

Abstract
Objective
Our goal was to perform detailed clinical and genomic analysis of a large multigenerational Chinese family with 21 individuals showing symptoms of Familial Cortical Myoclonic Tremor with Epilepsy (FCMTE) that we have followed for over 20 years.
Methods
Patients were subjected to clinical evaluation, routine EEG, and structural magnetic resonance imaging. Whole exome sequencing, repeat‐primed PCR, long‐range PCR, and PacBio sequencing were performed to characterize the disease‐causing mutation in this family.
Results
All evaluated patients manifested adult‐onset seizures and presented with progressive myoclonic postural tremors starting after the third or fourth decade of life. Seizures typically diminished markedly in frequency with implementation of antiseizure medications but did not completely cease. The electroencephalogram of affected individuals showed generalized or multifocal spikes and slow wave complexes. An expansion of TTTTA motifs with addition of TTTCA motifs in intron 4 of SAMD12 was identified to segregate with the disease phenotype in this family. Furthermore, we found that the mutant allele is unstable and can undergo both contraction and expansion by changes in the number of repeat motifs each time it is passed to the next generation. The size of mutant allele varied from 5 to 5.5 kb with 549‐603 copies of TTTTA and 287‐343 copies of TTTCA repeat motifs in this family.
Significance
Our study provides a detailed description of clinical progression of FCMTE symptoms and its management with antiseizure medications. Our method of repeat analysis by PacBio sequencing of long‐range PCR products does not require high‐quality DNA and hence can be easily applied to other families to elucidate any correlation between the repeat size and phenotypic variables, such as, age of onset, and severity of symptoms.
Keywords: epilepsy, FCMTE, myoclonus, SAMD12
Key Points.
Detailed clinical evaluation of FCMTE patients from a large multigenerational family demonstrated phenotypic adult‐onset seizures (partially responsive to antiseizure medications) and progressive myoclonic postural tremors.
Although most patients presented with generalized‐onset tonic‐clonic seizures, five patients from the same family presented with focal to bilateral tonic‐clonic seizures.
The disease occurs due to an expansion of intronic repeats in SAMD12 from < 1kb (in normal individuals) to 4.2‐4.7 kb in the affected individuals.
The expanded repeat region shows intergenerational instability.
1. INTRODUCTION
Familial Cortical Myoclonic Tremor with Epilepsy (FCMTE) is an autosomal dominant disorder characterized by progressive adult‐onset cortical myoclonic tremor and seizures that are partially responsive to antiseizure medication. Due to the heterogeneity of clinical symptoms, the disorder has been described under multiple names, for example, benign adult familial myoclonic epilepsy (BAFME), familial adult myoclonic epilepsy (FAME), and familial cortical tremor with epilepsy (FCTE). 1 , 2 , 3 , 4 Van Rootselaar and colleagues, 5 who first coined the term FCMTE, indicated that all these terms refer to a single disease entity with phenotypic and genetic heterogeneity. Earlier attempts at identifying the genetic basis of FCMTE mapped the candidate genes to multiple regions of the human genome, thus identifying at least five FCMTE loci. 1 , 4 , 6 Despite the identification of several candidate genes, such as, UBR5, NOL3, ACMSD, ADRA2B, CNTN2, CTNND2, PLA2G6, and SLC30A8, the underlying causative gene remained unknown for the majority of reported families. 1 , 4 , 6 , 7 , 8 Recently, Ishiura and colleagues 9 identified abnormal expansions of noncoding pentanucleotide repeat motifs (TTTCA and TTTTA) in three genes, SAMD12 (chromosome 8), TNRC6A (chromosome 16), and RAPGEF2 (chromosome 4), as the underlying cause of FCMTE in Japanese families. Of these, the repeat expansion in intron four of SAMD12 was the most frequent and was observed in 48 out of 51 FCMTE families. 9 These findings were subsequently validated in additional Japanese and Chinese families with FCMTE. 10 , 11 , 12 , 13 Here, we present a large multigenerational Chinese family with more than twenty individuals with FCMTE symptoms from five generations that we have been studying for 23 years. Using a combination of the repeat‐primed PCR, long‐range PCR, and PacBio sequencing, we identified a 4.2‐4.7 kb repeat with 549‐603 copies of TTTTA and 287‐343 copies of TTTCA repeat motifs in intron four of SAMD12 in affected individuals.
2. METHODS
2.1. Family collection and clinical evaluation
The proband, a 58‐year‐old woman (324 in Figure 1A) from suburban area of Beijing, China, was referred for intractable seizures. A detailed pedigree was constructed based on the clinical histories of all family members (Figure 2). At the initial evaluation, there were 21 affected individuals among over 130 family members spanning six generations. Detailed clinical evaluations were conducted on 12 patients by at least two of the authors (Table 1). Parts of the family including individuals who were young and asymptomatic at initial evaluation were recently examined again. Routine EEG and structural magnetic resonance imaging were performed for some of the family members. The electroclinical characterization of this family has been described previously. 14 Blood samples were obtained from 13 affected and 12 unaffected individuals in the family. High‐molecular‐weight genomic DNA was extracted from peripheral leukocytes by modified standard procedures. 15
FIGURE 1.
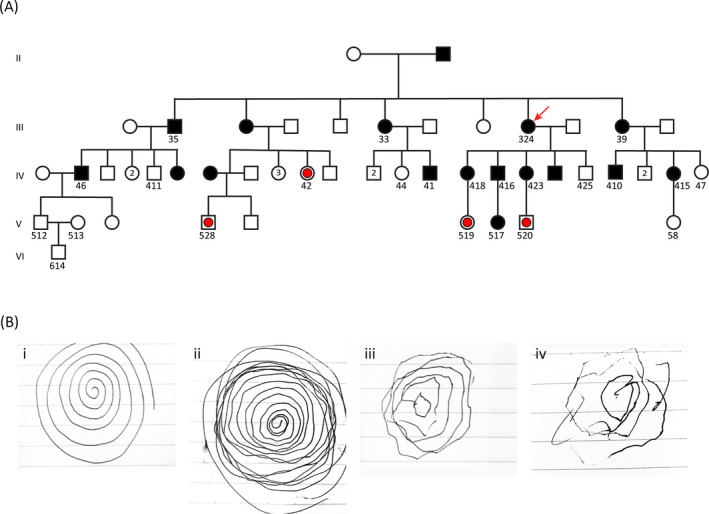
FCMTE pedigree and spiral drawing to show the range of tremor severity. A, Partial pedigree of the family showing the part we studied from generations II to VI. The proband (324) is marked by a red arrow. All individuals whose DNA was tested are identified by a number written underneath the male or female symbol. The filled symbols indicate clinically affected individuals; open symbols indicate clinically normal individuals, and open symbols with red dots indicate individuals who were clinically normal at the time of evaluation but carried the mutant allele as detected by repeat‐primed PCR data. B, Tremor as shown through the spiral drawing task. The examiner first drew a spiral to illustrate the task (i). The remaining drawings demonstrate the range of increasing severity of the tremor in three family members from mild (ii, 416 in panel A), moderate (iii, 418 in panel A), and to severe (iv, 324 in panel A)
FIGURE 2.
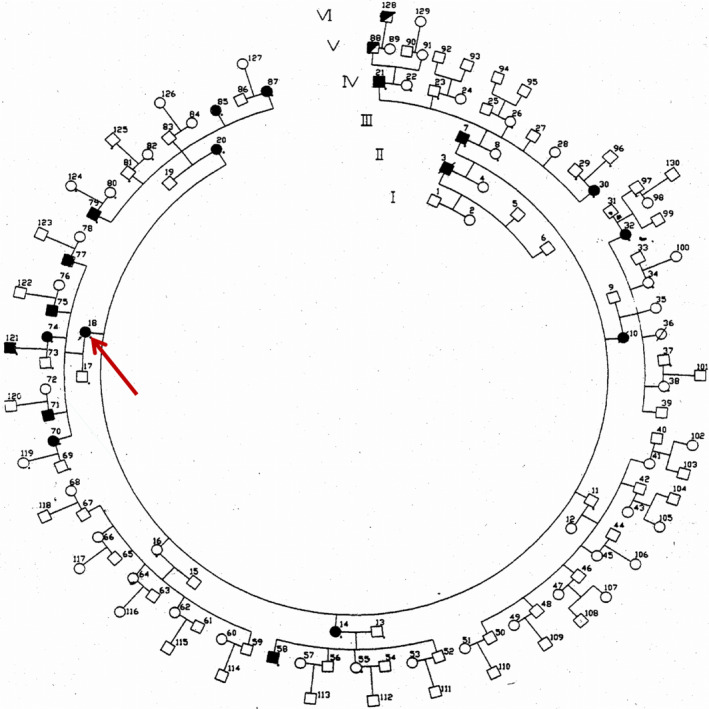
Complete pedigree of the six‐generation family at the time of clinical evaluation and sample collection. Proband is marked by the red arrow. Half‐filled symbols (patients 88 and 128) represented EEG epileptiform discharges without clinical seizure
TABLE 1.
Clinical findings in patients at the time of first evaluation in 1996
| Patient id | In Figure 2 | 18 | 70 | 71 | 74 | 75 | 20 | 79 | 14 | 58 | 32 | 7 | 21 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In Figure 1A | 324 | 418 | 416 | 423 | NA | 39 | 410 | 33 | 41 | NA | 35 | 46 | |
| Gender | F | F | M | F | M | F | M | F | M | F | M | M | |
| Age at evaluation | 58 | 40 | 37 | 34 | 32 | 55 | 36 | 64 | 28 | 52 | 74 | 56 | |
| Age of seizure onset | 28 | 25 | 25 | 25 | 26 | 28 | 26 | 26 | 25 | 28 | 35 | 30 | |
| Seizures | + | + | + | + | + | + | + | + | + | + | + | + | |
| Myoclonus | + | + | + | + | + | + | + | + | + | + | + | + | |
| Dementia | – | – | – | – | – | – | – | – | – | – | – | – | |
| Dysdiadochokinesia | + | – | + | + | + | + | + | ||||||
| Finger‐nose test | + | + | + | + | + | + | |||||||
| Heel‐shin coordination | + | + | + | + | + | + | |||||||
| Rombergism | ++ | + | ++ | + | ++ | + | |||||||
| Gait ataxia | + | + | + | + | + | + | |||||||
| Plantar reflexes flexor or Babinski reflex | + | + | + | + | + | + | |||||||
| EEG | SW | SW | SW | SW | SW | NA | NA | NL | NA | NL | NL | NL |
Abbreviations: NA, not available; NL, normal; SW, spikes and slow wave.
2.2. Ethics and patient consents
The study was fully approved by the Ethics Committee of the China‐Japan Friendship Hospital, Ministry of Health, China, 14 and written informed consent was obtained from all participants.
2.3. Whole exome sequencing (WES)
WES was performed on DNA from nine affected (Figure 1A, No. 33, 35, 39, 324, 41, 46, 410, 415, 423) and three unaffected individuals (Figure 1A, No. 411, 44, 425) using the xGen Exome Research Panel (IDT). Each sample was run on two lanes of HiSeq 4000 machine (Illumina). WES data were analyzed using Varsifter. 16 All variants unique to the family were identified and filtered by their effect on the protein function. The functional variants (stop‐gain/loss, splicing, nonsynonymous, frameshift, nonframeshift insertions/deletions) were then excluded as candidate variants by their presence in the 1000 Genome and ExAC databases.
2.4. Repeat‐primed fluorescent PCR
Repeat‐primed fluorescent PCR (RP‐PCR) was completed using the recently described primer sets, P1/P2 for TTTTA and P3/P4 for TTTCA repeats in intron four of SAMD12 9 and PCR conditions described previously with extension time of 1 min instead of 30 s. 17 Briefly, for each DNA sample, fluorescent PCR was performed using FAM‐M13R primer with the P1/P2 and P3/P4 primer sets. PCR products were separated by capillary electrophoresis on genetic analyzer ABI3130xL or 3730xL and analyzed by GeneMapper 5.
2.5. Long‐range PCR and PacBio sequencing
Long‐range PCR was performed using 200 ng of genomic DNA, PrimeSTAR GXL Premix (Takara Bio), and SAMD12LF/SAMD12LR primers and conditions as described recently. 10 The PCR products were analyzed by electrophoresis on 1% agarose gel and MassRuler DNA ladder mix (Thermo Fisher Scientific). For single‐molecule real‐time (SMRT) sequencing using PacBio technology, long‐range PCR was carried out in ten reactions/sample and pooled to generate 500 uL of PCR product. For each sample, two size‐selected libraries were created (~1 kb for the WT allele and ~5 kb for the mutant allele) and pooled by size. The pools were run on a single PacBio Sequel SMRTcell and demultiplexed to separate the reads for each size. For each sample, consensus sequences for wild‐type (WT) and mutant alleles were generated using the long amplicon analysis.
3. RESULTS
3.1. Clinical description of the proband and pedigree
At the time of initial evaluation, there were 21 clinically affected individuals in six generations in this extended family of over 130 members, with an inheritance pattern suggesting an autosomal dominant disease (Figure 1A and Figure 2). We performed detailed clinical evaluations on 12 affected individuals. Seizures in these 12 patients began in adulthood with onset between 25 and 35 years of age. Seizure semiology was typically generalized‐onset tonic‐clonic seizures, but three patients (Figure 1A, No. 324, 416, 418) presented with focal to bilateral tonic‐clonic motor seizures (3/12, 25%). Two individuals, who were asymptomatic at the time of initial evaluation due to their young age (Figure 1A, No. 42, 520), also manifested likely focal to bilateral tonic‐clonic motor seizures upon subsequent evaluations (5/14, 36%). All patients also presented with myoclonic postural tremors starting after the third or fourth decade of life (Figure 1B, Table 1). In comparison with progressive myoclonic epilepsies, seizures were partially responsive to medication and their frequency did not increase with time. Late‐onset ataxia was also present, but dementia was not observed in any patient (Table 1). The electroencephalogram of affected individuals showed generalized spikes or multifocal spike‐and‐slow wave complexes as described previously. 14 None of the evaluated patients had any significant CT or MRI abnormality. Valproic acid reduced seizure frequency in all patients, but did not ameliorate the tremor, which continued to worsen with age. Tremor also progressed over time even in a few patients who had discontinued valproic acid after long periods of seizure freedom. Together, this suggests that tremor progression was unrelated to the use of valproic acid. Detailed description of the clinical symptoms of the proband and her affected children is given below.
3.2. Proband (324 in Figure 1A)
Seizures began in this patient at age 28. On antiseizure medications, including carbamazepine and later valproic acid, seizures diminished from two per month to two or less per year. Semiologically, seizures began with palpitations, sweating, and blurry vision. This was followed by alteration of awareness, head deviation to the right, and often bilateral tonic‐clonic seizure with loss of bladder control and tongue biting. Postictal confusion and speech difficulty typically lasted 1‐2 hours. Her myoclonic jerks started at age 30, two years after seizure onset. Myoclonic jerks first appeared in the hands and arms and later involved the trunk and lower extremities. They were most pronounced in the extremities and were arrhythmical, asymmetrical, and exacerbated by emotions and voluntary movements. At times, myoclonus was severe enough to result in falls; on one occasion, she suffered nasal and facial fractures. An intention tremor then ensued and progressed in severity. Myoclonus and tremors were unresponsive to antiseizure medications and worsened with time showing transient improvement after alcohol intake. Examination at age 80 showed a 3‐7 Hz tremor that was more prominent in the arms than the legs. At the forearms, there were prominent supination/pronation movements and at the shoulders internal/external rotation. Tremor worsened with action and became particularly vigorous when she extended her arms. Tremor was worse when she drew a spiral using her right hand (Figure 1B‐iv). Tremor amplitude in the legs increased when she walked but was also observed when sitting.
3.3. Proband's son (416 in Figure 1A)
The proband's son reported a history of a head trauma and intracerebral hemorrhage at age 18. His seizures started at age 25 and tremors at age 35. Given the significant delay in seizure onset from the head trauma and with an age of onset and seizure semiology similar to other family members, the trauma was felt to be an unlikely etiology of his symptoms. Carbamazepine monotherapy followed by later introduction of Valproic acid resulted in diminished seizure frequency from 3‐4 seizures per month to 2‐3 seizures or less per year. His seizures were of the impaired awareness type at onset, usually with a preceding aura consisting of palpitations, sweating, and “feeling uncomfortable.” The auras were followed by alteration of awareness and head deviation to the right, lasting for a few minutes before postictal confusion ensued. Like his mother, tremors were lessened by alcohol intake. Examination at age 58 revealed subtle tremor in comparison with his mother (Figure 1B‐ii). Occasionally, the tremors manifested as internal/external rotation of the shoulders as he handled objects with his hands.
3.4. Proband's daughter (418 in Figure 1A)
The proband's daughter developed impaired awareness seizures and myoclonus at age 25. Her seizures initially occurred a couple of times per month and had a catamenial pattern. Her most common auras consisted of fear and a rising sensation in the chest. She also had an aura of visual hallucination on one occasion when she reported seeing her husband, who was not physically present, prior to a seizure. Rarely, there was jamais vu. Motor semiology, which began with right head deviation, was followed by a bilateral tonic‐clonic seizure. Examination at age 60 revealed left facial weakness, which reportedly occurred after a single lifetime febrile seizure at age 2 or 3. Of note, no other history of febrile seizure was reported in the family. She also reported herself to be seizure‐free for 3 years. Her tremor was mild, though obvious when she drew a spiral (Figure 1B‐iii).
Regarding the four individuals who were asymptomatic at the time of initial clinical evaluation due to their young age but found to carry the mutation by genomic analysis as described below (Figure 1 A, denoted with red dots), two of them were found to be affected during subsequent clinical evaluations. Individual #42 had developed an aura of fear, a rising sensation in the chest, and sometimes jamais vu; individual #520 had developed focal to bilateral tonic‐clonic motor seizures and tremor, while individuals No. #519 and 528 were still clinically normal.
3.5. Identification of expanded pentanucleotide repeat in intron 4 of SAMD12 as the causative mutation in this family
We selected 9 affected and 3 unaffected individuals from the 3rd and 4th generations of the pedigree for WES to identify the underlying mutation in this family. Analysis of the WES data revealed segregation of the disease phenotype with several variants from 8q22 to 8q24 region, indicating the causative gene may lie in this region (data not shown). At the same time, Ishiura and colleagues reported that expansion of TTTTA repeats to TTTTA and TTTCA repeats in intron 4 of SAMD12, which lies in the 8q24 region, causes FCMTE in multiple Japanese families. 9 This led us to examine our family for the presence of these intronic repeat expansions using repeat‐primed fluorescent PCR (Figure 3A). We tested DNA from 24 individuals (12 clinically affected and 12 unaffected at the time of initial evaluation) and detected the presence of expanded TTTTA and TTTCA motifs in intron 4 of SAMD12 in 16 out of 24 individuals. Eight individuals who were negative for the presence of expanded repeat were all clinically unaffected. However, of the 16 individuals who tested positive for the expanded repeat, only 12 individuals were known to be clinically affected at the time of DNA collection. The other four individuals may have been asymptomatic due to their young age at the time of evaluation (Figure 1A, denoted with red dots). As mentioned above, two of them presented with clinical symptoms at subsequent evaluations. These data led us to conclude that the expanded SAMD12 intronic repeat segregates with the disease phenotype and is the causative mutation for FCMTE in this family.
FIGURE 3.
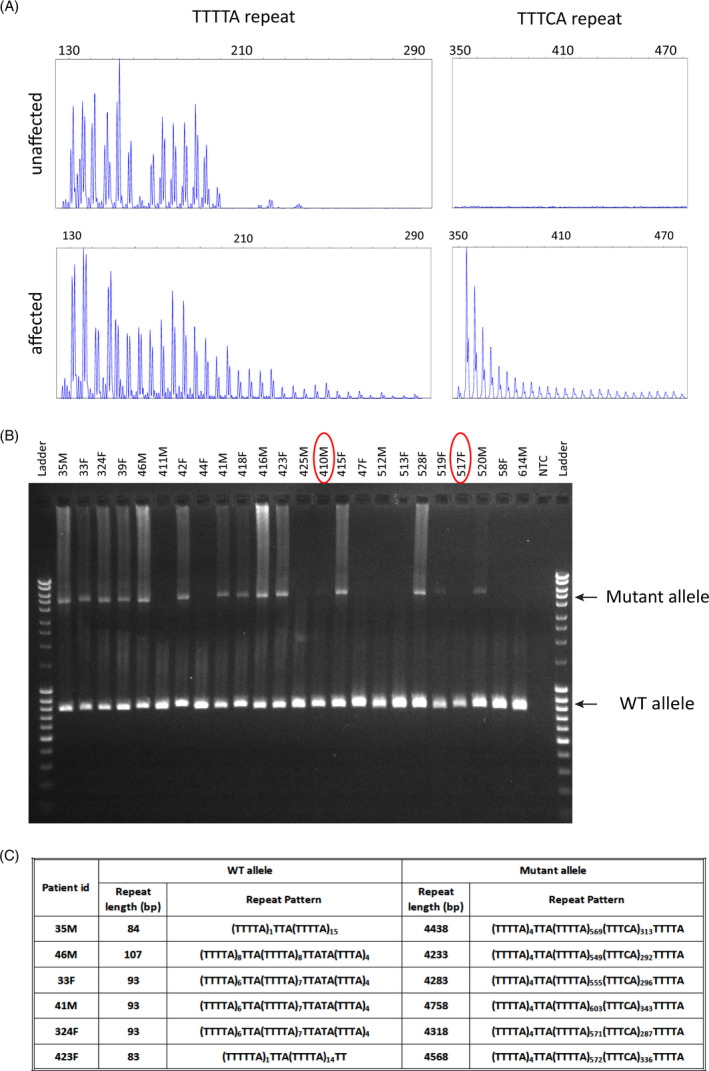
Characterization of the mutant allele by repeat‐primed and long‐range PCR and PacBio sequencing. A, Representative plots of repeat‐primed PCR for TTTTA and TTTCA repeats from an unaffected (411 in Figure 1A) and affected individual (46 in Figure 1A). B, Gel image showing the amplification of mutant and WT alleles by long‐range PCR from 24 family members whose DNA was available. Identity of each sample (corresponding to Figure 1A) is marked on the top. Red circles mark two samples where mutant allele is detected by RP‐PCR but not robustly by long‐range PCR due to the low‐quality DNA. The ladder used is Mass Ruler DNA ladder mix where the top band is 10 kb. C, Size and pattern of the SAMD12 intronic pentanucleotide repeat motifs in each patient demonstrating its unstable nature
3.6. Number of TTTTA and TTTCA motifs and the size of mutant allele changes each time it is transmitted
The size of mutant allele varies between tissues in the same individual, between generations in the same family and between families due to the variation in number of TTTTA and TTTCA motifs. 9 , 10 Therefore, we performed long‐range PCR to determine the size range of the mutant allele in this family. Our data showed that the mutant allele ranged in size between 5 and 5.5 kb, whereas the WT allele amplified as ~800 bp, thus suggesting the length of repeat expansion to be between 4.2 and 4.7 kb in this family (Figure 3B). To determine the exact nature of TTTTA and TTTCA motifs in the mutant allele and variation among patients from the same family, we performed targeted SMRT sequencing using PacBio technology with the long‐range PCR products from six patients. The sequenced patients included three parent‐child pairs where the three parents were also siblings, thus inheriting the mutation from the same parent (Figures 1A, 3C). Analysis of the sequence data revealed that the repeated region in the wild‐type (WT) allele varied from 83 to 107 bp with the following two patterns (TTTTA)1TTA(TTTTA)14‐15 or (TTTTA)6‐8TTA(TTTTA)7‐8TTATA(TTTA)4 (Figure 3C). The mutant allele carried between 4233 and 4758 bp of the repeated motifs with 549‐603 copies of the TTTTA motif and 287‐343 copies of the TTTCA motif (Figure 3C). Interestingly, the repeat length is closer in size for the three siblings from generation III: 35M (4438 bp), 33F (4283 bp), and 324F (4318 bp) who inherited the mutation from the same parent (Figure 3C). The repeat length was shorter in 46M compared with his father (35M), and larger in 41M and 423F compared with their mothers (33F transmitted to 41M, 324F transmitted to 423F), suggesting that the repeat can undergo contraction or expansion as it passes to the next generation (Figure 3C). Thus, our detailed analysis of the repeat structure of the mutant allele in this family revealed the instability of the expanded repeat. Overall, our study describes the use of targeted PacBio sequencing of amplified repeat region to determine the exact nature of repeat expansion and serves as a validation of the noncoding expanded repeat in intron 4 of SAMD12 as a common cause of FCMTE in Chinese families.
4. DISCUSSION
FCMTE is a unique disorder due to its clinical presentations of adult‐onset myoclonic tremor and seizures. Here, we present a large multigenerational FCMTE family from China that we first evaluated over 20 years ago and have followed up since then. Previous reports have implicated that the seizures in FCMTE patients are usually generalized tonic‐clonic seizures, although some can be of focal onset. 18 , 19 , 20 Interestingly, five of our patients presented with impaired awareness seizures, evolving into bilateral tonic‐clonic seizures. This raises the question of whether seizures in FCMTE could arise from the temporal lobe, as could be reasonably concluded from the detailed semiologic descriptions of probands 324, 416, and 418. However, EEG data did not suggest temporal lobe localization. One possibility is that simply due to the large number of patients examined, some focal findings were present. Aura and head version have been reported in patients with generalized epilepsy at rates greater than 10%. 21 , 22 If there is a mechanism by which FCMTE can result in focal onset seizures in addition to generalized‐onset seizures, perhaps this could in part be related to rapid secondary bilateral synchronization. Additional EEG data should be collected to further examine this possibility. With regard to medical treatment of seizures, individuals in this family were responsive to valproic acid as has been reported in some FCMTE patients 5 but also to a lesser extent, other antiseizure medications such as carbamazepine. Myoclonus and tremor appeared years after the seizure onset, became more severe with age, featured more prominently in the extremities, and had overlap with essential tremor, thus corroborating previous descriptions of FCMTE patients. 18 , 23 , 24
Recently, expanded intronic pentanucleotide repeats in SAMD12 (chromosome 8), TNRC6A (chromosome 16), RAPGEF2 (chromosome 4), STARD7 (chromosome 2), and MARCH6 (chromosome 5) were identified as the underlying cause of FCMTE in several Chinese, Japanese, and European families. 9 , 10 , 11 , 12 , 13 , 25 , 26 Using WES and RP‐PCR, we confirmed that the expansion of the intronic pentanucleotide repeat motif TTTTA to TTTTA and TTTCA motifs in SAMD12 is the causative mutation in our family. The reported size of the expanded repeat region varies within and between families and ranges from 2.2 to 18.4 kb as estimated by Southern blots or long‐range PCR. 9 , 10 , 12 Analysis of repeat length from peripheral blood and autopsied tissues (brain, liver, kidney) from several patients also revealed its somatic instability in the same patient. 9 In our family, we were unable to analyze the somatic variability due to lack of autopsied tissues. However, we demonstrated the interpatient and intergenerational instability and showed that the expanded repeat size varies between 4.2 and 4.7 kb. It has been proposed that the intergenerational instability of the expanded repeat region may correlate with specific clinical features, such as the age of onset of clinical symptoms, severity of disease, and progression of the disease. Therefore, it is important to determine the exact sequence of expanded repeats from several affected individuals from multiple families. So far, the exact repeat sequence has been determined for only a handful of affected individuals due to the technical challenges encountered in sequencing of DNA containing repetitive sequences. Ishiura and colleagues 9 sequenced BAC clones with repeat region of affected individuals and showed that the repeat undergoes contraction during BAC cloning. Whole genome long‐read sequencing using Oxford Nanopore or PacBio platforms works but is expensive and requires high‐molecular‐weight DNA that may not be available in most cases. 9 , 10 , 12 , 13 Here, we demonstrated that targeted PacBio sequencing of the mutant allele amplified by long‐range PCR is a cost‐effective method to determine its exact sequence. Our study also demonstrated that the mutant allele is unstable and can undergo both contraction and expansion of its repeat motifs during transmission to the progeny. Of the six affected individuals we sequenced, the three siblings who inherited the mutation from the same parent showed lesser variation in its size (35‐155bp), whereas their children showed higher variation (205‐475 bp). The number of repeat motifs decreased in one case (35M to 46M) and increased in two other cases (33F to 41M and 324F to 423F) of parental transmission to the next generation. However, we did not notice any correlation of repeat size with the age of seizure onset or other clinical symptoms.
Based on the expression data in the GTEx portal, SAMD12 is highly expressed in various regions of the brain (https://gtexportal.org/home/gene/SAMD12). However, not much is known about its role in cellular and biological processes and there are no published reports of animal models of its loss or gain of function. Thus, the exact mechanism of FCMTE with intronic repeat expansions in SAMD12 is not known. Identification of expanded intronic pentanucleotide repeats in five unrelated genes: SAMD12, 9 , 10 , 12 , 13 , 27 TNRC6A, 9 RAPGEF2, 9 STARD7, 25 and MARCH6 26 as the underlying cause of FCMTE in multiple ethnicities suggests that the expanded repeats themselves are pathogenic. Interestingly, more than thirty neurological, neurodegenerative, and neuromuscular diseases are known to be caused by expansions of coding or noncoding repeats. 28 , 29 , 30 , 31 The coding repeats often involve expansion of trinucleotide sequence leading to an expanded stretch of the same amino acid in the protein, for example, polyglutamine stretch in Huntington's disease and spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA17). 29 The noncoding repeats, however, are more variable in the size of an expanded motif as well as its location in the gene. They lie in the 5’ untranslated regions (Friedreich Ataxia), introns (SCA10, SCA31, SCA36, SCA37), or 3’ untranslated regions (SCA8). 30 , 31 , 32 , 33 , 34 The noncoding repeats can either lead to loss of function by transcriptional repression or cause RNA toxicity by being part of the transcript. 29 Various mechanisms have been proposed for RNA toxicity, for example, sequestration or depletion of essential RNA‐binding proteins, such as splicing factors, or generation of neurotoxic peptides by internal translation of the expanded repeats. 29 , 31 Ishiura and colleagues 9 suggested that the pathogenesis of FCMTE involves foci formation in neuronal cells of affected individuals, possibly by sequestration of RNA‐binding proteins. Future studies employing animal models and induced pluripotent cells containing the expanded repeats are needed to define exact mechanisms of FCMTE.
CONFLICTS OF INTEREST
None of the authors have any conflicts of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
We thank the family members for their active involvement with this work. The report is in memory of Dr Guoxiang Wang, who worked in China‐Japan Friendship Hospital and initiated the clinical study of this family. We would like to thank Dr Kenneth Fischbeck (NIH) for clinical evaluation and comments, Dr William Theodore (NIH) and Dr William Gaillard (CNMC) for case discussion and comments. We also appreciate the supports for this study from Dr Tricia Ting, Dr Carlo Tornatore, and Dr Stephen Michaels in MedStar Health.
Zhou Y, Sood R, Wang Q, et al. Clinical and genomic analysis of a large Chinese family with familial cortical myoclonic tremor with epilepsy and SAMD12 intronic repeat expansion. Epilepsia Open. 2021;6:102–111. 10.1002/epi4.12450
Yongxing Zhou, Raman Sood, and Qun Wang contributed equally.
Funding information
This work was supported by the Division of Intramural Research, Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA (to R Sood, B. Carrington, M. Park, A. Young, J. Mullikin, and P Liu), and supported in part by the National Key R&D Program of China (to Q Wang: 2017YFC1307500).
Contributor Information
Mohamad Z. Koubeissi, Email: mkoubeissi@mfa.gwu.edu.
Paul Liu, Email: pliu@mail.nih.gov.
DATA AVAILABILITY STATEMENT
Anonymized data from whole exome sequencing and PacBio sequencing will be shared by request from any qualified investigator.
REFERENCES
- 1. Cen ZD, Xie F, Xiao JF, Luo W. Rational search for genes in familial cortical myoclonic tremor with epilepsy, clues from recent advances. Seizure. 2016;34:83–9. [DOI] [PubMed] [Google Scholar]
- 2. Plaster NM, Uyama E, Uchino M, Ikeda T, Flanigan KM, Kondo I, et al. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology. 1999;12(53):1180–3. [DOI] [PubMed] [Google Scholar]
- 3. Sharifi S, Aronica E, Koelman JH, Tijssen MA, Van Rootselaar AF. Familial cortical myoclonic tremor with epilepsy and cerebellar changes: description of a new pathology case and review of the literature. Tremor Other Hyperkinet Mov (N. Y). 2012;2:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van den Ende T, Sharifi S, van der Salm SMA, van Rootselaar AF. Familial cortical myoclonic tremor and epilepsy, an enigmatic disorder: from phenotypes to pathophysiology and genetics. A Systematic Review. Tremor Other Hyperkinet Mov (N Y). 2018;8:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Rootselaar AF, van Schaik IN, van den Maagdenberg AM, Koelman JH, Callenbach PM, Tijssen MA. Familial cortical myoclonic tremor with epilepsy: a single syndromic classification for a group of pedigrees bearing common features. Mov Disord. 2005;20:665–73. [DOI] [PubMed] [Google Scholar]
- 6. van Rootselaar AF, Groffen AJ, de Vries B, Callenbach PMC, Santen GWE, Koelewijn S, et al. delta‐Catenin (CTNND2) missense mutation in familial cortical myoclonic tremor and epilepsy. Neurology. 2017;5(89):2341–50. [DOI] [PubMed] [Google Scholar]
- 7. Cen ZD, Xie F, Lou DN, Lu XJ, Ouyang ZY, Liu L, et al. Fine mapping and whole‐exome sequencing of a familial cortical myoclonic tremor with epilepsy family. Am J Med Genet B Neuropsychiatr Genet. 2015;168:595–9. [DOI] [PubMed] [Google Scholar]
- 8. Gao L, Li L, Ye J, Zhu X, Shen N, Zhang X, et al. Identification of a novel mutation in PLA2G6 gene in a Chinese pedigree with familial cortical myoclonic tremor with epilepsy. Seizure. 2016;41:81–5. [DOI] [PubMed] [Google Scholar]
- 9. Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581 – 90. [DOI] [PubMed] [Google Scholar]
- 10. Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain. 2018;1(141):2280–8. [DOI] [PubMed] [Google Scholar]
- 11. Lei XX, Liu Q, Lu Q, Huang Y, Zhou XQ, Sun HY, et al. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur J Neurol. 2019;26:513–8. [DOI] [PubMed] [Google Scholar]
- 12. Mizuguchi T, Toyota T, Adachi H, Miyake N, Matsumoto N, Miyatake S. Detecting a long insertion variant in SAMD12 by SMRT sequencing: implications of long‐read whole‐genome sequencing for repeat expansion diseases. J Hum Genet. 2019;64:191–7. [DOI] [PubMed] [Google Scholar]
- 13. Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, et al. Long‐read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. 2019;56:265–70. [DOI] [PubMed] [Google Scholar]
- 14. Gu W, Wang G, Xiao J, Wang M, Zhou Y. Clinical and gene mapping of autosomal dominant inherited adult‐onset of epilepsy, tremor and ataxia. Chinese J Neurol. 2006;39:399–402. [Google Scholar]
- 15. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual, 2nd edn. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 16. Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome‐scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Varshney GK, Carrington B, Pei W, Bishop K, Chen Z, Fan C, et al. A high‐throughput functional genomics workflow based on CRISPR/Cas9‐mediated targeted mutagenesis in zebrafish. Nat Protoc. 2016;11:2357–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crompton DE, Sadleir LG, Bromhead CJ, Bahlo M, Bellows ST, Arsov T, et al. Familial adult myoclonic epilepsy: recognition of mild phenotypes and refinement of the 2q locus. Arch Neurol. 2012;69:474–81. [DOI] [PubMed] [Google Scholar]
- 19. De Fusco M, Vago R, Striano P, Di Bonaventura C, Zara F, Mei D, et al. The alpha2B‐adrenergic receptor is mutant in cortical myoclonus and epilepsy. Ann Neurol. 2014;75:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guerrini R, Bonanni P, Patrignani A, Brown P, Parmeggiani L, Grosse P, et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: a newly recognized epilepsy syndrome with linkage to chromosome 2p11.1‐q12.2. Brain. 2001;124:2459–75. [DOI] [PubMed] [Google Scholar]
- 21. Chin PS, Miller JW. Ictal head version in generalized epilepsy Neurology. Neurology. 2004;63(2):370–2. [DOI] [PubMed] [Google Scholar]
- 22. Dugan P, Carlson C, Bluvstein J, Chong DJ, Friedman D, Kirsch HE, et al. Auras in generalized epilepsy. Neurology. 2014;14(83):1444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coppola A, Santulli L, DelGaudio L, Minetti C, Striano S, Zara F, et al. Natural history and long‐term evolution in families with autosomal dominant cortical tremor, myoclonus, and epilepsy. Epilepsia. 2011;52:1245–50. [DOI] [PubMed] [Google Scholar]
- 24. van Coller R, van Rootselaar AF, Schutte C,van der Meyden CH . Familial cortical myoclonic tremor and epilepsy: description of a new South African pedigree with 30 year follow up Parkinsonism. Relat Disord. 2017;38:35–40. [DOI] [PubMed] [Google Scholar]
- 25. Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019;29(10):4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Florian RT, Kraft F, Leitao E, Kaya S, Klebe S, Magnin E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat Commun. 2019;29(10):4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cen Z, Chen Y, Yang D, Zhu Q, Chen S, Chen X, et al. Intronic (TTTGA)n insertion in SAMD12 also causes familial cortical myoclonic tremor with epilepsy. Mov Disord. 2019;34:1571–6. [DOI] [PubMed] [Google Scholar]
- 28. Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–55. [DOI] [PubMed] [Google Scholar]
- 29. Loureiro JR, Oliveira CL, Silveira I. Unstable repeat expansions in neurodegenerative diseases: nucleocytoplasmic transport emerges on the scene. Neurobiol Aging. 2016;39:174–83. [DOI] [PubMed] [Google Scholar]
- 30. Todd PK, Paulson HL. RNA‐mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–4. [DOI] [PubMed] [Google Scholar]
- 33. Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, et al. Spinocerebellar ataxia type 31 is associated with "inserted" penta‐nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seixas AI, Loureiro JR, Costa C, Ordonez‐Ugalde A, Marcelino H, Oliveira CL, et al. A pentanucleotide ATTTC repeat insertion in the non‐coding region of DAB1, mapping to SCA37, Causes Spinocerebellar Ataxia. Am J Hum Genet. 2017;6(101):87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data from whole exome sequencing and PacBio sequencing will be shared by request from any qualified investigator.