

Abstract
Objective
It is often difficult to diagnose epilepsy syndromes in resource‐limited settings. This study was aimed to investigate the prospect of ascertaining the diagnosis, clinical profile, and treatment outcomes of epilepsy syndromes (ESs) among children in a resource‐limited setting.
Methods
This was a descriptive study done from 01/07/2009 to 15/06/2017 among children (1‐17 years of age) with unprovoked seizures presenting to the pediatric neurology clinic of a university hospital in eastern Nepal. Diagnosis, classification, and treatment of seizures were based upon International League Against Epilepsy guidelines.
Results
Of 768 children with unprovoked seizures, 120 (15.6%) were diagnosed as ES. The age of onset of seizure was unique for each ES. Developmental delay and cerebral palsy were present in 47.5% and 28.3% children, respectively. Common ESs were West syndrome (WS)‐26.7%, generalized tonic‐clonic seizures alone (GTCSA)‐21.7%, self‐limited childhood epilepsy with centrotemporal spikes (SLCECTS)‐12.5%, childhood absence epilepsy (CAE)‐10.0%, Lennox‐Gastaut syndrome (LGS)‐10.0%, other developmental and epileptic encephalopathies (DEE)‐5.8%, self‐limited familial infantile epilepsy (SLFIE)‐4.2%, and juvenile myoclonic epilepsy (JME)‐3.3%. Among children with known outcomes (87/120), overall response to pharmacotherapy and to monotherapy was observed in 72.4% (63/87) and 57.5% (50/87) children, respectively. All children with GTCSA, SLFIE, genetic epilepsy with febrile seizure plus (GEFS+), CAE, SLCECTS, and JME responded to pharmacotherapy and they had normal computerized tomography scans of the brain. Seizures were largely pharmaco‐resistant in progressive myoclonus epilepsy (PME)‐100.0%, LGS‐73.0%, WS‐52.0%, and other DEEs‐40%.
Significance
A reasonable proportion (15.6%) of unprovoked seizures could be classified into specific ES despite limited diagnostic resources. WS was the most common ES. GTCSA, SLCECTS, CAE, and LGS were other common ESs. GTCSA, SLFIE, CAE, SLCECTS, GEFS+, and JME were largely pharmaco‐responsive. PME, WS, and LGS were relatively pharmaco‐resistant. Electro‐clinical diagnosis of certain ES avoids the necessity of neuroimaging.
Keywords: child, epilepsy, epileptic syndromes, seizure, treatment outcome
Key Points.
In many children with unprovoked seizures, diagnosis of epilepsy syndrome could be established even with limited access to advanced diagnostic facilities.
Diagnosis of an epilepsy syndrome is more important in resource‐limited settings where neuroimaging facility is not widely available, because electro‐clinical diagnosis of certain epilepsy syndrome avoids the necessity of neuroimaging.
The age of onset of seizure was unique to each epilepsy syndrome.
West syndrome was the most common epilepsy syndrome. Other common epilepsy syndromes were generalized tonic‐clonic seizures alone, self‐limited childhood epilepsy with centrotemporal spikes, childhood absence epilepsy, and Lennox‐Gastaut syndrome.
Generalized tonic‐clonic seizures alone, self‐limited childhood epilepsy with centrotemporal spikes, self‐limited familial infantile epilepsy, childhood bsence epilepsy, genetic epilepsy with febrile seizure plus, and juvenile myoclonic epilepsy were essentially pharmaco‐responsive, whereas progressive myoclonus epilepsy, West syndrome, and Lennox‐Gastaut syndrome were relatively pharmaco‐resistant.
1. INTRODUCTION
Epilepsy syndromes (ESs) are a group of clinical entities that are reliably identified by a cluster of electro‐clinical characteristics. 1 , 2 An ES is an epileptic disorder characterized by a cluster of signs and symptoms customarily occurring together, such as the type of seizure, etiology, anatomical location, precipitating factors, age of onset, severity, chronicity, diurnal and circadian cycling, response to treatment, age of remission, and sometimes prognosis. 2 , 3 A major advance in recent epileptology is the recognition of ESs that allows an accurate diagnosis and thus guides management of seizure disorders. 2 It is important to identify the specific ES wherever possible, to refine the choice of medication, to maximize benefit and minimize adverse effects. In terms of epilepsy classification, the clinician starts by classifying the type of seizure. Then, the patient's type of epilepsy needs to be classified and, in many cases, a specific ES diagnosis is made. 2 , 4 Identifying ES and the syndrome classification of epilepsy provides invaluable prognostic, therapeutic, and, in the case of familial epilepsies, genetic information. 2 , 4 , 5
Recognizing ESs might be difficult in resource‐limited settings because of lack of professional training and diagnostic resources. Therefore, ESs might have been under‐recognized and thereby inappropriately managed in resource‐limited regions. There is paucity of data regarding ESs in Nepal and other underdeveloped areas. This study was aimed to investigate the prospect of diagnosis of ESs as well as to study the clinical profile and treatment outcomes of various ESs among children and adolescents presenting at a tertiary care university hospital in eastern Nepal. The International League Against Epilepsy (ILAE) paper on classification of the epilepsies emphasizes that classifying seizures is not always enough for treatment, there are three levels of diagnosis, and the third level of diagnosis is identification of epilepsy syndrome. 2 , 4 Hence, the findings of this study are also expected to encourage clinicians to start making the syndromic diagnosis of epilepsy rather than just classifying and treating the seizures without arriving at the third level of diagnosis.
2. METHODS
This was a descriptive study which included both the old patients and new patients during the study period. Study was done in the pediatric neurology clinic of BP Koirala Institute of Health Sciences, Dharan, Nepal (BPKIHS). BPKIHS is located in Province No. 1 in the eastern part of Nepal. BPKIHS is one of the largest university hospitals of Nepal and the main referral center for the province. Province No. 1 covers an area of 25,905 km2—about 17.5% of the country's total area. The province is bordered by the Tibet Autonomous Region of China to the north, the Indian states of Sikkim and West Bengal to the east and Bihar to the south and Province No. 2 to the west. There are around 4.5 million people in the province. 6 BPKIHS is the main treatment center for this population. But a lot of patients from bordering areas of Province No. 2 and India also come to this center for medical care.
Epileptic children are managed in the pediatric neurology clinic at BPKIHS. They are regularly followed up with well‐documented medical records at the clinic. Follow‐up is terminated when the child is free of seizure and the antiseizure medication (ASM) is stopped or if the child is lost to follow‐up. Medical records of 642 epileptic children managed at the clinic from July 1, 2009, to October 15, 2016, were reviewed. Additional 86 new children from October 16, 2016, to June 15, 2017 (study period), were enrolled. Children were kept on follow‐up from the date of enrolling until planned termination of follow‐up or until unplanned loss to follow‐up or until July 15, 2017 (date of starting data analysis). For the purpose of this study, children were enrolled only once. If a child evolved from one syndrome to another, the child was not enrolled again for another syndrome. Children were kept on follow‐up for treatment as clinically indicated even after the study completion. Data from subsequent follow‐up after the study period were not included in the analysis.
Inclusion criteria were children from one month to 17 years of age, presenting to the pediatric neurology clinic and presence of at least one unprovoked seizure. Neonates; children with febrile seizures; children with acute symptomatic and provoked seizures that occurred within the first 7 days of an event resulting from transient metabolic derangements, toxins, or side effects of medicines or an acute event such as stroke, traumatic brain injury, or central nervous system (CNS) infections; and children with psychogenic non epileptic seizures were excluded from the study. Children with seizures other than acute symptomatic and provoked seizures were considered to have unprovoked seizures. Children meeting the inclusion criteria were included in the study by consecutive sampling technique. Information of demographic profile, history, seizure semiology, clinical examination findings, developmental status, neuroimaging findings, family history of seizures, history of status epilepticus, previous treatment if any and other important parameters were recorded in predesigned data collection sheets. Presence of seizure in the first or the second degree relatives was considered as a positive family history of seizure.
Interictal electroencephalogram (EEG) was advised in all children with seizure. Neuroimaging was requested when clinically indicated according to the guideline laid out by the ILAE. 7 Although magnetic resonance imaging (MRI) was available in the study site, most of the parents could not financially afford it. Since children were not cooperative and required prolonged sedation for a long time in order to perform MRI, performing MRI was less practical for children who visited the neurology clinic as outpatients. Therefore, we requested a computerized tomography (CT) scan when neuroimaging was indicated. Both CT and MRI scans were done in five children because children were cooperative, parents could financially afford these investigations, and initial CT scans were inconclusive. In some children, neuroimaging was obtained because parents requested for it, although there was no clinical indication. EEG was reported by the principal investigator who is trained in clinical neurophysiology and regularly reports pediatric EEG at the institution. EEG was deemed as abnormal when there were generalized or focal abnormalities such as interictal epileptiform discharges or slow wave abnormalities.
Diagnosis and classification of seizures and ESs as well as usage of terminologies were based upon clinical features, EEG, and neuroimaging (whenever possible) in accordance with the recent reports of the ILAE commission on classification and terminology. 1 , 2 , 4 Seizure semiology was recorded based upon clinical description given by the witness and the child, when child could communicate. Home videos, if available, were also reviewed to aid the diagnosis. The concept of the developmental and epileptic encephalopathy (DEE) may be applicable to large numbers of heterogeneous epilepsies that can have genetic as well as acquired etiologies. 2 Therefore, to highlight the unique features of more common DDEs like West syndrome (WS) and Lennox‐Gastaut syndrome (LGS), or relatively unique DEE like epileptic encephalopathy with continuous spike‐and‐wave during sleep (CSWS), these were described separately. Other less common and homogenous DEEs, such as Rett syndrome and Landau‐Kleffner syndrome (LKS), were described together as “other DEEs” for the study purpose.
Children were treated with appropriately selected standard ASM based on evidence laid out by the ILAE guideline. 8 Prednisolone was used as the first‐line ASM for WS. Follow‐up period was calculated by adding retrospective (from onset of seizure until presentation) and prospective (from presentation until end of follow‐up or end of study period) follow‐up periods. There were three treatment outcomes—good seizure control, pharmaco‐resistant seizure, and unknown outcome. Seizure was considered to be under good control when the child remained seizure‐free either for at least three months or for two times the length of usual seizure‐free period between recurrences prior to treatment, whichever was longer. When seizure was not controlled with the use of 2 ASMs in combination, it was labeled as pharmaco‐resistant. When the follow‐up period was not sufficient to define outcome, children were considered to have unknown outcome.
Ethical clearance was obtained from the Institutional Review Committee of BPKIHS where this study was conducted. Data were entered and screened for error in MS Excel. The analysis was done using IBM SPSS Statistics 16.0 software.
3. RESULTS
During the study period, 768 (642 old and 86 new) children with unprovoked seizures were screened and evaluated. Of them, 120 (15.6%) were classified as ESs. Median age at presentation was 84 months (interquartile range, IQR, 14‐120). Median age of onset of seizure was 32 (IQR 6‐96) months. Median retrospective, prospective, and total follow‐up durations were 12 (IQR 4‐36), 4 (IQR 3‐17), and 28.5 (IQR, 9‐52) months, respectively.
3.1. Baseline characteristics
Baseline characteristics of children with ESs are presented in Table 1. There was male preponderance (69.2%), 47.5% children had developmental delay, 15.8% children had family history of seizure, interictal EEGs were abnormal in 90.0% children, whereas neuroimages were abnormal in 33 (27.5%) children out of 120 children with ESs.
TABLE 1.
Baseline characteristics of children (N = 120) with epilepsy syndromes
| Characteristics | N | % |
|---|---|---|
| Male gender | 83 | 69.2 |
| H/O hospital admission | 11 | 9.2 |
| H/O status epilepticus | 19 | 15.8 |
| H/O recurrent seizures | 115 | 95.8 |
| H/O past febrile seizures | 9 | 7.5 |
| Presence of developmental delay | 57 | 47.5 |
| Family H/O seizure | 19 | 15.8 |
| Abnormal neurological examination findings | 53 | 44.2 |
| Abnormal EEG | 108 | 90.0 |
| Abnormal neuroimaging a | 33 | 27.5 |
| Adequate follow‐up (outcome known) | 87 | 72.5 |
Abbreviations: CT, computerized tomography; EEG, electroencephalogram; H/O, history of; MRI, magnetic resonance imaging; N, number.
Neuroimaging done in 85 children. 31 abnormal CT scans and two abnormal MRI scans. Five children had both CT and MRI scans of brain. All five of them had normal CT scans and two had abnormal MRI scans.
3.2. Epilepsy syndromes and their characteristics
The ESs diagnosed during the study period are presented in Table 2. The most common ES was WS (26.7%). Other common ESs were generalized tonic‐clonic seizures alone (GTCSA) (21.7%), self‐limited childhood epilepsy with centrotemporal spikes (SLCECTS) (12.5%), childhood absence epilepsy (CAE) (10.0%), and LGS (10.0%). Less commonly diagnosed ESs were self‐limited familial infantile epilepsy (SLFIE) (4.2%), juvenile myoclonic epilepsy (JME) (3.3%), genetic epilepsy with febrile seizure plus (GEFS+) (1.7%), mesial temporal lobe epilepsy (MTLE) (1.7%), epilepsy with myoclonic absences (EMA) (0.8%), and progressive myoclonus epilepsies (PME) (0.8%). The most common DEEs were WS and LGS. Other DEEs were Rett syndrome (1.6%), epileptic encephalopathy with CSWS (0.8%), Dravet syndrome (0.8%), LKS (0.8%), and DEEs of unknown etiology (2.5%).
TABLE 2.
Epilepsy syndromes in children
| Epilepsy syndromes | N | % |
|---|---|---|
| WS | 32 | 26.7 |
| GTCSA | 26 | 21.7 |
| SLCECTS | 15 | 12.5 |
| CAE | 12 | 10.0 |
| LGS | 12 | 10.0 |
| SLFIE | 5 | 4.2 |
| JME | 4 | 3.3 |
| GEFS+ | 2 | 1.7 |
| MTLE | 2 | 1.7 |
| EMA | 1 | 0.8 |
| PME | 1 | 0.8 |
| CSWS | 1 | 0.8 |
| Other DEEs a | 7 | 5.8 |
| Total | 120 | 100.0 |
Abbreviations: CAE, childhood absence epilepsy; CSWS, epileptic encephalopathy with continuous spike‐and‐wave during sleep; DEE, developmental and epileptic encephalopathy; EMA, epilepsy with myoclonic absences; GEFS+, genetic epilepsy with febrile seizure plus; GTCSA, generalized tonic‐clonic seizures alone; JME, juvenile myoclonic epilepsy; LGS, Lennox‐Gastaut syndrome; MTLE, mesial temporal lobe epilepsy; N, number; PME, progressive myoclonus epilepsy; SLCECTS, self‐limited childhood epilepsy with centrotemporal spikes; SLFIE, self‐limited familial infantile epilepsy; WS, West syndrome.
Other DEEs, Rett syndrome (2, 1.6%), Dravet syndrome (1, 0.8%), Landau‐Kleffner syndrome (1, 0.8%), and unknown cause (3, 2.5%).
The age of onset of seizure was unique to each ES as shown in Figure 1. WS and SLFIE were infantile onset ESs, whereas LGS and other DEEs predominated in the second year of life. CAE, GTCSA, and SLCECTS had the age of onset mainly during mid‐childhood. JME had an age of onset during adolescence. Other various characteristics of ESs are presented in Table 3.
FIGURE 1.
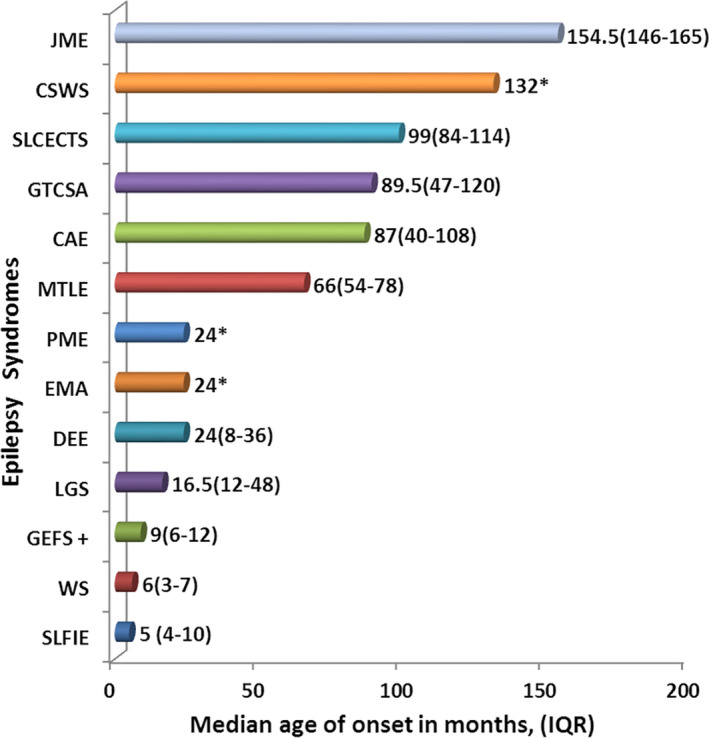
Median age of onset (months and IQR#) of seizure in various epilepsy syndromes. #‐IQR, interquartile range; *‐IQR not shown because there was a single child in each category. Abbreviations: JME, juvenile myoclonic epilepsy; CSWS, epileptic encephalopathy with continuous spike‐and‐wave during sleep; SLCECTS, self‐limited childhood epilepsy with centrotemporal spikes; GTCSA, generalized tonic‐clonic seizures alone; CAE, childhood absence epilepsy; MTLE, mesial temporal lobe epilepsy; PME, progressive myoclonus epilepsy; EMA, epilepsy with myoclonic absences; DEE, other developmental and epileptic encephalopathies; LGS, Lennox‐Gastaut syndrome; GEFS+, genetic epilepsy with febrile seizure plus; WS, West syndrome; and SLFIE, self‐limited familial infantile epilepsy
TABLE 3.
Clinical characteristics of various epilepsy syndromes
| Syndromes (N) | Causes (N, %) | Abnormal neurological examination (%) | Developmental delay (%) | Neuroimaging % done, % abnormal | Family H/O seizure (%) | SE (%) |
|---|---|---|---|---|---|---|
| WS (32) |
Perinatal asphyxia (16, 50.0%) CNS infections (7, 21.9%) Other causes a (9, 28.1%) |
100.0 | 100.0 | 78.1, 80.0 | 6.2 | 12.5 |
| GTCSA (26) | 3.8 | 0.0 | 73.0, 0.0 | 11.5 | 15.4 | |
| SLCECTS (15) | 0.0 | 0.0 | 60.0, 0.0 | 26.7 | 6.7 | |
| CAE (12) | 0.0 | 0.0 | 50.0, 0.0 | 16.7 | 8.3 | |
| LGS (12) | Perinatal asphyxia (7, 58.3%), meningoencephalitis (1, 8.3%), TS (1, 8.3%), neurodegenerative disease (1, 8.3%), unknown (2, 16.7%) | 100.0 | 100.0 | 75.0, 77.8 | 16.7 | 33.4 |
| SLFIE (5) | 0.0 | 0.0 | 40.0, 0.0 | 100.0 | 0.0 | |
| JME (4) | 0.0 | 0.0 | 50.0, 0.0 | 0.0 | 0.0 | |
| GEFS+ (2) | 50.0 | 50.0 | 50.0, 0.0 | 50.0 | 0.0 | |
| MTLE (2) | Hippocampal sclerosis (2, 100.0%) | 0.0 | 50.0 | 100.0/100.0 | 0.0 | 0.0 |
| CSWS (1) | 0.0 | 0.0 | 100.0/0.0 | 0.0 | 100.0 | |
| EMA (1) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| PME (1) | NCL2 (1, 100.0%) | 100.0 | 100.0 | 100.0/100.0 | 0.0 | 0.0 |
| Other DEEs (7) | Rett syndrome (2, 28.6%), Dravet syndrome (1, 14.3%), LKS (1, 14.3%), unknown (3, 42.9%) | 100.0 | 100.0 | 100.0/28.5 | 0.0 | 43.0 |
Abbreviations: CAE, childhood absence epilepsy; CNS, central nervous system; CSWS, epileptic encephalopathy with continuous spike‐and‐wave during sleep; DEE, developmental and epileptic encephalopathy; EMA, epilepsy with myoclonic absences; GEFS+, genetic epilepsy with febrile seizure plus; GTCSA, generalized tonic‐clonic seizures alone; H/O, history of; JME, juvenile myoclonic epilepsy; LGS, Lennox‐Gastaut syndrome; LKS, Landau‐Kleffner syndrome; MTLE, mesial temporal lobe epilepsy; N, number; NCL2, neuronal ceroid lipofuscinosis type 2; PME, progressive myoclonus epilepsy; SE, status epilepticus; SLCECTS, self‐limited childhood epilepsy with centrotemporal spikes; SLFIE, self‐limited familial infantile epilepsy; TS, tuberous sclerosis; WS, West syndrome.
Other causes: congenital hydrocephalus 1, tuberous sclerosis 1, neonatal stroke 1, and unknown 6 (18.7%).
All children of WS, LGS, PME, and other DEEs had abnormal neurological examination and developmental delay. CT scans of the brain were also abnormal in the majority of them (Table 3). CT scans of the brain were done in 85 (70.8%) children. The most common CT scan abnormality was diffuse cerebral atrophy with encephalomalacia (19 of 31 abnormal CT scans) suggestive of previous hypoxic‐ischemic brain injury. Details of CT scan findings of all epileptic children, but not limited to epilepsy syndromes, have been published elsewhere. 9 Of the children with ES and available neuroimaging, five children had both CT and MRI scans of the brain. All five children had normal CT scans and two had abnormal MRI scans. MRI abnormalities in those two children were features of mesial temporal lobe sclerosis suggesting the diagnosis of mesial temporal lobe epilepsy (MTLE). There was developmental delay in one child with MTLE but the other child was developmentally normal. Among children who had CT scans, all the children with GTCSA, SLCECTS, CAE, SLFIE, JME, GEFS+, and CSWS had normal CT scan findings. All the children with these ESs had normal neurological examination and normal development except for one child with GTCSA who had abnormal neurological examination (Table 3).
3.3. Treatment outcome
Follow‐up data required to define seizure control status were sufficient in 87 (72.5%) children with ESs. In the remaining 33 (27.5%) children, final seizure control status was not known because of various reasons, such as loss to follow‐up and inadequate duration of follow‐up to define seizure control status. Seizure control was achieved in 63 children (52.5% of total children with ESs and 72.4% of children with known outcome). Seizure was pharmaco‐resistant in 24 children (20.0% of total children and 27.6% of children with known outcome). Figure 2 shows final seizure control status.
FIGURE 2.
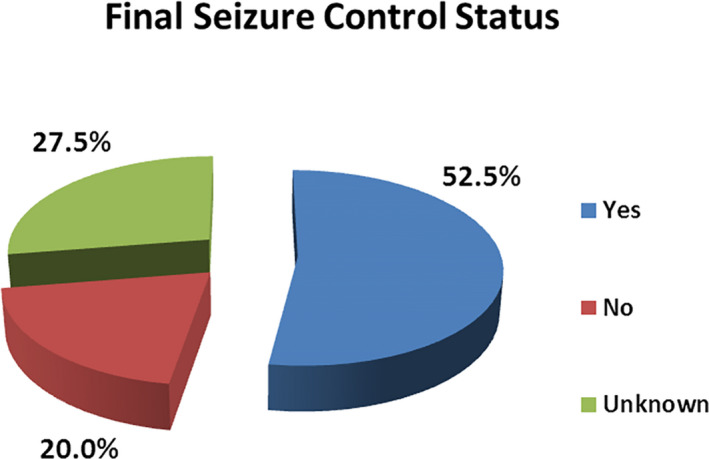
Final seizure control status in children with epilepsy syndromes
Table 4 shows treatment outcomes in various ESs. Seizure was controlled with single ASM in 50 children (57.5% of 87 children with known outcome and 79.4% of 63 children with pharmaco‐responsive seizures). There was complete cessation of seizures with polytherapy (2 or more ASMs) in additional 13 children (14.9% of children with known outcome and 20.6% of children with pharmaco‐responsive seizures). Children with SLFIE, GEFS+, EMA, CAE, SLCECTS, CSWS, JME, and GTCSA had an excellent treatment outcome, with no pharmaco‐resistant seizures. All of them responded to appropriately chosen single ASM except for one child with GTCSA, who required 2 ASMs for seizure control. There were 2 children with MTLE. One of them responded to monotherapy and the other child had pharmaco‐resistant seizures. Seizures were mostly pharmaco‐resistant in children with WS (52.2%), LGS (72.7%), PME (100.0%), and other DEEs (40.0%).
TABLE 4.
Response to antiepileptic drug treatment in various epilepsy syndromes
| Epilepsy syndrome | Total children | Seizure control status known |
Response to monotherapy N (% a ) |
Pharmaco‐resistant seizures N (% a ) |
|---|---|---|---|---|
| SLFIE | 5 | 2 | 2 (100.0) | 0 (0.0) |
| GEFS+ | 2 | 2 | 2 (100.0) | 0 (0.0) |
| EMA | 1 | 1 | 1 (100.0) | 0 (0.0) |
| CAE | 12 | 7 | 7 (100.0) | 0 (0.0) |
| SLCECTS | 15 | 12 | 12 (100.0) | 0 (0.0) |
| CSWS | 1 | 1 | 1 (100.0) | 0 (0.0) |
| JME | 4 | 2 | 2 (100.0) | 0 (0.0) |
| GTCSA | 26 | 18 | 17 (94.4) | 0 (0.0) |
| MTLE | 2 | 2 | 1 (50.0) | 1 (50.0) |
| Other DEEs | 7 | 5 | 2 (40.0) | 2 (40.0) |
| WS | 32 | 23 | 2 (8.7) | 12 (52.2) |
| LGS | 12 | 11 | 1 (9.1) | 8 (72.7) |
| PME | 1 | 1 | 0 (0.0) | 1 (100.0) |
| Total | 120 | 87 | 50 (57.5) | 24 (27.6) |
Abbreviations: CAE, childhood absence epilepsy; CSWS, epileptic encephalopathy with continuous spike‐and‐wave during sleep; DEE, developmental and epileptic encephalopathy; EMA, epilepsy with myoclonic absences; GEFS+, genetic epilepsy with febrile seizure plus; GTCSA, generalized tonic‐clonic seizures alone; JME, juvenile myoclonic epilepsy; LGS, Lennox‐Gastaut syndrome; MTLE, mesial temporal lobe epilepsy; N, number; PME, progressive myoclonus epilepsy; SLCECTS, self‐limited childhood epilepsy with centrotemporal spikes; SLFIE, self‐limited familial infantile epilepsy; WS, West syndrome.
% Of children with known outcome.
4. DISCUSSION
ESs have been widely studied over the years, classified by the ILAE, and reviewed in detail. 1 , 2 , 4 The identification of ESs continues to represent a vital approach to understanding the diagnosis, etiology, pathophysiology, prognosis, treatability, and comorbidities of large groups of children, even if their elucidation with ongoing discoveries will naturally lead to revisions of the syndromes over time. 2 , 3 , 10 , 11 The syndrome classification provides invaluable prognostic, therapeutic, and, in familial epilepsies, genetic information. 2 , 5 The classification of epilepsies and ESs has important practical implications when devising individual treatment plans and giving appropriate information to children and families. The likelihood of arriving at an ES diagnosis is very much more likely in children than in adults. 12
In this study, diagnosis of ES was established in 15.6% of the total children with unprovoked seizures. In a hospital‐based study conducted in India using the ILAE 2017 classification system, ES was diagnosed in 409 (56.1%) out of 726 children with epilepsy. 13 In a similar study done at a resource‐rich setup using the ILAE 1989 classification system, 21.0% children were classified to have specific ES. 12 In a population‐based study in Norway that used the ILAE 2017 classification system among 606 children with epilepsy, Aaberg et al found that 41.0% of children with epilepsy had a specific ES. 14 Therefore, proportion of childhood epilepsy that can be classified under definite ES depends upon the place of study, population and the system used to classify epilepsy. With limited access to MRI and use of single interictal EEG, identification of ESs in 15.6% epileptic children seems to be satisfactory in a resource‐limited setting where the present study was conducted. Therefore, in many children, diagnosis of ESs can be done even with limited access to diagnostic facilities. Male preponderance (69.2%) was seen in this study. Male preponderance (62.0%) has been also reported by other hospital‐based studies done in children with epilepsy in this region. 13 , 15 However, no such gender predilection has been reported in a population‐based study done in a developed country. 14 The reason for male preponderance is probably because male children are socially preferred and hence more likely to be referred to tertiary care centers in this region.
WS was the most common ES in this study. Other common ESs were GTCSA, SLCECTS, CAE, LGS, SLFIE, and JME. In a South Indian study, JME was the most common type of genetic generalized epilepsy (GGE), earlier termed as idiopathic generalized epilepsy (IGE), accounting for 4.9% of the total study population and 7.7% of children registered in the epilepsy clinic. 16 But that study included adult population and used the ILAE 1989 classification system. In a Spanish study among children, WS (34.1%) was reported to be the most common ES in infants. In early childhood, Doose syndrome (12.8%) was common. In school‐aged children, self‐limited epilepsies (27.3%) and absences (24.5%) were more prevalent. 17 In another Spanish study, two most common ESs were SLCECTS (29%) and CAE (9%). 18 In a hospital‐based study conducted by Sharma et al in India using the ILAE 2017 classification system, WS was the most common electro‐clinical syndrome, identified in 22.7% children with epilepsy and 40.3% children with ESs. Among children with ESs, SLCECTS (10.3%), GEFS+ (6.8%), JME (6.1%), LGS (5.6%), GTCSA (5.1%), CAE (4.4%) Dravet syndrome (2.0%), and EMA (0.7%) were the other common ESs. 13
In a population‐based study in Norway that used the ILAE 2017 classification system, SLCECTS was the most common (25.0%) ES reported among children with ESs. Other common ESs were WS (19%) CAE (16.5%), GEFS + (12.1%), LGS (4.8%), CSWS (2.8%), SLFIE (2.8%), and Dravet syndrome (2.0%). 14 JME was not reported probably because the study population was below 13 years of age. This demonstrates that the common ESs are common throughout the world, but their proportions in population vary slightly with respect to the place of study, availability of resources, study population, and the system used to classify epilepsy.
WS is the most common ES in infancy characterized by the triad of epileptic spasms, hypsarrhythmia on EEG, and neurodevelopmental arrest or regression. 19 WS is the manifestation of severe brain injury in early life and is a severe form of epilepsy of early infancy. Its diagnosis, evaluation, and management continue to pose many challenges. 20 As a developing country, many babies suffer from neonatal brain injury secondary to birth asphyxia, hypoxic‐ischemic encephalopathy (HIE), and CNS infections in our region. In the present study, 50.0% children with WS and 58.3% children with LGS had perinatal HIE as the underlying cause. Similarly, 21.9% children with WS and 8.3% children with LGS had CNS infection as the underlying cause. In a similar Indian study, WS was the most common ES. Perinatal HIE was the most common cause of epilepsy in children accounting for 265 (36.5%) of 726 epileptic children. Of these children, 129 (18.0%) had perinatal hypoxic‐ischemic brain injury. Sequela of CNS infection (26 neonatal infections and 37 postneonatal infections) was the cause of epilepsy in 63 (9%) of 726 children with epilepsy. 13 This reflects the scenario of higher prevalence of the conditions such as perinatal brain injury secondary to HIE and infectious etiologies in this region, causing long‐term morbidity like epilepsy. This might be the reason for WS being the most common ES in the present study and other similar studies in this region. 13 On the contrary, perinatal insults and infectious causes are less common in resource‐rich setup, as reported by a population‐based Norwegian study where etiologies were perinatal event in 13.3% and infectious in 2.0% of 606 children with epilepsy. 14
GGE is a relatively new category of disorders defined by strict clinical and EEG features. The general frequency of GGEs can be assessed at 15%–20% of all epilepsies. 21 Under the GGE category, GTCSA (21.7%), CAE (10.0%), and JME (3.3%) were the common ESs diagnosed in the present study. SLCECTS also known as Rolandic Epilepsy, early‐onset childhood occipital epilepsy (Panayiotopoulos syndrome), and late‐onset childhood occipital epilepsy (Gastaut type) are the principal pediatric focal ESs. 22 They share major common characteristics like age related onset and resolution, clinical course and EEG features. 22 SLCECTS was the third most common ES diagnosed in this study (12.5% of children with ESs). This was the second most common (10.3%) ES in a hospital‐based Indian study and was the most common (25.0%) ES in a population‐based Norwegian study. 13 , 14 This indicates that SLCECTS is a common ES among children all over the world.
DEE embodies the notion that the epileptic activity itself may contribute to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone. 1 , 2 Historically, prototypical DEEs have included Ohtahara syndrome, WS, LGS, severe myoclonic epilepsy of infancy (Dravet syndrome), and LKS. Now, with the explosion of identified genetic, structural, and autoimmune causes of severe early‐life epilepsy and intellectual disability, the list of presumed DEEs has expanded greatly. 23 According to recent position paper of the ILAE on classification of seizures and epilepsy, the concept of the DEE may be applicable to epilepsies at all ages and should be utilized more widely to include encephalopathies that have a genetic etiology, such as WS and epileptic encephalopathy with CSWS. Equally, such syndromes may have an acquired cause such as hypoxic‐ischemic encephalopathy or stroke, or may be associated with a malformation of cortical development that may also have a genetic or acquired etiology. 2 The epilepsies under DEEs are large number of heterogeneous epilepsies with variable clinical features and underlying etiologies. However, these epilepsies also share several important characteristics. 1 , 2 WS (26.7%) and LGS (10.0%) were the two most common DEEs detected in children in the present study. Perinatal asphyxia and CNS infection in early infancy were the common etiologies for underlying encephalopathies in WS and LGS. Epileptic encephalopathy with CSWS, Rett syndrome, Dravet syndrome, and LKS were other relatively less common DEEs diagnosed in this study. These DEEs have been reported as less common ESs also by other studies. 13 , 14 In three children with DEEs, underlying etiologies could not be established due to limited diagnostic resources.
For the classification purpose, the ILAE has organized ESs into different age groups according to their typical age of onset. 1 , 2 The distribution of seizures and epilepsies is strongly dependent on age of onset. 14 The age specificity of ES was also seen in our study (Figure 1). WS and SLFIE were infantile onset ESs, whereas LGS and various other DEEs predominated in the second year of life. CAE, GTCSA, and SLCECTS had an age of onset mainly during mid‐childhood. JME had an age of onset during adolescence. A child with epileptic encephalopathy with CSWS, unlike other DEEs, had an age of onset during adolescence (11 years of age).
MRI scan of the brain is the imaging modality of choice in epilepsy. CT scan is the alternative modality recommended where MRI is not available or less practical. 7 In this study; CT scan was performed in 85 (70.8%) children. In the rest of the children, CT scan was not performed either because of the reasons such as no clinical indication, loss to follow‐up, or financial constraints. Imaging is most often abnormal in children with focal or remote symptomatic epilepsy, whereas it is usually normal in the typical GGEs. 7 In this study, CT scan was abnormal in 31 children accounting for 36.5% of 85 children in whom CT scans were performed and 25.8% of 120 children with ESs. The details of CT scan findings of all these children with epilepsy, not limited to ES only, have been published elsewhere. 9 Among the children who performed CT scans in this study, the scans were abnormal in the majority of children with WS (80%) and LGS (77.8%); whereas the scans were normal in all the children with GTCSA, CAE, JME, GEFS+, SLFIE, and SLCECTS. Therefore, when electro‐clinical diagnosis of certain ES is made, for example, GTCSA, CAE, or SLCECTS, neuroimaging can be avoided, because neuroimaging is likely to be normal in these ESs. This is why diagnosis of ES is more significant in resource‐limited settings where neuroimaging, especially MRI is not widely available.
Most of the epilepsies will respond to the first or second ASM as monotherapy. 24 Response is even higher for childhood epilepsies. 25 Response to therapy also depends upon the type of epilepsy. 24 , 25 Out of 120 children with ESs, follow‐up data were sufficient to define seizure control status in 87 (72.5%) children. Among them, 50 children responded to monotherapy that accounted for 57.5% of 87 children with known outcomes and 79.4% of 63 children with pharmaco‐responsive seizures. Proportion of pharmaco‐resistant seizure was 24/87 (27.5%) among the children with known outcome. ESs under the category of GGEs such as GTCSA, CAE, JAE, JME, and GEFS + showed good response to ASMs. Most of them responded well to monotherapy. Under the category of focal epilepsies, SLCECTS showed good response to ASM. Most of the DEEs including WS and LGS were largely resistant to ASMs. Of the two children with MTLE, seizure control was achieved in a child. A child of PME secondary to neuronal ceroid lipofuscinosis type 2 had pharmaco‐resistant epilepsy.
4.1. Limitations
The main strengths of the present study were screening large numbers of children for ESs, use of the ILAE 2017 classification system, use of EEG in all the children with unprovoked seizure, and follow‐up to ascertain the treatment outcome. This study had some limitations too. As this was a hospital‐based study, the spectrum of ESs reported may not be a true reflection of the population characteristics. As the study was conducted in a tertiary care center, there may be a relatively greater proportion of pharmaco‐resistant and difficult to treat epilepsies. Home videos were available to aid the diagnosis for a limited number of children, and we did not record the exact number of videos available. The study was conducted in a resource‐limited setting. Although MRI of the brain is the investigation of choice in epilepsy, we were not able to perform MRI in most of the children. We were not able to obtain the metabolic profiles in many of the children. We might have missed some children with metabolic diseases presenting as ES. Repeated interictal EEGs and prolonged electrographic epilepsy monitoring were not done due to resource constraints. With a full investigation profile, more children of ESs could have been identified. Diagnosis of ES can be established in many children even with limited access to diagnostic facilities; however, in order to confirm that diagnosis, data on long‐term outcome would have been helpful to include. Although we had data on short‐term treatment outcome, we did not collect the data on long‐term treatment and cognitive outcomes as a part of this study.
4.2. Conclusion and clinical relevance
With the proper use of available basic resources such as EEG, CT scan, and clinical data, a reasonable proportion of childhood seizures can be classified into certain ESs even in a resource‐limited setting where advanced diagnostic facilities are limited. WS, GTCSA, SLCECTS, CAE, and LGS are the most commonly diagnosed childhood ESs. Frequent occurrence of WS reflects higher occurrence of perinatal brain insults such as birth asphyxia and neonatal sepsis in a developing country. There is an age‐specific pattern of onset of seizure in ESs. Syndromes such as GTCSA, CAE, SLCECTS, and JME are largely responsive to pharmacotherapy and most of them respond to monotherapy. WS, LGS, PME, and MTLE are largely pharmaco‐resistant. Neuroimaging is normal in most of the children with ESs under GGE category and self‐limited focal ESs such as SLCECTS. Electro‐clinical diagnosis of these syndromes might be helpful in reducing the need for neuroimaging, optimizing the treatment, prognosticating response to treatment, and advising long‐term prognosis of childhood epilepsies in resource‐constrained settings.
CONFLICT OF INTEREST
The authors declare that they have no competing interests. This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
ETHICAL APPROVAL
We (all authors) confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
The authors would like to acknowledge the children and parents for giving consent to participate in the research, EEG laboratory staff, radiologists, staff in the outpatient clinics of BPKIHS, and all the healthcare workers who were actively involved in the clinical management of these children. The authors also like to acknowledge Dr SM George for critically appraising and reviewing the manuscript for English‐language proofreading.
Poudel P, Kafle SP, Pokharel R. Clinical profile and treatment outcome of epilepsy syndromes in children: A hospital‐based study in Eastern Nepal. Epilepsia Open. 2021;6:206–215. 10.1002/epi4.12470
DATA AVAILABILITY STATEMENT
The authors state that the anonymized data on which the article is based will be shared by request of any qualified investigator.
REFERENCES
- 1. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde BW, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51(4):676–85. [DOI] [PubMed] [Google Scholar]
- 2. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989;30(4):389–99. [DOI] [PubMed] [Google Scholar]
- 4. Fisher RS, Cross JH, D’Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58(4):531–42. [DOI] [PubMed] [Google Scholar]
- 5. Duchowny M, Harvey AS. Pediatric epilepsy syndromes: an update and critical review. Epilepsia. 1996;37(Suppl 1):S26–40. [DOI] [PubMed] [Google Scholar]
- 6. Central Berau of Statistics (CBS) . National Population and Housing Census 2011(National Report). Government of Nepal, National Planning Commission Secretariat, Kathmandu, Nepal. 2012. https://unstats.un.org/unsd/demographic/sources/census/wphc/Nepal/Nepal‐Census‐2011‐Vol1.pdf. Accessed December 28, 2020.
- 7. Gaillard WD, Chiron C, Helen Cross J, Simon Harvey A, Kuzniecky R, Hertz‐Pannier L, et al. Guidelines for imaging infants and children with recent‐onset epilepsy. Epilepsia. 2009;50(9):2147–53. [DOI] [PubMed] [Google Scholar]
- 8. Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kälviäinen R, et al. ILAE Subcommission on AED Guidelines. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54(3):551–63. [DOI] [PubMed] [Google Scholar]
- 9. Poudel P, Gupta MK, Kafle SP. Computerized axial tomography findings in children with afebrile seizures: a hospital based study at Eastern Nepal. J Nepal Health Res Counc. 2017;15(35):61–6. [DOI] [PubMed] [Google Scholar]
- 10. Dulac O. Epileptic syndromes in infancy and childhood: recent advances. Epilepsia. 1995;36(Suppl 1):S51–S57. [DOI] [PubMed] [Google Scholar]
- 11. Pearl PL. Epilepsy syndromes in childhood. Continuum (Minneap Minn). 2018;24(1, Child Neurology):186‐209. [DOI] [PubMed] [Google Scholar]
- 12. Kellinghaus C, Loddenkemper T, Najm IM, Wyllie E, Lineweaver T, Nair DR, et al. Specific epileptic syndromes are rare even in tertiary epilepsy centers: a patient‐oriented approach to epilepsy classification. Epilepsia. 2004;45(3):268–75. [DOI] [PubMed] [Google Scholar]
- 13. Sharma S, Anand A, Garg D, Batra S, Mukherjee SB, Patra B, et al. Use of the international league against epilepsy (ILAE) 1989, 2010, and 2017 classification of epilepsy in children in a low‐resource setting: A hospital‐based cross‐sectional study. Epilepsia Open. 2020;5(3):397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aaberg KM, Surén P, Søraas CL, Bakken IJ, Lossius MI, Stoltenberg C, et al. Seizures, syndromes, and etiologies in childhood epilepsy: The International League Against Epilepsy 1981, 1989, and 2017 classifications used in a population‐based cohort. Epilepsia. 2017;58(11):1880–91. [DOI] [PubMed] [Google Scholar]
- 15. Chaudhary N, Gupta MM, Shrestha S, Pathak S, Kurmi OP, Bhatia BD, et al. Clinicodemographic profile of children with seizures in a tertiary care hospital: a cross‐sectional observational study. Neurol Res Int. 2017;2017:1524548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murthy JM, Yangala R, Srinivas M. The syndromic classification of the International League Against Epilepsy: a hospital‐based study from South India. Epilepsia. 1998;39(1):48–54. [DOI] [PubMed] [Google Scholar]
- 17. Durá‐Travé T, Yoldi‐Petri ME, Gallinas‐Victoriano F. Epilepsy in children in Navarre, Spain: epileptic seizure types and epileptic syndromes. J Child Neurol. 2007;22(7):823–8. [DOI] [PubMed] [Google Scholar]
- 18. Onsurbe Ramírez I, Hernández Rodríguez M, Aparicio Meix JM, Carrascosa RC. Incidencia de las epilepsias y síndromes epilépticos de la infancia en la provincia de Albacete [Incidence of epilepsy and epileptic syndromes in children in the province of Albacete]. An Esp Pediatr. 1999;51(2):154–8. [PubMed] [Google Scholar]
- 19. Brna PM, Gordon KE, Dooley JM, Wood EP. The epidemiology of infantile spasms. Can J Neurol Sci. 2001;28(4):309–12. [DOI] [PubMed] [Google Scholar]
- 20. Wheless JW, Gibson PA, Rosbeck KL, Hardin M, O'Dell C, Whittemore V, et al. Infantile spasms (West syndrome): update and resources for pediatricians and providers to share with parents. BMC Pediatr. 2012;12:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jallon P, Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46(Suppl 9):10–4. [DOI] [PubMed] [Google Scholar]
- 22. Sánchez Fernández I, Loddenkemper T. Pediatric focal epilepsy syndromes. J Clin Neurophysiol. 2012;29(5):425–40. [DOI] [PubMed] [Google Scholar]
- 23. Stafstrom CE, Kossoff EM. Epileptic encephalopathy in infants and children. Epilepsy Curr. 2016;16(4):273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwan P, Brodie MJ. Effectiveness of first antiepileptic drug. Epilepsia. 2001;42(10):1255–60. [DOI] [PubMed] [Google Scholar]
- 25. Rosati A, De Masi S, Guerrini R. Antiepileptic drug treatment in children with epilepsy. CNS Drugs. 2015;29(10):847–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors state that the anonymized data on which the article is based will be shared by request of any qualified investigator.