

Abstract
Innate immunity is associated with Alzheimer disease (AD)1, however, the influence of immune activation on Aβ production is unknown2,3. Here, we identify interferon-induced transmembrane protein 3 (IFITM3) as a γ-secretase modulatory protein, establishing a mechanism by which inflammation impacts Aβ generation. Inflammatory cytokines in neurons and astrocytes induce IFITM3 which binds to γ-secretase and upregulates its activity for Aβ production. IFITM3 expression is increased with aging and familial Alzheimer disease’s genes in mouse models. Furthermore, IFITM3 knock out reduces γ-secretase activity and the formation of amyloid plaques in 5XFAD mice. IFITM3 protein is upregulated in a subset of late onset AD (LOAD) patients that exhibit higher γ-secretase activity. The amount of IFITM3 in the γ-secretase complex has a strong and positive correlation with γ-secretase activity in LOAD. These findings reveal an unprecedented mechanism in which γ-secretase is modulated by IFITM3 by neuroinflammation and increases risks for AD.
γ-Secretase consists of four obligatory subunits, presenilin (PS), nicastrin (Nct), Aph-1 and, Pen-24. Increasing evidence suggests γ-secretase activity can be regulated by γ-secretase modulatory proteins (GSMPs)5. Identification of GSMPs is enormously challenging due to the nature of their modulatory, rather than essential, role in γ-secretase activity and the manner of cellular context-dependent interaction, such as Hif1α under hypoxia6. γ-Secretase modulators (GSMs) that selectively reduce the production of Aβ42, without affecting other cleavages, have emerged as a promising strategy for AD drug development7–10 and are valuable tools to study γ-secretase.
We have shown that E2012-BPyne, a GSM, interacts with PS1 with a target-based approach11. In the present study, we searched for E2012-BPyne binding proteins in an unbiased manner and identified IFITM3 (interferon-induced transmembrane protein 3). IFITM3 is an innate immune response protein known to restrict various viral infections12. We have determined that IFITM3 binds γ-secretase and regulates γ-secretase activity for Aβ production. This study reveals an unprecedented connection between innate immunity and Aβ and strongly suggests that neuroinflammation can modulate γ-secretase production of Aβ. Furthermore, IFITM3 increases γ-secretase activity for Aβ production in a subpopulation of LOAD patients and, therefore, could be a potential risk factor for AD. IFITM3 may function as an immune switch to increase γ-secretase activity for production of Aβ for its anti-microbial activity13,14, and consequently, contribute to AD pathogenesis.
IFITM3 part of the γ-secretase complex.
To identify GSM-binding proteins, E2012-BPyne (Fig. 1a) was incubated with cell membranes, photolabeled, clicked with TAMRA-azide and analysed by gel fluorescence (Fig. 1b). Two bands migrating at approximately 30 kDa and 15 kDa (Fig. 1b) were blocked by E2012. The 30 kDa protein is PS1-NTF as reported previously11, the 15 kDa protein was identified as IFITM3 by LC-MS/MS (ExtFig. 1a) and confirmed by Western blot analysis (Fig. 1c). Additionally, the photolabeling is partially suppressed by L-685,458 (L458) (Fig. 1c). The labeling of PS1-NTF is enhanced by L458 (Fig. 1c, ExtFig. 1b), consistent with previous report11. Different types of GSMs and γ-secretase inhibitors (GSIs) (ExtFig. 1c) have distinct effects on the labeling of IFITM3 (ExtFig. 1d).
Fig. 1. IFITM3 directly binds to γ-secretase.

(a) E2012 and E2012-BPyne structures. (b) Cell membranes were photolabeled with E2012-BPyne and then clicked with TAMRA-azide and analyzed (left: Coomassie blue; right: in-gel fluorescence). (c) Biotinylated proteins were captured by E2012-BPyne and analyzed by WB. (d) IFITM3 co-immunoprecipitated with γ-secretase subunits using anti-PS1. (e) IFITM3 was co-purified with γ-secretase subunits by GY6 or 163-BP-L-biotin. (f) An interaction (red) between PS1 and IFITM3 was determined by in situ PLA in mouse primary neurons (bottom panel). Negative controls using PS1 or IFITM3 alone (top two panels), dapi (blue), F-actin (green), scale bar = 50μm. (g) Labeling of IFITM3 by E2012-Bpyne in WT and dKO MEF cells. All WB and IF images are representative of three independent experiments (except b: 2 replicates).
IFITM3 immunoprecipitates with γ-secretase subunits when pulled-down with PS1-NTF antibodies but not with control IgG (Fig. 1d), indicating that IFITM3 interacts with the γ-secretase complex. Furthermore, GY6 (ExtFig. 1e) that only binds to active γ-secretase6 captures not only PS1-NTF and Nct, but also IFITM3 (Fig. 1e) under native conditions. Additionally, 163-BP-L-biotin (ExtFig. 1e), also captures PS1-NTF, Nct and IFITM3 (Fig. 1e), confirming that IFITM3 is associated with the active γ-secretase complex. PS1 interacts with IFITM3 in primary neurons as indicated by discrete red spots in a Proximity Ligation Assay (Fig. 1f). IFITM3 also interacts with PS2 (ExtFig. 1f), but not signal peptide peptidase (SPP)(ExtFig. 1g).
The labeling of IFITM3 by E2012-BPyne in γ-secretase deficient cells was examined. PS1-NTF is detected in wild-type (WT) but absent in dKO (PS1/2 KO) MEF cells (Fig. 1g). IFITM3 is present in both cell lines but is much lower in the dKO cells, indicating that PS1/PS2 affects the overall protein level of IFITM3 (Fig. 1g, ExtFig. 1h) but not mRNA levels (ExtFig. 1i). Both PS1-NTF and IFITM3 are labelled by E2012-BPyne in WT MEF but not in the dKO cells (Fig. 1g), demonstrating that IFITM3 must be part of the γ-secretase complex to be labelled.
IFITM3 modulates γ-secretase activity.
IFITM3 was knocked-down (KD) by siRNA in HEK-APP cells (Fig. 2a). KD of IFITM3 does not affect the levels of Nct, PS1-NTF and APP proteins (Fig. 2a, ExtFig. 2a) but reduces the production of Aβ40 and Aβ42 by 17.1 ± 2.4% and 24.6 ± 1.3%, respectively, compared to scrambled siRNA (SC) (Fig. 2b). Next, IFITM3 was knocked out (KO) in U138 astrocytoma cells (Fig. 2c, lane 3 and 4) and then reintroduced by transient overexpression (Fig. 2c, lane 2 and 4). γ-Secretase activity was determined using a recombinant APP substrate (ExtFig. 2b). IFITM3 KO reduced γ-secretase activity for both Aβ40 and Aβ42 cleavages as compared to the EV (empty vector guide RNA) cell line by 36% and 27%, respectively (Fig. 2d, bar 1 and 3). Importantly, IFITM3 overexpression in the KO cell line rescued the loss of γ-secretase activity (Fig. 2d, bar 3 and 4). Transient overexpression of IFITM3 in the EV cell line (Fig. 2c, lane 2) significantly increased γ-secretase activity for Aβ40- and Aβ42-cleavage (Fig. 2d, bar 1 and 2). Taken together, the level of IFITM3 is associated with γ-secretase activity for Aβ cleavage.
Fig. 2. Effect of IFITM3 on γ-secretase activity for APP cleavage.

(a) IFITM3 KD by siRNA (n=3) in HEK-APPWT cells was confirmed by WB. Scramble siRNA (SC, n=3), a negative control. (b) IFITM3 KD reduces secreted Aβ40 (n=6, ****p<0.0001) and 42 (n=6, ****p<0.0001). Aβ levels were calculated as % of SC. (c) IFITM3 was KO of U138 cells, and empty vector (EV) was used as control. IFITM3 was reintroduced by transient transfection in both EV and KO cell lines. IFITM3 expression was confirmed by WB. (d) Effect of KO and rescue of IFITM3 on γ-secretase activity for Aβ40 (n=3, *p= 0.0249, ****p<0.0001) and 42 (n=3, *p=0.0359, **p=0.0025) cleavage. (e) Comparison IC50 of GSM E2012-BPyne against γ-secretase for Aβ40 (n=6, **p= 0.0017) and Aβ42 (n=6, ns) cleavages in U138 EV and KO cells. All WB images and graphs are representative of three independent experiments (except; e: contains data from 2 experimental replicates of n=3). Graphs are mean ± SD. Ns, not significant, two-sided Student’s t-test (except; d: one-way ANOVA and Fishers LSD test).
Next, we determined how IFITM3 affects the potency and selectivity of GSMs and L458, which bind to the different sites of γ-secretase7 (ExtFig. 2c). E2012-BPyne selectivity (Aβ40/Aβ42) is 21-fold in γ-secretase from EV membranes compared to 57-fold in KO membrane (Fig. 2e). IFITM3 KO has an opposite effect on two types of GSMS and no effect on L458 (ExtFig. 2d–f), indicating that IFITM3 not only affects γ-secretase activity, but also alters the selectivity to GSMs. Surprisingly, KD (ExtFig. 2g–h) and KO of IFITM3 increases γ-secretase activity for Notch1 cleavage in cell-based assays (ExtFig. 2h, i) and γ-secretase in in vitro activity assay (ExtFig. 2j), demonstrating that IFITM3 exhibits distinct modulatory effects on γ-secretase processing of APP and Notch1.
Aging and FAD upregulates IFITM3.
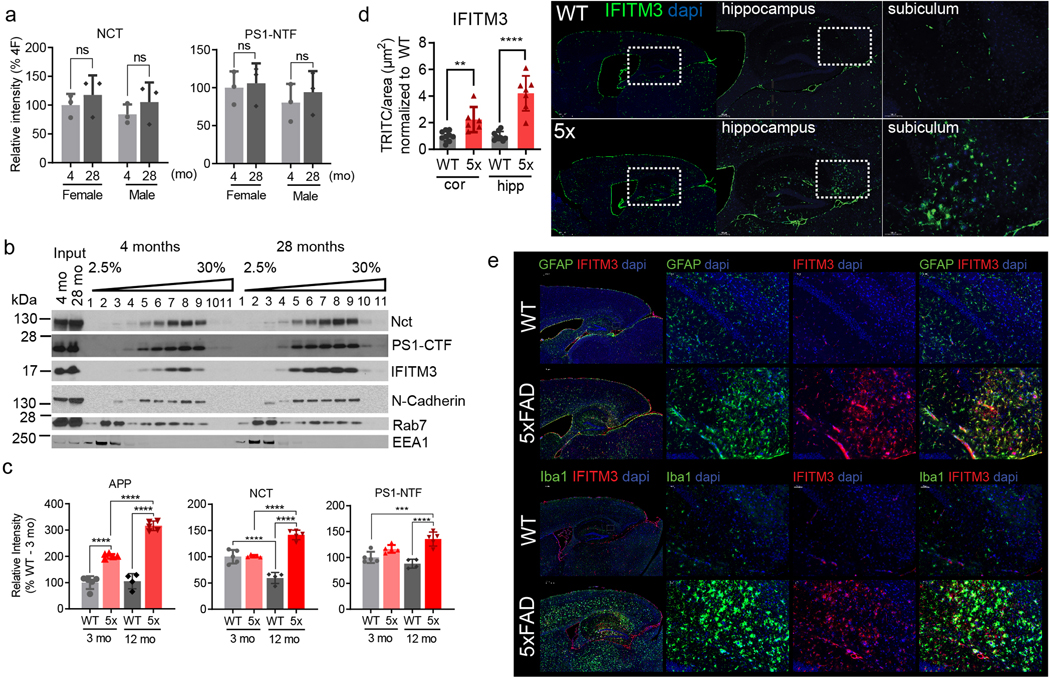
The expression of IFITM3 significantly increases with age (2.9- and 2.3- fold at 28-months compared to 4-months in females and males, respectively) whereas Nct and PS1-NTF levels are relatively consistent (Fig. 3a, ExtFig. 3a). Furthermore, the subcellular distribution of IFITM3 and γ-secretase components does not change with age (ExtFig. 3b). γ-Secretase activity in vitro for both Aβ40 and Aβ42 cleavages is higher in the 28-month old mouse brains compared to 4-months (Fig. 3b). Capture of solubilized γ-secretase by 163-BP-L-biotin showed that no detectable IFITM3 co-existed with PS1-NTF despite the existence of IFITM3 in solubilized membranes at 4 months, however, IFITM3 is clearly part of the γ-secretase complex in 28-month-old mice of both genders (Fig. 3c). To look directly at the impact of IFITM3 on γ-secretase, we assayed γ-secretase activity in cortical tissue of 18-month-old IFITM3 KO (IFITM3−/−) and WT mice. IFITM3−/− does not impact the amount of γ-secretase complex, however, IFITM3−/− reduces γ-secretase activity for the Aβ40- and Aβ42-site by 15.3% and 24.3%, respectively (Fig. 3d). Collectively, these results indicate that the increase in γ-secretase activity and Aβ production with aging is at least partially due to the increased IFITM3 protein.
Fig. 3. Effect of aging and the expression of APP/PS1 on IFITM3 and γ-secretase.

(a) Protein levels of γ-secretase subunits and IFITM3 in pooled membranes from 4- and 28-month-old WT mouse brains (n=5 mice per age and sex group except n=4 for 28 female, female: **p=0.0014, male: **p=0.0065). Quantified as percent of 4-month female level. (b) γ-Secretase activity for Aβ40 (female: p=***0.0002, male: **p=0.0095) and Aβ42 (female: p=**0.0024, male: **p=0.0088) production in vitro from pooled membrane. (c) Solubilized membranes were captured by 163-BP-L-biotin and then analyzed for γ-secretase and IFITM3 levels. (d) WB for PS1-NTF and IFITM3 (left panel, representative mice shown) in membranes of 18-month-old WT (n=4) and IFITM3−/− (n=5) mouse brains. γ-Secretase activity for Aβ40 (***p=0.0004) and 42 (****p=<0.0001) cleavage (right panels). (e) APP, Nct, PS1-NTF and IFITM3 in membranes from 3- and 12-month-old WT and 5xFAD mice. Quantified as percent of 3-months-old WT (n=5 per group except n=4 for WT at 12 months)(3moWT-12mo5X: ****p<0.0001, 12moWT-12mo5X: ***p=0.004, 3mo5X-12mo5X: ***p=0.0003). (f) WB for PS1-NTF and IFITM3 (left panel) in membranes from 4-month-old 5xFAD (n=3) and IFITM3−/−; 5xFAD (n=3) mouse brains. γ-Secretase activity in vitro for Aβ40 (***p=0.0003) and 42 (**p=<0.0011) cleavage (right panels). (g) Fluorescence microscopy of amyloid plaques in cortex and hippocampus (Thioflavin-S, green) in 4-month-old PFA perfused mice (5xFAD: n=4, IFITM3−/−; 5xFAD: n=5). Scale bar = 300μm (left), 200μm (right). Number of plaques per mm2 of tissue was calculated throughout the brain and averaged (cor (cortex): *p=0.0109, hip (hippocampus): **p=0.0026). All WB and IF images and graphs are representative of three independent experiments (except; c and quantifications in e: 2 replicates and g: every 1/6th section throughout the brain). Bar graphs are mean ± SD. Violin plots represent median (middle line) and interquartile range (outer lines). Ns, not significant, two-sided Student’s t-test (except; e: two-way ANOVA followed by Tukey).
Next, we examined IFITM3 levels in 5XFAD mice15. The 5XFAD brain has elevated levels of both APP, Nct and PS1 at 3 and 12 months compared to WT (Fig. 3e, ExtFig. 3c). Similarly, IFITM3 protein is higher in 5XFAD mice than WT at 12 months, suggesting that overexpression of APP and PS1 leads to an increase in IFITM3 (Fig. 3e). IFITM3 upregulation in the 5XFAD brain at 12 months is most evident in regions with dense Aβ burden such as the hippocampus and cortex (ExtFig. 3d). Within the WT brain, IFITM3 expression is primarily located within the meninges and blood vessels, whereas, IFITM3 heavily colocalizes with GFAP and Iba1 in the 5XFAD brain suggesting that FAD genes induce IFITM3 in glia (ExtFig. 3e). The level of IFITM3 also increases with age in 5XFAD mice, indicating that aging and FAD genes have either a synergistic or additive effect on IFITM3 expression.
IFITM3−/− does not alter the amount of γ-secretase complex in 4-month-old 5XFAD mice (Fig. 3f), but significantly reduces γ-secretase activity for Aβ40 and Aβ42 cleavages (by 30% and 22%, respectively) indicating that IFITM3 expression affects Aβ40 and Aβ42 production (Fig. 3f). Consequently, at 4-months-old, IFITM3−/− dramatically reduces amyloid plaque deposition in 5XFAD mice by 54.2% in the hippocampus and 81.9% in the cortex (Fig. 3g). These findings demonstrate that IFITM3 regulates γ-secretase activity for Aβ production with aging and PS1 mutations in vivo.
Association of IFITM3 and γ-secretase in AD patients.
Analysis of gene expression patterns in the Mayo Clinic large cohort dataset16 revealed that IFITM3 expression is significantly higher in AD patients than control in the temporal cortex (Fig. 4a). Moreover, the expression of IFITM3 is positively correlated with age in both the cortex and hippocampus of human brains in the Genotype-Tissue Expression (GTEx) cohort17 (ExtFig. 4a). Next, expression of IFITM3 in the frontal cortex of postmortem tissues from human LOAD (n=18) and non-demented subject control (n=10) (Table S1) was examined. The mRNA level of IFITM3 is higher in LOAD samples than in controls (Fig. 4b). Interestingly, GFAP (astrocyte marker) is also elevated in LOAD whereas MAP2 (neurons) and AIF1 (microglia) levels are similar to control (ExtFig. 4b), which is consistent with the Mayo Clinic large cohort dataset data (ExtFig. 4c)16. Protein levels of Nct and PS1-NTF are higher in the control than in LOAD samples (ExtFig. 4d). IFITM3 protein is more highly expressed in LOAD samples, and while expression is similar throughout the control brains, there is large variation between LOAD samples (Fig. 4c). Since IFITM3 level is associated with γ-secretase activity, and IFITM3 protein is significantly elevated in some LOAD patients (Fig. 4c), LOAD brain samples were divided into two groups. Samples with IFITM3 level greater than two standard deviations from the mean of control were defined as LOAD-H (High, 8 patients), while the remaining samples were defined as LOAD-L (Low, 10 patients) (Fig. 4d, left panel) (Table S1). Strikingly, LOAD-L samples have similar γ-secretase activity as the control brains, whereas LOAD–H samples exhibit 127.6% and 133.6% activity compared to control for Aβ40 and Aβ42, respectively. In addition, the expression of IFITM3 is correlated with Aβ load in the brain regions of BA22 (rho=0.149, p=0.0497) and BA36 (rho=0.170, p=0.0204) in the Mount Sinai Brain Bank dataset18 (Table S2). These findings demonstrate that there is an association between IFITM3 protein and γ-secretase activity for Aβ production and suggests that IFITM3 contributes to Aβ production in a subset of LOAD.
Fig. 4. The association of IFITM3 with the γ-secretase complex in human brains.

(a) The expression profiles of IFITM3 in human control (n=76) and LOAD (n=80) from temporal cortex (**p=0.002)(Mayo Clinic cohort). (b) IFITM3 mRNA expression levels in control and LOAD samples (LOAD: n=18 and control: n=10, **p=0.0056). (c) The protein levels of γ-secretase subunits and IFITM3 in human brain membranes (LOAD: n=18 and control: n=10, *p=0.0127). IFITM3 quantified as relative intensity. (d) IFITM3 expression (left) between control (n=10), LOAD-L (n=10) and LOAD-H (n=8)(****p<0.0001). γ-Secretase activity for Aβ40 (**p=0.0037, ***p=0.0007) and 42 (**p=0.0036, ***p=0.0002) cleavage between groups. (e) Primary mouse neurons were treated with control (n=2), 10ng/mL (n=3, ***p=0.0005) or 100 ng/mL (n=3, ***p=0.0003) of IFN-γ, membranes were probed for γ-secretase and IFITM3 (left panel, quantified: right panel). (f) Quantification of secreted Aβ40 from 10ng/mL (n=8, **p=0.0010) and 100 ng/mL (n=8, **p=0.0026) IFN-γ treated neurons Aβ42 from 10ng/mL (n=8, ***p=0.0006) and 100 ng/mL (n=8, ***p=0.0003) IFN-γ (n=8). (g) γ-Secretase activity for Aβ40 (n=6, ***p=0.0007) and 42 (n=6, ***p=0.006) cleavage from IFN-γ treated neuron membranes. (h) Photolabeled PS-NTF in neuronal membranes and quantified as % of control (control: n=10, 10ng/ml: n=5, **p=0.0023, 100ng/ml: n=7,****p<0.0001). (i) WB for IFITM3 and PS1-NTF primary human astrocytes treated with PBS, IL-6, or IL-1β (control: n=6, IL-6: n=4, **p=0.0014, IL-1β: n=3, *p=0.0103), data normalized to PBS. (j) γ-Secretase activity for Aβ40 (control: n=8 IL-6: n=7, **p=0.0098, IL-1β: n=9,****p<0.0001) and 42 (control: n= IL-6: n=7, *p=0.0467, IL-1β: n=9, ****p<0.0001) in astrocyte membrane. All WB images and graphs are representative of three independent experiments (except; b: 1 replicate, c/g: 2 replicates, d: γ-secretase activity graph contains data from two independent replicates, h: 5 replicates). Graphs are mean ± SD. Ns, not significant, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, two-sided Student’s t-test.
Single nucleotide polymorphisms (SNPs) in IFITM3 have been associated with the severity of viral infection and gene expression level19. We genotyped for IFITM3 SNP variants rs12252 and rs34481144 in the human samples (Table S3, ExtFig. 4e–f). R analysis indicates T allele dosage for rs34481144 doesn’t affect the relationship between AD status (control, LOAD-L, LOAD-H) and IFITM3 protein expression in this sample set.
Induction of IFTM3 in neurons and astrocytes.
The expression of IFITM3 is detected in mouse hippocampal neurons, human iPSC-derived neurons and primary human astrocytes (ExtFig. 4g–j). The effect of interferon-γ (IFN-γ) on IFITM3 and γ-secretase in mouse primary neuronal culture was investigated. IFN-γ (at 10 and 100 ng/mL) significantly induces IFITM3 but has little effect on Nct and PS-NTF (Fig. 4e). Furthermore, IFN-γ treatment leads to an increase in secreted Aβ40 and Aβ42 (Fig. 4f), potentiates γ-secretase for Aβ40 and Aβ42 cleavage by 1.69- and 1.7-fold, respectively (Fig. 4g), and increases photolabeling of active γ-secretase (Fig. 4h). IFN-α exhibits similar effects to IFN-γ (ExtFig. 4k–m). As astrocytes are another source of Aβ20, dissociated human astrocytes (hAST) were treated with IL-6 and IL-1β which also upregulated IFITM3 expression (Fig. 4i) and increased γ-secretase activity for Aβ40 and Aβ42 cleavages (Fig. 4j).
Association of IFITM3 with cytokines is supported by gene expression data from the Harvard Brain Tissue Resource Center21, in which there is a correlation between transcription of IFITM3 and IL-6, IL-1β, IL-10 and IL-8 and type I IFN responsive genes (IFITM1 and IFITM2) (Table S4–5). Furthermore, the expression level of IFITM3 is positively correlated with the expression level of the human herpes virus 6B (HHV-6B, rho=0.248, P=0.044) and hepatitis C virus genotype 4 (rho=0.255, P=0.033) in the Mount Sinai Brain Bank dataset18 (ExtFig. 4n), suggesting that cytokine induction of IFITM3 could be part of the immune response to such viruses.
IFITM3 is near the active site of γ-secretase.
To examine whether IFITM3 is near the catalytic site of γ-secretase (Fig. 5a), four photoaffinity active site directed inhibitors (ExtFig. 5a) were used. Since all four probes specifically labelled PS1-NTF and IFITM3 (Fig. 5b), we conclude that IFITM3 interacts with γ-secretase near the active site. In addition, 11bt that was used to labeling the docking site of γ-secretase22 is blocked by pep 11, but not E2012, suggesting the E2012 binding site does not overlap with the docking site (ExtFig. 5b).
Fig. 5. IFITM3 is located near the active site of γ-secretase.

(a) Schematic model showing L458 binding to the subsites (S2-S3’) in the active site of γ-secretase. (b) Photolabeling of IFITM3 and PS1 by four inhibitors. (c) Photolabeling of IFITM3 and PS1 by L505 in human brains (control: n=4, LOAD-L: n=5, LOAD-H: n=5). (d) Pearson’s correlation between γ-secretase activity (Fig. 4d) and L505 labeled IFITM3 (Fig. 5c) in LOAD samples (n=10). (e) Double cross-linking of γ-secretase and IFITM3 by the dual probe, L631. WB reveals multiple protein complex species containing IFITM3, PS1-NTF, PS1-CTF, IFITM3 homodimer, IFITM3-PS1 and PS1-NTF-CTF heterodimers. (f) Schematic representation of the interaction between IFITM3 and γ-secretase, IFITM3 is near the active site and can be crosslinked with PS1-NTF. (g) IFITM3 connects infections and innate immunity with Aβ production and AD risk. (A) Pathogenic challenge, or other inflammatory conditions, induce the release of proinflammatory cytokines from astrocytes and microglia. (B) Cytokines upregulate IFITM3 expression in neurons and astrocytes that potentiates γ-secretase, increasing Aβ production. (C) As part of an innate immune response, Aβ acts as an antimicrobial or antiviral peptide. In turn, Aβ accumulation also triggers AD pathology. All WB images and graphs are representative of three independent experiments (except; c: 2 replicates).
Next, membranes isolated from human samples were labeled with L505. L505 labels both PS1-NTF and IFITM3 (Fig. 5c), indicating that IFITM3 is engaged in the γ-secretase complex in human brains as well. There is a positive correlation between the L505 labeled PS1-NTF and γ-secretase activity for Aβ40 cleavage (R=0.67) and Aβ42 cleavage (R=0.66) (ExtFig. 5c). Furthermore, the correlation coefficients between the L505 labeled IFITM3 and γ-secretase activity for Aβ40 and Aβ42 are 0.71 and 0.73, respectively (Fig. 5d). Taken together, the strong correlations between the L505 labeled IFITM3 and γ-secretase activity in LOAD brains highlights IFITM3 as a marker of γ-secretase activity.
To investigate whether IFITM3 is cross-linked with PS1, we used L631, which contains two photoreactive groups allowing for crosslinking of two labeled species (Fig. 5e). L631 labels three species: PS1-NTF, PS1-CTF and cross-linked PS1-NTF/CTF (Fig. 5e). When cross-linked species were probed by anti-IFITM3 antibody, three species that co-migrate at molecular mass of approximately 15, 30 and 45 kDa (Fig. 5e) were detected, corresponding to IFITM3 monomer, dimer and crosslinked IFITM3/PS1-NTF (recognized by both IFITM3 and PS1-NTF antibodies), respectively. Although, IFITM3 dimer has been observed, whether IFITM3 exists as monomer or dimer needs to be determine by structural studies. The crosslinking of IFITM3 and PS1-NTF by L631 further confirms that IFITM3 binds near the active site of γ-secretase (Fig. 5f).
Discussion
Our work identifies IFITM3 as a GSMP associated with aging and AD and directly connects Aβ production with an emerging field in AD research: innate immunity and neuroinflammation. IFITM3 has a broad spectrum of antiviral activities and serves as in the first line of defense against infection12. IFITM3 is basally expressed in many cells and highly upregulated by both type I and II IFNs12. IFITM3 has also previously been suggested as one of hundreds of potential PS1- and PS2-interacting proteins23. However, whether IFITM3 regulates γ-secretase and is involved in AD was previously unknown.
Neuroinflammation has emerged as a critical component of AD pathogenesis1,24–26. Multiple studies have shown that TREM2 and CD33 regulate microglia-mediated clearance and uptake of Aβ2,3. In contrast, our work reveals that neuroinflammation can also contribute to the production of Aβ through an IFITM3-γ-secretase complex. It has been reported that aging, the biggest risk factor for AD, induces type I IFNs that modulate brain function27. Therefore, aging induced neuroinflammation could lead to an increase in the level of IFITM3 which potentiates γ-secretase activity for Aβ production in human (Fig. 5g).
Increasing evidence indicates that Aβ peptides have antimicrobial and antiviral activities13,14 as part of the innate immune response in the brain. This raises the possibility that pathogenic challenge stimulates Aβ production as part of an innate immune response and a secondary consequence is AD risk (Fig. 5g). Discovery that IFITM3 upregulates γ-secretase activity for Aβ production (Fig. 5g, B) offers a mechanism as to how Aβ production is regulated in an innate immune response (Fig. 5g, C). Pathogenic challenge results in cytokine production by microglia, astrocytes and possibly other cell types (Fig. 5g, A). Proinflammatory cytokines induce IFITM3 expression which in turn augments γ-secretase in neurons and astrocytes, resulting in increased Aβ secretion (Fig. 5g, B). This has two consequences: anti-infection and increased risk for AD (Fig. 5g, C).
We identified IFITM3 as a novel GSMP and demonstrated that IFITM3 binds to PS1 in the proximity of the active site and upregulates γ-secretase for Aβ production. The strong correlation between the level of IFITM3 associated with γ-secretase and enzymatic activity for Aβ cleavage suggests that IFITM3 protein could be an indicator of γ-secretase activity in LOAD. Importantly, the differential level of IFITM3 protein within LOAD patients suggests that IFITM3 could potentially serve as a biomarker and therapeutic target for a specific subpopulation of LOAD. LOAD is a multifactorial disease; therefore, identification of AD subpopulations will aid in studying the underlying mechanisms and developing precision medicine treatments.
Materials and Methods
Chemical compounds
Structures and characterization of γ-secretase modulators (GSMs) and inhibitors (GSIs) including E2012, E2012-BPyne, E2212, NGP-97555, GSM-1, GSM-25, GY6, 163-BP-L-biotin, LY450139, BMS708163, L458, JC8, L505, L646, GY4, and L631 have been described previously10,11,28–36. Structures of compounds were drawn by using ChemDraw (PerkinElmer). Docking site inhibitor, pep 11Bt (11Bt) and its parent compound, pep11 have been described previously22.
Cell line culture
The following cells were cultured in DMEM medium with high glucose, penicillin, streptomycin and 10 % FBS: MEF (WT and PS1/2 double KO, a kind gift from Dr. Huaxi Xu, Sanford Burnham Prebys Medical Discovery Institute); HEK293-APP695 cells (a kind gift from Dr. Elizabeth Chen, Merck Research Laboratories) and HEK293-NotchΔE cells (a kind gift from Dr. Douglas S. Johnson, Pfizer); U138 cells (U-138 MG human grade IV glioblastoma cells, HTB-16, ATCC). All cells were cultured at 37 °C with 5% CO2/95% air.
Primary cell culture
Neuronal culture:
Mouse embryos at E16 (Embryonic day 16) were isolated from WT C57BL/6J or C57BL/6NJ mice as described previously37. Briefly, cortices were collected from dissected brains and meninges were removed. Cortices were trypsinized and cell were dissociated and plated onto PDL/PLL (Sigma) coated chamber slides (Corning) or cell culture plates. Primary mouse neurons were seeded in seeding medium (neurobasal medium/L-glutamax/10% FBS). At DIV1 (1 day in vitro), cell culture media was changed for 100% to NBM complete medium (neurobasal medium/L-glutamax/B-27/N2/FDU) and media was changed for 50% thereafter.
Human primary astrocytes (hAST):
hAST were isolated from cerebral cortex and cryopreserved were obtained from ScienCell (1800). hAST were cultured in basal astrocyte medium (1801, ScienCell) containing 2% fetal bovine serum (0010, ScienCell), 1% astrocyte growth supplement (1852, ScienCell) and 1% penicillin/streptomycin solution (0503, ScienCell). hAST were confirmed by immunofluorescence staining with GFAP and S100β (data not shown).
Generation of iNeurons from Human iPSCs:
Homogeneous population of excitatory neurons were generated from neural progenitor cells (NPC) derived from human induced pluripotent stem cells (iPSCs)38. NPCs were dissociated with accutase (Innovative Cell Technologies) and plated at 250,000 cells per well of a 24-well plate on day 2. Cells were plated on matrigel (BD Biosciences)-coated coverslips or plates in NPC medium (DMEM/F12/N2/B27/FGF2, Invitrogen). On day 1, hNGN2 lentivirus (TetO-hNGN2-P2A-PuroR (79049, Addgene) with CAGGs-rtTA lentivirus at 1×106 pfu/ml per plasmid (multiplicity of infection (MOI) of 2) was added in fresh NPC medium and spinfected at 1,000 X g for 1 hr. After 3–4 hrs, the medium was replaced with fresh NPC medium. Doxycycline (2 μg/l, Clontech) was added on day 0 to induce TetO gene expression and retained in the medium until the end of the experiment. On day 1, a 48 hr puromycin selection (1 μg/ml) period was started. On day 2, the culture medium was replaced by neuron medium: DMEM/F12 medium supplemented with N2/B27/Glutamax (Invitrogen) containing BDNF (20 ng/ml, Peprotech), GDNF (10 ng/ml, PeproTech), Dibutyryl cyclic-AMP (250 μg/ml, Sigma-Aldrich), and L-ascorbic acid (200 nM, Sigma-Aldrich); Ara-C (2 μg/l, Sigma-Aldrich) was added for 48 hrs to the medium to inhibit proliferation of any non-neuronal cells. Beginning at day 4, half of the medium in each well was replaced every other day. hNGN2-induced neurons were assayed on day 14.
Animals
All procedures were carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by Research Animal Resource Center of Memorial Sloan Kettering Cancer Center and the Animal Care and Use Committees of the Rockefeller University. B6.CgTg (APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax) (5XFAD) mice were obtained from the Jackson Laboratory. 5XFAD mice were maintained hemizygous by breeding with C57BL/6J. Frozen IFITM3−/− embryos were obtained from the European Mutant Mouse Archive and rederived in pseudopregnant host mice. IFITM3−/− mice were bred with 5XFAD mice to generate IFITM3−/−; 5XFAD mice. All mice were genotyped by PCR amplification of DNA extracted from tail.
For primary neuronal culture, wild type C57BL/6J or C57BL/6NJ mice with timed pregnancy were obtained from the Jackson Laboratory and Charles River Laboratories, respectively. Translating ribosome affinity purification (TRAP) knock-in mice were generated by crossing mice bearing a loxP-stop-loxP-EGFPRPL10a sequence in the Eef1α1 promoter (EEF1A1–LSL.EGFPL10)39 with CCK-Cre (CCKtm1.1(cre)Zjh/J), Glutamate decarboxylase 2 (65 kDa)-Cre (B6N.Cg-Gad2tm2(cre)Zjh /J), Cortistatin-Cre (Tg(Cortcre)IM42Gsat) or parvalbumin-Cre (Pvalbtm1(cre)Arbr/J) mice. RNA isolation and qPCR analysis were done as previously described40.
Mouse brain samples
Brains of C57BL/6 female and male mice at different age groups (mature adult (4-month-old) and old (28-month-old)) were collected from NIA Aged Rodent Tissue Bank and stored at −80 °C before use. Brains of female WT and 5XFAD Tg mice at 3- (mature adult) and 12-month-old (middle aged) were kindly provided by Dr. Robert J Vassar (Northwestern University). Brains were extracted from WT and IFITM3−/− at 18-month-old, then the cortex dissected and flash frozen and stored at −80 °C before use. Cortical tissue from 4-month-old 5XFAD and IFITM3−/−; 5XFAD mice was extracted in the same way.
Human brain smples
Frontal cortices of human brains from control subjects and LOAD cases were provided by the UCSD ADRC Neuropathology Core after review and approval by the ADRC Biospecimen Review Committee. All human samples were obtained from subject who consented to autopsy for the UCSD ADRC. All tissues provided were deidentified and contained a number code only.
LOAD samples include 6 females and 11 males with an average age of 81 years and controls include 7 females and 3 males (average age = 83.2 years) (Table S1). Based on the quantification of IFITM3 protein levels by WB, LOAD brain samples were separated into two groups: LOAD-L (Low, 10 patients) and LOAD-H (High, 8 patients). Samples with IFITM3 level greater than two standard deviations from the mean of control were defined as LOAD-H, while the remaining samples were defined as LOAD-L.
Human IFITM3 SNP genotyping
We determined if IFITM3 single nucleotide polymorphism (SNP) impact IFITM3 protein expression levels in human brain samples (control, LOAD-L, and LOAD-H groups). Briefly, genomic DNA was extracted from frozen post mortem human brain tissue using the KingFisher Flex System (ThermoFisher Scientific) according to manufacturer’s instructions. TaqMan SNP genotyping assays (ThermoFisher Scientific) were used to interrogate the human IFITM3 SNPs rs12252 (C_175677529_10) and rs34481144 (C__26288451_10) on the QuantStudio 7 Flex Real-Time PCR System (ThermoFisher Scientific) according to manufacturer’s instructions. Molecular grade water and a standard in-house DNA sample were used as non-template control and positive control, respectively. Genotype calls and allelic discrimination plots were generated using QuantStudio Software v1.1 (ThermoFisher Scientific). Genotyping data were reported in REF/ALT format, which is consistent with the human reference genome. The effect of SNP allele dosage on the relationship between AD status and IFITM3 protein expression levels was analyzed using the lm() function in R (version 3.6.2)41. All samples have the same A/A genotype by IFITM3 SNP rs12252 genotyping, indicating that this SNP doesn’t influence IFITM3 protein expression levels in our cohort (Table S3). The allelic discrimination plot for rs34481144 showed control contains homozygous T/T (equivalent to A/A on the negative strand of DNA) and heterozygous C/T (G/A) while LOAD-L and LOAD-H are homozygous C/C (G/G), heterozygous C/T (G/A), and homozygous T/T (A/A) (ExtFig. 4e). Allele frequency of C and T in control and LOAD was calculated (ExtFig. 4f). R analysis indicates T allele dosage for rs34481144 doesn’t affect the relationship between AD status (control, LOAD-L, LOAD-H) and IFITM3 protein expression in our sample set.
Photolabeling with clickable probe, LC-MS/MS and protein identification
Photolabeling with E2012-Bpyne was prepared as described previously11,33. Briefly, cell membranes were incubated with clickable probe, E2012-BPyne in the absence or presence of the indicated compounds (GSMs or GSIs). Samples were cross-linked at UV 350 nm and clicked with TAMRA-Azide for In-gel fluorescence imaging or Biotin-Azide for Western blotting (click chemistry). SDS-PAGE gels were stained with Coomassie Blue for a total protein loaded or an In-gel fluorescence image was scanned by Typhoon Trio Variable Mode Imager (GE Healthcare). For Western blotting analysis, streptavidin beads were added to solubilized samples. For protein identification, HeLa membranes were photolabeled with E2012-BPyne, concentrated with Microcon YM-3, run on SDS-PAGE gel, and stained with Coomassie Blue. Gels were cut, digested by trypsin, and analyzed by LC-MS/MS. Photolabeling with GSI probes (JC8, L505, L646, and GY4)28,32,42,43 and L63144 with two photoreactive groups were performed as described. Photolabeling with the substrate binding site inhibitor, pep 11Bt was performed as described22. Whole cell photolabeling with JC8 was done as described previously6 in primary mouse neurons.
Preparation of membranes and solubilization
HeLa cell membranes (membrane fraction) were prepared from HeLa cell pellet (BioVest International)28. All procedures were carried out on ice. MEF (WT and PS1/2 double KO) cell pellets were resuspended in 5mM Tris, pH 7.4, protease inhibitor cocktail, and PMSF, and incubated on ice for 1 hr. After homogenization with a mechanical pestle homogenizer by 20 strokes, cells were centrifuged at 1,000 X g for 30 min and the post-nuclear supernatants were centrifuged at 100,000 X g for 1 hr to yield the pellet. The pellet was washed with 1X MES (pH = 6.0) protease inhibitor, and PMSF and the final pellet was collected after 100,000 X g for 1 hr, resuspended in buffer, and stored at −80 °C.
Brain tissue membranes were prepared as described previously with some modifications45. Whole brains (or dissected cortices) were homogenized by using TissueLyser II (Qiagen). Briefly, brains were placed with 5 mm stainless steel bead in lysis buffer and homogenized by high-speed shaking at 30/sec for 2 min. Cell and tissue membranes were solubilized with 1% SDS and protein concentrations were determined by the Lowry protein assay (Bio-Rad).
Membranes extracted from brain tissue were solubilized at a protein concentration of 5 mg/ml in lysis buffer containing 1% CHAPSO for 2 hrs at 4 °C with rotation46. Samples were centrifuged at 100,000 X g for 1 hr and CHAPSO solubilized γ-secretase fraction was collected and protein concentration was measured by the Lowry protein assay. HeLa cell membranes were CHAPSO solubilized as described previously28,47,48.
Capture of γ-secretase complex
CHAPSO solubilized γ-secretase was pre-incubated with parent compounds (L458 or BMS708163) at 37 °C for 20 min in the buffer containing 0.25% CHAPSO, followed by the capture with GY6 or 163-BP-L-Biotin at 37 °C for 1 hr6,31,47. Pre-washed streptavidin beads were added and incubated with at 4 °C for overnight. Beads were washed with 0.25% CHAPSO and eluted in SDS sample buffer and analyzed by Western blotting.
Co-immunoprecipitation
CHAPSO solubilized HeLa cell membranes (400 μg of protein/sample) were immunoprecipitated by anti-PS1-NTF or anti-IFITM3 antibody in immunoprecipitation buffer (20 mM Hepes (pH 7.5), 50 mM KCl, 2 mM EGTA, 0.25% CHAPSO and protease inhibitor mixture)6,48. In brief, protein A/G magnetic beads (Pierce) were washed then added to samples for 30 min at 4 °C with rotation as a pre-clear step. The supernatant from samples were incubated with 5 μg of purified PS1-NTF antibody, IFITM3 antibody, monoclonal IFITM3 antibody (9D11, generated in our lab), ImmunoPure rabbit IgG (Pierce) or mouse IgG isotype control (Invitrogen) for overnight at 4 °C with rotation. Beads were added to samples for 1 hr at RT with rotation. Beads were washed three times with 1 ml of immunoprecipitation buffer containing 1% CHAPSO and the beads were eluted in 30 μl of SDS-PAGE sample buffer with DTT for 10 min at RT. The eluted samples were subjected to SDS-PAGE and Western blotting (for source gels see Supplementary Figure 1).
Subcellular fractionation
Male wild-type C57BL/6 mouse hemibrains at 4 and 28 months were homogenized and layered on iodixanol gradient for subcellular fractionations according to Frykman et al49 with some modifications. Briefly, brains were homogenized by 20 strokes on ice using a mechanical pestle homogenizer (Eberbach Corporation), followed by 10 passages through a 21-gauge needle (BD). Homogenates were centrifuged at 1000 X g for 10 min then the post-nuclear supernatant was centrifuged at 10,000 X g for 15 min. The supernatant was layered on the top of 2.5 – 30% (w/v) iodixanol gradients (OptiPrep density gradient medium, Sigma-Aldrich) and were centrifuged at 126,000 X g for 3 hrs (TH-641 rotor, Thermo Scientific). All centrifugation procedures were carried out at 4 °C. 1ml fractions were collected from the top and γ-secretase, IFITM3 and different subcellular markers were analyzed by SDS-PAGE and WB (for source gels see Supplementary Figure 1).
Cytokine induction in neurons and astrocytes
Primary mouse neuronal culture was treated with IFN-γ (R&D Systems, 485-MI-100/CF) or IFN-α (R&D Systems, 12100–1) at the final concentrations of 10 ng/ml and/or 100 ng/ml was added to neuronal culture at DIV12 for 24 hrs. hAST cultures were treated with either PBS, 1 ng/mL IL-6 (R&D Systems, 206-IL-010/CF) or 10 ng/mL IL-1β (R&D Systems, 201-LB-005) for 48 hrs. After cytokine treatment, mouse neurons and hAST were either solubilized with RIPA buffer for WB or membranes were extracted for γ-secretase activity assay.
Western blot analysis
Protein samples were subjected to SDS-PAGE gels (Bio-Rad), electrophoresed, transferred to Immobilon-P PVDF (Millipore) or Immobilon-FL PVDF membranes (Millipore) and Western blotting (WB). The following primary antibodies were used: PS1-NTF (a kind gift of Merck Research Laboratories); PS1-CTF (MAB5232, Millipore); PS2-CTF (1987–1, Epitomics); nicastrin was generated in our laboratory; Aph-1aL (38–3600, Invitrogen); Pen-2 (ab18189, Abcam); human IFITM3 (anti-Fragilis, ab109429, Abcam); mouse IFITM3 (anti-Fragilis, ab15592, Abcam); APP (22C11, MAB348, Millipore); SPP (317) was generated in our laboratory; cleaved Notch1 (Val1744), Cell Signaling Technology and SM320, generated in our lab); c-myc (9E10, Roche Life Science); N-cadherin (D4R1H, #13116, Cell Signaling Technology); Rab7 (B-3, sc-376362, Santa Cruz Biotechnology); EEA1 (Ab2900, Abcam); β-actin (C4, sc-47778, Santa Cruz Biotechnology); β-tubulin III (T8578, Sigma-Aldrich); tubulin (ab56676, Abcam). HRP-conjugated anti-rabbit and mouse secondary antibodies (NA9340V, NXA931V, GE Healthcare) for ECL substrate (Pierce) and IRDye 800CW goat anti-rabbit IgG (H+L) or anti-mouse IgG (H+L) secondary antibodies (925–32211, 925–32210, LI-COR) for Odyssey CLx Imaging (LI-COR) were used. For quantification, ImageJ and Image Studio Lite (LI-COR) were used, respectively (for source gels see Supplementary Figure 1).
RNA Interference
Silencer® Select pre-designed siRNA oligonucleotides targeting IFITM3 were purchased from Ambion and tested: siRNA ID #s195033, #s195034, and #s195035 (sense sequence 5΄-CCCACGUACUCCAACUUCCtt −3΄ and antisense sequence 5΄-GGAAGUUGGAGUACGUGGGat-3΄). IFITM3 siRNA (#s195035) and scrambled siRNA (negative control) were used. siRNAs were transfected to HEK293-APP695 cells in triplicates as described previously50. The conditioned medium was measured for secreted Aβ40 and 42 levels by using Aβ peptide multiplex kits (6E10, MSD) according to manufacturer’s instructions. To check cell viability, Alamar Blue (AbD Serotec) was added to the cells and incubated50. Transferred medium in a 96-well black polystyrene microplate (Costar) was read for fluorescence (530–560 nm excitation/ 590 nm emission) by EnVision Plate Reader (PerkinElmer). Protein knock-down levels were measured in cell lysates by WB. siRNA transfection in HEK293-NotchΔE cells was performed as described above. Levels of protein knock-down, NotchΔE and NICD were measured in cell lysates by WB.
CRISPR/Cas9n and rescue
CRISPR constructs were purchased from the RNAi core at Memorial Sloan Kettering Cancer Center and purified with the HiSpeed Maxiprep kit (Qiagen). U138 cells were transfected using Lipofectamine LTX according to the manufacturer’s instructions with px459 vector containing Cas9 and guide RNAs (either an empty vector or one of three IFITM3 guides). Transfected cells were incubated for 48 hrs prior to puromycin selection. Single cells were extracted and expanded. IFITM3 KO was confirmed by sequencing and WB.
Plasmid DNAs for human IFITM3 (OriGene) were cloned into the pcDNA3.1 vector. U138 EV and U138 IFITM3−/− cells were transfected with either pcDNA3.1 empty vector of pcDNA3.1 IFITM3 construct using Lipofectamine LTX according to the manufacturer’s instructions and allowed to incubate for 48 hrs prior to membrane preparation.
Secreted Aβ level measurement
To measure γ-secretase activity in cells, cell culture media were harvested and Aβ40 and 42 levels were measured by Aβ peptide multiplex kits (4G8, MSD) according to the manufacturer’s instructions. γ-Secretase activity (secreted Aβ) was shown in % Control or pg per mg lysate (pg/mg lysate).
In vitro γ-secretase activity assay (Aβ40 and 42 cleavage rate)
In vitro γ-secretase activity assay (Aβ40 and 42 cleavage rate) was carried as described previously51–53 and is illustrated (ExtFig 2b). Briefly, to detect Aβ production by the cleavage of γ-secretase, cells were lysed with 0.25% CHAPSO and incubated with a recombinant APP substrate, Sb4 at 37 °C for 3 hrs. 20 μl of lysates were incubated with 20 μl detection buffer (for Aβ40; anti-mouse IgG AlphaLISA acceptor beads (PerkinElmer), G2–10 antibody and streptavidin-conjugated Alpha donor beads (PerkinElmer), for Aβ42; Protein A AlphaLISA acceptor beads (PerkinElmer), 10G-3 antibody and streptavidin-conjugated Alpha donor beads (PerkinElmer)) for overnight at room temperature. For solubilized γ-secretase, 0.25% CHAPSO solubilized membranes from cells (U138 EV and KO cells, and hAST) and mouse brain tissues (WT, IFITM3−/−, 5XFAD and IFITM3−/−;5XFAD) were used to incubate with a recombinant APP substrate. Aβ40 and Aβ42 levels were measured as AlphaLISA signals by EnVision Plate Reader (PerkinElmer). γ-Secretase activity (Aβ cleavage rate) was shown in AlphaLISA Units/min per mg of protein (AU/min, mg) for U138 EV and KO cells or Normalized Units (normalized to the average of WT for 18-month-old WT and IFITM3−/−, to the average of 5XFAD for 4-month-old 5XFAD and IFITM3−/−; 5XFAD, and to the average of PBS treatment for PBS, IL-6 and IL-1β treated hAST). To measure IC50 for Aβ cleavage, in vitro γ-secretase activity assay was carried out in the absence or presence of GSMs or GSIs with different dilutions.
To measure γ-secretase activity in 4 and 28-month-old C57BL/6 mouse brains, human brains, and primary neuronal culture: membranes were incubated in the same conditions as above except that C100-ΔID-FLAG54,55 was used as a substrate. Aβ40 and 42 levels were measured by Aβ peptide multiplex kits (6E10, MSD) according to the manufacturer’s instructions. γ-Secretase activity (Aβ cleavage rate) was shown in pg/min per mg of protein (pg/min, mg).
In vitro γ-secretase activity assay (NICD production level)
In vitro γ-secretase activity assay (NICD production level) was carried as described previously42 and illustrated (ExtFig 2i). Briefly, to detect NICD production by the cleavage of γ-secretase, HEK293-NotchΔE cells were lysed with 0.25% CHAPSO after siRNA transfection. 5μl of lysates were incubated with 20μl detection buffer (Protein A-conjugated acceptor beads (PerkinElmer), NICD antibody (SM320), 9E10 anti-human c-myc conjugated to biotin antibody and streptavidin-conjugated donor beads (PerkinElmer)) for overnight at room temperature. NICD levels were measured as AlphaLISA signals by EnVision Plate Reader (PerkinElmer).
To detect NICD production by the cleavage of γ-secretase in IFITM3 KO cells, cell membranes were lysed with 0.25% CHAPSO and incubated with a recombinant Notch substrate, N1-Sb1 at 37 °C for 2 hrs. 20 μl of lysates were incubated with 20 μl detection buffer (Protein A-conjugated acceptor beads (PerkinElmer), NICD antibody (SM320, generated in our lab) and streptavidin-conjugated donor beads (PerkinElmer)) for overnight at room temperature42. NICD levels were measured as described above.
Gene expression analysis
To measure mRNA expression levels of genes (IFITM3, MAP2, GFAP and AIF1) in LOAD and control, RNA was extracted from human brain tissues using QIAzol Lysis Reagent (Qiagen) and RNAneasy Mini kit (Qiagen). RNA concentrations were measured by using NanoDrop 8000 spectrophotometer (Thermo Scientific) and cDNAs were synthesized by reverse transcription (High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor, Life Techonologies). cDNA was mixed with TaqMan assays for human IFITM3 (Hs04194512_g1, Life Technologies), MAP2 (Hs00258900_m1, Life Technologies), GFAP (Hs00909233_m1, Life Technologies) and AIF1 (Hs00610419_g1, Life Technologies) in TaqMan Universal master mix II (Applied Biosystems). GAPDH (Hs04420632_g1, Life Technologies) was used as a reference gene.
To measure IFITM gene expression in human iPS-derived neurons, human primary astrocyte culture, and MEF (WT and PS1/2 double KO) cells, RNA was extracted from cells with RNeasy mini kit (Qiagen) with on-column DNase I digestion (Qiagen) and cDNA was synthesized by reverse transcription according to the manufacturer’s instructions (iScript™ cDNA Synthesis kit, Bio-Rad). Total gene expression levels were measured by real time PCR (ABI-7900HT Fast Real-Time PCR System or 7500 Fast Real-Time PCR System, Applied Biosystems). The comparative CT method was used to calculate total mRNA expression levels of genes. Gene expression levels were normalized to the ones of GAPDH56–58. For MEF cells, mouse IFITM3 (Mm00847057_s1, Life Technologies) and GAPDH (Mm99999915_g1, Life Technologies) probes were used.
AD Datasets
To better understand the role of IFITM3 in AD pathogenesis, we examined IFITM3 mRNA expression changes in AD and correlations between IFITM3 and genes known to be involved in AD progression in 5 transcriptomic datasets from 3 large cohorts studying AD. The three cohorts include 1) the Mount Sinai Brain Bank (MSBB) which contains 2 brain regions – superior temporal gyrus (Brodmann Area 22) and entorhinal area (Brodmann Area 36)18; 2) the Harvard Brain Tissue Resource Center (HBTRC) which contains 2 brain regions – prefrontal cortex and visual cortex59; 3) the Mayo clinic cohort which contains 1 brain region – temporal cortex16. The AD status of each sample was defined by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD).
Differential gene expression and correlation analyses
We performed differential gene expression analysis between AD and control samples using R package limma (V3.34.0) with default settings60. Multiple tests were adjusted using the Benjamini–Hochberg’s (BH) FDR method. Genes with an FDR less than 0.05 and fold change (FC) greater than 1.2 were considered significant.
Correlation analysis of IFITM3 and Age
Correlation analyses of IFITM3 and age in cortex and hippocampus were performed based on the Genotype-Tissue Expression (GTEx) brain data. GTEx brain RNA-Seq data were downloaded from the GTEx Portal17. Lowly expressed genes with expression levels at least 1 count per million in less than 20% of samples were removed. A total of 16,494 genes were included in the correlation analysis. Expression data were then normalized, log2 transformed and corrected for race and BMI. Spearman correlation coefficient and p-value were calculated between expression levels and age. Adjusted p-values were calculated based on a null distribution from 1,000 permutations.
Correlation analysis of IFITM3 and viruses
To study the effect of viruses on the expression level of IFITM3 in AD, we used the virus data from a previous study61. Raw reads were normalized by library size of each sample, and further corrected for covariants (i.e. age of death, race, RIN value, sex, batch, and postmortem interval), based on a linear regression model. Spearman correlation coefficient and p-value were calculated between expression levels of IFITM3 and human herpes virus 6B (HHV-6B) or hepatitis C virus genotype 4, and nominal p-values were reported.
Immunofluorescence staining and microscopy
Proximity Ligation Assay (PLA):
Interaction of PS1 and IFITM3 was determined by PLA in mouse primary neurons. Primary neurons at DIV13 were permeabilized in 0.4% CHAPSO62 and PLA (Sigma-Aldrich) was performed according to manufacturer’s instructions. After final washes, neurons were stained with Alexa Fluor 488 phalloidin (Invitrogen) for F-actin, mounted with a mounting medium containing DAPI (Invitrogen) as described63, and PLA signals were imaged by Axio Imager. Z2 (ZEISS).
Human iPSC-derived neurons and human primary astrocytes staining:
Human iPSC-derived neurons and human primary astrocytes at passage 4 (ScienCell, 1800) were fixed using 4% PFA (Electron Microscopy Sciences) or 10% formalin (Sigma-Aldrich), permeabilized with 0.01% Triton X-100, blocked with 10% donkey serum (Sigma-Aldrich) for 1 hr, and incubated with primary antibodies (MAP2 (Sigma-Aldrich), S100β (Sigma-Aldrich)) for overnight at 4˚C and Alexa Fluor 488 and 594 conjugated secondary antibodies (Life Technologies) for 1–2 hrs at room temperature. Cells were counterstained using DAPI (Invitrogen), mounted with AquaPolymount mounting solution (Polysciences Inc.), and imaged with EVOS microscope.
Mouse perfusion:
12-month-old WT and 5XFAD mice and 4-month-old 5XFAD and IFITM3−/−; 5XFAD mice were anesthetized with a single dose of ketamine/xylazine (100 mg/kg/5.0 mg/kg) and transcardially perfused with 50mL of PBS followed by 50mL of 4% paraformaldehyde (PFA). Brains were removed and post fixed in 4% PFA overnight at 4 °C.
IFITM3 staining:
12-month-old WT and 5XFAD samples were then processed for paraffin embedding with tissue processor (Leica Biosystems, ASP6025) and 8-micron paraffin sections were obtained and mounted on slides for IHC. The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 minutes with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 minutes (Ventana Medical Systems). For IFITM3 staining, sections were incubated with anti-Fragilis antibody (Abcam, ab15592, 1μg/ml) for 5 hrs, followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG (Vector labs, PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa 488 (Invitrogen, B40953).
For IFITM3 and GFAP or Iba1 colocalization staining, sections were first incubated with anti-GFAP antibody (DAKO, Z0334, 1μg/ml) or anti-Iba1 antibody (Abcam, cat# ab178847, 0.1μg/ml) for 5 hrs, followed by 60 minutes incubation with biotinylated goat anti-rabbit IgG (Vector labs, PK6101) at 1:200 dilution. The detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with Tyramide Alexa 488 (Invitrogen, B40953). Next, sections were incubated with anti-Fragilis antibody as described above except the incubation with Tyramide Alexa Fluor 568 (Invitrogen, T20948). After staining, slides were counterstained with DAPI (Sigma Aldrich, D9542, 5 μg/ml) for 10 min and coverslipped with Mowiol. Slides were imaged with MIRAX SCAN (Zeiss) and IF was quantified using CaseViewer and ImageJ.
Thioflavin-S staining:
4-month-old 5XFAD and IFITM3−/−; 5XFAD samples were then washed 3 times in PBS and incubated in 30% sucrose solution overnight at 4°C. Brains were then flash frozen in O.C.T. compound and stored at −80 °C. Samples were serially sectioned coronally at 30μm and stored at −20 °C in tissue storage solution (30% sucrose, 30% ethylene glycol in 0.1M phosphate buffer). Every 6th section was stained with thioflavin-S (0.1% in ddH20 for 8 minutes) and washed 3 times in PBS with a fourth wash overnight at 4 °C (samples were protected from light at all times). Stained sections were mounted on charged slides and imaged with MIRAX SCAN (Zeiss) and immunoflouresence (FITC) was visualized and quantified using CaseViewer and ImageJ. Number of plaques per section was counted and divided by total tissue area (in mm2), the average number of plaques/area across all sections was reported for each samples.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 7 and GraphPad Prism 8 softwares. Results were presented as Mean ± SD bar graphs or violin plots show the frequency distribution of the data. Violin plots represent median (middle line) and interquartile range outer lines. p values were calculated with two tailed unpaired Student’s t-test, one-way ANOVA followed by Fisher’s LSD or two-way ANOVA followed by Tukey). Significance was set as P < 0.05 and expressed as * < 0.05, ** < 0.01, *** < 0.001, and **** < 0.0001.
Extended Data
Extended Data Figure 1. Identification of IFITM3 as γ-secretase binding protein.

(a) LC-MS/MS analysis of the 15 kDa band identified four peptides that match with human IFITM3. (b) WB analysis of E2012-BPyne (500 nM) labeled PS1-NTF protein. (c) Structures of imidazole GSMs, acid GSM and GSIs. (d) WB analysis of E2012-BPyne labeled proteins in the absence or presence of imidazole GSMs, acid GSM and GSIs. Labeled proteins were captured and analyzed by WB for IFITM3. (e) Structures of GY6 and 163-BP-L-biotin. (f) IFITM3 co-immunoprecipitates with γ-secretase subunits. CHAPSO solubilized cell membranes were immunoprecipitated with anti-IFITM3 antibody and probed with antibodies against PS2-CTF and Pen-2. Rabbit IgG was used as a negative control. (g) IFITM3 does not co-immunoprecipitate with SPP. CHAPSO solubilized cell membranes were immunoprecipitated with a monoclonal anti-IFITM3 antibody (9D11) and probed with antibodies against SPP and IFITM3. Mouse IgG was used as a negative control. (h) Analysis of the total protein level in WT MEF or PS1/2 double KO MEF cells. The same amount of membrane proteins was loaded and analyzed by Western blotting. (i) IFITM3 mRNA expression levels were measured by RT-PCR in WT MEF or PS1/2 double KO MEF cells (n=6). All WB images and graphs are representative of three independent experiments (except; a/d: 2 replicates). Graphs are mean ± SD. ns, not significant, two-sided Student’s t-test.
Extended Data Figure 2. Effect of IFITM3 knockdown and knockout on γ-secretase.

(a) Quantification of WB (Fig. 2a) showed that IFITM3 KD did not change protein expression levels of APP, Nct and PS1-NTF in HEK-APPWT cells (n=3). (b) Schematic representation of cell-free γ-secretase assay. γ-Secretase is incubated with a recombinant APP substrate in the presence of 0.25% CHAPSO. Cleaved Aβ40 and 42 species are measured with cleavage specific antibodies and AlphaLISA technology. (c) Schematic model showing different GSM and GSI binding sites in γ-secretase: E2012 (imidazole GSM), GSM-1 (acid GSM), and L458 (transition state analogue inhibitor, GSI). (d-f) Comparison of IC50 of (d) GSM-25 (EV: n=9, KO: n= 8) for Aβ40 (****p<0.0001) and Aβ42 (ns), (e) GSM-1 (n=6) for Aβ40 (**p=0.0039) and Aβ42 (ns), and (f) L458 (n=3) for Aβ40 (ns) and Aβ42 (ns) cleavages in the U138 EV or KO cell lines (n≥3). (g) IFITM3 knockdown (KD) does not affect expression of γ-secretase subunits. IFITM3 was knocked-down by siRNA (6 pmol, n=3) in HEK-NotchΔE cells and scramble siRNA (SC, n=3) was used as a negative control. Cell lysates were probed by antibodies against Nct, PS1-NTF and IFITM3. β-Actin was used as a loading control. (h) Effect of IFITM3 KD on γ-secretase activity. IFITM3 KD increased γ-secretase cleaved product NICD, analyzed by WB. Cell lysates were probed by antibodies against c-myc (NotchΔE) and NICD and a representative quantification of NICD (n=8, ***p=0.001) is shown (lower panel). (i) Cell based NICD AlphaLISA assay (left panel) revealed an increase in NICD production with IFITM3 KD. Quantification of NICD (n=8, ***p=0.001) is shown in the right panel. (j) Effect of IFITM3 KO on γ-secretase activity. KO cells lines have increased γ-secretase activity as compared to the EV cell line. The NICD cleavage in vitro was measured by AlphaLISA assay (n=3,**p=0.0096). All WB images and graphs are representative of three independent experiments Graphs are mean ± SD. ns, not significant, two-sided Student’s t-test.
Extended Data Figure 3. Effect of aging and the expression of APP/PS1 on the level of γ-secretase and IFITM3.
(a) WBs for Nct and PS1-NTF (Fig. 3a) were quantified by Odyssey imaging (n=5 mice pooled per group, except n=4 for 28F, all=ns). (b) Effect of aging on subcellular localizations of IFITM3. A hemibrain from male wild-type C57BL/6 mouse at 4 and 28 months (n=1 per group) were homogenized and layered on iodixanol gradient (2.5 – 30 %). Fractions were collected from the top and resolved by WBs for γ-secretase, IFITM3 and different subcellular markers. (c) WBs for APP, Nct, and PS1-NTF (Fig. 3e) were quantified by Odyssey imaging (n=5 mice per group except n=4 for WT at 12 months). APP: 3moWT-3mo5X: ****p<0.001, 3mo5X-12mo5X: ****p<0.0001, 12moWT-12mo5X: ****p<0.0001). Nct: 3moWT-12moWT: ****p<0.001, 3mo5X-12mo5X: ****p<0.001, 12moWT-12mo5X: ****p<0.001. PS1-NTF: 3moWT-12mo5X: ***p=0.0004, 12moWT-12mo5X: ****p<0.001. (d) Immunostaining of IFITM3 in mouse brains. Fluorescence microscopy of IFITM3 expression in 12-month-old PFA perfused mice (WT, upper panel, 5XFAD, lower panel). Representative images of cortex, hippocampus and subiculum (left to right) show IFITM3 (green) and DAPI (blue). Scale bar = 1000μm, 200μm, 100μm (left to right). Total IFITM3 fluorescence area within the hippocampus and cortex of WT and 5XFAD was quantified using FIJI. Total IFITM3 was divided by tissue area and 5XFAD expression was normalized to average of WT (WT: n=7, 5XFAD: n=9)(cor (cortex): **p=0.0035, hip (hippocampus): ****p<0.0001 ). (e) IFITM3 expression in astrocytes and microglia is upregulated in 5XFAD mice compared to WT mice. Fluorescence microscopy of IFITM3, GFAP (top) and Iba1 (bottom) expression in 12-month-old PFA perfused mice (WT, upper panel, 5XFAD, lower panel). Representative images of the hippocampus and cortex show IFITM3 (red), GFAP (green – top), Iba1 (green - bottom), and DAPI (blue), scale bar = 500μm. Inset panels (left to right) show GFAP or Iba1 (green), IFITM3 (red) and merge. Scale bar = 50μm. All WB images and graphs are representative of three independent experiments (except; b: 2 replicates). Graphs are mean ± SD. ns, not significant, , two-sided Student’s t-test, (except; c: one-way ANOVA followed by Tukey).
Extended Data Figure 4. Expression profile of IFITM3 and other markers in LOAD and age-matched controls.

(a) Spearman’s correlation of mRNA expression of human IFITM3 gene with age was analyzed in the cortex (n=158) and hippocampus (n=123) of normal human brains using the Genotype-Tissue Expression (GTEx) cohort.. (b) mRNA expression in non-demented subject control (n=10) and LOAD samples (n=18) of MAP2 (ns), GFAP (**p=0.0046), and AIF1 (ns) were measured, which were used in Fig. 4c–d. (c) Expression profiles of MAP2 (ns), GFAP (****p<0.0001), and AIF1 (ns) in the temporal cortex of human control (n=76) and LOAD samples (n=80) using the Mayo Clinic cohort data. Correlation analyses were carried out and p values were calculated. (d) The protein levels of Nct (****p<0.0001) and PS1-NTF (**p=0.0042) in human brain membranes (control and LOAD). The samples were analyzed by WB and quantified (n=10 and 18, respectively). Signal was normalized to HeLa cell membrane. (e-f) IFITM3 SNP Genotypes. (e) Allelic discrimination plot depicting rs34481144 genotype calls for control (n=9), LOAD-L (n=10), and LOAD-H (n=8) brain samples. The axes show delta Rn values obtained from TaqMan SNP genotyping analysis. Samples without genomic DNA were used as non-template controls (shown as black squares in the left lower quadrant, n=2). (f) Allele frequency of rs34481144 genotype in control (n=9) and LOAD (n=18). (g) mRNA level of IFITM3 gene in four types of EGFP/L10a-expressing mouse hippocampal neurons (GAD2 (glutamate decarboxylase 2), CCK (cholecystokinin), PV (parvalbumin), and CORT (cortistatin) expressing GABAergic neurons)(n=4 per group, GAD2-PV: ***p=0.0003, CCK-PV: ****p<0.0001, CCK-Cort *p=0.0363, PV-Cort: **p=0.0059). (h) mRNA levels of IFITM3 in human iPSC-derived neurons (n=4) and human primary astrocytes (n=3) were measured by qPCR (****p<0.0001). (i-j) Human iPSC-derived neurons (i) and human primary astrocytes (j) were stained for IFITM3 with MAP2 (neuronal marker) or S100β (astrocyte marker). DAPI was used for nucleus staining. Scale bar = 200 and 500 μm. (k) Induction of IFITM3 by IFN-α in primary neurons. E16 mouse primary neurons were treated with 100 ng/ml of IFN-α at DIV12 for 24 hours. The protein levels of γ-secretase and IFITM3 were analyzed by WB (n=4 per group). β-Tubulin III was used as a loading control. (l) Effect of IFITM3 induction on γ-secretase activity for Aβ40 (*p=0.0116) and Aβ42 (*p=0.0319) activity. Membranes from primary neurons were incubated with the recombinant APP substrate C100-ΔID-FLAG and γ-secretase activity (Aβ cleavage rate) was assayed by human Aβ three-plex MSD kits (n=12, 10). (m) JC8 whole cell photolabeling. Neuronal membranes were photolabeled with JC8 in the absence or presence of L458 and analyzed by anti-PS1-NTF antibody. Photolabeled PS1-NTF protein level was quantified by Odyssey imaging (n=3, *p=0.0210). (n) Spearman’s correlation between the expression level of IFITM3 and viruses. In the Brodmann Area 22 (BA-22 region) in the Mount Sinai Brain Bank (MSBB) cohort, the expression level of IFITM3 is positively correlated with the expression level of the human herpesvirus-6B (HHV-6B) (rho=0.248, p=0.044, n=66). In the Brodmann Area 36 (BA-36 region), the expression level of IFITM3 is positively correlated with the expression of hepatitis C virus genotype 4 (rho=0.255, p=0.033, n=70). All WB images and graphs are representative of two independent experiments (except; b/e: 1 replicate, h: data pooled from 2 experiments, k: 3 replicates, l: data pooled from 4 experiments). Bar graphs are mean ± SD. Violin plots represent median (middle line) and interquartile range (outer lines). ns, not significant, * P < 0.05, ** P < 0.01, **** P < 0.0001, two-sided Student’s t-test (except; g: One-Way ANOVA followed by Tukey).
Extended Data Figure 5. Correlation between L505 labeled protein and γ-secretase activity.

(a) Structures of JC8, L505, L646, GY4 and L631. (b) WB analysis of 11Bt labeled proteins in the absence or presence of its parent compound, a substrate binding site inhibitor pep11 and imidazole GSM, E2012. Labeled proteins were captured and analyzed by WB for PS1-NTF (n=1). (c) Pearson’s correlation between γ-secretase activity (Aβ40, Aβ42 cleavage rates in Fig. 4d) and L505 labeled PS1-NTF (in Fig. 5c) in LOAD samples (n=10). Linear regression analysis was used to calculate R and p values.
Supplementary Material
Acknowledgments:
We thank Drs Alex Kentsis and Ronald Hendrickson (Memorial Sloan Kettering Cancer Center (MSKCC)) for LC-MS/MS and proteomic analysis. We thank Heidi Brogdon and Dr. Nancy L. Nadon (National Institute on Aging) for mouse brains. We thank Elizabeth Sikora (Mount Sinai) for ApoE genotyping, Dmitry Yarilin for immunostaining (MSKCC Molecular Cytology Core), Sho Fujisawa and Yevgeniy Romin for helping microscopy (MSKCC Molecular Cytology Core), and Sara Shuldberg for human brain samples (UCSD). We thank Dr. Steve Wagner for providing NGP-97555. This work is supported by the JPB Foundation (YML, PG and AG), the Fisher Center for Alzheimer’s Research Foundation (PG), Cure Alzheimer’s Fund (YML), the National Institutes of Health R01NS096275 (YML), RF1AG057593 (YML) R01AG061350(YML), R01AG046170 (BZ), RF1AG057440 (BZ), R01AG057907 (BZ). Authors also acknowledge the MSK Cancer Center Support Grant/Core Grant (Grant P30 CA008748) the ADRC parent grant (P30 AG062429), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
Footnotes
Data availability: Source data for Figs. 1–5 and Extended Data Figs. 1–5 and table S1 – S5 are available with the paper. All other data are available from the corresponding authors upon reasonable request.
Authors declare no competing financial interests.
COI
LYM is co-inventor of intellectual property (assay for gamma secretase activity and screening method for gamma secretase inhibitors) owned by MSKCC and licensed to Jiangsu Continental Medical Development.
References and Notes:
- 1.Shi Y & Holtzman DM Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nature reviews. Immunology 18, 759–772, doi: 10.1038/s41577-018-0051-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griciuc A et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643, doi: 10.1016/j.neuron.2013.04.014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071, doi: 10.1016/j.cell.2015.01.049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 38, 9–12 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Gertsik N, Chiu D & Li YM Complex regulation of gamma-secretase: from obligatory to modulatory subunits. Frontiers in aging neuroscience 6, 342, doi: 10.3389/fnagi.2014.00342 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villa JC et al. Nontranscriptional Role of Hif-1alpha in Activation of gamma-Secretase and Notch Signaling in Breast Cancer. Cell reports 8, 1077–1092, doi: 10.1016/j.celrep.2014.07.028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crump CJ, Johnson DS & Li Y-M Development and Mechanism of γ-Secretase Modulators for Alzheimer’s Disease. Biochemistry 52, 3197–3216, doi: 10.1021/bi400377p (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner SL et al. Soluble gamma-secretase modulators selectively inhibit the production of the 42-amino acid amyloid beta peptide variant and augment the production of multiple carboxy-truncated amyloid beta species. Biochemistry 53, 702–713, doi: 10.1021/bi401537v (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner SL et al. Pharmacological and Toxicological Properties of the Potent Oral gamma-Secretase Modulator BPN-15606. The Journal of pharmacology and experimental therapeutics 362, 31–44, doi: 10.1124/jpet.117.240861 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kounnas MZ et al. Modulation of γ-Secretase Reduces β-Amyloid Deposition in a Transgenic Mouse Model of Alzheimer’s Disease. Neuron 67, 769–780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pozdnyakov N et al. gamma-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. The Journal of biological chemistry 288, 9710–9720, doi: 10.1074/jbc.M112.398602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey CC, Zhong G, Huang IC & Farzan M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annual review of virology 1, 261–283, doi: 10.1146/annurev-virology-031413-085537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar DK et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8, 340ra372, doi: 10.1126/scitranslmed.aaf1059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eimer WA et al. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 100, 1527–1532, doi: 10.1016/j.neuron.2018.11.043 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Oakley H et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 10129–10140, doi: 10.1523/JNEUROSCI.1202-06.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen M et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data 3, 160089, doi: 10.1038/sdata.2016.89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Consortium GT Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660, doi: 10.1126/science.1262110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang M et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data 5, 180185, doi: 10.1038/sdata.2018.185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen EK et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat Med 23, 975–983, doi: 10.1038/nm.4370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frost GR & Li YM The role of astrocytes in amyloid production and Alzheimer’s disease. Open biology 7, doi: 10.1098/rsob.170228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang B et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamura Y et al. Inhibition of gamma-secretase activity by helical beta-peptide foldamers. Journal of the American Chemical Society 131, 7353–7359, doi: 10.1021/ja9001458 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Wakabayashi T et al. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nature cell biology 11, 1340–1346, doi: 10.1038/ncb1978 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England journal of medicine 368, 107–116, doi: 10.1056/NEJMoa1211103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. The New England journal of medicine 368, 117–127, doi: 10.1056/NEJMoa1211851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertram L et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet 83, 623–632, doi: 10.1016/j.ajhg.2008.10.008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baruch K et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93, doi: 10.1126/science.1252945 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 28.Li YM et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Chun J, Yin YI, Yang G, Tarassishin L & Li YM Stereoselective Synthesis of Photoreactive Peptidomimetic gamma-Secretase Inhibitors. J Org Chem 69, 7344–7347 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Gertsik N, Chau DM & Li YM gamma-Secretase Inhibitors and Modulators Induce Distinct Conformational Changes in the Active Sites of gamma-Secretase and Signal Peptide Peptidase. ACS Chem Biol, doi: 10.1021/acschembio.5b00321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crump CJ et al. Development of Sulfonamide Photoaffinity Inhibitors for Probing Cellular gamma-Secretase. ACS Chem Neurosci 7, 1166–1173, doi: 10.1021/acschemneuro.6b00127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crump CJ et al. Piperidine acetic acid based gamma-secretase modulators directly bind to Presenilin-1. ACS Chem Neurosci 2, 705–710, doi: 10.1021/cn200098p (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crump CJ et al. BMS-708,163 Targets Presenilin and Lacks Notch-Sparing Activity. Biochemistry 51, 7209–7211, doi: 10.1021/bi301137h (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang G et al. Stereo-controlled synthesis of novel photoreactive gamma-secretase inhibitors. Bioorg Med Chem Lett 19, 922–925, doi:S0960–894X(08)01493–5 [pii] 10.1016/j.bmcl.2008.11.117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M et al. gamma-Secretase: characterization and implication for Alzheimer disease therapy. Neurobiol Aging 23, 1023–1030 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Pettersson M et al. Discovery of indole-derived pyridopyrazine-1,6-dione gamma-secretase modulators that target presenilin. Bioorganic & medicinal chemistry letters 25, 908–913, doi: 10.1016/j.bmcl.2014.12.059 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Sung JY et al. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc Natl Acad Sci U S A 105, 3112–3116, doi: 10.1073/pnas.0712180105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho SM et al. Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 101, 113–124, doi: 10.1016/j.ymeth.2015.11.019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanley S et al. Profiling of Glucose-Sensing Neurons Reveals that GHRH Neurons Are Activated by Hypoglycemia. Cell Metab 18, 596–607, doi: 10.1016/j.cmet.2013.09.002 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Heiman M, Kulicke R, Fenster RJ, Greengard P & Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat Protoc 9, 1282–1291, doi: 10.1038/nprot.2014.085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R. Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. . URL https://www.R-project.org/. (2017). [Google Scholar]
- 42.Chau DM, Crump CJ, Villa JC, Scheinberg DA & Li YM Familial Alzheimer Disease Presenilin-1 Mutations Alter the Active Site Conformation of gamma-secretase. The Journal of biological chemistry 287, 17288–17296, doi: 10.1074/jbc.M111.300483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shelton CC et al. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci U S A 106, 20228–20233, doi:0910757106 [pii] 10.1073/pnas.0910757106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong E et al. GSAP modulates gamma-secretase specificity by inducing conformational change in PS1. Proc Natl Acad Sci U S A 116, 6385–6390, doi: 10.1073/pnas.1820160116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hur JY et al. Active gamma-secretase is localized to detergent-resistant membranes in human brain. The FEBS journal 275, 1174–1187, doi: 10.1111/j.1742-4658.2008.06278.x (2008). [DOI] [PubMed] [Google Scholar]
- 46.Placanica L, Zhu L & Li YM Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS ONE 4, e5088, doi: 10.1371/journal.pone.0005088 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Placanica L et al. Pen2 and Presenilin-1 Modulate the Dynamic Equilibrium of Presenilin-1 and Presenilin-2 {gamma}-Secretase Complexes. The Journal of biological chemistry 284, 2967–2977, doi:M807269200 [pii] 10.1074/jbc.M807269200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li YM et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A 97, 6138–6143 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frykman S et al. Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS ONE 5, e8948, doi: 10.1371/journal.pone.0008948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hur JY et al. Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. J Biol Chem 287, 11991–12005, doi: 10.1074/jbc.M111.246074 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn K et al. Activation and intrinsic gamma-secretase activity of presenilin 1. Proc Natl Acad Sci U S A 107, 21435–21440, doi: 10.1073/pnas.1013246107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shelton CC, Tian Y, Frattini MG & Li YM An exo-cell assay for examining real-time gamma-secretase activity and inhibition. Mol Neurodegener 4, 22, doi:1750-1326-4-22 [pii] 10.1186/1750-1326-4-22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Placanica L et al. Pen2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 gamma-secretase complexes. J Biol Chem 284, 2967–2977, doi: 10.1074/jbc.M807269200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian Y, Bassit B, Chau D & Li YM An APP inhibitory domain containing the Flemish mutation residue modulates gamma-secretase activity for Abeta production. Nat Struct Mol Biol 17, 151–158, doi: 10.1038/nsmb.1743 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Tian Y, Crump CJ & Li YM Dual role of {alpha}-Secretase Cleavage in the regulation of {gamma}-secretase activity for amyloid production. J Biol Chem, doi:M110.128439 [pii] 10.1074/jbc.M110.128439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller PY, Janovjak H, Miserez AR & Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques 32, 1372–1374, 1376, 1378–1379 (2002). [PubMed] [Google Scholar]
- 57.Wang JC et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet 18, 3125–3135, doi: 10.1093/hmg/ddp231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang JC et al. Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Mol Psychiatry 14, 501–510, doi: 10.1038/mp.2008.42 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang B et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720, doi: 10.1016/j.cell.2013.03.030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smyth GK Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3, Article3, doi: 10.2202/1544-6115.1027 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Readhead B et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 99, 64–82 e67, doi: 10.1016/j.neuron.2018.05.023 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Inoue M et al. Human brain proteins showing neuron-specific interactions with gamma-secretase. FEBS J 282, 2587–2599, doi: 10.1111/febs.13303 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Teranishi Y et al. Proton myo-inositol cotransporter is a novel gamma-secretase associated protein that regulates Abeta production without affecting Notch cleavage. FEBS J 282, 3438–3451, doi: 10.1111/febs.13353 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.