 547: 99–103 [DOI] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA et al (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184: 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JD, Kwon YC, Park S, Zhang H, Corr N, Ljumanovic N, Adedeji AO, Varfolomeev E, Goncharov T, Preston J et al (2020) RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J Leukoc Biol 107: 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JD, Vucic D (2020) The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front Cell Dev Biol 8: 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ (2000) tBID, a membrane‐targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14: 2060–2071 [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel K, Scott NE, Tompson DJ, Votta BJ, Madhavan S, Povey K, Wolstenholme A, Simeoni M, Rudo T, Richards‐Peterson L et al (2017) Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect 5: e00365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez‐Majada V, Ermolaeva M, Kirsch P, Sterner‐Kock A, van Loo G, Pasparakis M (2011) FADD prevents RIP3‐mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477: 330–334 [DOI] [PubMed] [Google Scholar]
- Weng D, Marty‐Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA et al (2014) Caspase‐8 and RIP kinases regulate bacteria‐induced innate immune responses and cell death. Proc Natl Acad Sci U S A 111: 7391–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NS, Dixit V, Ashkenazi A (2009) Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 10: 348–355 [DOI] [PubMed] [Google Scholar]
- Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, Rodriguez Martinez M, Lopez G, Mattioli M, Realubit R et al (2015) Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 162: 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113: E4966–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol 15: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chang HY, Baltimore D (1998) Autoproteolytic activation of pro‐caspases by oligomerization. Mol Cell 1: 319–325 [DOI] [PubMed] [Google Scholar]
- Yang Y, Jiang G, Zhang P, Fan J (2015) Programmed cell death and its role in inflammation. Mil Med Res 2: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Kubes P (2013) NETosis: how vital is it? Blood 122: 2784–2794 [DOI] [PubMed] [Google Scholar]
- Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, Simon HU (2019) Untangling "NETosis" from NETs. Eur J Immunol 49: 221–227 [DOI] [PubMed] [Google Scholar]
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M (2006) Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 103: 4952–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP‐ribose) polymerase‐1‐dependent cell death by apoptosis‐inducing factor. Science 297: 259–263 [DOI] [PubMed] [Google Scholar]
- Yuan J, Amin P, Ofengeim D (2019) Necroptosis and RIPK1‐mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20: 19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Ganley IG (2017) The mammalian ULK1 complex and autophagy initiation. Essays Biochem 61: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakeri ZF, Quaglino D, Latham T, Lockshin RA (1993) Delayed mternucleosomal DNA fragmentation in programmed cell death. Faseb J 7: 470–478 [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH (1994) Nitric oxide activation of poly(ADP‐ribose) synthetase in neurotoxicity. Science 263: 687–689 [DOI] [PubMed] [Google Scholar]
- Zhang X, Dowling JP, Zhang J (2019) RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis 10: 245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Fan C, Zhang H, Zhao Q, Liu Y, Xu C, Xie Q, Wu X, Yu X, Zhang J et al (2016) MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep 16: 3247–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, Guo SQ, Wang S, Guo T, Wang ZY et al (2018) alpha‐Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol 14: 535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X (1997) Apaf‐1, a human protein homologous to C. elegans CED‐4, participates in cytochrome c‐dependent activation of caspase‐3. Cell 90: 405–413 [DOI] [PubMed] [Google Scholar]