

Summary
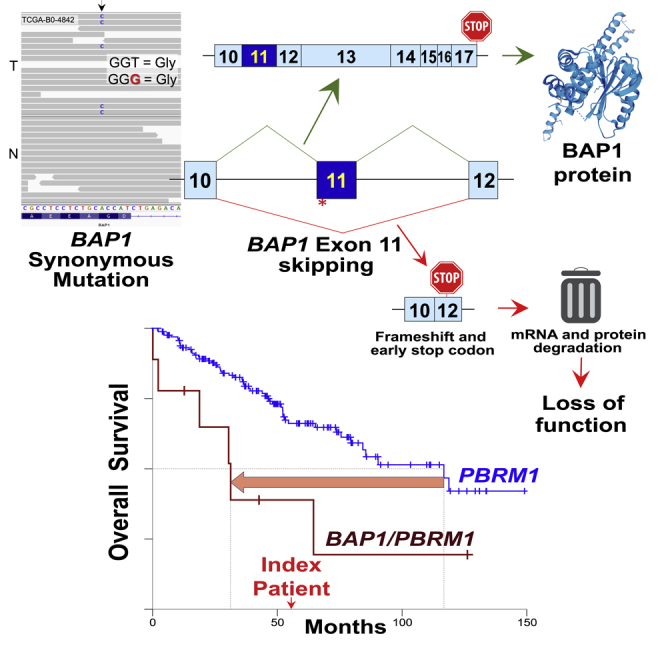
Synonymous mutations are generally disregarded by genomic analyses because they are considered non-pathogenic. We identified and characterized a somatic synonymous mutation in the epigenetic modifier and tumor suppressor BAP1, resulting in exon skipping and complete protein inactivation. This radically altered the prognosis of a clear-cell renal cell carcinoma patient from The Cancer Genome Atlas (TCGA) with a PBRM1 mutation (a predictor biomarker for positive responses to immune checkpoint inhibitors) from good (an estimated overall survival of 117 months) to a very bad prognosis (an estimated overall survival of 31 months), emphasizing the importance of scrutinizing synonymous mutations near acceptor splice sites of cancer genes for accurate precision medicine.
Subject areas: human genetics, genetics, cancer, KIRC-TCGA, personalized medicine
Graphical abstract
Highlights
-
•
First synonymous BAP1 mutation that leads to exon skipping and loss of function
-
•
Exon 11 skipping is a hotspot for BAP1 inactivation
-
•
First synonymous mutation reported to reduce fourfold the expected patient survival
-
•
Synonymous mutations can inactivate cancer genes and affect patient prognosis
Human genetics; genetics; cancer; KIRC-TCGA; personalized medicine
Introduction
Cancer can be broadly defined as a collection of remarkably complex diseases caused by the accumulation of genomic and epigenetic modifications, such as mutations and chromatin alterations, which can be fatal when metastatic. For each tumor type, and preferably for each patient, it is essential to identify the ‘driver’ mutations that lead to tumor development among the ‘passenger’ mutations. Synonymous mutations alter the DNA sequence without changing the encoded amino acid of the resulting protein and, thus, are assumed as ‘silent’ and overlooked in many genomic studies.
Recent studies point out that synonymous mutations might affect the translation kinetics, mRNA stability, miRNA binding sites, or splicing machinery and ultimately alter protein function (Jayasinghe et al., 2018; Kimchi-Sarfaty et al., 2007; Sharma et al., 2019; Supek et al., 2014). In addition, synonymous mutations represent about 6–8% of all driver mutations by single-nucleotide variants in oncogenes (Supek et al., 2014). Overall, synonymous mutations (1) were found to be enriched in known cancer genes, (2) show a negative correlation of their frequency with the mutational load, indicating a selective pressure resulting in highly recurrent synonymous mutations, (3) are non-randomly distributed along the coding sequence and within internal exons and (4) differentially affect codons for specific amino acids (Sharma et al., 2019).
Kidney cancer is diagnosed annually in over 400,000 individuals and causes more than 175,000 deaths worldwide, being clear-cell renal cell carcinoma (ccRCC) the most frequent subtype (∼75%) (Ricketts et al., 2018). An early event in ccRCC development is the inactivation of the pVHL pathway, followed by inactivating mutations of chromatin remodelers, such as SETD2 (Dalgliesh et al., 2010) and PBRM1 (Varela et al., 2011). We previously established that the deubiquitinase and epigenetic modifier BRCA1-associated protein 1 (BAP1) is a major driver of tumor development in ccRCC (Peña-Llopis et al., 2012) and its mutations are mutually exclusive with PBRM1 (Peña-Llopis et al., 2012, 2013). Thus, tumors with BAP1 mutations exhibit dismal prognosis and are characterized by aggressive features, including high tumor grade, mTORC1 activation (Peña-Llopis et al., 2012), rhabdoid and sarcomatoid histologies, tumor necrosis, and poor patient survival (Kapur et al., 2013). In contrast, patients with PBRM1 mutations only (without BAP1 mutations) have good prognosis and are characterized by low tumor grade, low mTORC1 activity (Peña-Llopis et al., 2012), and good overall survival (Kapur et al., 2013). Tumors with loss of BAP1 and PBRM1 showed the highest aggressiveness and shorter patient survival (Kapur et al., 2013; Peña-Llopis et al., 2012). Notably, several other research teams independently confirmed these discoveries (Creighton et al., 2013; Hakimi et al., 2013; Ricketts et al., 2018; Sato et al., 2013; Turajlic et al., 2018) and enabled the molecular genetic classification of this tumor type based on inactivating mutations in BAP1 and PBRM1 (Kapur et al., 2013; Peña-Llopis et al., 2012), providing the rationale for precision medicine using subtype-specific therapies.
Results
A somatic synonymous BAP1 mutation leads to its loss of function and worsens patient prognosis
We identified a patient from The Cancer Genome Atlas (TCGA) kidney renal clear cell carcinoma (KIRC) (Creighton et al., 2013) whose tumor harbored 3p chromosomal loss (Figure S1) and a somatic nonsense PBRM1 mutation. This patient (TCGA-B0-4842), a 73-year-old Caucasian woman, died 56 months after diagnosis. This was considerably rapid for a ccRCC patient with a PBRM1 mutation, since an analysis of TCGA data showed a median overall survival of 117 months (95% confidence interval [CI]: 84–150 months) (Figure 1A). This relatively short survival was more similar to those patients with a BAP1 mutation (median overall survival of 73 months [95% CI: 20–127 months]) or patients with mutations in both BAP1 and PBRM1 (median overall survival of 31 months [95% CI: 15–48 months]) (Figure 1A). Indeed, this patient revealed a somatic synonymous BAP1 mutation near the acceptor splice site of exon 11 (c.936T>G, p.G312G) (Figure 1B).
Figure 1.

A somatic synonymous BAP1 mutation leads to its loss of function and worsens patient prognosis
(A) Kaplan-Meier curves for the indicated groups from the KIRC-TCGA data set. The green arrow indicates the index patient. The blue arrow depicts the worsening of the median overall survival from tumors with PBRM1 mutations to tumors with mutations in both BAP1 and PBRM1 (B/P).
(B) Genomic DNA sequence of the KIRC-TCGA patient displaying the somatic synonymous c.936T>G, p.G312G mutation in exon 11 of BAP1 in the tumor (T) but not in the normal (N) kidney.
(C) Log2-transformed RNA-Seq Expectation-Maximization (RSEM) normalized gene expression for BAP1 from KIRC-TCGA.
(D) Reverse phase protein array (RPPA) analysis for BAP1 from KIRC-TCGA stratifying for patients with BAP1 and PBRM1 mutations individually or for both genes (B/P). Index patient was compared with each group by the one-sample t test.
(E and F) Ribosomal protein S6 phosphorylation at S235/S236 (E) and at S240/S244 (F). For all plots, the green dot or dashed line indicates the index patient.
Surprisingly, the tumor displayed low BAP1 gene expression (Figure 1C) and one of the lowest BAP1 protein expressions among KIRC-TCGA patients (Figure S2B). A one-sample t test showed that the BAP1 protein expression of the index patient was closer to those of patients with mutations in BAP1 or both BAP1 and PBRM1 (p = 10−10 and 0.004, respectively) than patients with mutations in PBRM1 (p = 2·10−103) or wild-type for these genes (p = 3·10−112) (Figure 1D). In addition, mTORC1 was highly active, as assessed by very high ribosomal protein S6 phosphorylation levels (Figures 1E and 1F). These levels, together with the highest tumor grade (grade 4) and pathologic stage III, are typically seen in patients with inactivating mutations in both BAP1 and PBRM1, or only in BAP1, rather than just in PBRM1 (Kapur et al., 2013; Peña-Llopis et al., 2012). Indeed, these levels were closer to tumors with BAP1 or BAP1/PBRM1 mutations than PBRM1 mutations (Figure S2). Thereby, these data strongly suggest that the p.G312G synonymous BAP1 mutation results in loss of BAP1 function and, consequently, higher tumor aggressiveness.
The synonymous BAP1 mutation do not affect protein stability at the cDNA level
To understand the mechanism of inactivation of BAP1 expression caused by the synonymous mutation, we cloned the full-length cDNA of wild-type BAP1 (or a catalytically inactivating p.C91S BAP1 mutation) into pBABE-hygro vector to ensure a constitutive basal expression and we generated the c.936T>G, p.G312G mutation by site-directed mutagenesis (Figure 2A). We then reconstituted the BAP1-null ccRCC cell line UMRC-6 (or UM-RC-6) with wild-type BAP1, the p.G312G BAP1 mutant or the p.C91S BAP1 mutant constructs (and corresponding empty vector control). We observed similar total BAP1 protein levels (Figure 2B) and BAP1 protein stability upon blocking the protein synthesis with cycloheximide treatment in both wild-type BAP1 and the p.G312G mutant (Figure 2C). We found similar results in a cholangiocarcinoma cell line, TFK-1, which had a nonsense mutation in BAP1 (Figures 2C and 2E).
Figure 2.

The synonymous BAP1 mutation do not affect protein stability at the cDNA level
(A) Schema depicting the c.936T>G mutation in the full-length BAP1 cDNA.
(B) UMRC-6 cells stably expressing the p.G312G BAP1 mutant show by western blot similar total protein levels as cells reconstituted with wild-type (WT) BAP1 or the p.C91S mutant. EV, empty vector.
(C) The decay over time of the protein encoded by the cDNA of the synonymous BAP1 mutation is similar to the protein decay from the wild-type BAP1 cDNA in UMRC-6 cells treated with 80 μg/ml of cycloheximide.
(D) BAP1-deficient cholangiocarcinoma TFK-1 cells reconstituted with the p.G312G BAP1 mutant show similar levels of protein expression as wild-type or the p.C91S mutant BAP1.
(E) Western blot of the same stable TFK-1 cell lines treated with 10 μg/ml of cycloheximide for the indicated time shows similar protein decay for the p.G312G mutation as the wild-type BAP1.
(F) Reconstitution of UMRC-6 cells with the p.G312G mutation in BAP1 decreases the ubiquitination of histone H2A to a similar level as to wild-type BAP1, but different than the p.C91S mutant or empty vector.
(G) UMRC-6 cells reconstituted with the p.G312G mutation show similar mTORC1 activation readouts in response to starvation as wild-type and the p.C91S mutant BAP1.
BAP1 targets the deubiquitination of histone H2A (Peña-Llopis et al., 2012; Scheuermann et al., 2010), and we observed that the synonymous p.G312G BAP1 mutation deubiquitinated histone H2A to the same extent as the wild-type BAP1 (Figure 2F). However, UMRC-6 cells lacking BAP1 or reconstituted with the p.C91S BAP1 mutant showed strong ubiquitination of histone H2A, suggesting that the synonymous c.936T>G mutation in the full-length cDNA is not affecting the BAP1 deubiquitinating function.
We previously showed that the correlation of BAP1 loss with mTORC1 activation was not direct (Peña-Llopis et al., 2012). We analyzed several mTORC1 readouts, such as phosphorylated ribosomal protein S6 kinase (S6K), phosphorylated S6, and phosphorylated eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1, also known as 4E-BP1), observed by changes in mobility shift by western blotting (Figure 2G). As expected, cells reconstituted with the p.G312G BAP1 mutant showed similar mTORC1 activation as the wild-type and the p.C91S BAP1 mutant in UMRC-6 (Figure 2G) and TFK-1 (Figure S3A) cells. Furthermore, cell proliferation of the p.G312G BAP1 mutant cells was comparable to the wild-type BAP1 and the p.C91S mutant, as well the empty vector control in UMRC-6 and TFK-1 cells (Figure S3B).
Taking together, these data suggest that the c.936T>G, p.G312G synonymous mutation is not affecting BAP1 at the cDNA level and the change of codon is not responsible for its inactivation.
The synonymous BAP1 mutation leads to exon skipping
Since the c.936T>G mutation is located 4 base pairs away from the acceptor splice site (Figure 1B), we next considered whether the synonymous mutation might affect the alternative splicing machinery, presumably by generating a new binding site for an exonic splicing silencer (Giulietti, 2013). Thus, we cloned exon 11 of BAP1 into a bichromatic splicing minigene reporter (Orengo et al., 2006) to examine exon inclusion and exon skipping events in an in vivo splicing assay (Figure 3A). As a control, we cloned a BAP1 synonymous mutation found in lung adenocarcinoma 6 base pairs away from the donor splice site in exon 7 (c.576C>T, p.D192D) obtained from the SynMICdb database (Sharma et al., 2019). These plasmids were transiently expressed in HeLa cells. Controls showed similar levels of exon integration and skipping, as assessed by western blotting (Figure S5A), RT-PCR (Figure S5B) and fluorescence microscopy (Figure S5C). The co-expression of CELF2 (also known as ETR-3) induced exon integration, whereas the co-expression of MBNL3 induced exon skipping, as expected (Orengo et al., 2006).
Figure 3.

The synonymous BAP1 mutation leads to exon skipping
(A) Diagram of the bichromatic alternative splicing reporter system to assess exon inclusion or skipping.
(B–D), BAP1 exon 11 is skipped in HeLa cells transiently transfected with a minigene reporter containing the c.936T>G, p.G312G synonymous mutation whereas it is more inclusive with the wild-type exon 11, as indicated by western blotting of Flag-tagged proteins (B), RT-PCR (C) and fluorescence microscopy (D). Quantifications are the average ± SD of three independent experiments. ∗, p<0.05; ∗∗, p<0.01; ∗∗∗, p<0.001. Scale bar represents 200 µm.
Replacement of the synthetic exon by BAP1 exon 7 resulted in exon integration, which was not affected by the c.576C>T mutation and only partially altered by the co-expression of CELF2 and MBNL3 (Figure S4). However, the synonymous p.G312G mutation near the acceptor splice site at exon 11 showed striking exon skipping, quantified as 92% by western blotting (Figure 3B), 96% by RT-PCR (Figure 3C), and 92% by fluorescence microscopy (Figure 3D). Exon skipping of the p.G312G mutant BAP1 was significantly higher than the wild-type BAP1 exon 11 (74% [p = 2·10−7], 84% [p = 5·10−7], and 60% [p = 2·10−11], respectively). The degree of exon skipping difference between the wild-type and mutant sequence was then between 12 and 32%, which is similar to the 10-23% reduction of exon-skipping frequency previously reported for several oncogenes and TP53 (Supek et al., 2014). Therefore, these data demonstrate that the synonymous c.936T>G, p.G312G BAP1 mutation leads to exon 11 skipping. The fact that exon 11 was virtually completely excluded in the mutant and partially included in the wild-type BAP1 suggests that exon 11 might be a potential bottleneck for BAP1 synthesis.
Clinical significance of BAP1 exon 11 skipping
To get further insight into the clinical significance of BAP1 exon 11 skipping, we analyzed the raw RNA-Seq data from the index patient. Unfortunately, the coverage was very poor and only two wild-type reads were observed between exon 10 and exon 11 and three reads between exon 11 and exon 12 of BAP1 from two RNA-Seq tumor samples (Figure S6A). However, these reads did not contain the c.936T>G, p.G312G synonymous mutation, indicating that there is an extensive mRNA degradation of BAP1, in consonance with its low gene expression (Figure 1C). Thus, the wild-type reads proceed from the mixture of the RNA from the tumor with normal cells, such as stroma and infiltrating lymphocytes, as reflected at DNA level in Figures 1B and S1, and as we previously described in solid tumors but not in patient-derived xenografts, where human stroma is replaced by mouse stroma, which do not interfere and dilute the tumor DNA and RNA (Peña-Llopis et al., 2012). In addition, BAP1 exon 11 skipping is predicted to result in protein frameshift and a premature stop codon (Figure S7), which can enhance mRNA decapping and the nonsense-mediated mRNA decay to selectively degrade imperfect mRNAs with early translation termination codons (Couttet and Grange, 2004; Muhlrad and Parker, 1994). Consistent with this notion, similar wild-type reads were observed in PBRM1, which harbored a nonsense mutation, but not in adjacent genes (Figure S6B). Therefore, the lack of BAP1 mutant mRNA reads in the index patient (Figure S6A) agrees with the nonsense-mediated mRNA decay caused by premature translation termination codon due to exon 11 skipping.
Unexpectedly, a close examination of the eight KIRC-TCGA patients with splice-site mutations in BAP1 revealed that two patients had mutations in the acceptor splice site of BAP1 exon 11. TCGA-BP-4798 showed a c.IVS932-2A>G mutation that was considered likely pathogenic according to dbSNP database (rs112194987) and resulted in exon 11 skipping, as evidenced by RNA-Seq reads between exons 10 and 12 (Figure 4A). TCGA-CZ-5985 showed a c.IVS932-1G>T mutation (rs9848343 in dbSNP) and solid evidence for exon 11 skipping with 4 reads between exons 10 and 12 (Figure 4B). Similar to the index patient, both splice-site mutations resulted in low BAP1 gene expression (Figure 4C) and BAP1 protein expression (Figure 4D), especially for TCGA-CZ-5985, which displayed the second lowest BAP1 levels from KIRC-TCGA. In addition, ribosomal protein S6 expression (Figure 4E) and phosphorylation levels (Figure 4F) were high, as expected to be found in patients with BAP1 loss.
Figure 4.

Mutations in the acceptor splice site of BAP1 exon 11 lead to exon skipping and loss of function
(A) Patient TCGA-BP-4798 has a somatic mutation in the acceptor splice site of exon 11 (c.IVS932-2A>G) as depicted by whole-genome sequencing (WES) and RNA-Seq of the tumor (T) but not normal kidney (N) WES. Exon 11 skipping for patient TCGA-BP-4798 is evidenced by RNA-Seq reads spanning from exon 10 to exon 12 and highlighted in red. The red arrow indicates the splice-site mutation.
(B) Patient TCGA-CZ-5985 has a somatic mutation in the acceptor splice site of exon 11 (c.IVS932-1G>T) as depicted by whole-genome sequencing (WES) and RNA-Seq of the tumor (T) but not normal kidney (N). Exon 11 skipping for patient TCGA-CZ-5985 is indicated by RNA-Seq reads spanning from exon 10 to exon 12 and highlighted in red. The red arrow indicates the splice-site mutation.
(C–F) Log2-transformed RNA-Seq RSEM normalized gene expression for BAP1 (C), RPPA for BAP1 (D), total ribosomal protein S6 expression (E) and S6 phosphorylation at S235/S236 (F) from KIRC-TCGA for patients TCGA-BP-4798 (in cyan) and TCGA-CZ-5985 (in orange).
(G) Diagram illustrating the summary of this study, where a synonymous mutation near the acceptor splice site of exon 11 of BAP1 leads to exon skipping, frameshift and premature stop codon, inducing its mRNA and protein degradation and loss of function.
Considering that there are 32 splicing sites among the 17 exons of BAP1, the fact of finding 2 of 8 patients with splice-site mutations in the acceptor splice site of exon 11 is very unlikely by chance alone (p = 0.0015), according to a cumulative binomial distribution. This suggests that exon 11 skipping might be a hotspot for BAP1 inactivation.
The clinical significance of exon 11 skipping is, however, more complicated. Patient TCGA-BP-4798 harbored an additional missense mutation in TP53 and showed distant metastasis at diagnosis, dying almost 11 months thereafter, whereas patient TCGA-CZ-5985 was still alive 65 months after diagnosis. This suggests that despite having a similar BAP1 mutation and inactivation, other alterations contribute decisively in the patient prognosis, such as an additional PBRM1 mutation in the index patient and a TP53 mutation in TCGA-BP-4798.
In summary, we have strong evidence to claim that the somatic synonymous mutation in BAP1 of the index patient results in exon 11 skipping, frameshift and premature stop codon, leading to mRNA and protein degradation and ultimately, together with chromosome 3p loss, to the complete loss of function for BAP1 (Figure 4G). The inactivation of BAP1 contributed to a shorter survival than the one that would be expected for a patient with ccRCC with a PBRM1 mutation.
Discussion
We describe here a patient with renal cell carcinoma with presumably good prognosis due to a PBRM1 mutation, who experienced a relatively short survival by harboring an additional unacknowledged inactivating synonymous mutation in BAP1.
The tumor with the synonymous p.G312G mutation in BAP1 showed low gene expression and one of the lowest protein expressions from KIRC-TCGA, suggesting loss of function, as evidenced by phosphorylation of ribosomal protein S6, which is associated with BAP1 loss (Kapur et al., 2013; Peña-Llopis et al., 2012). However, reconstitution of BAP1-deficient ccRCC and cholangiocarcinoma cell lines with the c.936T>G BAP1 mutation in the full-length cDNA showed similar protein levels, stability and histone H2A deubiquitination as the wild-type BAP1, proving that the synonymous mutation is not disrupting BAP1 at the cDNA level. Interestingly, the synonymous mutation was near the acceptor splice site of exon 11. Thus, we cloned this exon into a sophisticated bichromatic in vivo splicing system (Orengo et al., 2006) and demonstrated by western blotting, RT-PCR, and fluorescent microscopy that the BAP1 synonymous mutation resulted in exon 11 skipping. Remarkably, two of eight patients with splice-site BAP1 mutations from KIRC-TCGA showed mutations in the acceptor splice site of exon 11, suggesting that exon 11 skipping might be a hotspot for BAP1 inactivation. It is well known that frameshift mutations leading to premature termination of translation can cause rapid mRNA degradation to prevent the production of non-functional or toxic truncated proteins (Couttet and Grange, 2004; Muhlrad and Parker, 1994). Hence, we provide here strong evidence to support that the c.936T>G, p.G312G synonymous mutation in BAP1 results in exon 11 skipping, frameshift, premature stop codon, mRNA degradation, protein loss and, eventually, loss of function, which contributed negatively to decrease almost fourfold the expected patient survival (from 117 to 31 months).
This study has clinical relevance, since PBRM1 mutations were reported to be predictive biomarkers for positive responses to immune checkpoint inhibitors in renal cell carcinoma (Braun et al., 2019; Miao et al., 2018). However, recent clinical trials in advanced renal cell carcinoma showed no association of PBRM1 mutations with progression-free survival (Motzer et al., 2020). Furthermore, a recent study failed to associate the inactivation of several genes of the mammalian Switch/Sucrose Non-Fermentable (mSWI/SNF) with clinical outcomes in patients with cancer treated with systemic immune checkpoint inhibitors, except PBRM1 in ccRCC (Abou Alaiwi et al., 2020). Therefore, the clinical benefits observed by treatment with immune checkpoint inhibitors in patients with ccRCC with PRBM1 mutations, most likely are simply because, as we previously described, they are mutually exclusive of BAP1 mutations (Peña-Llopis et al., 2012, 2013), which displayed poor overall patient survival (Kapur et al., 2013). Thus, PBRM1 mutation should not be taken solely as a predictive marker in ccRCC but rather in combination with potential BAP1 inactivation (by DNA sequencing and/or immunohistochemistry) (Peña-Llopis et al., 2012) to have a more complete picture for each patient, fostering an accurate identification of the individual cancer driver genes and facilitating precision oncology.
A BAP1 missense mutation (p.N78S), but not a synonymous mutation, was recently found to lead to alternative splicing (Jayasinghe et al., 2018). By comparing immunohistochemistry with mutation data, we previously showed that all frameshift mutations (11/11) and 85% (11/13) of point mutations in BAP1 were unable to be translocated to the nucleus in ccRCC tumors, indicating loss of function (Peña-Llopis et al., 2012). The two missense mutations that showed nuclear BAP1 expression had mutations in the catalytic site of binding to ubiquitin and the binding to the ULD domain, respectively, and thus unlikely to be functional (Peña-Llopis et al., 2012). In addition, all tumors with BAP1 mutations are accompanied by chromosomal 3p loss. Thus, the only allele left is the one with the mutation. Therefore, to our knowledge, this study provides the first evidence of a synonymous mutation in BAP1 that leads to exon skipping and protein inactivation. This work also delivers insight into an unappreciated mechanism of inactivation of a very important tumor suppressor, which is frequently mutated and drives tumorigenesis in many cancer entities besides ccRCC (Peña-Llopis et al., 2012), including uveal melanoma (Harbor et al., 2010), mesothelioma (Bott et al., 2011), and cholangiocarcinoma (Jiao et al., 2013). Remarkably, synonymous mutations toward the 5′ end of coding sequences were shown to be depleted but exhibited more pronounced predicted structural impact, suggesting higher selective pressure in this region (Sharma et al., 2019). The database of synonymous mutations in cancer (SynMICdb) recently generated (Sharma et al., 2019) could be a good resource for assessing the potential impact of specific synonymous mutations. Hence, synonymous mutations identified in clinical samples should not be systematically discarded but carefully analyzed in cancer genes, especially near the acceptor splice sites.
Limitations of the study
A limitation of this study is that we are analyzing the expression of an exogenous minigene. However, it was previously shown to be a reliable system for claiming that certain mutations affected splicing (Jayasinghe et al., 2018; Supek et al., 2014), observing similar ratios of exon inclusion/exclusion as we report here.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Samuel Peña-Llopis (Samuel.Pena-Llopis@dkfz.de).
Materials availability
Plasmids generated in this study were deposited to Addgene (https://www.addgene.org/browse/article/28211439). Stable cell lines generated in this study will be deposited at the German Collection of Microorganisms and Cell Cultures (DSMZ) upon manuscript acceptance.
Data and code availability
This study did not generate any unique data sets or code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank TCGA for sharing their data and Prof. Benedikt Brors, Dr. Martina Fröhlich, and Dr. Jules Kerssemakers for facilitating access to genomic raw data, as well as Prof. Stefan Fröhling and Dr. Stephanie Rössler for kindly providing cells. This project has received funding from the German Cancer Consortium (DKTK), the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. 798637 and a grant from the German Research Foundation (DFG) (PE 2696/1-1) to S.P.-L. S.V.-R. was supported by an NCT-DKTK School of Oncology Fellowship and a DFG grant (VE1153/1-1). J.T.S. was supported by a DFG grant (SI1549/3-1, Clinical Research Unit KFO337), through the Collaborative Research Center SFB824 (project C4), and the German Cancer Aid (70112505; PIPAC consortium).
Author contributions
J.N., A.B., and S.M. performed experiments. S.V.-R. designed and performed experiments and revised the manuscript. V.J. and J.T.S. provided resources, feedback, and revised the manuscript. S.P.-L. conceived the study, designed experiments, performed the bioinformatics and statistical analyses, supervised the project, and wrote the manuscript.
Declaration of interests
J.T.S. reports personal fees from AstraZeneca, Baxalta, Immunocore, Novartis, Shire; research grants (institutional) and personal fees from Bristol-Myers Squibb, Celgene, Roche; ownership from FAPI holding (<3%); board membership from Pharma15, outside the submitted work.
Published: March 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102173.
Supplemental information
References
- Abou Alaiwi S., Nassar A.H., Xie W., Bakouny Z., Berchuck J.E., Braun D.A., Baca S.C., Nuzzo P.V., Flippot R., Mouhieddine T.H. Mammalian SWI/SNF complex genomic alterations and immune checkpoint blockade in solid tumors. Cancer Immunol. Res. 2020;8:1075–1084. doi: 10.1158/2326-6066.CIR-19-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M., Brevet M., Taylor B.S., Shimizu S., Ito T., Wang L., Creaney J., Lake R.A., Zakowski M.F., Reva B. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D.A., Ishii Y., Walsh A.M., Van Allen E.M., Wu C.J., Shukla S.A., Choueiri T.K. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 2019;5:1631–1633. doi: 10.1001/jamaoncol.2019.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couttet P., Grange T. Premature termination codons enhance mRNA decapping in human cells. Nucleic Acids Res. 2004;32:488–494. doi: 10.1093/nar/gkh218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton C.J., Morgan M., Gunaratne P.H., Wheeler D.A., Gibbs R.A., Gordon Robertson A., Chu A., Beroukhim R., Cibulskis K., Signoretti S. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh G.L., Furge K., Greenman C., Chen L., Bignell G., Butler A., Davies H., Edkins S., Hardy C., Latimer C. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulietti M. SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013 doi: 10.1093/nar/gks997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi A.A., Ostrovnaya I., Reva B.A., Schultz N., Chen Y.B., Gonen M., Liu H., Takeda S., Voss M.H., Tickoo S.K. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin. Cancer Res. 2013;19:3259–3267. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour J.W., Onken M.D., Roberson E.D., Duan S., Cao L., Worley L.A., Council M.L., Matatall K.A., Helms C., Bowcock A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe R.G., Cao S., Gao Q., Wendl M.C., Vo N.S., Reynolds S.M., Zhao Y., Climente-Gonzalez H., Chai S., Wang F. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018;23:270–281 e273. doi: 10.1016/j.celrep.2018.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Pawlik T.M., Anders R.A., Selaru F.M., Streppel M.M., Lucas D.J., Niknafs N., Guthrie V.B., Maitra A., Argani P. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur P., Peña-Llopis S., Christie A., Zhrebker L., Pavia-Jimenez A., Rathmell W.K., Xie X.J., Brugarolas J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol. 2013;14:159–167. doi: 10.1016/S1470-2045(12)70584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C., Oh J.M., Kim I.W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- Miao D., Margolis C.A., Gao W., Voss M.H., Li W., Martini D.J., Norton C., Bosse D., Wankowicz S.M., Cullen D. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer R.J., Robbins P.B., Powles T., Albiges L., Haanen J.B., Larkin J., Mu X.J., Ching K.A., Uemura M., Pal S.K. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020;26:1733–1741. doi: 10.1038/s41591-020-1044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D., Parker R. Premature translational termination triggers mRNA decapping. Nature. 1994;370:578–581. doi: 10.1038/370578a0. [DOI] [PubMed] [Google Scholar]
- Orengo J.P., Bundman D., Cooper T.A. A bichromatic fluorescent reporter for cell-based screens of alternative splicing. Nucleic Acids Res. 2006;34:e148. doi: 10.1093/nar/gkl967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S., Christie A., Xie X.J., Brugarolas J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res. 2013;73:4173–4179. doi: 10.1158/0008-5472.CAN-13-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-Llopis S., Vega-Rubín-de-Celis S., Liao A., Leng N., Pavía-Jiménez A., Wang S., Yamasaki T., Zhrebker L., Sivanand S., Spence P. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012;44:751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts C.J., De Cubas A.A., Fan H., Smith C.C., Lang M., Reznik E., Bowlby R., Gibb E.A., Akbani R., Beroukhim R. The cancer genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:313–326. doi: 10.1016/j.celrep.2018.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Yoshizato T., Shiraishi Y., Maekawa S., Okuno Y., Kamura T., Shimamura T., Sato-Otsubo A., Nagae G., Suzuki H. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013;45:860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- Scheuermann J.C., de Ayala Alonso A.G., Oktaba K., Ly-Hartig N., McGinty R.K., Fraterman S., Wilm M., Muir T.W., Muller J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–247. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma Y., Miladi M., Dukare S., Boulay K., Caudron-Herger M., Gross M., Backofen R., Diederichs S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019;10:2569. doi: 10.1038/s41467-019-10489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Minana B., Valcarcel J., Gabaldon T., Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- Turajlic S., Xu H., Litchfield K., Rowan A., Horswell S., Chambers T., O'Brien T., Lopez J.I., Watkins T.B.K., Nicol D. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell. 2018;173:595–610. doi: 10.1016/j.cell.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I., Tarpey P., Raine K., Huang D., Ong C.K., Stephens P., Davies H., Jones D., Lin M.L., Teague J. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique data sets or code.