

Abstract
There remain no approved therapies for rare but devastating neuronopathic glyocosphingolipid storage diseases, such as Sandhoff, Tay-Sachs, and Gaucher disease type 3. We previously reported initial optimization of the scaffold of eliglustat, an approved therapy for the peripheral symptoms of Gaucher disease type 1, to afford 2, which effected modest reductions in brain glucosylceramide (GlcCer) in normal mice at 60 mg/kg. The relatively poor pharmacokinetic properties and high Pgp-mediated efflux of 2 prompted further optimization of the scaffold. With a general objective of reducing topological polar surface area, and guided by multiple metabolite identification studies, we were successful at identifying 17 (CCG-222628), which achieves remarkably greater brain exposure in mice than 2. After demonstrating an over 60-fold improvement in potency over 2 at reducing brain GlcCer in normal mice, we compared 17 with Sanofi clinical candidate venglustat (Genz-682452) in the CBE mouse model of Gaucher disease type 3. At doses of 10 mg/kg, 17 and venglustat effected comparable reductions in both brain GlcCer and glucosylsphingosine. Importantly, 17 achieved these equivalent pharmacodynamic effects at significantly lower brain exposure than venglustat.
Keywords: Glucosylceramide synthase, eliglustat tartrate, Gaucher disease, blood—brain barrier
Graphical Abstract
INTRODUCTION
Glycosphingolipid storage diseases, although rare, represent a significant fraction of all lysosomal storage disorders by prevalence.1 In most cases, these disorders result from the loss of an enzyme or enzyme activator that is required for the catabolism of a glucosylceramide based glycolipid. These diseases include Gaucher and Fabry disease, Tay-Sachs and Sandhoff disease, and gangliosidosis GM1. Gaucher disease type 1 and Fabry disease, in their classic clinical phenotypes, lack central nervous system manifestations, although Parkinson’s disease may be a long-term complication of Gaucher, and stroke is common in Fabry due to vascular accumulation of glycolipids.2 As such, the standard of care has been the use of enzyme replacement therapy in which a recombinant enzyme, β-glucocerebrosidase or α-galactosidase A, is effective in lowering lysosomal glycolipid storage due to the ability of tissues to bind, endocytose, and traffick lysosomal enzymes through recognition of mannose or mannose-6-phosphate groups. Unfortunately, enzyme replacement has not proven suitable for the treatment of diseases with central nervous system (CNS) involvement due to the failure to deliver recombinant enzyme across the blood brain barrier.
A more recent therapeutic strategy has been the use of small molecular entities that reversibly inhibit enzymes central to glycolipid synthesis.3 This approach, termed substrate reduction therapy, has primarily targeted glucosylceramide (GlcCer) synthase. This enzyme uses ceramide and UDP-glucose as substrates to form GlcCer which accumulates in Gaucher disease and which is the base cerebroside in glycolipids that accumulate in Fabry disease and the gangliosidoses. In 2014, eliglustat tartrate4 (1, Figure 1) was approved as the first standalone oral agent for the treatment of Gaucher disease type 1 based on clinical studies that demonstrated comparable efficacy in preventing and reversing key clinical features of the disease including splenomegaly, hepatomegaly, anemia, and thrombocytopenia and its noninferiority in patients previously stabilized on imiglucerase.5,6 The safety profile of eliglustat tartrate in clinical studies was favorable as well. Despite its low nanomolar potency against GlcCer synthase, eliglustat exhibits a high degree of selectivity vs a number of closely related enzymes, including lysosomal glucocerebrosidase (GBA1) and nonlysosomal glucosylceramidase (GBA2).7 Unfortunately, eliglustat fails to achieve significant exposure in the brain and thus is unsuited for the treatment of those glycosphingolipidoses with CNS involvement which include Gaucher disease types 2 and 3, the GM2 gangliosidoses (Tay-Sachs and Sandhoff disease), and GM1 gangliosidosis.
Figure 1.

Lead glucosylceramide synthase inhibitors.
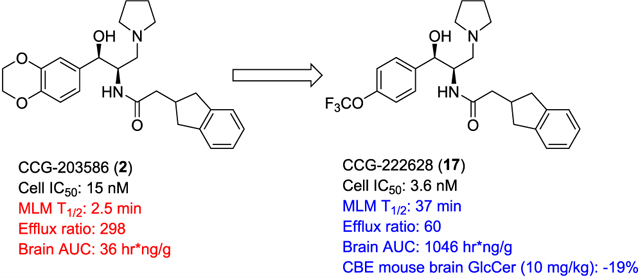
We previously reported on the use of the eliglustat scaffold as the basis for the design of CNS-permeant glucosylceramide synthase inhibitors.8 Based on a computational comparison of eliglustat to CNS-active FDA approved compounds, modifications to the carboxamide N-acyl group of eliglustat were made to lower topological polar surface area (TPSA) and rotatable bond number. One analogue CCG-203586 (2, Figure 1) was identified that inhibited glucosylceramide synthase at low nanomolar concentrations and was able to lower glucosylceramide in wild-type mouse brain. We subsequently demonstrated that this compound could partially prevent the accumulation of ganglioside GM2 in a mouse model of Sandhoff disease.9 While conceptually promising, 2 is characterized by rapid clearance and relatively poor brain exposure in mice. The present work reports on the further optimization of eliglustat analogues with a continued focus on properties that determine CNS exposure, combined with a metabolite-guided optimization of pharmacokinetics. We now report the identification of an analogue of eliglustat with significantly greater CNS exposure and activity.
Sanofi/Genzyme has reported CNS-active GlcCer synthase inhibitors based on a novel pharmacophore.10 One compound, venglustat (3, Figure 1), has been shown to reduce brain glycolipids and increase lifespan in murine models of both Gaucher disease type 3 and Sandhoff.11,12 Compound 3 has progressed to Phase II clinical trials for Fabry, and recruitment is underway for Gaucher type 3 patients. Compound 3 is used as a comparator in the present studies, and we were able to successfully demonstrate in this work that our new lead has comparable pharmacodynamic efficacy in vivo in a mouse model of Gaucher type 3.
RESULTS AND DISCUSSION
Chemistry.
All new compounds were prepared by the route shown in Scheme 1, which has been previously reported.13 Commercial benzaldehydes 4 were condensed with (S)-5-phenylmorpholin-2-one under Dean−Stark conditions to produce aminals 5 as single diastereomers. Lactone opening with a secondary amine, followed by acidic hydrolysis provided amides 6. After hydride-mediated reduction of the amide, the chiral auxiliary was removed by catalytic hydrogenation to give primary amines 8. Final N-acylation with N-hydroxysuccinimidyl esters or carbodiimide-mediated coupling to carboxylic acids afforded the desired final compounds 9, which were isolated as single diastereomers within the limits of NMR detection, thereby ruling out racemization of either chiral center.
Scheme 1.

Synthetic Route to New Eliglustat Analoguesa
aReagents and conditions: (a) (S)-5-phenylmorpholin-2-one, toluene, azeotropic reflux, 24 h; (b) R3R4NH, heat; (c) aq HCl; (d) LiAlH4; (e) BH3; (f) H2, Pd/C; (g) R5CH2COO-Suc; (h) R5CH2COOH, EDC.
Assessment of In Vitro Biological Activity.
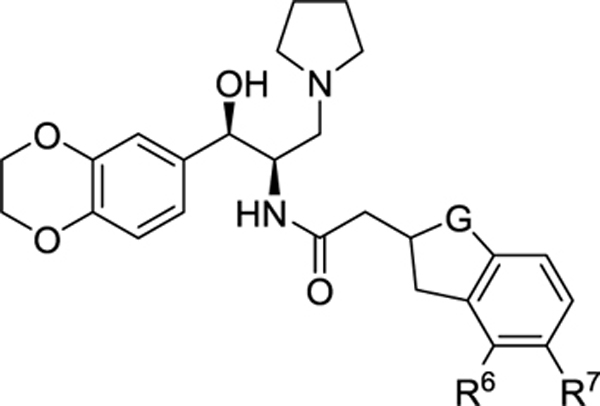
All new analogues were assayed for inhibitory activity against GlcCer synthase in two different formats: broken cell membrane preparations (“crude enzyme”) and intact MDCK cells, and results are reported in Tables 1–3. The broken cell assay is a direct measurement of the glycolipid synthase activity in an assay optimized for the detection of glucosylceramide synthesis. The Table 1. Modification of the Indane MDCK cell assay measures the ability of a compound to lower the cellular content of glucosylceramide over 24 h in a viable cell in the setting of its endogenous glycolipid metabolism. We have previously described these assays in detail.8,14
Table 1.
Modification of the Indane
 | ||||||
|---|---|---|---|---|---|---|
| no. | G | R6 | R7 | crude enzyme IC50 (nM) | MDCKs cell IC50 (nM) | MLM T1/2 (min)a |
| 2 | CH2 | H | H | 27 | 15.3 | 2.5 |
| 10 | C=O | H | H | 193 | >30b | |
| 11 | CH2 | H | F | 14 | 1.3 | 1.1 |
| 12 | CH2 | F | H | 36.5 | 7.2 | 2.6 |
Half-life when incubated with mouse liver microsomes.
28% inhibition at 30 nM.
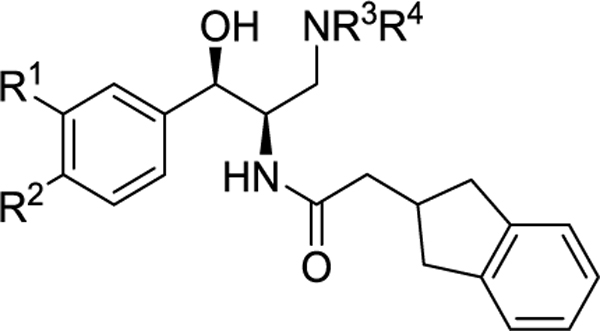
Table 3.
Modification of the Amine
 | ||||||
|---|---|---|---|---|---|---|
| no. | R1 | R2 | NR3R4 | crude enzyme IC50 (nM) | MDCK cells IC50 (nM) | MLM T1/2 (min)a |
| 2 | -OCH2CH2O- | pyrrolidine | 27 | 15.3 | 2.5 | |
| 23 | -OCH2CH2O- | (R)-3-F-pyrrolidine | 43.6 | 6.6 | 1.4 | |
| 24 | -OCH2CH2O- | azetidine | 112 | 21.1 | ||
| 17 | H | OCF3 | pyrrolidine | 110 | 3.6 | 37 |
| 25 | H | OCF3 | (R)-3-F-pyrrolidine | >300 | 23.2 | |
| 26 | H | OCF3 | azetidine | >300 | 23.9 | >60 |
Half-life when incubated with mouse liver microsomes.
Structure−Activity Relationships.
Although in our previous work we demonstrated that 2 can induce measurable decreases of GlcCer in the brains of normal mice and prevent the accumulation of ganglioside GM2 in a mouse model of Sandhoff disease, subsequent pharmacokinetic studies revealed that systemic clearance in mice is rapid and overall brain exposure low (Table 4). To guide the design of more metabolically stable analogues, we subjected 2 to a metabolite identification study to pinpoint the specific sites at which CYP450-mediated oxidative metabolism was occurring (Figure 2). After incubation with mouse liver microsomes (MLM) for 60 min, analysis by LC/MS/MS indicated that the two major routes of metabolism entail oxidation of the benzodioxane ring, along with a minor route of indane oxidation.
Table 4.
In Vitro and In Vivo Pharmacokinetic Data
| MDR1-MDCK cell permeability | Mouse PK (IP, 10 mg/kg) | ||||
|---|---|---|---|---|---|
| no. | Papp A-B (×10−6 cm/s) | Papp B-A (×10−6 cm/s) | efflux ratio | brain AUC (h·ng/g) | plasma AUC (h·ng/mL) |
| 2 | 0.14 (9.8)a | 40.8 (6.9)a | 298 | 36 | 317 |
| 17 | 0.95 (15.8)a | 56.6 (8.6)a | 60 | 1046 | 1308 |
| 49 (13.4)a | |||||
| 3 | 0.58 (19.8)a | 84 | 6188 | 7164 | |
Values in parentheses were measured in the presence of the Pgp inhibitor, valspodar.
Figure 2.

Identification of metabolites of 2 formed by incubation with mouse liver microsomes.
Based on these results, we first explored modification of the indane (Table 1). Blocking oxidation of the cyclopentyl ring of the indane (10) with a carbonyl resulted in an unacceptable loss of potency. Fluorination of the aromatic ring at the two possible locations (11, 12) enhanced potency, especially at the meta position, but did not improve metabolic stability.
We then turned our focus to the left-hand aromatic ring (Table 2). Because the ethylenedioxy moiety of the benzodioxane ring donates significant electron density into the aromatic ring, as well as being highly susceptible to metabolism itself, we first replaced it with simple regioisomeric (and lower TPSA) monomethoxys (13 and 14). Although both of these analogues retained excellent potency, they did not significantly improve the half-life in MLM. We next added ortho-fluorines to the methoxy regioisomers (15 and 16). Consistent with SAR precedent,15−17 the para-methoxy analogue 15 was more potent than the para-fluoro 16. Unfortunately, the addition of the fluorine failed to improve stability to metabolism, suggesting that the majority of metabolism was due to O-demethylation. A metabolite identification study on 15 indeed confirmed this hypothesis (Figure 3). Interestingly, this study also showed that a minor metabolite is the dealkylated primary amine, with no indication of indane oxidation occurring.
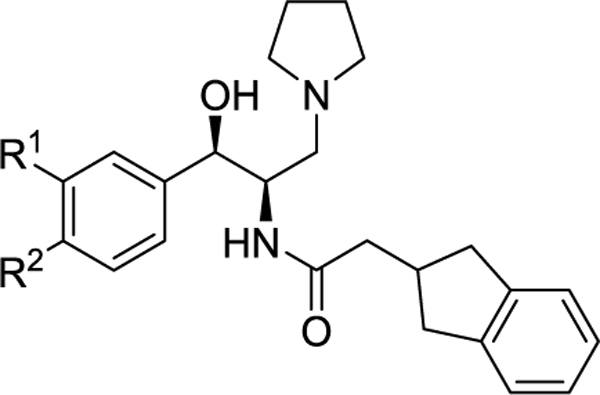
Table 2.
Modification of the Aromatic Ring
 | ||||||
|---|---|---|---|---|---|---|
| no. | R1 | R2 | crude enzyme IC50 (nM) | MDCK cells IC50 (nM) | MLM T1/2 (min) a | HLM T1/2 (min)b |
| 2 | -OCH2CH2O- | 27 | 15.3 | 2.5 | 28 | |
| 13 | H | OMe | 47.6 | 3.4 | 5.5 | |
| 14 | OMe | H | 54.1 | 5.5 | 4 | |
| 15 | F | OMe | 27 | 2.7 | 2.7 | |
| 16 | OMe | F | >300 | 72.7 | ||
| 17 | H | OCF3 | 110 | 3.6 | 37 | 84 |
| 18 | H | OCHF2 | 123 | 9.6 | 18 | 33 |
| 19 | H | OCH2CF3 | 56 | 0.87 | 10 | 21 |
| 20 | H | OCH2c-Pr | 46 | 1.0 | <3 | |
| 21 | OCF3 | H | >1000 | 52.4 | ||
| 22 | H | CF3 | >300 | 35.0 | ||
| 3 | NA | NA | 240 | 37 | >60 | >60 |
Half-life when incubated with mouse liver microsomes.
Half-life when incubated with human liver microsomes.
Figure 3.

Identification of metabolites of 15 formed by incubation with mouse liver microsomes.
Following up on these results, we examined several analogues that replace the O-methyl with more metabolically stable moieties (Table 2). Gratifyingly, the simple 4-trifluoromethoxy analogue 17 retained excellent potency in our whole cell assay while improving the half-life in mouse liver microsomes by over 10-fold (37 min vs 2.5 min). Importantly, this improvement extended to human liver microsomes (HLM), where we observed a 3-fold increase in half-life. Closely related analogues difluoromethoxy (18) and 2,2,2-trifluoroethoxy (19) also retained potency, but were not as stable to metabolism in MLM or HLM as 17. This could be due to either oxidation of the available CH moieties on the side chains, or to increased electron density on the aromatic ring, or both. The poor metabolic stability of the cyclopropylmethyl analogue 20 is consistent with this hypothesis. Moving the trifluoromethoxy group to the meta position (21) greatly diminished activity, as did replacement by trifluoromethyl (22). Overall the SAR results in Table 2 are consistent with the well-established requirement for an electron donating atom at the 4-position (R2) of the aromatic ring for good inhibition of GlcCer synthase.15,17
Further follow up of the metabolite id results in Figure 2 entailed replacement of the pyrrolidine of 2 with 3-fluoropyrrolidine (23) and azetidine (24), both of which were expected to be less susceptible to N-dealkylation (Table 3). The more active of the two new analogues, fluoro-pyrrolidine 23 unfortunately offered no improvement in stability to MLM, confirming that N-dealkylation is secondary to oxidation of the benzodioxane ring.
We performed one final metabolite id study on 17 to determine if the major site of metabolism had shifted due to oxidation of the aromatic ring being largely blocked by the trifluoromethoxy group (Figure 4). Although all metabolites were minor relative to the parent drug under the conditions of the assay, both of the major metabolites were the result of oxidation proximal to the pyrrolidine nitrogen, indicating a clear shift away from the alkoxy aromatic ring. We therefore also prepared the azetidine and 3-fluoropyrrolidine analogues of 17 to determine if stability could be improved even further (25 and 26, Table 3). Although azetidine 26 ultimately proved to be our most stable analogue to date in MLM, both of these new amine analogues suffered unacceptable losses in potency.
Figure 4.

Identification of metabolites of 17 formed by incubation with mouse liver microsomes.
Included in Table 2 for comparison to 17 is the Sanofi/Genzyme clinical lead 3. Our optimized compound is 10-fold more potent in our MDCK cell assay (3.6 nM vs 37 nM), and now has more comparable stability in liver microsomes, in contrast to our original lead 2. It is also important to note that 17 has stability in HLM (T1/2 = 84 min) that is much greater than that of the approved oral drug from this class, eliglustat HLM (T1/2 = 12.5 min).
Pharmacokinetic Profiles.
Prior to in vivo efficacy testing in mice, 17 was compared with 2 and 3 in mouse pharmacokinetics (PK) studies (Table 4). Following single IP doses of drug (10 mg/kg), 17 exhibited a nearly 30-fold increase in overall brain exposure vs 2 (AUC = 1046 h·ng/g vs 36 h·ng/g). Furthermore, the cumulative B/P ratio (as estimated from the simple ratio of brain AUC/plasma AUC) improved from 0.11 to 0.80. The B/P AUC value of 17 is now comparable to 3 (0.86), although the overall brain exposure of 17 remains much less.
There are at least two rationales for why 17 is so much more brain-permeable than 2. First, its physical properties are more aligned with those of CNS-permeable drugs, namely, a higher ClogP (4.8 vs 2.9) and lower TPSA (62 vs 71), although its MW is somewhat higher (463 vs 437). We also hypothesized that 17 might be less susceptible to efflux by P-glycoprotein (Pgp). This was tested using a standard MDR1-MDCK cell monolayer (Table 4), and indeed revealed a markedly lower susceptibility of 17 to efflux than 2 (efflux ratio 60 vs 298). Confirmation that efflux was primarily by the Pgp transporter was achieved by parallel experiments in the presence of Pgp inhibitor valspodar, which resulted in nearly equivalent apical (A-B) and basal (B-A) permeabilities. Since these two analogues possess the same basic amine, a known Pgp recognition element, it is possible that the ethylendioxy moiety of 2 is also recognized by Pgp, perhaps due to its high TPSA. Furthermore, the TPSA of the single remaining aryl oxygen atom of 17 may be effectively masked by the strongly electron-withdrawing trifluoromethyl moiety. Interestingly, despite its high brain exposure in mice, clinical comparator 3 also exhibited an efflux ratio in our assay that is much higher than typical CNS permeant drugs and that was comparable to 17 (84, Table 4). This suggests that this particular attribute may not itself be a barrier to clinical development of our series for neuronopathic disease.
To assess the potential for 17 to be an oral drug, bioavailability was determined through standard IV/PO PK studies in CD1 mice, and compared against venglustat and eliglustat, both of which are effective orally (Tables S-1−S-3 in the Supporting Information). At a PO dose of 10 mg/kg, the bioavailability of 17 was 34%, lower than that of venglustat (74%) but markedly superior to that of eliglustat (<1%) in this model. The IV half-life of 17 is also about 4 times that of eliglustat, consistent with reduced clearance and a greater volume of distribution at steady state (Vss). Although the Vss of 17 is high (7549 mL/kg), it is comparable to that of venglustat (6673 mL/kg). Importantly, in separate studies in mice, we demonstrated the achievement of significant brain exposure by 17 through the oral route (Table S-4). In this model, the B/P ratio of 17 (0.88) was actually higher than that of venglustat (0.64).
Pharmacodynamic Studies in Mice.
To compare the in vivo efficacy of 17 with our previous lead 2, we first evaluated it in normal C57BL/6 mice. Mice (6−8-weeks old) were injected IP with 17 or vehicle for 3 days in two separate dose response assays (Figure 5). Collectively, these data indicate a significant reduction in brain GlcCer of about 34% at 1 mg/kg, which did not substantially increase in magnitude up to a dose of 30 mg/kg (−41%). By contrast, previous lead 2 in this model was only able to achieve a 17% reduction in brain GlcCer at 60 mg/kg,8 indicating an over 60-fold improvement in in vivo potency by 17 in this model.
Figure 5.

Effect of 17 on brain glucosylceramide in wild-type mice. Compound or vehicle was administered IP at the indicated doses for 3 days. Data shown is a merge of two separate dose response experiments (Exp 1: 30, 10, 1 mg/kg; Exp 2: 1.0, 0.30, 0.10, 0.03 mg/kg). ns: change is not significant.
As a more physiologically relevant in vivo measure of efficacy, an inducible model of Gaucher disease type 3 was next employed using D409 V/D409 V mice of C57BL/6 background.18 Beginning at postpartum day 15, mice were injected intraperitoneally with conduritol B epoxide (CBE), an irreversible inhibitor of lysosomal β-glucocerebrosidase, at a dose of 25 mg/kg, to induce a Gaucher disease phenotype characterized by the accumulation of the glycosphingolipid substrates: GlcCer and glucosylsphingosine (GlcSph). Daily injections continued for 14 days total. In addition to 17, eliglustat and Genz-682452 were included as comparators. GlcCer and GlcSph levels in brain were quantified by LC/MS-MS and expressed as concentration of pmol/mg of tissues. Results are summarized in Figure 6.
Figure 6.

Effect of 17 (CCG-222628) on brain glycosphingolipids in CBE mice. Eliglustat and venglustat (3, Genz-682452) were included as comparators. Compounds were all coadministered IP at a dose of 10 mg/kg qd for 14 days.
From these data, it is clear that 17 was able to achieve reductions in brain GlcCer that were comparable to those of Genz-682452 (3) (19% and 26%, respectively). Reductions in brain GlcSph were likewise similar (39% and 35%, respectively). As expected, eliglustat was inactive in this assay at 10 mg/kg. It is important to note that 17 achieved these reductions at significantly lower brain drug levels than Genz-682452. Drug levels in the brains of the mice at sacrifice (2 h post final dose) were 134 ng/g for 17 and 493 ng/g for Genz-682452. Our PK studies at the same IP dose levels (Table 4) are consistent with this observation, indicating that the total brain exposure (AUC) for 17 after a single 10 mg/kg dose is only about 20% that of Genz-682452. This suggests that differences in total brain exposure (AUC) in the CBE mouse study were likely even more pronounced than the 2 h drug levels. On the other hand, drug exposures in the peripheral organs, estimated by spleen drug levels at sacrifice, were similar (4880 and 4570 ng/g for 17 and 3, respectively). It is also important to note that 17 apparently retains the selectivity of eliglustat for GlcCer synthase vs galactosylceramide synthase as evidenced by a lack of effect on brain levels of galactosylceramide and galactosylsphingosine (data not shown).
CONCLUSIONS
Eliglustat is an approved therapy for Gaucher disease type 1, but has not been pursued for neuronopathic glycosphingolipid storage diseases. We previously demonstrated, however, that the eliglustat scaffold can be modified (2) to reduce glycosphingolipid levels in the brains of mice. Subsequent in vivo pharmacokinetic studies revealed only modest brain penetration and rapid clearance, suggesting that further optimization was warranted. In this work, we used multiple metabolite identification studies to define primary and secondary sites for CYP-mediated oxidation, guiding the design of more stable analogues. In parallel, we endeavored to lower TPSA where possible to facilitate brain penetration. General SAR observations were that fluoro- or oxygen-substitution of the indane did not improve metabolic stability; modification of the pyrrolidine ring was unacceptably detrimental to potency; and electron-density on the left-hand aromatic ring was critical to both potency and metabolic stability. Ultimately, we found that replacement of the 3,4-ethylenedioxy moiety of the benzodioxane ring with 4-trifluoromethoxy (17, CCG-222628) afforded one of our most stable analogues when incubated with mouse liver microsomes, and also retained excellent cell potency. In vivo pharmacokinetics in mice confirmed that brain exposure had improved by nearly 30-fold, which translated into almost 60-fold greater potency at reducing glucosylceramide in the brains of wild-type mice. One likely contributor to the increased brain exposure is reduced susceptibility to efflux by Pgp, which was confirmed in an MDR1-MDCK cell monolayer assay. A logical rationale for the markedly lower efflux is a reduction in TPSA of the ethylendioxy moiety realized by replacement with trifluoromethoxy. Finally, we evaluated 17 in a CBE mouse model of Gaucher disease type 3 and compared its efficacy directly against clinical candidate Genz-682452 (venglustat) at equal doses. In this assay, 17 and venglustat induced comparable reductions in brain glycosphingolipids after 14 days of dosing. Importantly, 17 was able to effect these changes at significantly lower total brain drug exposure than venglustat. We believe the in vitro and in vivo activity of 17 firmly establish the potential for the eliglustat class of compounds to treat neuronopathic glycosphingolipid storage diseases.
METHODS
Chemistry.
General Experimental Information.
All reagents and solvents were used in the condition received from commercial sources. Chromatographic purifications were performed using a Teledyne ISCO Combiflash RF with Redisep columns or by standard flash chromatography using silica gel (220−240 mesh) obtained from Silicycle. 1H NMR were taken in CDCl3 or DMSO-d6 at room temperature on Varian Inova 400 or 500 MHz instruments. Reported chemical shifts are expressed in parts per million (ppm) on the δ scale from an internal standard of tetramethylsilane (0 ppm). Mass spectra were recorded on a Micromass LCT time-of-flight instrument utilizing the electrospray ionization mode. An Agilent 1100 series HPLC with an Agilent Zorbax Eclipse Plus−C18 column (3.5 um, 4.6 × 100 mm) was used to determine “HPLC purity” of biologically tested compounds. All tested compounds were assayed for purity using a 6 min gradient of 10−90% acetonitrile in water followed by a 2 min hold at 90% acetonitrile with detection at 254 nm.
Eliglustat tartrate used in these studies was obtained from Genzyme Corporation. Genz-682452 (3, venglustat) was prepared by the method described by Zhao et al.19
Exemplary Procedures (from Scheme 1; Preparation of 15). (1R,3S,5S)-1,3-Bis(3-fluoro-4-methoxyphenyl)-5-phenyltetrahydro-3H,8H-oxazolo[4,3-c][1,4]oxazin-8-one (5).
(S)-5-Phenylmorpholin-2-one (2.1 g, 11.9 mmol) and 3-fluoro-4-methoxybenzaldehyde (5.5 g, 35.6 mmol) were placed in a 250 mL round-bottom flask and dissolved in toluene (80 mL). The flask was fitted with a magnetic stirrer bar and a Dean−Stark trap. The reaction mixture was heated to reflux for 24 h under nitrogen and the solvent removed in vacuo to yield a pale yellow oil, which solidified under drying under high vacuum. The material was purified by flash chromatography (EtOAc/hexane) as follows: Silica gel was placed in a 500 mL sintered glass funnel and slurried with hexane. The material was placed on top of the silica gel and eluted with 1000 mL of hexane, 1000 mL of 10% EtOAc/hexane, 1000 mL of 20%, and 1000 mL of 30% 1000 mL 40%. The appropriate fractions were concentrated. The resulting solid was triturated in ether, filtered, and dried in a vacuum oven overnight at room temperature, leaving an off-white solid. The title compound was obtained (1.9 g, 4.1 mmol, 34.3% yield). 1H NMR (400 MHz, chloroform-d) δ 7.36−7.15 (m, 7H), 7.16−6.96 (m, 3H), 6.92 (t, J = 8.6 Hz, 1H), 6.75 (t, J = 8.4 Hz, 1H), 5.54−5.16 (m, 2H), 4.53−4.23 (m, 2H), 4.23−3.98 (m, 2H), 3.87 (s, 2H), 3.82 (s, 3H).
(2S,3R)-3-(3-Fluoro-4-methoxyphenyl)-3-hydroxy-2-(((S)-2-hydroxy-1-phenylethyl)amino)-1-(pyrrolidin-1-yl)propan-1-one (6).
To a solution of (1R,3S,5S)-1,3-bis(3-fluoro-4-methoxyphenyl)-5-phenyltetrahydrooxazolo[4,3-c][1,4]oxazin-8(3H)-one (1 g, 2.1 mmol) in CH2Cl2 was added pyrrolidine (0.89 mL, 10.7 mmol). The resulting mixture was stirred overnight at room temperature. After warming to 50 °C for 2 h, the reaction was cooled and concentrated in vacuo. The residue was dissolved in MeOH, treated with 2 M HCl, and heated to reflux for 2 h. Completion of the reaction required adding more 2 M HCl and stirring 2 more hours. The reaction was cooled and concentrated in vacuo. The crude material was diluted with water and extracted with ether. The aqueous layer was treated with saturated NaHCO3 until it was slightly basic. The mixture was extracted with 3 × 1:1 EtOAc/ether. The combined organic extracts were washed with water and dried over MgSO4. Filtration through flash silica gel with 5% MeOH/CH2Cl2 eluent provided the title compound (0.6 g, 1.5 mmol, 69.7% yield). 1H NMR (400 MHz, chloroform-d) δ 7.43−7.16 (m, 4H), 7.13 (m, 1H), 7.00 (m, 1H), 6.83 (t, J = 8.5 Hz, 1H), 4.50 (d, J = 8.6 Hz, 1H), 3.83 (s, 3H), 3.79−3.59 (m, 2H), 3.14−2.95 (m, 2H), 2.95−2.76 (m, 2H), 2.35−2.12 (m, 1H), 2.04−1.68 (m, 2H), 1.51−0.99 (m, 4H).
(1R,2R)-1-(3-Fluoro-4-methoxyphenyl)-2-(((S)-2-hydroxy-1-phenylethyl)amino)-3-(pyrrolidin-1-yl)propan-1-ol (7).
To a 0 °C solution of (3R)-3-(3-fluoro-4-methoxyphenyl)-3-hydroxy-2-(((S)-2-hydroxy-1-phenylethyl)amino)-1-(pyrrolidin-1-yl)propan-1-one (0.4 g, 0.9 mmol) in dry THF (15 mL) was added lithium aluminum hydride (0.10 g, 2.9 mmol). The resulting mixture was stirred overnight at room temperature. After heating to reflux for 1.5 h to complete the reaction, it was cooled to °0 C and treated dropwise with 0.11 mL of H2O followed by 0.11 mL of 15% NaOH. The mixture was stirred for 20 min and treated with 3.3 mL of H2O. After stirring for 1 h, the mixture was filtered through Celite with ether eluent. The eluent was concentrated and purified by flash chromatography (MeOH/CH2Cl2) to obtain the title compound (0.3 g, 0.6 mmol, 62.5% yield). 1H NMR (400 MHz, chloroform-d) δ 7.36−7.21 (m, 5H), 7.21−7.12 (m, 1H), 7.12−6.98 (m, 1H), 6.92 (t, J = 8.6 Hz, 1H), 4.51 (d, J = 4.5 Hz, 1H), 3.88 (s, 3H), 3.69 (dd, J = 8.8, 4.3 Hz, 1H), 3.62−3.44 (m, 4H), 3.08−2.79 (m, 1H), 2.62 (dd, J = 12.5, 8.6 Hz, 1H), 2.50−2.30 (m, 3H), 2.30−2.13 (m, 1H), 1.81−1.52 (m, 3H).
(1R,2R)-2-Amino-1-(3-fluoro-4-methoxyphenyl)-3-(pyrrolidin-1-yl)propan-1-ol (8).
To a solution of (1R)-1-(3-fluoro-4-methoxyphenyl)-2-(((S)-2-hydroxy-1-phenylethyl)amino)-3-(pyrrolidin-1-yl)-propan-1-ol (0.3 g, 0.7 mmol) in methanol (15 mL) and 1 M HCl 10 mL was added palladium on carbon (10% Degussa) (0.08 g, 0.8 mmol). The resulting mixture was bubbled with N2 for 5 min then placed on a Parr Hydrogenator, placed briefly under vacuum then filled with hydrogen gas. The reaction was shaken overnight at room temperature. The mixture was filtered through Celite with methanol eluent and concentrated in vacuo. The crude product was purified by flash chromatography (7% conc ammonia in methanol/CH2Cl2) to obtain the title compound as an oil (0.11 g, 0.41 mmol, 53% yield). 1H NMR (500 MHz, chloroform-d) δ 7.62−7.61 (m, 1H), 7.03−6.71 (m, 2H), 4.59 (m, 1H), 3.79 (m, 2H), 3.14 (m, 1H), 2.75−2.26 (m, 5H), 1.78 (br s, 4H).
2-(2,3-Dihydro-1H-inden-2-yl)-N-((1R,2R)-1-(3-fluoro-4-methoxyphenyl)-1-hydroxy-3-(pyrrolidin-1-yl)propan-2-yl)acetamide (9, Compound 15).
To a solution of (1R,2R)-2-amino-1-(3-fluoro-4-methoxyphenyl)-3-(pyrrolidin-1-yl)propan-1-ol (0.10 g, 0.36 mmol) in THF 10 mL was added 2-(2,3-dihydro-1H-inden-2-yl)acetic acid (0.08 g, 0.45 mmol), HOBt (0.06 g, 0.47 mmol), EDC (0.103 g, 0.54 mmol) followed by DIPEA (0.14 mL, 0.83 mmol). The resulting mixture was stirred overnight at room temperature. Saturated NaHCO3 and EtOAc were added, and the separated aqueous layer was extracted again. The combined organic layers were washed with saturated NaCl (3×) and dried (MgSO4). Purification by flash chromatography (gradient of 2.5% MeOH/CH2Cl2 to 5% to 10% MeOH (with 7% NH3)/CH2Cl2) afforded pure 15 (0.05 g, 0.11 mmol, 28% yield). 1H NMR (400 MHz, chloroform-d) δ 7.26 (s, 2H), 7.14 (d, J = 10.3 Hz, 3H), 7.04 (d, J = 8.5 Hz, 1H), 6.92 (t, J = 8.4 Hz, 1H), 6.11 (s, 1H), 5.06 (d, J = 2.8 Hz, 1H), 4.27 (m, 1H), 3.97−3.61 (m, 3H), 3.13−2.60 (m, 7H), 2.49 (dd, J = 15.5, 6.7 Hz, 1H), 2.43−2.08 (m, 3H), 1.87 (s, 4H), 1.25 (s, 4H). MS m/z (EI): 426.2 [M + H]. HPLC purity: 97%.
2-(2,3-Dihydro-1H-inden-2-yl)-N-((1R,2R)-1-hydroxy-3-(pyrrolidin-1-yl)-1-(4-(trifluoromethoxy)phenyl)propan-2-yl)acetamide (17).
Compound 17 was prepared by procedures analogous to those described for the preparation of 15. 1H NMR (400 MHz, chloroform-d) δ 7.40−7.35 (m, 2H), 7.21−7.19 (m, 2H), 7.12 (m, 4H), 6.03 (d, J = 7.8 Hz, 1H), 5.12 (d, J = 2.7 Hz, 1H), 4.30−4.20 (m, 1H), 2.97−2.72 (m, 8H), 2.49 (dd, J = 15.6, 6.4 Hz, 1H), 2.29−2.16 (m, 3H), 1.83 (br s, 4H). MS m/z (APCI): 463.3 [M + H]. HPLC purity: 96%.
Preparation of Monooxalate Salt of 17.
To a solution of 2-(2,3-dihydro-1H-inden-2-yl)-N-((1R,2R)-1-hydroxy-3-(pyrrolidin-1-yl)-1-(4-(trifluoromethoxy)phenyl)propan-2-yl)acetamide (17, 0.28 g, 0.605 mmol) in isopropanol (10 mL) was added a solution of oxalic acid (0.055 g, 0.605 mmol) in methanol (5 mL). The resulting mixture was stirred for 30 min, and then concentrated on a rotary evaporator. The crude solid was triturated with EtOAc/ether, and stirred for 30 min and then allowed to set overnight. The mixture was filtered and washed with ether. After a second trituration with isopropyl alcohol, the white solid was dried overnight under high vacuum. Elemental analysis calcd for C25H29N2O3·C2H2O4·0.5H2O: C, 57.75; N, 5.74; N, 4.98. Found: C, 57.77; H, 5.55; N, 5.00.
Broken Cell Assay for Glucosylceramide Synthase Inhibition.
Enzyme activity was measured as described previously.14 Madin-Darby canine kidney (MDCK) cell homogenates (120 μg of protein) were incubated with uridine diphosphate-[3H]glucose (100 000 cpm) and liposomes consisting of 85 μg of octanoylsphingosine, 570 μg of dioleoylphosphatidylcholine, and 100 μg of sodium sulfatide in a 200 μL reaction mixture and kept for 1 h at 37 °C. Glucosylceramide synthase inhibitors dissolved in dimethyl sulfoxide (final concentration < 1%, which did not affect enzyme activity) were dispersed into the reaction mixture after adding the liposomes. Seven concentrations of each analogue were assayed in duplicate and compared to control activity using buffer alone.
MDCK Cell Assay for Reduction in Glucosylceramide.
Following inhibitor treatment at seven concentrations (assayed in duplicate), whole cellular lipids of MDCK cells were extracted as previously described in detail.8 Briefly, cells were washed with ice-cold PBS, fixed by methanol and collected with a rubber scraper. Chloroform was then added to yield a theoretical ratio of chloroform−methanol−water at 1:2:0.8 (v/v/v) to form a monophase. Cell debris and proteins were removed by centrifugation at 2200 g for 30 min. The supernatants were portioned by adding chloroform and 0.9% NaCl. The lower organic phases containing neutral glycosphingolipids lipids were washed with methanol and 0.9% NaCl and subjected to base- and acid-hydrolysis. A portion of purified glycosphingolipids normalized to 100 nmol of total phospholipids was analyzed by high-performance TLC. The TLC separations were processed twice. The plate pretreated with 1% sodium borate was first developed in a solvent system consisting of chloroform−methanol (98:2, v/v). After air drying, the plate was then developed in a solvent system containing chloroform−methanol−water (70:30:4, v/v/v). The levels of glucosylceramide were detected by charring with 8% CuSO4·5H2O in 6.8% phosphoric acid and quantified by densitometric scanning using ImageJ, NIH Image. Image data was analyzed, and the IC50 of each inhibitor was calculated using GraphPad Prism.
Determination of Stability to Liver Microsomes.
Metabolic stability was assessed using CD-1 mouse or human liver microsomes. A concentration of 1 μM of each compound was incubated with 0.5 mg/mL microsomes and 1.7 mM cofactor β-NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 5, 10, 15, 30, 45, and 60 min of incubation, 40 μL of the reaction mixture were taken out, and the reaction was quenched by adding 3-fold excess of cold acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 15 000 rpm for 10 min to collect the supernatant for LC−MS/MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Identification of Metabolites Induced by Mouse Liver Microsomes.
The parent compound (10 μM) was incubated with mouse liver microsomes (MLM) and β-NADPH at 37 °C for 20 and 40 min. Then the reaction was quenched by adding a 3-fold volume of ice-cold acetonitrile. The mixture was centrifuged at 15,000 rpm for 10 min, and the supernatant was saved under −80 °C for analysis. The negative control samples were prepared by a similar procedure without NADPH or using boiled microsome. The general approach for metabolite identification using an AB Sciex QTrap 5500 mass spectrometer involves the following steps: (1) Obtain a product ion spectrum of the parent compound to establish fragmentation. (2) Interpret the spectrum to identify major fragment ion and possible neutral loss. (3) Collect spectra of samples using both established precursor ion scan and neutral loss scan, EMS scan of both control and samples were acquired also. (4) Run product ion scans and MRM scans for all possible metabolite identified from step 3 plus expected metabolite. (5) Interpret the spectrum of the metabolites and determine the structure with their logical fragmentation pattern.
Determination of Efflux by MDR1-MDCK Cells.
Permeability through and efflux from an MDR1-MDCK cell monolayer were determined by Absorption Systems, 436 Creamery Way, Suite 600, Exton, PA 19341−2556.
Pharmacokinetic Studies in Mice.
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM). The pharmacokinetics of compounds were determined in female CD-1 mice following intraperitoneal (IP) injection of 10 mg/kg, respectively. Compounds were first dissolved in citrate buffer at 2 mg/mL and then diluted with sterile saline (1:1) for injection volumes of 1 mg/mL. Blood samples were collected from individual cohorts of mice (n = 3) using heparinized calibrated pipets following sacrifice at the given time point over 24 h (at 0.083h, 0.25h, 0.5h, 1.5h, 4h, 8h and 24h), centrifuged at 15000 rpm for 10 min. Subsequently, blood plasma was collected from the upper layer. The plasma was frozen at −80 °C for later analysis. Brains were taken out at the same time and weighed out immediately. Brains were frozen at −80 °C for later preparation and analysis, Plasma concentrations of the compounds were determined by the LC−MS/MS method developed and validated for this study. The LC−MS/MS method consisted of a Shimadzu HPLC system and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
In Vivo Reduction of Brain Glycosphingolipids in Normal Mice.
C57BL/6 mice were maintained on regular chow in specific-pathogen-free facilities. All animal studies were performed under the review of the University of Michigan Committee on the Use and Care of Animals and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Injection solutions were prepared from inhibitor-ethanol stock solution (100 mM). A portion of the stock solution was evaporated under a stream of N2 gas. Dried eliglustat tartrate was directly dissolved into 1× PBS. Compound 17 was dissolved with 250 μL of water plus 13.7 μL of 0.5 N HCl by hand shaking and gentle vortexing resulting in a 1 or 3 mg/mL solution. The acidic solution was neutralized by mixing 100 μL of 10× PBS with 636.3 μL of water to bring the total volume to 1 mL. Inhibitor solutions were sterilized by passage through a 0.2 μM filter. Inhibitor recovery after filtration was confirmed by UV spectrometry and exceeded 99%. For control injections, PBS containing the same amount of HCl was used. Inhibitors were given to 6−8 week old female or male C57BL/6 mice by intraperitoneal injection volume at 1% of body weight.
Lipid extractions of brain were performed as previously described.20 Briefly, frozen whole brain (∼0.4 g) was individually homogenized in sucrose buffer (250 mM sucrose, pH 7.4, 10 mM Hepes and 1 mM EDTA), at 0.2 g tissue/1 mL of sucrose buffer, with a Tri-R homogenizer. Each 0.8 mL of homogenate was mixed with 2 mL of methanol and 1 mL of chloroform, bath sonicated for 1 min, and incubated at room temperature for 1 h. Tissue debris was removed by centrifugation at 2400g for 30 min. The pellets were re-extracted by mixing with 1 mL of methanol, 0.5 mL of chloroform, and 0.4 mL of 0.9% NaCl (chloroform/methanol/0.9% NaCl, 1:2:0.8), incubated at room temperature for 1 h, and centrifuged at 2400g for another 30 min. Two extracts were combined and mixed with 4.5 mL of chloroform and 1.2 mL of 0.9% NaCl (chloroform/methanol/0.9% NaCl, 2:1:0.8). After centrifugation at 800g for 5 min, the lower layer was washed with 3 mL of methanol and 2.4 mL of 0.9% NaCl. A second washing was carried with 3 mL of methanol, 2 mL of water, and 0.4 mL of 0.9% NaCl followed by a 5 min centrifugation at 800g. The resultant lower phase was collected and dried under a stream of N2 gas.
The analysis of neutral glycosphingolipids from mouse brain was processed after alkaline methanolysis. Brain lipids were incubated with 2 mL of chloroform and 1 mL of 0.21 N NaOH in methanol for 7.5 h at room temperature. The lipid extract was normalized to 2 μmol of total phospholipid phosphate for high performance TLC analysis. After alkaline methanolysis, the brain lipids were passed through a silica gel column.20 Borate-impregnated TLC plates were developed in a two solvent system. Plates were first developed in chloroform/methanol (98:2, v/v). The plates were then developed in chloroform/methanol/water (64:24:4, v/v/v) and were further separated in chloroform/methanol/water (60:30:6, v/v/v). GlcCer levels were quantified by comparison to known standards.
In Vivo Reduction of Brain Glycosphingolipids in CBE Mice.
An inducible model of Gaucher disease type 3 was employed using Gba D409V/D409V mutant mice of C57BL/6 genetic backgrounds.18 The Cincinnati Children’s Hospital Research Foundation (CCHRF) Institutional Animal Care and Use Committee (IACUC) reviewed and approved these studies under protocol IACUC2015–0050. All mice were housed under pathogen-free conditions in the barrier animal facility and according to IACUC standard procedures at CCHRF. Beginning at postpartum day 15, D409V/D409V mice were injected intraperitoneally with conduritol B epoxide (CBE), an irreversible inhibitor of lysosomal β-glucocerebrosidase at a dose of 25 mg/kg. Daily injections of CBE continued for 14 days total. Test compounds (17 and 3; 10 mg/kg) were coadministered by IP in citrate-saline buffer or buffer alone. Mice were monitored and weighed daily. GlcCer and GlcSph levels in brain from D409V/D409V mice treated with CBE with or without test compounds were separated from galactosylceramide and galactosylsphingosine, respectively, and quantified by LC/MS-MS (Lipidomics Shared Resource, Medical University of South Carolina). The glycolipid levels were normalized to tissue weight and expressed as the concentration (pmol/mg tissue) of the GlcCer or GlcSph of matched samples from citrate-saline-injected vehicle control mice. Student’s t test was used for statistical comparisons. Each group consisted of 8−11 mice with mixed gender.
Supplementary Material
Acknowledgments
Funding
This work was supported by NINDS and NICHD of the National Institutes of Health under Award Numbers R21 NS065492 (S.D.L.) and R01 HD076004 (S.D.L. and J.A.S).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00558.
IV/PO Pharmacokinetic data for 17, 3, and 1; brain/plasma ratio by the oral route in mice for 17 and 3; 1H NMR, MS, and HPLC purity of compounds 10−14, 16, and 18−26 (PDF)
The contents of the work are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
The authors declare no competing financial interest.
Contributor Information
Michael W. Wilson, Vahlteich Medicinal Chemistry Core, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States
Liming Shu, Department of Internal Medicine - Nephrology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Vania Hinkovska-Galcheva, Department of Internal Medicine - Nephrology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Yafei Jin, Vahlteich Medicinal Chemistry Core, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Walajapet Rajeswaran, Vahlteich Medicinal Chemistry Core, College of Pharmacy, University of Michigan, Ann Arbor, Michigan 48109, United States.
Akira Abe, Department of Internal Medicine - Nephrology, University of Michigan, Ann Arbor, Michigan 48109, United States.
Ting Zhao, Pharmacokinetics Core, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Ruijuan Luo, Pharmacokinetics Core, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Lu Wang, Pharmacokinetics Core, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Bo Wen, Pharmacokinetics Core, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Benjamin Liou, Division of Human Genetics, Cincinnati Children’s Hospital, Cincinnati, Ohio 45229, United States.
Venette Fannin, Division of Human Genetics, Cincinnati Children’s Hospital, Cincinnati, Ohio 45229, United States.
Duxin Sun, Pharmacokinetics Core, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor, Michigan 48109, United States.
Ying Sun, Division of Human Genetics, Cincinnati Children’s Hospital, Cincinnati, Ohio 45229, United States.
James A. Shayman, Department of Internal Medicine - Nephrology, University of Michigan, Ann Arbor, Michigan 48109, United States
Scott D. Larsen, Vahlteich Medicinal Chemistry Core, College of Pharmacy and Department of Medicinal Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Platt FM, d’Azzo A, Davidson BL, Neufeld EF, and Tifft CJ (2018) Lysosomal storage diseases. Nat. Rev. Dis. Primers 4 (1), 27. [DOI] [PubMed] [Google Scholar]
- (2).Miller JJ, Kanack AJ, and Dahms NM (2020) Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta, Gen. Subj. 1864 (1), 129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shayman JA, and Larsen SD (2014) The development and use of small molecule inhibitors of glycosphingolipid metabolism for lysosomal storage diseases. J. Lipid Res. 55 (7), 1215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shayman JA (2013) Eliglustat tartrate, a prototypic glucosylceramide synthase inhibitor. Expert Rev. Endocrinol. Metab. 8 (6), 491–504. [DOI] [PubMed] [Google Scholar]
- (5).Cox TM, Drelichman G, Cravo R, Balwani M, Burrow TA, Martins AM, Lukina E, Rosenbloom B, Ross L, Angell J, and Puga AC (2015) Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet 385 (9985), 2355–62. [DOI] [PubMed] [Google Scholar]
- (6).Mistry PK, Lukina E, Ben Turkia H, Amato D, Baris H, Dasouki M, Ghosn M, Mehta A, Packman S, Pastores G, Petakov M, Assouline S, Balwani M, Danda S, Hadjiev E, Ortega A, Shankar S, Solano MH, Ross L, Angell J, and Peterschmitt MJ (2015) Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA 313 (7), 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang W-L, Hutto E, Shayman JA, Grabowski GA, Aerts JMFG, Cheng SH, Copeland DP, and Marshall J. (2007) A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol. Genet. Metab. 91 (3), 259–267. [DOI] [PubMed] [Google Scholar]
- (8).Larsen SD, Wilson MW, Abe A, Shu L, George CH, Kirchhoff P, Showalter HD, Xiang J, Keep RF, and Shayman JA (2012) Property-based design of a glucosylceramide synthase inhibitor that reduces glucosylceramide in the brain. J. Lipid Res. 53 (2), 282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Arthur JR, Wilson MW, Larsen SD, Rockwell HE, Shayman JA, and Seyfried TN (2013) Ethylenedioxy-PIP2 oxalate reduces ganglioside storage in juvenile Sandhoff disease mice. Neurochem. Res. 38 (4), 866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bourque E, Celatka C, Hirth B, Metz M, Zhao Z, Skerlj R, Xiang Y, Jancisics K, Marshall J, Cheng S, Scheule R, Cabrera-Salazar M, and Good A. (2012) Quinuclidine derivatives as glucosylceramide synthase inhibitors and their preparation and use in the treatment of lysosomal storage diseases and cancer. WO2012129084A2. [Google Scholar]
- (11).Marshall J, Nietupski JB, Park H, Cao J, Bangari DS, Silvescu C, Wilper T, Randall K, Tietz D, Wang B, Ying X, Leonard JP, and Cheng SH (2019) Substrate Reduction Therapy for Sandhoff Disease through Inhibition of Glucosylceramide Synthase Activity. Mol. Ther. 27 (8), 1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Marshall J, Sun Y, Bangari DS, Budman E, Park H, Nietupski JB, Allaire A, Cromwell MA, Wang B, Grabowski GA, Leonard JP, and Cheng SH (2016) CNS-accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol. Ther. 24 (6), 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hirth BH, and Siegel C. (2003) Preparation of ceramide analogs as UDP-glucose: N-acylsphingosine glucosyltransferase inhibitors and intermediates thereof. WO2003008399A1. [Google Scholar]
- (14).Shayman JA, and Abe A. (2000) Glucosylceramide synthase: assay and properties. Methods Enzymol. 311, 42–9. [DOI] [PubMed] [Google Scholar]
- (15).Abe A, Inokuchi J, Jimbo M, Shimeno H, Nagamatsu A, Shayman JA, Shukla GS, and Radin NS (1992) Improved inhibitors of glucosylceramide synthase. J. Biochem. 111 (2), 191–6. [DOI] [PubMed] [Google Scholar]
- (16).Abe A, Radin NS, Shayman JA, Wotring LL, Zipkin RE, Sivakumar R, Ruggieri JM, Carson KG, and Ganem B. (1995) Structural and stereochemical studies of potent inhibitors of glucosylceramide synthase and tumor cell growth. J. Lipid Res. 36 (3), 611–21. [PubMed] [Google Scholar]
- (17).Lee L, Abe A, and Shayman JA (1999) Improved inhibitors of glucosylceramide synthase. J. Biol. Chem. 274 (21), 14662−9. [DOI] [PubMed] [Google Scholar]
- (18).Xu YH, Quinn B, Witte D, and Grabowski GA (2003) Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am. J. Pathol. 163 (5), 2093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhao J, Gimi R, Katti S, Reardon M, Nivorozhkin V, Konowicz P, Lee E, Sole L, Green J, and Siegel CS (2015) Process Development of a GCS Inhibitor Including Demonstration of Lossen Rearrangement on Kilogram Scale. Org. Process Res. Dev. 19 (5), 576–581. [Google Scholar]
- (20).Abe A, Gregory S, Lee L, Killen PD, Brady RO, Kulkarni A, and Shayman JA (2000) Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. J. Clin. Invest. 105 (11), 1563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.