

Summary
Objective:
Neuroinflammation is a major theme for epilepsy, which has been characterized in acquired epilepsy but is poorly understood in genetic epilepsy. GABAA receptor subunit gene mutations are significant causes of epilepsy, and we have studied the pathophysiology directly resulting from defective receptor channels. Here we determined the proinflammatory factors in a genetic mouse model, the Gabrg2+/Q390X knockin (KI). We have identified increased cytokines in multiple brain regions of the KI mouse throughout different developmental stages and propose that accumulation of the trafficking deficient mutant protein may increase neuroinflammation, which would be a novel mechanism for genetic epilepsy.
Methods:
We used enzyme-linked immunosorbent assay (ELISA), immunoprecipitation, nuclei purification, immunoblot, immunohistochemistry, and confocal microscopy to characterize increased neuroinflammation and its potential causes in a Gabrg2+/Q390X knockin mouse and a Gabrg2+/− knockout (KO) mouse, each associated with a different epilepsy syndrome with different severities.
Results:
We found that proinflammatory cytokines such as tumor necrosis factor alpha (TNF), interleukin 1-beta (IL-1β) and IL-6 were increased in the KI mice but not in the KO mice. A major underlying basis for the discrepancy in cytokine expression between the two mouse models is likely due to chronic mutant protein accumulation and endoplasmic reticulum (ER) stress. The presence of mutant protein dampened cytokine induction upon further cellular stimulation or external stress such as elevated temperature. Pharmacological induction of ER stress upregulated cytokine expression in the wildtype and KO but not in the KI mice. The increased cytokine expression was independent of seizure occurrence since it was upregulated in both mice and cultured neurons.
Significance:
Together, these data demonstrate a novel pathophysiology for genetic epilepsy, increased neuroinflammation which is common mechanism for acquired epilepsy. The findings thus provide the first link of neuroinflammation between genetic epilepsy associated with an ion channel gene mutation and acquired epilepsy.
Keywords: GABAA receptors, epilepsy, Gabrg2+/Q390X knockin (KI) mice, neuroinflammation, proinflammatory cytokines, ER stress
Introduction
Mounting evidence indicates a role for neuroinflammation in human epilepsy while reducing neuroinflammation inhibits seizures and improves seizure outcome1–3. However, studies on neuroinflammation have been focused on acquired epilepsies such as those due to head trauma, viral encephalitis and other external insults, or in epilepsy mouse epilepsy models with non-genetic causes4;5. Although these mouse models may not precisely represent the pathophysiology in human epilepsy, common pathways of neuroinflammation have been established and also identified in human resected tissues3;4, providing strong converging evidence in favor of the contribution of neuroinflammation in epileptogenesis.
Genetic epilepsy is caused by gene mutations that are present from conception to adulthood. Mutation knockin mouse models of the human mutation would provide a great opportunity to understand the pathophysiology of disease due to the nature of disease starting from a very clean background instead of massive tissue injury as seen in head trauma. Many of the affected epilepsy genes are ion channels and transporters, thus previous studies have been logically focused on the function of the mutant ion channels and transporters6;7. However, it is unclear if alteration of the ion channel or transporter function is the whole story of the basis for the epilepsy or if other factors such as neuroinflammation, which is commonly identified in acquired epilepsy, may also play a role in the pathogenesis of genetic epilepsy.
GABRG2 is an established epilepsy gene, and mutations in GABRG2 are associated with a wide spectrum of epilepsy syndromes, including childhood absence epilepsy and Dravet syndrome. GABRG2(Q390X) is a mutation associated with GEFS+ and Dravet syndrome8. Gabrg2+/Q390X knockin (KI) mice show spontaneous generalized tonic clonic seizures (GTCS), myoclonic jerks, sudden unexplained death in epilepsy (SUDEP), anxiety, impaired social activity and cognition, thus representing a mouse model of severe epilepsy9. Gabrg2+/− knockout (KO) mice have been reported to have anxiety without seizures or with the mild epilepsy syndrome generalized absence epilepsy10;11, thus representing a mouse model of mild epilepsy12.
Our previous work has extensively compared these two mouse models from mutant gene expression to protein, channel function, GABAergic neurotransmission, seizure phenotype and comorbidities9;11;13. We determined that mutant protein accumulation in Gabrg2+/Q390X mice contributed to exacerbated epilepsy phenotype and could present a therapeutic target. Here we have characterized the major proinflammatory factors in the mouse model and propose neuroinflammation as mechanistic link between genetic and acquired epilepsy. We compared the proinflammatory profile of the two mouse models and focused on the severe epilepsy in the Gabrg2+/Q390X mouse harboring a trafficking deficient protein-generating mutation.
Materials and Methods
Mice
The Gabrg2+/Q390X KI mouse line was recently developed9, and the Gabrg2+/− KO mouse line was reported before14. Mice used in the study were crossed with C57BL/6J mice for at least 8 generations and were between postnatal day 0 to 8 months old. Both sexes were included. All experimental procedures were approved by Vanderbilt University Division of Animal Care.
GABAA receptor subunit cDNAs
The cDNAs encoding human GABAA receptor α1, β2 and γ2 subunits were as described previously6;15. GABRG2(Q390X) mutation was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and was confirmed by DNA sequencing. FLAG tagged γ2 subunit plasmids were generated as previously described16.
Cell cultures
Mouse cortical neurons were prepared as previously described17;18. Mouse cortical neurons were cultured from postnatal day 0 mouse pups. The neurons were plated at a density of 0.5–1×105 for immunohistochemistry and 2×105 for immunoblot in plating media that contained DMEM 420ml, F12 40ml, fetal bovine serum 40ml, penicillin and streptomycin 1ml and L-Glutamine (200mM) 0.2ml for 4 hrs and then maintained in Neurobasal media that contained B27 supplement (50:1), L- Glutamine (200mM) and penicillin and streptomycin 1ml. The medium was refreshed by half every other day. The experiments were conducted on day 15–16 in dish. For culture of HEK293T cells and mouse L929 cells, Polyethylenimine (PEI) transfection, as well as immunoprecipitation and immunoblot were detailed in Supplementary Methods.
Isolation of cell nuclei from mouse brain homogenates
We developed a protocol to prepare cell nuclei from freshly dissected mouse brain based on previously described19. Detailed procedures were provided in Supplementary Methods.
Enzyme-linked immunosorbent assay (ELISA)
A standard sandwich ELISA was performed to measure tumor necrosis factor-alpha (TNF), interleukin-1 beta (IL-1β) and IL-6 levels in mouse brain and blood plasma. The ELISA kits for measuring mouse TNF, IL-1β and IL-6 were purchased from ThermoFisher and all the cytokine standards were included in the kits. The blood was drawn from mouse tail and plasma was separated. 10–30μg protein of brain tissues and 50 μl of undiluted plasma was used for reaction but the data presented were from reactions of 30μg protein. The optical density (OD) of each well was read at 450 nm with an absorbance-based microplate reader. The final concentration was calculated by converting the OD readings against a standard curve. The quantifications of the mutant mice were normalized to the same brain region of the wildtype littermates.
Mouse body temperature elevation
The heating procedure was adapted from a previous study20 and detailed in Supplementary Methods.
Lipopolysaccharide (LPS) treatment
Adult mice were injected with a single dose of LPS (L2630, Escherichia coli O111:B4, Sigma-Aldrich, St. Louis, MO,) 1 mg/kg i.p. to induce neuroinflammation. LPS is a cell-wall immunostimulatory component of gram-negative bacteria that was first identified as a Toll-like receptor 4 (TLR-4) ligand17. TLR-4 is primarily expressed on microglia18 in the central nervous system, which once activated, produces proinflammatory cytokines, such as TNF, IL-1β, prostaglandin E2 (PGE2) and NO. The mice were sacrificed 6 hrs after LPS administration. We chose 6hrs post-LPS treatment because it has been established that the progression of proinflammatory response in macrophage upon LPS stimulation is initiated at 2hrs and peaked at 6hrs21–23.
Brain slice immunohistochemistry and related quantifications
The experimental procedures were as previously described9 and detailed in Supplementary Methods.
Experimental design and statistical analysis
For ELISA, the optical density value for each condition was calculated based on the formulae of the standard. For biochemistry experiment, subunit integrated density values (IDVs) were quantified on immunoblots by using the Quantity One or Odyssey fluorescence imaging system (Li-Cor). The fluorescence intensity values were quantified by using ImageJ. Statistical analysis was performed using Prism 8 software (GraphPad Software). Details on statistical analysis and experimental design, including tests performed, exact p values, and sample sizes are provided in the result section describing each figure, or within the legend of each figure. All data were expressed as mean ± S.E.M values. Analysis of variance (ANOVA), including one-way and two-way ANOVA, unpaired Student t tests, one sample t test were used. Post hoc and a priori Bonferroni comparisons or Newman-Keuls Multiple Comparison test were conducted to evaluate individual mean comparisons where appropriate. All analyses used an alpha level of 0.05 to determine statistical significance.
Results
Proinflammatory cytokines increase in multiple brain regions in Gabrg2+/Q390X KI mice in an age dependent manner.
TNF, IL-1β and IL-6 are major cytokines produced in the CNS, and these cytokines may have overlapping functions. We first chose these three proinflammatory factors because it has been established that they are all expressed in the CNS24–26. We first determined TNF, IL-1β and IL-6 levels in four major brain regions including somatosensory cortex, cerebellum, hippocampus, and thalamus, because γ2 subunit is globally expressed based on our biochemical and immunohistochemical studies9. We first studied the cytokines in 6–8 month old mice because this was the time point that Gabrg2+/Q390X mice start to show mutant protein aggregates9. However, the spontaneous seizures was first identified at postnatal day 16–17 in Gabrg2+/Q390X mice9. We used ELISA and determined the cytokine levels in brain lysates from tissues of each brain region. We found that the cytokines were increased in all brain regions(Figure 1A–C). For TNF, the expression was highest in the hippocampus of Gabrg2+/Q390X mice (1.31 ± 0.05 for cortex (cor), 1.005 ± 0.04 for cerebellum (cb), 1.53 ± 0.067 for hippocampus (hip) and 1.37 ± 0.04 for thalamus (thal), with the wildtype levels for each brain region arbitrarily taken as 1). IL-1β had a low level of expression in cerebellum than in, hippocampus while other regions had similar levels (1.198 ± 0.045 for cor, 1.14 ± 0.035 for cb, 1.353 ± 0.054 for hip and 1.26 ± 0.057 for thal). The expression levels of IL-6 (1.74 ± 0.14 for cor, 1.638 ± 0.283 for cb, 1.74± 0.17 for hip, and 1.62± 0.16 for thal) were similar across brain regions.
Figure 1. Increased proinflammatory cytokines tumor-necrosis factor alpha (TNF), interleukin 1β (IL-1β) and interleukin 6 (IL-6) in Gabrg2+/Q390X, a genetic mouse model of epileptic encephalopathy.
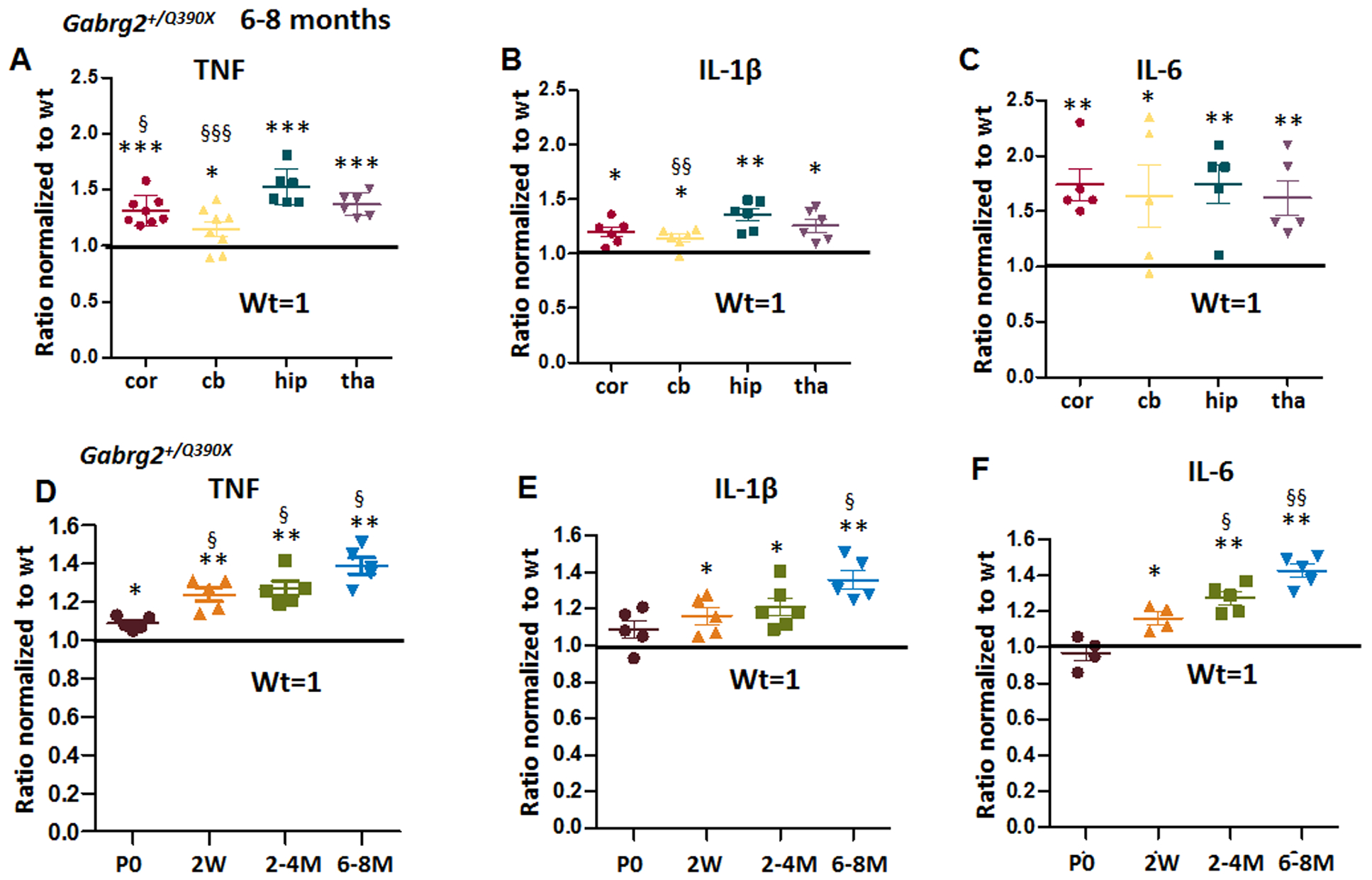
A-C. The brains from 6–8 months old Gabrg2+/Q390X mice were dissected and processed for measurement of pro-inflammatory cytokines. Equal amounts of protein lysates (30μg) from each brain region were determined for cytokines including tumor-necrosis factor alpha (TNF) (A), interleukin 1β (IL-1β) (B) and interleukin 6 (IL-6) (C) with enzyme-linked immunosorbent assay (ELISA). The measurements in the heterozygotes (het) were normalized to the same brain region of their own wildtype (WT) littermates. D-F. The forebrain cortex from the Gabrg2+/Q390X mice at different ages (P0=postnatal day 0, 2W=2 weeks, 2–4M=2–4 months, 6–8M=6–8 months) were dissected and processed for measurement of TNF (D), IL-1β (E) and IL-6 (F) with ELISA. Equal amounts of protein lysates (30μg) from the mouse cortex of different ages were determined. In D-F, the measurements in the het were normalized to the cortex of their own wildtype littermates. (In A-F, *p < 0.05; ** p < 0.01; *** p < 0.001 vs wt, In A-B, § P< 0.05; §§ P< 0.01 §§§ P< 0.001 vs hip in het; in D-F, § P< 0.05; §§P< 0.01 vs P0). Data were presented as mean ± S.E.M. In A-C, N=5–8 mice. In D-F, N=4–6 mice.
Because genetic mutations are present from beginning of life, the mutant protein may be produced in early brain long before seizure onset at approximately postnatal day16–19 old27. We thus measured the cytokines levels at different ages during development and adulthood. We included mice at multiple ages including postnatal day 0, 2 weeks, 2–4 months, and 6–8 months. IL-6 showed a clear increasing trend. At P0, although the levels of TNF were low (1.09 ± 0.015 for P0, 1.24 ± 0.036 for 2 weeks, 1.27 ± 0.04 for 2–4 months, and 1.39 ± 0.042 for 6–8 months), it was higher in the mutant Gabrg2+/Q390X mice than the wildtype mice. For IL-6: 0.97 ± 0.04 for P0, 1.163 ± 0.034 for 2 weeks, 1.274 ± 0.0347 for 2–4 months, and 1.428 ± 0.037 for 6–8 months (wildtype for P0 was arbitrarily taken as 1). Likewise, IL-1β showed an increasing trend with age (1.088 ± 0.049 for P0, 1.162 ± 0.05 for 2 weeks, 1.196 ± 0.043 for 2–4 month, and 1.37 ± 0.05 for 6–8 months, while the wildtype for P0 was arbitrarily taken as 1). Because these cytokines are also expressed in the periphery, the cytokines in CNS could be released from monocytes from blood circulation. The increased in cytokines could be from peripheral macrophages. We then measured the cytokines in plasma and found that there was no difference for any of the three cytokines in plasma between wildtype and heterozygous mice (Supplementary Figure 1). By contrast, there were no changes in these cytokine levels in the Gabrg2+/− mice11,28 (Figure 2A–C, Supplementary Table 1).
Figure 2. There was no increase of proinflammatory cytokines TNF, IL-1β and IL-6 in Gabrg2+/− knockout, a mouse model of infrequent absence epilepsy.
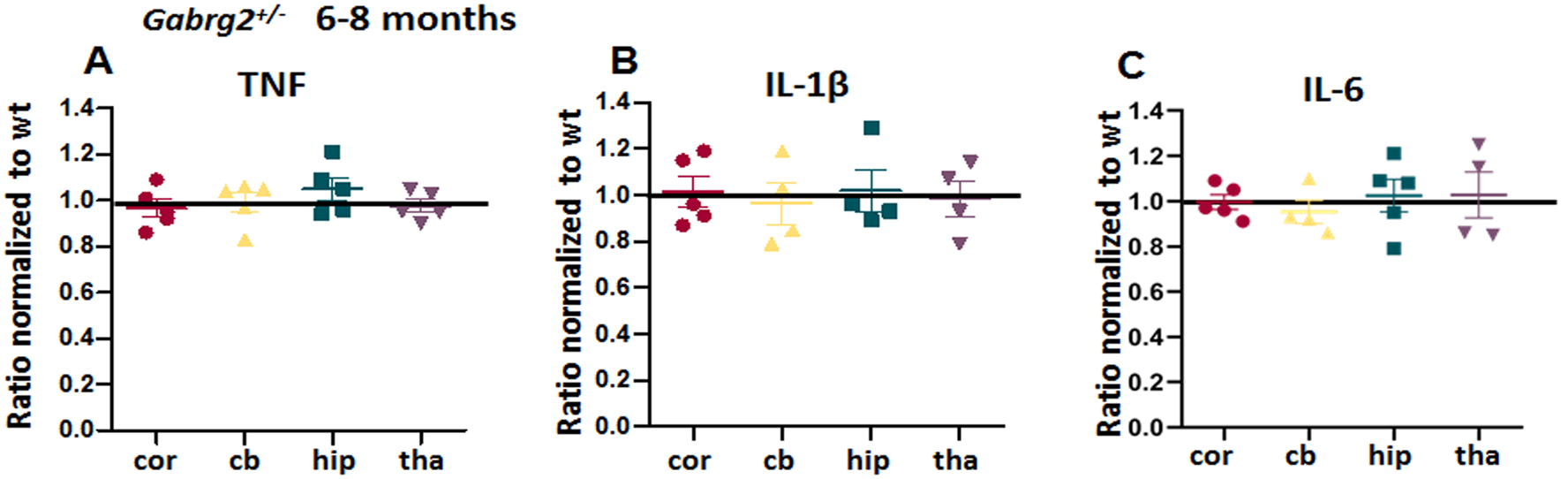
A-C. The brains from 6–8 months old Gabrg2+/− mice were dissected and processed for measurement of pro-inflammatory cytokines. Equal amounts of protein lysates (30μg) from each brain region were determined for cytokines including tumor-necrosis factor alpha (TNF) (A), interleukin 1β (IL-1β) (B) and IL-6 (C) with enzyme-linked immunosorbent assay (ELISA). The measurements in the het were normalized to the same brain region of their own wildtype littermates. Data were presented as mean ± S.E.M. N=5 mice for each group.
External stimulation such as elevated temperature or LPS treatment increased proinflammatory cytokines in the wildtype but not in Gabrg2+/Q390X KI mice.
Febrile seizures are often the presenting symptom for children with many of the epilepsy mutations28;29. We thus determined the effects of short temperature elevations on Gabrg2+/Q390X mice using a temperature-controlled heating lamp. We avoided longer heat exposure because sustained high temperature exposure may cause cellular injury, and even cell death. The temperature induction protocol was modified based on previously described studies in both Gabrg2+/Q390X and Scn1a+/− knockout mice30,20 (Figure 3A). Heating stopped once the mouse’s core temperature reached 42.5 °C but maintained for 30 min before tissue harvest (Figure 3B). In addition to heat exposure, we determined the effect of a commonly used proinflammatory factor inducer, LPS. We administered LPS (ip) for 6 hrs to induce an inflammatory response in mice (Figure 3C). Both heat and LPS treatment increased cytokines in the wildtype mice in all surveyed regions. Surprisingly, neither heat nor LPS further increased the cytokines in the Gabrg2+/Q390X mice. In fact, TNF was reduced or trended downward in the cortex and hippocampus of the heterozygous Gabrg2+/Q390X mice (Figure 3D) while the level of TNF in other regions were plateaued compared with the baseline levels (Figure 3E) (taking cortex as example: For heat: 1.25 ± 0.08 for het, 1.34 ± 0.15 for wt+heat, 1.18± 0.021 for het+heat; For LPS: 1.35 ± 0.24 for het, 1.37 ± 0.07 for wt+LPS, 1.21 ± 0.017 for het+LPS). By contrast, both elevated temperature and LPS increased cytokines in Gabrg2+/− mice (Figure 3F and 3G) (taking cortex as example: For heat: 1.08 ± 0.11 for het, 1.43 ± 0.06 for wt+heat, 1.29± 0.11 for het+heat; For LPS: 1.09 ± 0.05 for het, 1.49 ± 0.11 for wt+LPS, 1.46 ± 0.21 for het+LPS), although the magnitude of increase of TNF in Gabrg2+/− was less than in wildtype mice in hippocampus upon heat challenge (in hippocampus, for heat: 1.06 ± 0.19 for het, 1.54 ± 0.12 for wt+heat, 1.32± 0.03 for het+heat). A pattern similar to TNF was obtained for IL-1β and IL-6 expression in Gabrg2+/Q390X and Gabrg2+/− mice.
Figure 3. Treatment with elevated temperature or lipopolysaccharide had dampened response in Gabrg2+/Q390X mice.
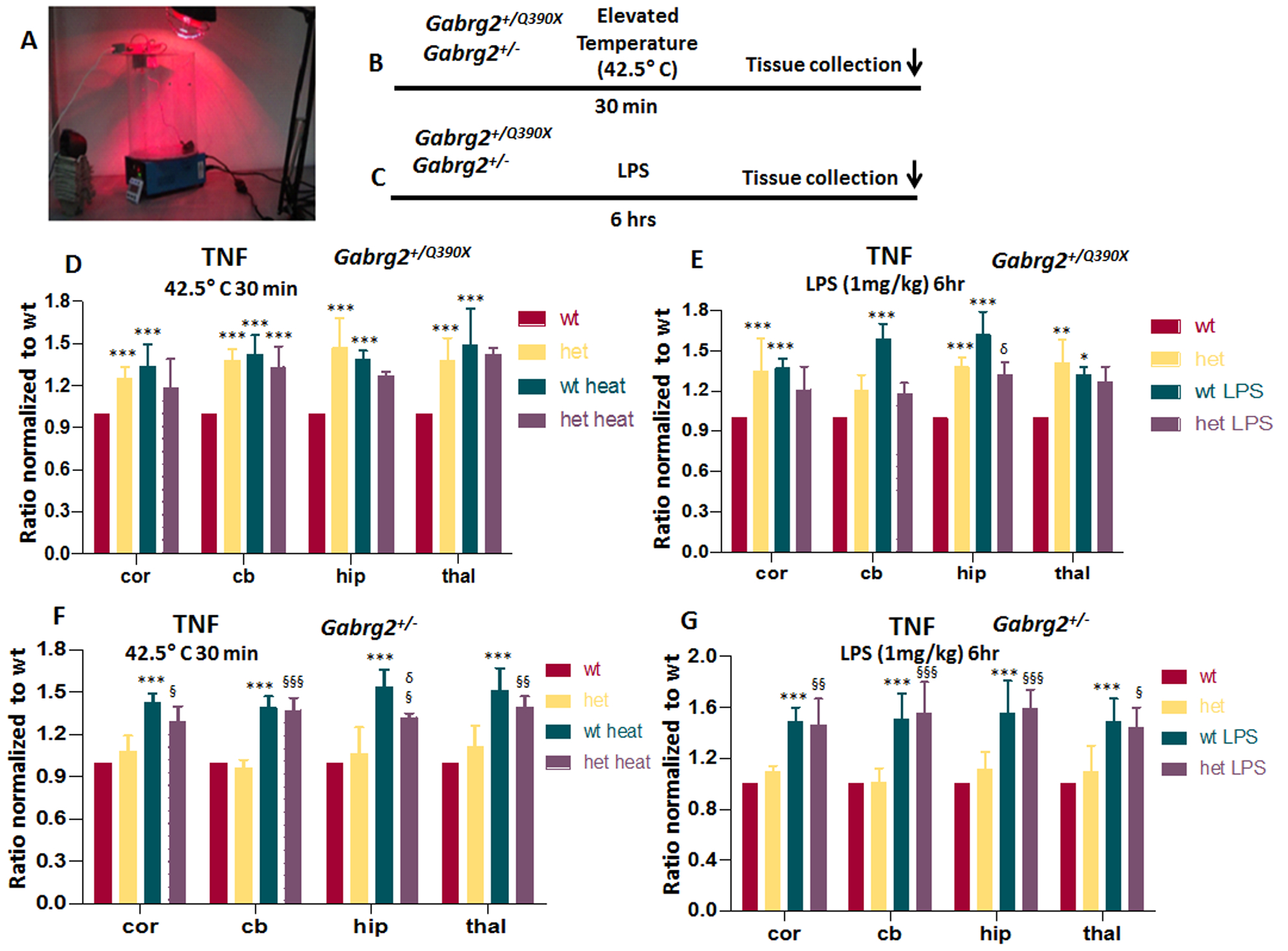
(A) The temperature induction apparatus and heating setup are shown. (B, C) A diagram of stress with temperature elevation (B) or lipopolysaccharide (LPS) treatment (C) procedure in mice is shown. In B, the mice were heated up with a heating lamp and the core temperature was maintained for 30 min followed by tissue harvest. In C, a single dose of LPS (1 mg/kg i.p) was injected to induce neuroinflammation and the brain was harvested 6hr after drug administration. D-G. The brains from 6–8 months old Gabrg2+/Q390X (D, E) or Gabrg2+/− mice (F, G) treated with elevated temperature (D, F) or LPS (E, G) were dissected and processed for measurement of pro-inflammatory cytokines. Equal amounts of protein lysates (30μg) from each brain region were determined for TNF with enzyme-linked immunosorbent assay (ELISA). The measurements in the het was normalized to the same brain region of the wt without temperature elevation (D, F) or LPS treatment (E, G). Data were presented as mean ± S.E.M (*p < 0.05, **p < 0.01, ***p < 0.001 vs wt, δ P< 0.05 vs wt heat, § p<0.05, §§ P<0.01, §§§ P<0.001 vs het heat). In D, N=4–5 mice for each group. In E, F, G, N=4 mice for each group.
The Gabrg2+/Q390X mice had accumulation of the mutant protein which may cause ER stress but this accumulation was not presented in the Gabrg2+/− mice
We have extensively studied the trafficking of γ2 subunits and identified that trafficking deficiency is a major pathology for GABRG2 mutations associated with epilepsy. The chronic accumulation of the mutant protein may disturb intracellular signaling including pro-inflammation molecules. To determine the underlying mechanisms for the increased proinflammatory cytokine expression in Gabrg2+/Q390X and the difference in cytokine expression in Gabrg2+/Q390X and Gabrg2+/− mice, we compared the γ2 subunit expression in the somatic region of neurons in layers IV-VI of somatosensory cortex of both Gabrg2+/Q390X KI mice and Gabrg2+/− KO mice.
We co-labeled the γ2 subunits with the cell nuclei marker TO-PRO-3. We surveyed the whole brain but specifically investigated layers IV-VI in the somatosensory cortex for both Gabrg2+/Q390X KI mice and Gabrg2+/− KO mice (Figure 4A–C). Compared with the wildtype littermates, the het KI mice had increased γ2 subunits in neuronal soma (52.1± 1.6, n = 12 for het vs 29.4 ± 4.9, n = 12 for wt). By contrast, compared to the wildtype mice, the het KO mice had reduced fluorescence signal of γ2 subunits in neuronal soma (28.6 ± 2.1, n = 14 for wt vs 19.3 ± 4.1, n = 11 for het) (Figure 4D). It is worth noting that previous studies have demonstrated that the synaptic γ2 subunits were reduced in both het KI and het KO mice11. This study indicates that the somatic distribution of the mutant protein is different although both had reduced synaptic functional γ2 protein.
Figure 4. The Gabrg2+/Q390X mice, but not the Gabrg2+/− mice, had toxic accumulation of the trafficking deficient γ2 subunits.
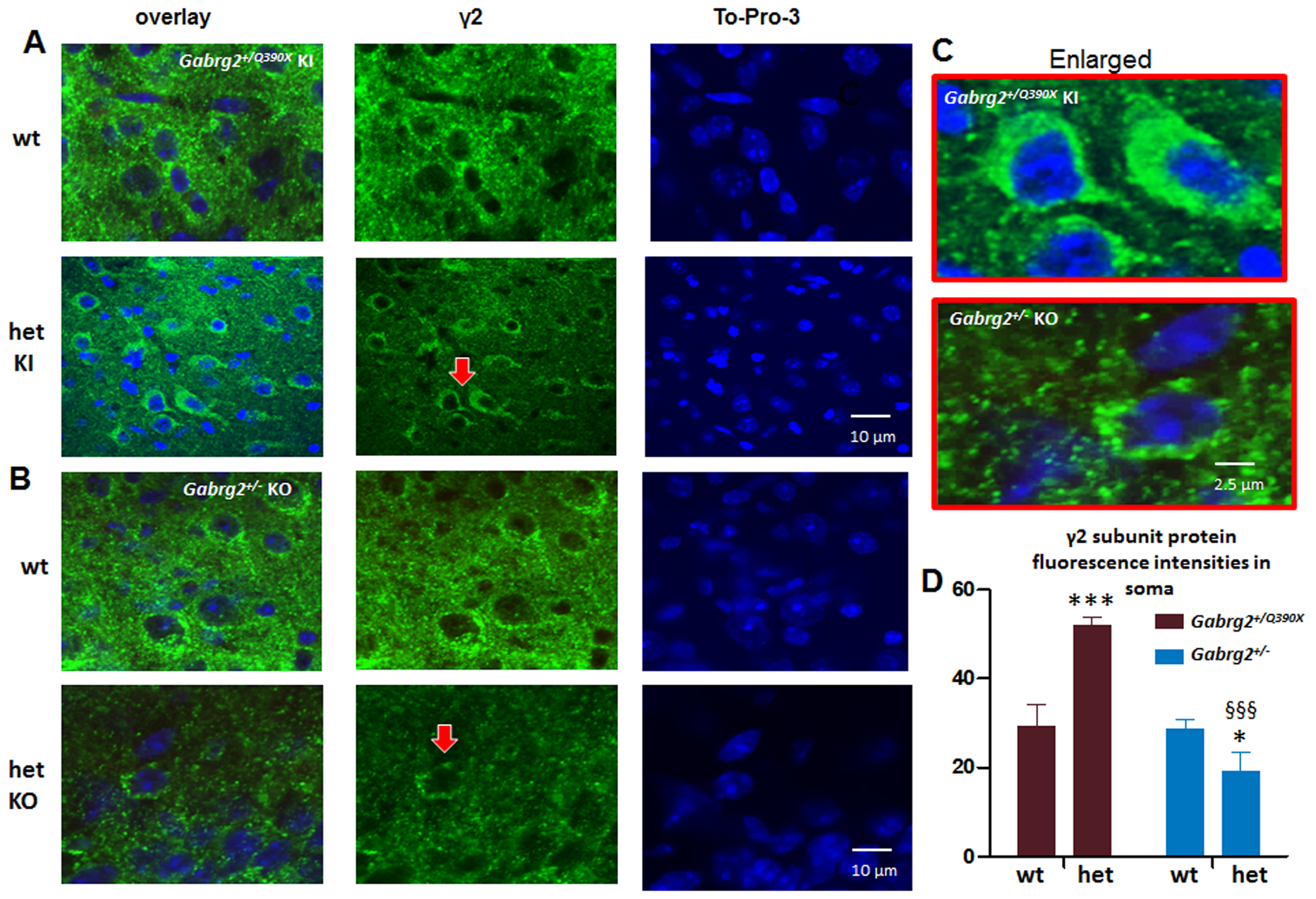
A, B. Brains from Gabrg2+/Q390X knockin (KI) (A) or Gabrg2+/− knockout mice (KO) (B) were short fixed in 4% PFA for 30 min and then processed for immunostaining. Images from cortical layers 5–6 in the somatosensory region of brain section were stained with rabbit anti-γ2 subunit (green) antibody and the nuclei marker TO-PRO-3. C. The arrow pointed regions in A and B were enlarged for a better visualization. D. The γ2 subunit fluorescence intensities were analyzed by ImageJ. The γ2 subunit fluorescence signals in somatic region of each individual neurons in the whole sampled region were measured by subtracting the background value in the nuclei region from the total raw. Data were presented as mean ± S.E.M (***p < 0.05; ***p < 0.001 vs wt; §§§ P< 0.05 vs het Gabrg2+/Q390X, In Gabrg2+/Q390X , N=12 sections for wt and het. In Gabrg2+/− , N=14 sections for wt and 11 for het. Brain sections were obtained from 4 mice in each group.
The mutant γ2(Q390X) protein had increased interaction with ER chaperone proteins and ER stress in cells and mice
The presence of the mutant protein causes the unfolded protein response (UPR) and ER stress31. To adapt to an increased protein load inside the ER, cells will increase protein degradation to remove the mutant protein by engaging the ER resident chaperones such as GRP78 and calnexin. Compared with the wildtype, the mutant γ2(Q390X) subunits had increased steady state level of protein expression (Figure 5A, E) (1 ± 2.494; wt vs mut) and increased conjugation with either GRP78 (1 ± 2.425; wt vs mut) or calnexin (1 ± 2.725; wt vs mut) (Figure 5B, F). We then determined the ER stress of the cells expressing the mutant protein. GADD153 (CHOP) is a recognized ER stress marker. Mouse L929 cells are a recognized tool for studying ER stress6 and were mock transfected or transfected with α1β2γ2S or α1β2γ2S(Q390X) subunits for 48 hr and tunicamycin (10 μg/ml) was applied 16 hours before harvest. The mouse L929 cells have baseline GADD153 expression. When normalized to the baseline GADD153 expression from mock transfected cells, the GADD153 expression was substantially increased with tunicamycin treatment. When transfected with the wildtype α1β2γ2S receptor subunits, the GADD153 expression level was similar to baseline but with transfection of the mutant α1β2γ2S(Q390X) receptor subunits, the GADD153 expression level was higher (Figure 5C, G) (1 for con, 4.32 ± 0.36 for con+Tuni.; 1.09 ± 0.12 for wt; 2.56 ± 0.21 for mut).
Figure 5. The mutant γ2(Q390X) subunit protein increased conjugation with ER chaperones and caused ER stress.
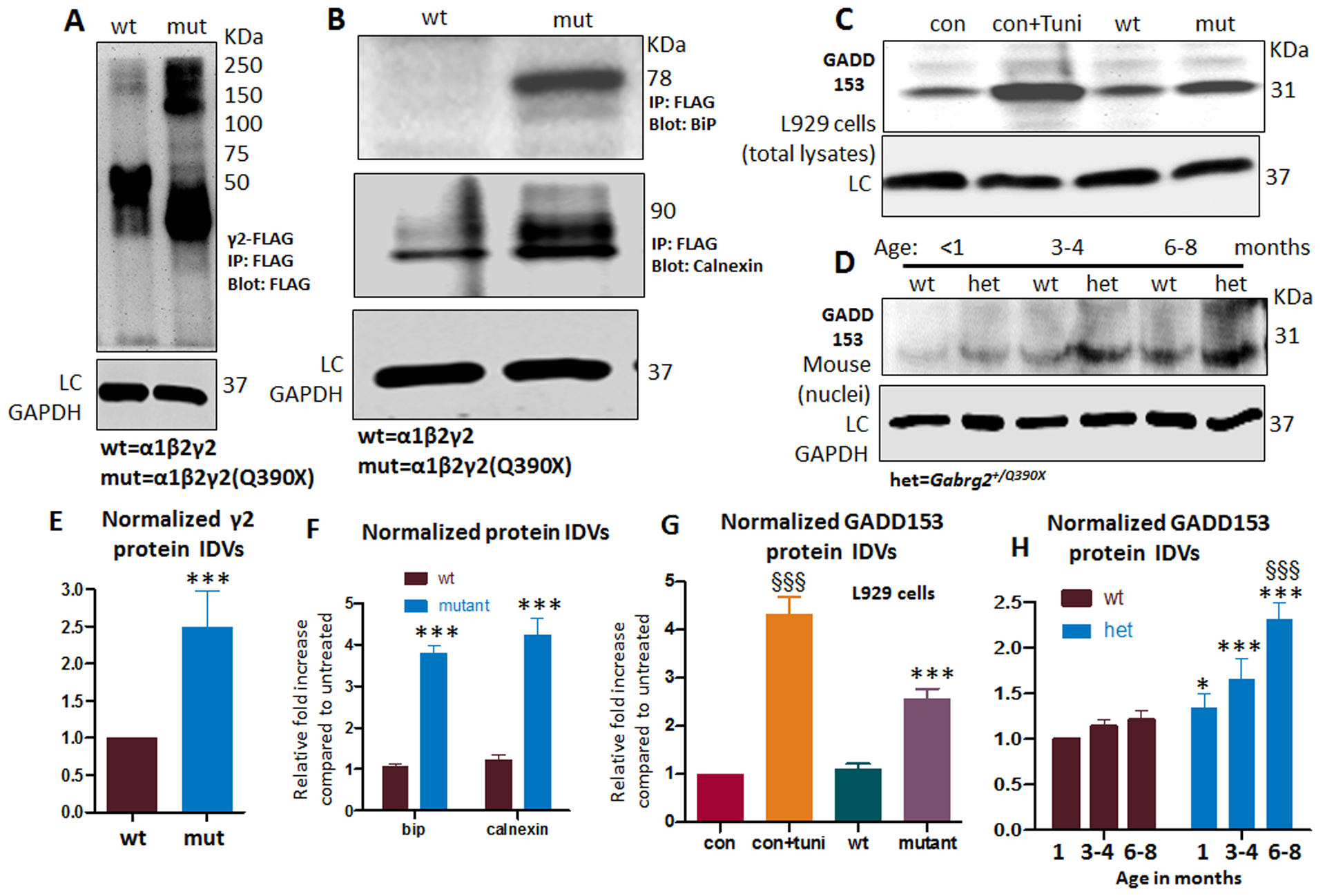
(A-B) Total lysates from HEK293T cells expressing γ2FLAG subunits (cDNA 3μg) were immunoprecipitated with FLAG beads and immunoblotted with anti-FLAG antibody (A) or GRP78 (B, upper panel) or calnexin (B, lower panel) antibody. The γ2FLAG subunits, GRP78, and calnexin were normalized to the wildtype condition. In A, the γ2FLAG subunits were measured from about 75 KDa to 250 KDa. Individual wildtype γ2FLAG subunits were predicted to be 55 KDa and migrated at about 50 KDa, and individual FLAG-tagged truncated mutant γ2(Q390X) subunits migrated at lower molecular masses predicted to be about 40 KDa. (C). Total lysates from mouse L929 cells expressing wildtype or mutant γ2 subunits in combination with the wildtype partnering α1 and β2 subunits were analyzed by SDS-PAGE and immunoblotted with mouse monoclonal anti-GADD153 antibody. In A-D, LC stands for loading control GAPDH. (D). The nuclei portion of the total forebrain of Gabrg2+/Q390X knockin mice at different ages was purified and lysed. Equal amounts of the protein were analyzed by SDS-PAGE and immunoblotted with GADD153. (E-F). Total mutant subunit band IDVs of γ2FLAG subunits (E) or the conjugated chaperone GRP78 and calnexin (F) were normalized to the wildtype conditions. (G) The total amounts of endogenous GADD153 were normalized to untreated controls (con). (H) The endogenous GADD153 in the nuclei of forebrain in the Gabrg2+/Q390X knockin mice at different ages were normalized to wt in the youngest group (<1 month). Seizure onset is around P19 while mutant protein aggregates become detectable around 6mo. (In E, F and G, *p < 0.05, **p < 0.01, ***p<0.001 vs wt. In G, §§§ P< 0.001 vs con, †† vs P< 0.01 vs con+Tuni, N=5 batches of cells. In H, §§§ P< 0.001 vs 1 month old. In E to G, n=5 batch of cells, in H, N=4 mice for each group.)
We next wanted to determine if the mutant γ2S(Q390X) subunits cause ER stress in the heterozygous mutant mice. Because GADD15 is enriched in cell nuclei, we isolated the cell nuclei of the total forebrain tissues from mice at ages of 1 month, 3–4 months and 6–8 months. Compared with the wildtype, the Gabrg2+/Q390X KI mice at each age group had increased expression of GADD153 (Figure 5D and H). It is interesting that compared with mice at 1–2 month old, the baseline expression of GADD153 in the wildtype mice at older ages trended toward increased levels of expression (Figure 5D and H) (for wt: 1 for 1–2 month; 1.14 ± 0.08 for 3–4 months; 1.21 ± 0.11 for 6–8 months; for het: 1.34 ± 0.16 for 1–2 month; 1.66 ± 0.22 for 3–4 months; 2.31 ± 0.18 for 6–8 months).
ER stress increased proinflammatory cytokines independent of seizures.
To exclude the effect of seizures on the expression of proinflammatory cytokines, we determined the cytokine levels in cultured neurons from postnatal day 0 wildtype and Gabrg2+/Q390X KI or Gabrg2+/− KO pups. TNF was measured in the total lysates of cultured neurons. The measurements of the mutant neurons were normalized to the sister cultures of the wildtype pups in the same litters. Consistent with our findings in mouse tissue, the neurons from the heterozygous KI but not the KO pups had increased TNF (Figure 6A). However, the wildtype and the het KO but not the het KI mouse cultures had increased cytokines with tunicamycin treatment (wt = 1, 1.882 ± 0.114 for wt+Tuni, 1.476 ±0 .104 for het KI and 1.37 ± 0.05 for het KI+Tuni; 1.048 ± 0.07 for het KO and 1.59 ± 0.08 for het KO+Tuni), suggesting a ceiling effect of cytokine expression in the mutant neurons. We then determined if ER stress itself can induce the expression of proinflammatory cytokines in mouse L929 cells expressing the wildtype γ2 and the mutant γ2(Q390X) subunits in combination with the GABAA receptor α1 and β2 subunits for 48 hrs. We treated the cultured cells with tunicamycin. Administration of tunicamycin increased the TNF expression in the cells expressing the wildtype receptors. However, the treatment of tunicamycin failed to increase the TNF expression in the cells expressing the mutant γ2(Q390X) subunits. This suggested a ceiling effect of TNF induction in neurons or non-neuronal cells expressing the mutant γ2(Q390X) subunits (Figure 6B) (wt = 1, 1.876 ± 0.098 for wt+Tuni, 1.470 ± 0.09 for mut and 1.646 ± 0.085 for mut+Tuni). We also determined the expression of IL-1β and IL-6 in the same cultures treated with tunicamycin. Similar to TNF, IL-1β and IL-6 expression failed to increase as in the wildtype neurons or cells transfected with the wildtype α1β2γ2 receptors. It is worth noting that spontaneous seizures were observed in mice around postnatal day 16–19 while the mutant γ2(Q390X) subunit protein aggregates were observed in mice at around 6–8 months old. However, a germline mutation like GABRG2(Q390X) may exist from conception on, producing abundant mutant protein in the young brain. Importantly, TNF in the newborn Gabrg2+/Q390X knockin mouse cortex was increased. All three cytokines measured were increased at postnatal day 14, suggesting that neuroinflammation exists prior to seizure onset (Figure 6C) . The data indicated that ER stress increased proinflammatory cytokines independent of seizures and there exists a complex interplay between neuroinflammation and seizures in the brain.
Figure 6. ER stress increased TNF in cultured cells but the cells expressing the mutant γ2(Q390X) had dampened response.
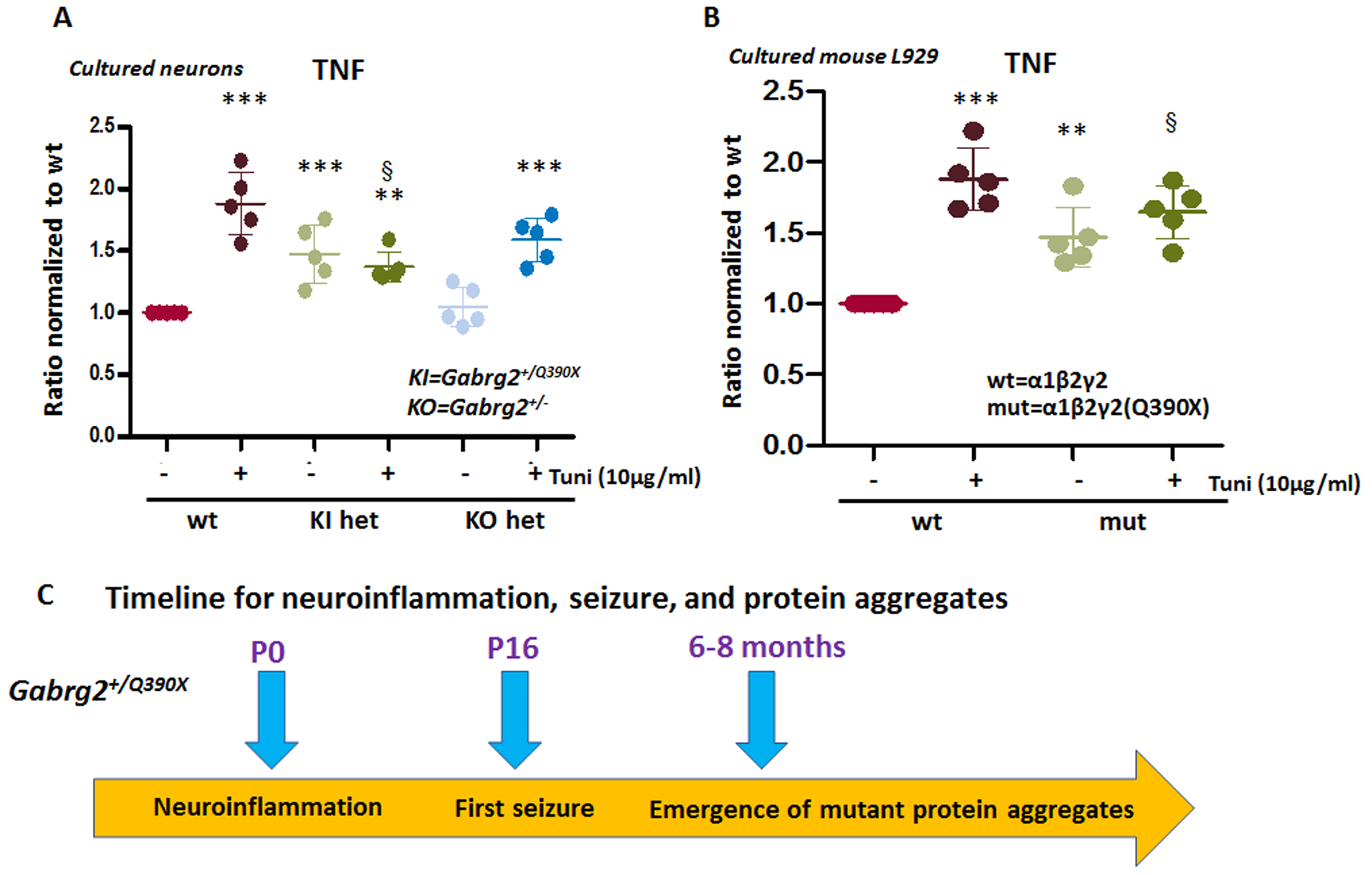
A. The forebrain cortex of postnatal day 0 old pups of Gabrg2+/Q390X and Gabrg2+/− mouse lines were dissected, disassociated and the neurons were cultured for 15 days in dish before harvest. Equal amounts of cell protein lysates (30μg) from the wildtype or the heterozygous mice were determined for TNF with enzyme-linked immunosorbent assay (ELISA). The measurements in the het were normalized to the wildtype sister cultures. B. Mouse L929 cells were transfected with the wildtype or the mutant γ2(Q390X) subunits in combination with the wildtype α1 and β2 subunits for 48 hrs. Sister cultures were treated with or without tunicamycin (10μg/ml) for 16 hrs before harvest. Equal amounts of cell protein lysates (30μg) from each group were determined for TNF with ELISA. (**p < 0.01 vs wt, ***p < 0.001 vs wt, § p<0.05 vs Wt+Tuni. n = 4–5 batches of cultures. Data were presented as mean ± S.E.M).
Discussion
Inflammatory cytokines in the brain are emerging as important components in processes of major neurological diseases including ischemia, neurodegenerative diseases and demyelinating diseases32. In epilepsy, brain inflammation has been identified as a contributor to seizure susceptibility and epileptogenesis in various types of seizures and epilepsies2;3. It is established that cytokines and their receptors are distributed widely in the CNS as well as in PNS, and their expression is influenced by changes in tissue homeostatsis33. In CNS, expression of cytokines such as TNF, IL-6, and IL-β is thought to originate predominantly from glia and microglia cells34 but can also be expressed from neuronal-like cells or neurons35;36. In CNS, cytokines can exert their function through both traditional engagement of their receptors37 and through less traditional means such as modulation of neurotransmitter receptor function38;39.
This is the first time to our knowledge that increased expression of proinflammatory cytokines in a mouse model of genetic epilepsy like the Gabrg2+/Q390X knockin, has been identified, though increased inflammation has been previously characterized in acquired epilepsy4. Cytokines in the brain, especially TNF, have been suggested to play both a pathogenic role40 or protective roles24. In neurological autoimmune diseases such as multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE), TNF exerts damaging effects on oligodendrocytes, the myelin-producing cell of the central nervous system (CNS), and myelin itself3;40. However, it has been proposed that TNF may protect neurons via enhancing calbindin production and increase calcium homeostasis and prevent glutamate-mediated neuronal injury41. Additionally, TNF can provide neuronal protection against β-amyloid toxicity, possibly through suppressing the accumulation of reactive oxygen intermediates42. Together, this suggests TNF plays a dual role of neuronal injury and neuroprotection43;44.
Increased neuroinflammation in Gabrg2+/Q390X mice is likely before seizure onset. Based on the profiling of cytokines during different developmental stages, the expression of cytokines including both TNF and IL-1β in the forebrain cortex were increased by P14, although the magnitude was less compared with the expression at later developmental stages (Figure 1D, E). In fact, TNF (Figure 1D) was increased in the newborn mouse cortex, suggesting that neuroinflammation could exist even in embryonic brain. By contrast, the spontaneous seizures are first observed over 2 weeks later which is normally around postnatal day 16–199. It is not surprising given the fact that a germline mutation like GABRG2(Q390X) exists from conception on. The mutant protein would chronically accumulate inside host cells over time depending on the cell capacity of protein disposal. This may help explain why spontaneous seizures are only observed in Gabrg2+/Q390X mice but not in Gabrg2+/− mice that has no mutant protein produced. However, it is unknown as to what extent the seizures are contributed by increased cytokines vs reduced GABAA receptor channel function nor the dynamic interplay between seizure activity and ER stress during a chronic process of recurring seizures and constant presence of the mutant protein.
Increased expression of proinflammatory cytokines in the Gabrg2+/Q390X knockin mice is unlikely to receive contributions from peripheral sources such as macrophages, T lymphocytes and B lymphocytes. Seizures or fever may increase blood brain barrier permeability (BBB)45 so it is possible that the increased cytokine expression could result from impaired BBB function. However, the expression of cytokines in plasma were the same between the wildtype and the heterozygous Gabrg2+/Q390X knockin mice, suggesting that the increased cytokines in the mouse brain tissues were unlikely resulted from peripheral sources or due to the stimulation of peripheral cytokines upon neurons or astrocytes46. Additionally, the baseline level of the cytokines in blood was very low compared with the level of cytokines in the brain. The studies from the cultured neurons and L929 cells transiently expressing the mutant GABRG2(Q390X) subunits also indicated increased cytokines in the mutation-bearing cells, consistent with the findings from Gabrg2+/Q390X KI mice, thus limiting the impact from seizures and infiltration of myeloid cells from peripheral blood system, and showing a more direct inflammatory effect from the mutant protein.
The difference in expression of proinflammatory cytokines in the Gabrg2+/Q390X knockin and Gabrg2+/− knockout mice is likely due to the production of the mutant protein. It is unequivocal that the γ2(Q390X) subunit protein causes mutant protein misfolding and aggregation due to increased protein stability and slow degradation47. However, the mutant protein accumulation is absent in the Gabrg2+/− knockout mice because there is no mutant protein produced11. Previous studies indicate the Gabrg2+/Q390X mice had a more severe epilepsy phenotype than Gabrg2+/− KO mice. However, it merits more studies to elucidate the contribution of increased neuroinflammation in disease phenotype manifestation.
Our findings indicate that in Gabrg2+/Q390X mice, ER stress alone could increase cytokine expression. The misfolded mutant γ2(Q390X) subunit protein was retained inside the ER, increased interaction with ER chaperone proteins and increased ER stress in both Gabrg2+/Q390X mice and cells expressing the mutant γ2(Q390X) subunit (Figure 5). This in vitro data does not exclude that in vivo the occurrence of seizures may contribute to the cytokine changes in Gabrg2+/Q390X vs Gabrg2+/− mice. However, the data indicate more complicated cellular cascades could occur in the Gabrg2+/Q390X mice with severe epilepsy due to an ER retention-causing mutation but not in Gabrg2+/− mice11. Pharmacological increase of ER stress with tunicamycin increased cytokine expression in the wildtype cultures further validated this notion. This thus establishes a link of ER stress due to mutant protein accumulation and cytokine increase in genetic epilepsy.
It is of note that pharmacological upregulation of ER stress failed to further increase cytokine expression in the mutant neuronal cultures. This ceiling effect of cytokine induction was also observed with both heat stress and LPS treatment (Figure 3). This suggests the capacity of cytokine production upon further cellular stress is dampened in the Gabrg2+/Q390X mice. This finding is in line with recent studies that some endogenous anti-inflammatory pathways and resolving factors such as n-3 docosapentaenoic acid-derived protectin D148 and glucocorticoid receptor (GR)-annexin A149 are reduced in experimental or human epileptic tissues whereas external supplementation improved seizure outcome. This suggests anti-inflammation could be beneficial for some forms of genetic epilepsy with increased neuroinflammation. However, the production of proinflammatory cytokines and their role in modulating neurotransmitter receptors such as GABAA receptors merits more in-depth characterizations. It has been reported that GABAA receptor currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy50, implicating that increased neuroinflammation can directly affect GABAergic neurotransmission and alter brain excitability. Nevertheless, this study for the first time reported increased neuroinflammation—a major theme mechanism for acquired epilepsy—in a genetic mouse model with an ion channel gene mutation. This thus proposes a concrete link between acquired and genetic epilepsy, for epileptogenesis in a chronic process.
Supplementary Material
Supplementary Figure 1. The proinflammatory cytokines were unchanged in the blood plasma of Gabrg2+/Q390X KI mice. A. The proinflammatory cytokines tumor necrosis factor alpha (TNF), interleukin-1beta (IL-1β) and IL-6 in 50 μl plasma from periphery blood of the wildtype (wt) or the heterozygous Gabrg2+/Q390X mice untreated or treated at 40° C for 30 min were measured with enzyme-linked immunosorbent assay (ELISA). The measurements in the het were normalized to the wt.
Supplementary table 1. The data showing that proinflammatory cytokines were unchanged in the brain of Gabrg2+/− KO mice.
Key Points.
The Dravet syndrome epilepsy mouse model Gabrg2+/Q390X had increased neuroinflammation.
Proinflammatory cytokines such as TNF, IL−−1β and IL-6 were increased in the brain but not in the plasma of Gabrg2+/Q390X mice.
The proinflammatory cytokines were not increased in the Gabrg2+/− knockout mice.
Further cellular stress induced cytokine production in wildtype but not Gabrg2+/Q390X mice.
The increase of proinflammatory cytokines was likely caused by ER stress in Gabrg2+/Q390X mice.
Acknowledgements:
Research was supported by grants from Citizen United for Research in Epilepsy (CURE), Dravet Syndrome Foundation (DSF), Vanderbilt Brain Institute Award and Vanderbilt Clinical and Translation Science Award and NINDS R01 NS082635 to J.Q.K. Special thanks to Dr. Lan Wu from Department of Pathology, Microbiology and Immunology for generous sharing reagents and providing critical consultation on experimental design and data interpretation. Imaging data were performed in part through the VUMC Cell Imaging Shared Resource.
Footnotes
Conflict of Interest statement: None of authors declared any conflict of interest.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines
Reference List
- 1.McElroy PB, Liang LP, Day BJ, Patel M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp Neurol 2017;298:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker-Haliski ML, Loscher W, White HS, Galanopoulou AS. Neuroinflammation in epileptogenesis: Insights and translational perspectives from new models of epilepsy. Epilepsia 2017;58 Suppl 3:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol 2019;15:459–72. [DOI] [PubMed] [Google Scholar]
- 4.Aronica E, Bauer S, Bozzi Y et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia 2017;58 Suppl 3:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufer C, Chhatbar C, Broer S et al. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc Natl Acad Sci U S A 2018;115:E8929–E8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang JQ, Shen W, Macdonald RL. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann Neurol 2013;74:547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai K, Wang J, Eissman J et al. A missense mutation in SLC6A1 associated with Lennox-Gastaut syndrome impairs GABA transporter 1 protein trafficking and function. Exp Neurol 2019;320:112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harkin LA, Bowser DN, Dibbens LM et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 2002;70:530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neurosci 2015;18:988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reid CA, Kim T, Phillips AM et al. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology 2013;80:1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warner TA, Shen W, Huang X, Liu Z, Macdonald RL, Kang JQ. DIfferential molecular and behavioral alterations in mouse models of GABRG2 haploinsufficiency versus dominant negative mutations associated with human epilepsy. Hum Mol Genet 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry 2010;68:512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang CQ, McMahon B, Dong H et al. Molecular basis for and chemogenetic modulation of comorbidities in GABRG2-deficient epilepsies. Epilepsia 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunther U, Benson J, Benke D et al. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A 1995;92:7749–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delahanty RJ, Kang JQ, Brune CW et al. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol Psychiatry 2011;16:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang JQ, Shen W, Macdonald RL. The GABRG2 mutation, Q351X, associated with generalized epilepsy with febrile seizures plus, has both loss of function and dominant-negative suppression. J Neurosci 2009;29:2845–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci 2010;30:13895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang JQ, Shen W, Macdonald RL. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABA(A) receptor GABRA1 PTC mutation, 975delC, S326fs328X. J Neurosci 2009;29:2833–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.SPORN MB WANKO T, DINGMAN W. The isolation of cell nuclei from rat brain. J Cell Biol 1962;15:109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oakley JC, Kalume F, Frank HY, Scheuer T, Catterall WA. Temperature-and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proceedings of the National Academy of Sciences 2009;106:3994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katsuno M, Sang C, Adachi H et al. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 2005;102:16801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsson R, Bajic VB, Suzuki H et al. Transcriptional network dynamics in macrophage activation. Genomics 2006;88:133–42. [DOI] [PubMed] [Google Scholar]
- 23.Gilchrist M, Thorsson V, Li B et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 2006;441:173–8. [DOI] [PubMed] [Google Scholar]
- 24.Gahring LC, Carlson NG, Kulmar RA, Rogers SW. Neuronal expression of tumor necrosis factor alpha in the murine brain. Neuroimmunomodulation 1996;3:289–303. [DOI] [PubMed] [Google Scholar]
- 25.Toulmond S, Parnet P, Linthorst AC. When cytokines get on your nerves: cytokine networks and CNS pathologies. Trends Neurosci 1996;19:409–10. [DOI] [PubMed] [Google Scholar]
- 26.Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther 1996;69:85–95. [DOI] [PubMed] [Google Scholar]
- 27.Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neurosci 2015;18:988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang JQ, Macdonald RL. Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol 2016;73:1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boillot M, Morin-Brureau M, Picard F et al. Novel GABRG2 mutations cause familial febrile seizures. Neurol Genet 2015;1:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warner TA, Liu Z, Macdonald RL, Kang JQ. Heat induced temperature dysregulation and seizures in Dravet Syndrome/GEFS+ Gabrg2+/Q390X mice. Epilepsy Res 2017;134:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol 2009;39:81–9. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez AI, de HR, Salobrar-Garcia E et al. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front Aging Neurosci 2017;9:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothwell NJ, Hopkins SJ. Cytokines and the nervous system II: Actions and mechanisms of action. Trends Neurosci 1995;18:130–6. [DOI] [PubMed] [Google Scholar]
- 34.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol 1993;150:2659–67. [PubMed] [Google Scholar]
- 35.Tchelingerian JL, Quinonero J, Booss J, Jacque C. Localization of TNF alpha and IL-1 alpha immunoreactivities in striatal neurons after surgical injury to the hippocampus. Neuron 1993;10:213–24. [DOI] [PubMed] [Google Scholar]
- 36.Tchelingerian JL, Vignais L, Jacque C. TNF alpha gene expression is induced in neurones after a hippocampal lesion. Neuroreport 1994;5:585–8. [DOI] [PubMed] [Google Scholar]
- 37.Schobitz B, Voorhuis DA, De Kloet ER. Localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Neurosci Lett 1992;136:189–92. [DOI] [PubMed] [Google Scholar]
- 38.Miller LG, Galpern WR, Dunlap K, Dinarello CA, Turner TJ. Interleukin-1 augments gamma-aminobutyric acidA receptor function in brain. Mol Pharmacol 1991;39:105–8. [PubMed] [Google Scholar]
- 39.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015;96:70–82. [DOI] [PubMed] [Google Scholar]
- 40.Chung IY, Benveniste EN. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J Immunol 1990;144:2999–3007. [PubMed] [Google Scholar]
- 41.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron 1994;12:139–53. [DOI] [PubMed] [Google Scholar]
- 42.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U S A 1995;92:9328–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinberg MS, Blake BL, McCown TJ. Opposing actions of hippocampus TNFalpha receptors on limbic seizure susceptibility. Exp Neurol 2013;247:429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balosso S, Ravizza T, Aronica E, Vezzani A. The dual role of TNF-alpha and its receptors in seizures. Exp Neurol 2013;247:267–71. [DOI] [PubMed] [Google Scholar]
- 45.Claudio L, Martiney JA, Brosnan CF. Ultrastructural studies of the blood-retina barrier after exposure to interleukin-1 beta or tumor necrosis factor-alpha. Lab Invest 1994;70:850–61. [PubMed] [Google Scholar]
- 46.Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 1999;163:3963–8. [PubMed] [Google Scholar]
- 47.Kang JQ, Shen W, Lee M, Gallagher MJ, Macdonald RL. Slow degradation and aggregation in vitro of mutant GABAA receptor gamma2(Q351X) subunits associated with epilepsy. J Neurosci 2010;30:13895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frigerio F, Pasqualini G, Craparotta I et al. n-3 Docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain 2018;141:3130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zub E, Canet G, Garbelli R et al. The GR-ANXA1 pathway is a pathological player and a candidate target in epilepsy. FASEB J 2019;33:13998–4009. [DOI] [PubMed] [Google Scholar]
- 50.Roseti C, van Vliet EA, Cifelli P et al. GABAA currents are decreased by IL-1beta in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis 2015;82:311–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The proinflammatory cytokines were unchanged in the blood plasma of Gabrg2+/Q390X KI mice. A. The proinflammatory cytokines tumor necrosis factor alpha (TNF), interleukin-1beta (IL-1β) and IL-6 in 50 μl plasma from periphery blood of the wildtype (wt) or the heterozygous Gabrg2+/Q390X mice untreated or treated at 40° C for 30 min were measured with enzyme-linked immunosorbent assay (ELISA). The measurements in the het were normalized to the wt.
Supplementary table 1. The data showing that proinflammatory cytokines were unchanged in the brain of Gabrg2+/− KO mice.