

TO THE EDITOR:
The dysregulation of RNA and protein levels, in comparison with the counterpart normal tissues, is a constant molecular feature of all human malignancies. Many causes can be invoked as culprits of the altered transcriptome and proteome of cancer cells, but recently, the emergence of disrupted chemical modifications of the RNA molecule (that can affect its stability, targeting, or translation) is gaining momentum as a primary cause. In this regard, the emergence of a so-termed altered epitranscriptome,1,2 including malignant hematopoiesis, is starting to be described in tumorigenesis.3 More than 100 distinctly modified nucleotides have been described in RNA molecules.4,5 Although some of these, such as cytosine methylation, have also been described in DNA, DNA has a smaller repertoire of modified nucleotides, whereas RNA molecules can show a more diverse spectrum of modifications, including, among others, pseudouridine or queuosine. However, the most abundant modification of RNA is the methylation of adenosine (A) in the form of m6A and to a lesser extent of m1A. m6A is not only the most common internal modification of messenger RNA (mRNA), affecting various facets of RNA metabolism,5,6 but it is also relevant to microRNAs, controlling their maturation and expression levels (as has been described for the tumor-suppressor microRNA let-7).7 The other methylated adenosine present in RNA, m1A, is typically found in abundant noncoding RNAs, and is also observed around the start codon upstream of the first splice site and in the 5′ untranslated regions (UTRs) of mRNAs.8-10 The m1A location is opposed to m6A, which is enriched in 3′ UTRs and near stop codons.11,12
The discovery of an m6A eraser, FTO,13 provided the first evidence of reversible posttranscriptional modifications in mRNAs, further generating interest in the epitranscriptome and its possible impact in cancer biology. Since then, complex pathways of highly regulated enzymes that add (writers) or remove (erasers) the RNA-modification marks have been dissected at the physiological level, but little is still know about their possible alterations in the cancer arena. Focusing on the methylation of adenosine, the most prevalent type of RNA modification, and knowing that promoter cytosine guanine dinucleotide (CpG) island hypermethylation-associated transcriptional silencing is a common mechanism of gene inactivation in cancer cells,14,15 we interrogated the presence of this type of epigenetic inactivation in the genes encoding the writers and erasers of the m6A and m1A marks. Thus, herein, we have investigated cancer-specific DNA-methylation changes at the 5′-end promoter regions of the METTL3 and METTL14 core subunits of the m6A writer complex; the m6A erasers ALKBH5 and FTO; the m1A writers TRMT6, TRMT61A, TRMT10C, and TRMT61B; and the m1A eraser ALKBH3.5 To uncover candidate epigenetic changes in these enzymes, we first data-mined a collection of close to 1000 human cancer cell lines in which we have characterized the DNA-methylation profiles.16 The promoter-associated CpG islands of METTL3, METTL14, ALKBH5, FTO, TRMT6, TRMT61A, TRMT10C, and TRMT61B were unmethylated in all of the assessed cancer cell lines (supplemental Data Set 1, available on the Blood Web site). However, the ALKBH3 promoter CpG island was commonly methylated in lymphoma cell lines (35%; 23 of 66) among different cancer cell line types (Figure 1A; supplemental Data Set 1). The second tumor type with the highest percentage of ALKBH3 hypermethylation was breast cancer (10.20%; 5 of 49), where this epigenetic alteration has recently been described.17 Although ALKBH3 has been reported to be upregulated in head and neck cancer,18 data-mining of the available transcriptional patterns in the studied cancer cell line set16 showed that ALKBH3 hypermethylation was associated with mRNA downregulation (Figure 1B). This genomic locus was found unmethylated in all of the different normal tissue samples analyzed from the TCGA data set (n = 730; supplemental Data Set 2). Further detailed in silico analyses of the hematological malignancies according to their subtype identified that ALKBH3 promoter CpG island hypermethylation was most frequent in Hodgkin lymphoma cell lines (44%; 4 of 9), followed by Burkitt lymphoma (38%; 5 of 13), other non-Hodgkin lymphoma cell lines (37%; 12 of 32), and anaplastic large cell lymphoma (33%; 1 of 3) (Figure 1C). Data-mining of the available microarray-expression results16 demonstrated that ALKBH3 hypermethylation was linked to transcript downregulation in the cell lines derived from hematological malignancies (Figure 1D). Further in silico analyses showed that the 5′ end of of ALKBH3 was unmethylated in all tested samples of peripheral blood mononuclear cells, naive B lymphocytes, and naive T lymphocytes obtained from healthy donors (supplemental Data Set 3). Thus, the cancer-specific DNA-methylation event at the ALKBH3 promoter became our focus of interest and was herein further studied in the context of Hodgkin lymphoma, the lymphoma type exhibiting the highest hypermethylation frequency.
Figure 1.
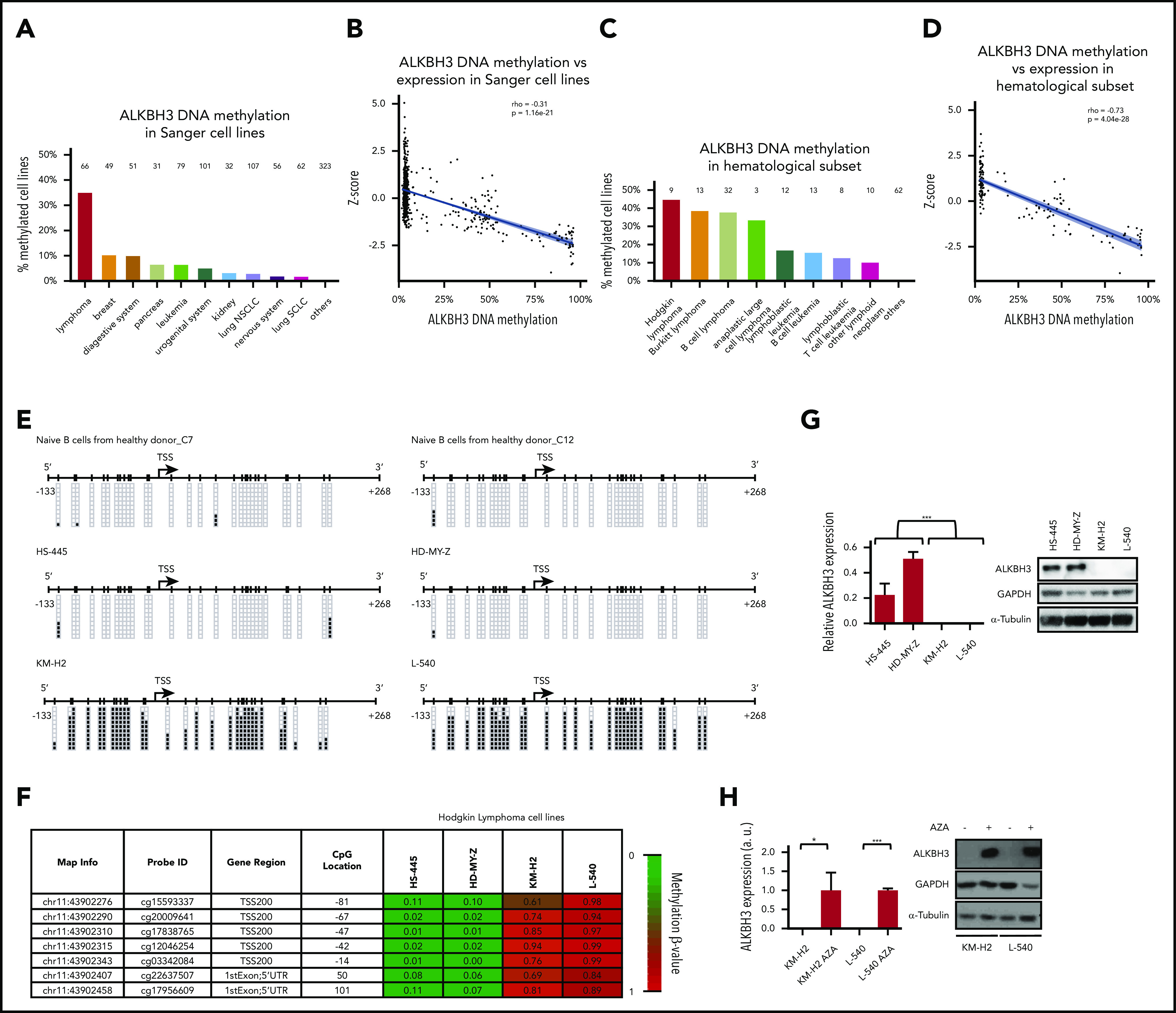
ALKBH3 promoter CpG island hypermethylation and transcriptional silencing in Hodgkin lymphoma cells. (A) Percentage of ALKBH3 methylation in the Sanger set of cancer cell lines according to tumor type. Number of cell lines studied for each tumor type is shown on top of each column. (B) ALKBH3 hypermethylation is associated with loss of the mRNA in cell lines from the Sanger panel (n = 957). (C) Percentage of ALKBH3 methylation in the Sanger set of cell lines derived from hematological malignancies according to subtype. Number of cell lines studied for each tumor type is shown on top of each column. (D) ALKBH3 methylation is associated with loss of the transcript in the Sanger set of cell lines derived from hematological malignancies (n = 162). (E) Bisulfite genomic sequencing of the ALKBH3 promoter CpG island in Hodgkin lymphoma cell lines and naive B cells from healthy donors. CpGs are represented as short vertical lines; the transcription start site (TSS) is represented as a black arrow. Single clones are shown for each sample. Presence of an methylated or unmethylated cytosine is indicated by a black or white square, respectively. (F) DNA-methylation profile of the ALKBH3 promoter CpG island analyzed using the 450 000 DNA-methylation microarray. Single CpG-methylation levels (0-1) are shown. Red, methylated; green, unmethylated. Data from the 4 studied Hodgkin lymphoma cell lines. (G) Expression levels of ALKBH3 in Hodgkin lymphoma cell lines assessed by quantitative reverse transcription PCR (data shown represent mean plus or minus standard deviation [SD] of biological triplicates) (left) and western blot (right). (H) Expression of the ALKBH3 RNA transcript (data shown represent the mean plus or minus SD of biological triplicates) and protein was recovered in the ALKBH3-hypermethylated and -silenced KM-H2 and L540 cells upon use of the DNA-demethylating agent 5-aza-2′-deoxycytidine (AZA). *P < .05; **P < .01; ***P < .001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NSCLC, non–small cell lung carcinoma; SCLC, small cell lung carcinoma.
Having found the ALKBH3 CpG island–methylation patterns shown herein, we assessed in detail the possible association of the hypermethylation event with the loss of ALKBH3 gene expression at the RNA and protein levels. We performed bisulfite genomic sequencing of multiple clones in the Hodgkin lymphoma cell lines KM-H2, L540, HS445, and HD-MY-Z using primers that encompassed the transcription start site–linked CpG island (supplemental Methods). We observed that the 5′-end CpG island of ALKBH3 in the L540 and KMH2 cell lines was hypermethylated (Figure 1E), whereas the HD-MY-Z and HS445 cells were unmethylated at these sites (Figure 1E). Naive B cells were also found unmethylated (Figure 1E). These data corroborated the DNA-methylation profiles obtained by the DNA-methylation microarray platform (Figure 1F). The methylated ALKBH3 cell lines KM-2 and L540 did not express the ALKBH3 RNA transcript and protein, as determined by quantitative reverse transcription polymerase chain reaction (PCR) and western blot, respectively (Figure 1G). Expression of ALKBH3 RNA and protein was found in the unmethylated cell lines HD-MY-Z and HS445 (Figure 1G). Treatment of the ALKBH3-hypermethylated cell lines with the DNA-demethylating agent 5′-aza-2′-deoxycytidine restored ALKBH3 RNA and protein expression (Figure 1H). The association between ALKBH3 promoter hypermethylation and gene silencing was further validated in 2 additional Hodgkin cell lines not included in our original screening, SUP-HD1 and HS611-T (supplemental Figure 1). Overall, these results indicate the presence of cancer-specific promoter CpG island hypermethylation–associated loss of ALKBH3 gene expression in Hodgkin lymphoma cells. DNA methylation–associated silencing of ALKBH3 was also validated in 13 cell lines derived from Burkitt lymphoma and other non-Hodgkin lymphomas (supplemental Figure 2).
Once we had shown the presence of ALKBH3 CpG island hypermethylation–linked transcriptional inactivation in human Hodgkin lymphoma cell lines, we then wondered about the molecular targets of ALKBH3 loss in these cells. Beyond its role in DNA repair,19-21 ALKBH3 exhibits an RNA-demethylase activity for m1A mRNA.9 Thus, the epigenetic inactivation of ALKBH3 in Hodgkin lymphoma cells hinders a downstream m1A RNA-demethylating event and this can contribute to the biology of these malignancies. To prove this idea, we performed RNA high-throughput sequencing using an m1A antibody to enrich m1A-modified mRNA fragments8 in HD-MY-Z cells, unmethylated at the ALKBH3 promoter CpG island (Figure 1E-F), and expressing the transcript and protein (Figure 1G), transfected with a doxycycline-inducible short harpin RNA (shRNA) against ALKBH3 (Figure 2A). Read-mapping statistics from the m1A-sequencing samples are shown in supplemental Table 1.
Figure 2.
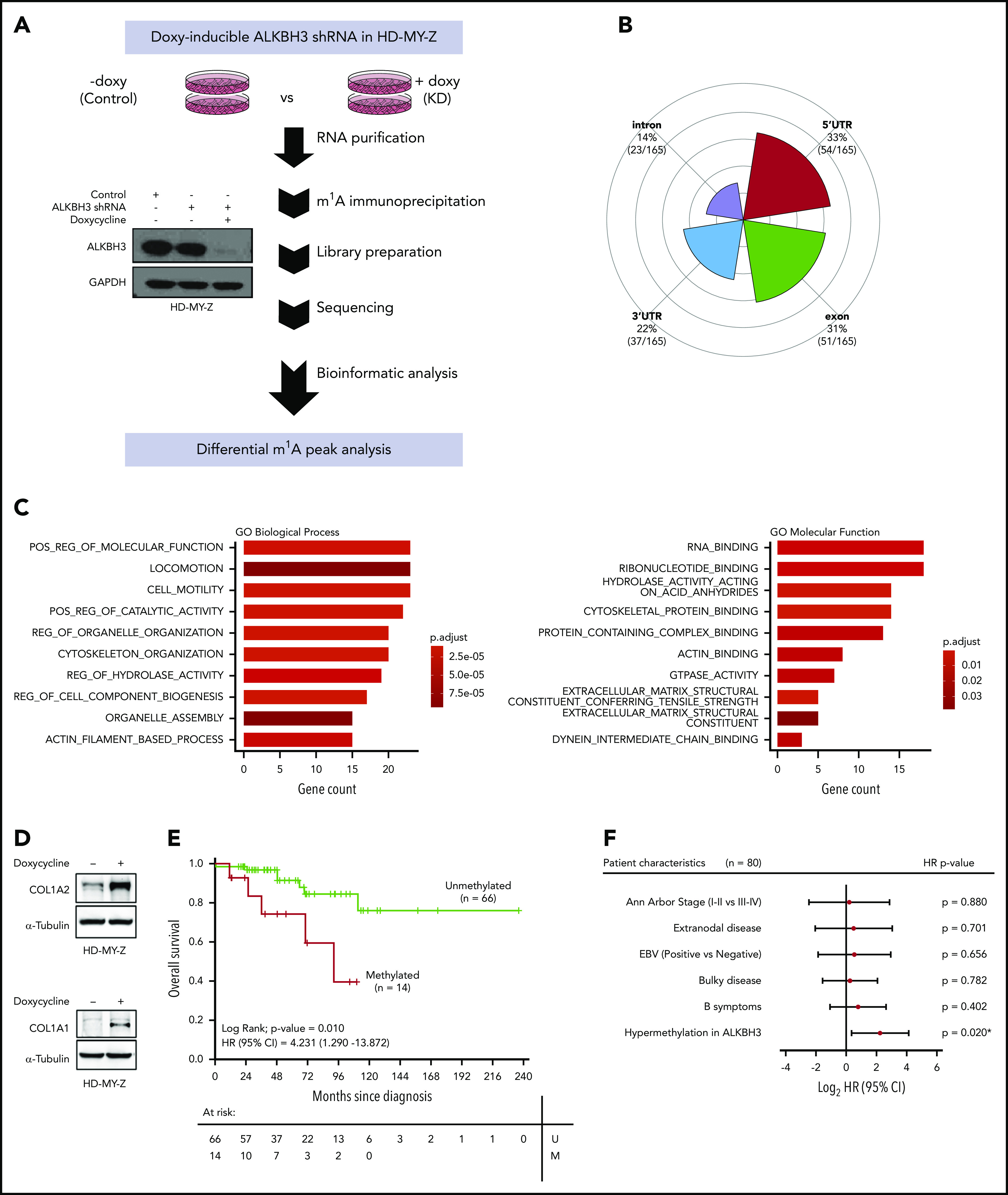
ALKBH3 loss induces a gain of m1A in the transcriptome of Hodgkin lymphoma cells and is associated with poor clinical outcome. (A) Western blot validation of the efficient shRNA-mediated depletion of ALKBH3 in HD-MY-Z cells upon the addition of doxycycline and workflow of the RNA high-throughput sequencing using an m1A antibody to enrich m1A-modified mRNA-fragment analysis developed to detect changes in m1A peaks upon ALKBH3 depletion in HD-MY-Z cells. (B) Distribution of RNA location sites for the m1A peaks undergoing changes in upon ALKBH3 depletion in HD-MY-Z cells. (C) GO analysis (GO) of the genes with differential m1A content in HD-MY-Z cells upon ALKBH3 depletion (hypergeometric test with a false discovery rate [FDR] adjusted P < .05). (D) Western blot of COL1A2 and COL1A1 protein levels upon shRNA-mediated depletion of ALKBH3 in HD-MY-Z cells. (E) Kaplan-Meier analysis of OS in primary Hodgkin lymphomas according to ALKBH3-methylation status determined by pyrosequencing. The P value corresponds to the log-rank test. Results of the univariate Cox regression analysis are represented by the HR and 95% CI. (F) Multivariate Cox regression analysis of OS, represented by a forest plot, considering the clinical characteristics of the cohort of primary Hodgkin lymphoma patients. ALKBH3 promoter hypermethylation is an independent prognostic factor for OS. Values of P < .05 were considered statistically significant. *P < .05. EBV, Epstein-Barr virus; M, methylated; U, unmethylated.
Upon efficient shRNA-mediated depletion of ALKBH3 in the HDMYZ cells (Figure 2A; supplemental Figure 3), our epitranscriptomic approach identified that, whereas 2511 m1A peaks (corresponding to 1522 transcripts) were unchanged in both conditions (supplemental Table 2), the shRNA-mediated depletion of ALKBH3 in HD-MY-Z cells caused a change of 165 m1A peaks (corresponding to 147 transcripts; supplemental Table 3). Most importantly, the induced downregulation of ALKBH3 caused a gain of m1A, being observed in 159 m1A sites (96% of the total 165 sites with distinct m1A content) corresponding to 141 transcripts (supplemental Table 3), a finding that it is consistent with the described role of ALKBH3 as an m1A RNA demethylase.9 The most frequent location of the differential m1A peaks was the 5′ UTR of the transcripts (Figure 2B), in agreement with previous reports for this epitranscriptomic mark,8,9,22 although they were also observed in other transcript regions such as exons, 3′ UTR, and introns. To better characterize the identified set of 147 genes with significantly distinct m1A content upon ALKBH3 depletion, we performed a gene functional annotation by gene-set enrichment analysis using gene ontology (GO) signature collections. We observed an overrepresentation of GO biological processes, molecular function, cellular component, and reactome pathways related to cell migration (such as ”locomotion” and “cell motility”), the cytoskeleton (such as “cytoskeleton organization” and “actin filament based process”), and the microenvironment (such as “extracellular matrix structural component”, “extracellular matrix organization,” and collagen formation, degradation, and trimerization) (Figure 2C; supplemental Figure 4). These processes are highly relevant in Hodgkin lymphoma, in which the malignant cells are surrounded by a large tumor microenvironment that exerts a critical role in the disease23 and the cytoskeleton, through association with extracellular connective tissues, acts as a “guardian” for tissue stabilization and the prevention of cell migration. In this regard, the 2 transcripts with the highest number of gained m1A peaks upon ALKBH3 depletion corresponded to 2 collagens, type I α 2 (COL1A2) and type I α 1 (COL1A1) (supplemental Table 4), critical noncellular components of the Hodgkin lymphoma microenvironment.24,25 Genome browser screenshots for differential m1A peaks in COL1A2 and COL1A1 are shown in supplemental Figure 5. Presence of m1A in 5′ UTRs correlates with increased protein expression,8,9 and we indeed observed that the shRNA-mediated depletion of ALKBH3 in HD-MY-Z cells induced high protein-expression levels of both collagens (Figure 2D). We also performed the reverse experiment, in which we restored ALKBH3 expression by transduction in the hypermethylated/silenced cell line L540 (supplemental Figure 6). Herein, we found the opposite scenario compared with the shRNA model (Figure 2D): the recovery of ALKBH3 expression reduced m1A content in the COL1A2 mRNA and decreased COL1A2 protein levels (supplemental Figure 6). The link between ALKBH3 epigenetic silencing and collagen expression was further reinforced by showing how restoration of ALKBH3 expression in hypermethylated KM-H2 and L540 cells, upon the use of the DNA-methylating agent, decreased COL1A2 levels (supplemental Figure 7).
To further improve the real representation of the disease beyond the limitations of established cell lines, we also studied the relevance of ALKBH3 aberrant CpG island methylation in human primary Hodgkin lymphomas. To achieve this goal, we determined the ALKBH3 promoter-methylation status by bisulfite-coupled pyrosequencing (supplemental Methods) in a collection of 80 well-characterized primary Hodgkin lymphoma cases whose clinicopathological features are shown in supplemental Table 5. We detected ALKBH3 promoter hypermethylation in 18% (14 of 80) of the primary Hodgkin lymphoma cases. The presence of ALKBH3 aberrant methylation was not associated with any of the studied clinicopathological parameters (supplemental Table 5). We also analyzed ALKBH3 expression by western blot in 4 samples of the studied primary Hodgkin lymphoma cases, observing that ALKBH3 CpG island hypermethylation was associated with protein loss (supplemental Figure 8). Importantly, ALKBH3 epigenetic silencing was also associated with COL1A2 and COL1A1 overexpression, whereas an unmethylated ALKBH3 CpG island was linked to the absence of collagen expression (supplemental Figure 8), mimicking the results found in the cell line models (Figure 2D). Most importantly, we wondered whether ALKBH3 hypermethylation also conferred any clinical outcome value. We observed that ALKBH3 hypermethylation was associated with shorter overall survival (OS) (log-rank; P = .010; hazard ratio [HR] = 4.231; 95% confidence interval [CI] = 1.290 - 13.872) (Figure 2E) in the studied Hodgkin lymphoma cohort. In the multivariate analysis, ALKBH3 promoter hypermethylation was shown to be an independent prognostic factor for reduced OS (HR = 4.752; 95% CI = 1.277-17.683; P = .020) (Figure 2F).
Overall, the described research provides an illustrative example that aberrations of the so-called epitranscriptome occur in human lymphomas, herein shown by the epigenetic silencing of the m1A RNA demethylase ALKBH3 in Hodgkin lymphoma. The epigenetic event we describe shifts the m1A targeting of migration and cytoskeleton-related genes, and provides an independent biomarker of poor OS in this disease of otherwise good prognosis.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Centres de Recerca de Catalunya (CERCA) Programme/Generalitat de Catalunya for institutional support.
This work was supported by the Health Department Pla Estratègic de Recerca i Innovació en Salut (PERIS) project no. SLT/002/16/00374 and Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) projects no. 2017SGR1080 of the Catalan Government (Generalitat de Catalunya), Ministerio de Ciencia e Innovación (MCI), Agencia Estatal de Investigación (AEI) and European Regional Development Fund (ERDF) project no. RTI2018-094049-B-I00, the Cellex Foundation; “la Caixa” Banking Foundation (LCF/PR/GN18/51140001), the Instituto de Salud Carlos III (PT17/0015/0024), the Xarxa de Bancs de Tumors de Catalunya (XBTC) sponsored by Pla Director d’Oncologia de Catalunya (XBTC) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Grant agreement No. 743168). P.S.-O. is a fellow of the Severo Ochoa Program (Bp17-165). V.D. is supported by the Spanish Association Against Cancer (AECC).
Footnotes
Raw data reported in this article have been deposited in the Sequence Read Archive (SRA) BioProject (accession number PRJNA602695; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA602695).
Authorship
Contribution: R.E.-P. and M. Esteller conceived and designed the study, and wrote the manuscript with contributions and approval from all authors; R.E.-P., A.R., M.S., M.L., V.O.-B., P.S.-O., A.F.F., and M.F.F. performed molecular analyses; D.P. analyzed multiomics data; F.C., E.D.-D., V.D., M. Encuentra, N.E.-H., G.T., J.-T.N., J.C., and A.S. provided primary samples and analyzed clinical data; L.F., A.V., I.C., R.M., and M.A.P. provided and studied cellular models; and N.K., C.A., S.M.-M., and G.R. performed the RNA high-throughput sequencing using the m1A antibody.
Conflict-of-interest disclosure: M. Esteller is a consultant of Ferrer International and Quimatryx. The remaining authors declare no competing financial interests.
Correspondence: Anna Sureda, Clinical Hematology Department, Catalan Institute of Oncology (ICO), Hospital Duran i Reynals, Av. Gran Via de L’Hospitalet 199-203, 08909 L’Hospitalet, Barcelona, Catalonia, Spain; e-mail: asureda@iconcologia.net; or Manel Esteller, Josep Carreras Leukaemia Research Institute (IJC), Carretera de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Catalonia, Spain; e-mail: mesteller@carrerasresearch.org.
REFERENCES
- 1.Esteller M, Pandolfi PP. The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017;7(4):359-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lian H, Wang QH, Zhu CB, Ma J, Jin WL. Deciphering the Epitranscriptome in cancer. Trends Cancer. 2018;4(3):207-221. [DOI] [PubMed] [Google Scholar]
- 3.Vu LP, Cheng Y, Kharas MG. The Biology of m6A RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 2019;9(1):25-33. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Science. 2016;352(6292):1408-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davalos V, Blanco S, Esteller M. SnapShot: messenger RNA modifications. Cell. 2018;174(2):498-498.e1. [DOI] [PubMed] [Google Scholar]
- 6.Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293-306. [DOI] [PubMed] [Google Scholar]
- 7.Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311-316. [DOI] [PubMed] [Google Scholar]
- 10.Zhou H, Rauch S, Dai Q, et al. Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat Methods. 2019;16(12):1281-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201-206. [DOI] [PubMed] [Google Scholar]
- 12.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8(9):a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109-127. [DOI] [PubMed] [Google Scholar]
- 16.Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166(3):740-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stefansson OA, Hermanowicz S, van der Horst J, et al. CpG promoter methylation of the ALKBH3 alkylation repair gene in breast cancer. BMC Cancer. 2017;17(1):469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pilžys T, Marcinkowski M, Kukwa W, et al. ALKBH overexpression in head and neck cancer: potential target for novel anticancer therapy. Sci Rep. 2019;9(1):13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ringvoll J, Nordstrand LM, Vågbø CB, et al. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 2006;25(10):2189-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ringvoll J, Moen MN, Nordstrand LM, et al. AlkB homologue 2-mediated repair of ethenoadenine lesions in mammalian DNA. Cancer Res. 2008;68(11):4142-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvo JA, Meira LB, Lee CY, et al. DNA repair is indispensable for survival after acute inflammation. J Clin Invest. 2012;122(7):2680-2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Xiong X, Zhang M, et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68(5):993-1005.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Sattarzadeh A, Diepstra A, Visser L, van den Berg A. The microenvironment in classical Hodgkin lymphoma: an actively shaped and essential tumor component. Semin Cancer Biol. 2014;24:15-22. [DOI] [PubMed] [Google Scholar]
- 24.Jaspars LH, Bloemena E, Bonnet P, Van der Valk P, Meijer CJ. Distribution of extracellular matrix components and their receptors in human lymphoid tissue and B-cell non-Hodgkin lymphomas. Histopathology. 1995;26(2):113-121. [DOI] [PubMed] [Google Scholar]
- 25.Cader FZ, Vockerodt M, Bose S, et al. The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood. 2013;122(26):4237-4245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.