

In this study, Cohen et al. sought to understand the functional contribution of PRC1 and PRC2, which largely overlap in their genomic binding and cooperate to establish repressive chromatin domains demarcated by H2AK119ub and H3K27me3, to gene repression. By using the developing murine epidermis as a paradigm, they uncovered a previously unappreciated functional redundancy between Polycomb complexes, and their findings show how PRC1 and PRC2 function as two independent counterparts, providing a repressive safety net that protects and preserves lineage identity.
Keywords: Polycomb, PRC1, PRC2, skin, epidermis, stem cell, epigenetics, H2AK119ub, H3K27me3
Abstract
Polycomb repressive complex 1 (PRC1) and PRC2 are critical epigenetic developmental regulators. PRC1 and PRC2 largely overlap in their genomic binding and cooperate to establish repressive chromatin domains demarcated by H2AK119ub and H3K27me3. However, the functional contribution of each complex to gene repression has been a subject of debate, and understanding of its physiological significance requires further studies. Here, using the developing murine epidermis as a paradigm, we uncovered a previously unappreciated functional redundancy between Polycomb complexes. Coablation of PRC1 and PRC2 in embryonic epidermal progenitors resulted in severe defects in epidermal stratification, a phenotype not observed in the single PRC1-null or PRC2-null epidermis. Molecular dissection indicated a loss of epidermal identity that was coupled to a strong derepression of nonlineage transcription factors, otherwise repressed by either PRC1 or PRC2 in the absence of its counterpart. Ectopic expression of subsets of PRC1/2-repressed nonepidermal transcription factors in wild-type epidermal stem cells was sufficient to suppress epidermal identity genes, highlighting the importance of functional redundancy between PRC1 and PRC2. Altogether, our studies show how PRC1 and PRC2 function as two independent counterparts, thereby providing a repressive safety net that protects and preserves lineage identity.
One of the main challenges in modern biology is to understand the mechanisms by which stem cells and progenitor cells control tissue development and homeostasis (Ge and Fuchs 2018). Epigenetic factors are critical regulators of stem cells, exerting their function through the modification of DNA, histones, or nucleosome remodeling (Allis and Jenuwein 2016; Atlasi and Stunnenberg 2017).
The Polycomb group (PcG) proteins are evolutionarily conserved chromatin modifiers that function in diverse systems ranging from plants to mammals and are considered to be part of the paradigm for epigenetic regulation (Simon and Kingston 2009; Aloia et al. 2013). PcG proteins are present in two multi-subunit complexes, Polycomb repressive complex (PRC) 1 and PRC2 (Simon and Kingston 2009; Schuettengruber et al. 2017). The canonical PcG proteins function to repress transcription (Schuettengruber et al. 2017), although PRC2-independent roles of PRC1 in promoting gene expression have been reported (Frangini et al. 2013; Gao et al. 2014; Cohen et al. 2018, 2019). Mammalian PRC2 contains EED, SUZ12, and EZH1/2 core subunits and establishes trimethylation on histone H3 lysine 27 (H3K27me3) (Cao et al. 2002; Czermin et al. 2002; Kuzmichev et al. 2002; Healy et al. 2019). Mammalian PRC1 complexes are diverse in their subunit composition (Gao et al. 2012). However, all PRC1 complexes contain an E3 ubiquitin ligase, RING1A or RING1B, which catalyzes monoubiquitination on histone H2A lysine 119 (H2AK119ub) (de Napoles et al. 2004; Wang et al. 2004a), a histone mark critical for Polycomb-mediated gene repression (Endoh et al. 2012; Cohen et al. 2018, 2020; Blackledge et al. 2020).
At silent genes, PRC1 and PRC2 largely overlap in their chromatin binding in many developmental systems (Boyer et al. 2006; Tolhuis et al. 2006; Ku et al. 2008). This overlap is thought to be a consequence of the ability of PRC1 to recognize the PRC2-mediated histone modification and vice versa (Fischle et al. 2003; Min et al. 2003; Wang et al. 2004a,b; Blackledge et al. 2014; Cooper et al. 2014). This interplay and mutual recruitment of Polycomb complexes suggest that loss of one of these complexes would affect the stability of its counterpart's binding, yielding similar phenotypes of PRC1 and PRC2 mutants. Indeed, in flies, loss of either PRC1 or PRC2 leads to similar homeotic transformations through derepression of homeotic genes (Simon et al. 1992). In the developing murine skin, loss of PRC2- or PRC1-mediated H2AK119ub catalysis results in a similar phenotype of ectopic Merkel cell formation (Bardot et al. 2013; Dauber et al. 2016; Cohen et al. 2018). However, studies in murine embryonic stem cells (mESCs) have shown that PRC1 and PRC2 may also possess redundant functions. Indeed, the loss of both PRC1 and PRC2 complexes impaired mESC self-renewal and differentiation, phenotypes that were not observed in single knockouts (Leeb et al. 2010). It is not known whether functional redundancy between PRC1 and PRC2 in transcriptional repression exists in vivo. Also unknown is the physiological significance of PRC1/2 corepression for the somatic stem cell identity and tissue development.
Here, we used the developing murine epidermis as a paradigm for studying redundant functions between Polycomb complexes. The epidermis provides an essential protective barrier against external insults and prevents dehydration. To carry out these functions, a monolayer of embryonic epidermal stem cells (EpSCs) must execute a precise developmental program, resulting in epidermal stratification. Using in vivo functional genomics combined with genetic loss-of-function studies, we discovered that PRC1 and PRC2 each provide a compensatory mechanism required for epidermal stratification by redundantly repressing unwanted lineage genes.
Results
Repression of Polycomb targeted genes is largely maintained in the absence of PRC1 or PRC2 activity
Immunofluorescence analysis showed that maintenance of PRC1 or PRC2 histone marks is not mutually dependent (Supplemental Fig. S1A,B). This observation prompted us to test whether PRC1 and PRC2 function redundantly in gene silencing, using the murine epidermis as a somatic stem cell developmental system. By analyzing chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) data for H2AK119ub and H3K27me3 in FACS-purified EpSCs from control newborn skins (Cohen et al. 2018), we defined a total of 2652 genomic loci comarked by PRC1 and PRC2 (Supplemental Table S1), as well as loci negative to these marks (Supplemental Fig. S1C, bottom). These regions varied in their epigenetic profiles and were divided using k-means clustering into four distinct clusters (Supplemental Fig. S1C, top). Cluster 1 was characterized by abundant and broad ChIP-seq signals for H2AK119ub and H3K27me3, low and diffused signals for H3K4me3 and for assay for transposase-accessible chromatin (ATAC)-seq, and no H3K27ac signal (Supplemental Fig. S1C, top). Cluster 2 was similar to cluster 1 but had slightly narrower and lower H2AK119ub and H3K27me3 signals than cluster 1. Overall, clusters 1 and 2 share an epigenetic signature of transcriptionally silent loci. Cluster 3 had focal ChIP-seq signals for H2AK119ub and H3K27me3, with moderate H3K4me3 and ATAC-seq signals and low H3K27ac signal, indicative of bivalency (Bernstein et al. 2006; Zhou et al. 2011). Finally, cluster 4 had low and focal H2AK119ub and H3K27me3 ChIP-seq signals, weak and diffused H3K4me3 signal, and no H3K27ac or ATAC signals (Supplemental Fig. S1C, top). Transcriptional analysis of control EpSCs using high-throughput RNA sequencing (RNA-seq) showed that genes associated with clusters 1–2 were typically silent with mean expression levels of less than one transcript per million (TPM). Genes in cluster 3, despite bivalency, were also expressed at low levels, but their expression levels were slightly higher than that of genes of clusters 1 and 2. Genes in cluster 4 were expressed at slightly higher levels than genes in clusters 1–3, with mean TPM <2.5 (Supplemental Fig. S1D). Gene ontology (GO) analysis revealed that clusters 1 and 2, in which H2AK119ub and H3K27me3 signals were abundant, were mostly enriched for cell fate regulators, nonepidermal lineage genes, and transcription factors (Supplemental Fig. S1E; Supplemental Table S2). Clusters 3 and 4, however, were enriched for genes related to cell communication, cell–cell signaling, and transport, as well as nonepidermal lineage genes (Supplemental Fig. S1E; Supplemental Table S2). Overall, PRC1 and PRC2 comarked genes in each cluster vary in their epigenetic landscape and molecular pathways unique to each cluster.
We next performed functional studies of PRC1-null and PRC2-null EpSCs. To do this, we used mice in which the essential PRC1 core (RING1A and RING1B) or PRC2 core (EED) subunits were conditionally ablated by Krt14-Cre, a Cre recombinase expressed in epidermal progenitors starting embryonic day (E) 12.5 (Krt14-Cre; Ring1a−/− Ring1bflox/flox = Ring1a/b 2KO, and Krt14-Cre; Eedflox/flox = Eed cKO) (Dassule et al. 2000). ChIP-seq analysis of PRC1/2 comarked loci showed that H2AK119ub mark was reduced but overall maintained in Eed cKO EpSCs compared with control EpSCs (Fig. 1A,C; Supplemental Fig. S1F). A similar trend was also observed for the H3K27me3 mark in Ring1a/b 2KO EpSCs compared with control EpSCs (Fig. 1B,C; Supplemental Fig. S1G). RNA-seq analysis revealed that <12% of PRC1/2 coregulated genes were significantly up-regulated in Eed cKO EpSCs, with subtle changes in their mean mRNA expression levels (Supplemental Fig. S1H,J,K; Supplemental Table S3). We observed stronger derepression in Ring1a/b 2KO EpSCs compared with PRC2-null EpSCs, with up-regulation of ∼25% of the shared PRC1/2 targets and a slightly higher increase in the mean mRNA expression level, suggesting a dominant repressive role of PRC1 in EpSCs (Supplemental Fig. S1I,L,M; Supplemental Table S3). While genes up-regulated in Eed cKO and Ring1a/b 2KO showed a decrease in H2AK119ub or H3K27me3 signal compared with control EpSCs, the retention of PRC1-dependent H2AK119ub in Eed cKO and PRC2-dependent H3K27me3 in Ring1a/b 2KO did not strongly correlate with the level of gene derepression in these mutants (Supplemental Fig. S1H,I). In addition, we did not observe any marked differences between genes in the various PRC1/2 clusters, in terms of tendency for up-regulation upon PRC1 or PRC2 loss (Supplemental Fig. S1J–M). Collectively, these data show that in EpSCs, only a small portion of PRC1/2 coregulated genes was derepressed in the absence of either PRC1 or PRC2 function, further supporting the notion of functional redundancy in gene repression between PRC1 and PRC2.
Figure 1.

Repressive marks of PRC1 and PRC2 are largely maintained in the absence of their counterparts in FACS-purified EpSCs. (A) Read densities of H2AK119ub ChIP-seq peaks in P0 control, Eed cKO, and Ring1a/b 2KO FACS-purified EpSCs, ±10 kb from peak centers. (B) Read densities of H3K27me3 ChIP-seq peaks in P0 control, Ring1a/b 2KO, and Eed cKO FACS-purified EpSCs, ±10 kb from peak centers. (C) Integrative Genomics Viewer (IGV) browser view of ChIP-seq tracks for H2AK119ub and H3K27me3 in P0 control, Eed cKO, and Ring1a/b 2KO FACS-purified EpSCs on the HoxA gene cluster. Y-axis normalized by sequencing depths.
Activity of PRC1 and PRC2 is required for epidermal differentiation and tissue development
To functionally test whether PRC1 and PRC2 play redundant roles in epidermal development, we ablated both PRC1 and PRC2 functions in EpSCs (K14-Cre; Eedflox/flox; Ring1a−/−Ring1bflox/flox = Eed; Ring1a/b 3KO). Eed; Ring1a/b 3KO mice displayed a severely impaired skin appearance (Fig. 2A) and died shortly after birth. Using immunofluorescence (IF), we confirmed the loss of both H2AK119ub and H3K27me3 histone marks in the Eed; Ring1a/b 3KO epidermis (Fig. 2B,C). Hematoxylin and eosin (H&E) staining revealed that the epidermis of Eed; Ring1a/b 3KO mice was thin and poorly developed (Fig. 2D). IF analysis of the basal layer marker keratin 5 (KRT5) and the early suprabasal layer differentiation marker of KRT10 showed weak and dramatically reduced expression in the suprabasal layers (Fig. 2E). IF staining for the late differentiation marker loricrin (LOR) was almost absent in the Eed; Ring1a/b 3KO epidermis (Fig. 2F), suggesting that the epidermal differentiation program was impaired in the absence of both PRC1 and PRC2 functions. These pronounced developmental defects were not observed in the Eed cKO or Ring1a/b 2KO epidermis, although a slight reduction in differentiated layers was also apparent in the Ring1a/b 2KO epidermis (Fig. 2D–F). Cell proliferation was reduced, but no aberrant apoptosis was observed in the Eed; Ring1a/b 3KO epidermis (Supplemental Fig. S2A–D). Interestingly, the Eed; Ring1a/b 3KO epidermis also completely lacked Merkel cells (Supplemental Fig. S2E,F).
Figure 2.

Epidermal development is arrested in PRC1/2-null mutants. (A) Gross appearance of newborn (P0) pups for all indicated genotypes. Note that the Eed; Ring1a/b 3KO pup displays red skin, indicating epidermal defects. (B,C) IF staining for H2AK119ub (B; green) and H3K27me3 (C; green) confirming complete loss of these marks in the Eed; Ring1a/b 3KO epidermis. The basement membrane is labeled by Integrin β4 (ITGB4). Scale bar, 50 µm. (D) Hematoxylin and eosin (H&E) analysis of newborn skins. Scale bar, 50 µm. (E) IF staining for basal layer marker KRT5 (green) and suprabasal marker KRT10 (red). Scale bar, 50 µm. (F) IF staining for the late differentiation marker Loricrin (LOR; red). Scale bar, 50 µm.
Transcriptional analysis of FACS-purified Eed; Ring1a/b 3KO EpSCs using RNA-seq showed increased transcriptional dysregulation compared with Eed cKO and Ring1a/b 2KO EpSCs (Fig. 3A,B,D). Focusing on significantly up-regulated genes, we observed 551 genes in Eed cKO, 1353 genes in Ring1a/b 2KO, and 2492 genes in Eed; Ring1a/b 3KO versus control EpSCs (Fig. 3B; Supplemental Tables S3, S4). The majority of genes up-regulated in Eed cKO or Ring1a/b 2KO EpSCs were also up-regulated in Eed; Ring1a/b 3KO EpSCs (Fig. 3A,B). The expression levels of up-regulated genes were higher in Eed; Ring1a/b 3KO EpSCs compared with single complex knockouts, suggesting that both Polycomb complexes contribute to their repression (Supplemental Fig. S3A). Functional annotation of Eed; Ring1a/b 3KO up-regulated genes showed enrichment in GO terms related to nonepidermal lineage developmental genes and transcription factors (TFs) (Fig. 3C; Supplemental Table S5). We observed a smaller overlap between the three different mouse lines for the down-regulated genes. Only 89 genes were down-regulated in Eed cKO EpSCs compared with 436 genes in Ring1a/b 2KO EpSCs and 1129 genes in Eed; Ring1a/b 3KO EpSCs (Fig. 3D; Supplemental Tables S3, S4). The expression levels of the down-regulated genes were lower in Eed; Ring1a/b 3KO compared with Eed cKO and Ring1a/b 2KO (Supplemental Fig. S3B). Genes down-regulated in Eed; Ring1a/b 3KO EpSCs were related to cell proliferation and DNA repair, as well as skin and epidermis developmental processes (Fig. 3E; Supplemental Table S5). Altogether, we conclude that coablation of both PRC1 and PRC2 activity in EpSCs exacerbates the molecular and developmental phenotypes of the single PRC1-null and PRC2-null EpSCs.
Figure 3.

Loss of both PRC1 and PRC2 in EpSCs exacerbates transcriptional changes in single complex mutants. (A) Heat map showing that the DEGs from 3KO were significantly less affected in Eed cKO or Ring1a/b 2KO. The DEGs from 3KO versus control were used. (B) Venn diagram showing overlap of up-regulated genes in P0 Eed cKO, Ring1a/b 2KO, and Ring1a/b; Eed 3KO epidermis. (C) Gene ontology (GO) analysis of up-regulated genes in the Eed; Ring1a/b 3KO epidermis. (D) Venn diagram showing overlap of down-regulated genes in P0 Eed cKO, Ring1a/b 2KO, and Eed; Ring1a/b 3KO epidermis. (E) GO analysis of down-regulated genes in the Eed; Ring1a/b 3KO epidermis.
Indirect PRC1 and PRC2 functions preserve epidermal identity
To gain insight into the observed PRC1/2-null epidermal phenotype, we analyzed genes down-regulated in Eed; Ring1a/b 3KO EpSCs. By performing reverse transcription followed by qPCR (RT-qPCR), we confirmed our RNA-seq analysis and observed that deletion of both Polycomb complexes had an additive effect on the expression levels of epidermal cell identity genes, epidermal TFs, and cell proliferation genes (Fig. 4A). Among epidermal genes significantly down-regulated in Eed; Ring1a/b 3KO EpSCs, there were key epidermal lineage TFs such as P63 and SATB1, as well as the signal transducer FGFR2, essential for epidermal development (Fig. 4A–C; Supplemental Fig. S4A; Mills et al. 1999; Petiot et al. 2003; Yang et al. 2010; Fessing et al. 2011). To test whether epidermal genes down-regulated in Eed; Ring1a/b 3KO EpSCs are directly coregulated by PRC1 and PRC2, we analyzed our ChIP-seq data. There was practically no overlap between PRC1/2 comarked loci and genes down-regulated in Eed; Ring1a/b 3KO EpSCs (Fig. 4D; Supplemental Fig. S4B), and the vast majority of down-regulated epidermal genes were not targeted by the PRC1 subunit RING1B (Fig. 4D; Supplemental Fig. S4C). For the 18% of down-regulated genes that were bound by RING1B (Supplemental Fig. S4C), we did not observe any significant changes in their expression between Ring1a/b 2KO and Eed; Ring1a/b 3KO EpSCs, suggesting no additive effect between PRC1 and PRC2 complexes in promoting their expression (Supplemental Fig. S4D,E). This is in line with previous observations that noncanonical PRC1 promotes gene expression independently of PRC2 (Gao et al. 2014; Cohen et al. 2018, 2019). Taken together, we conclude that the changes in epidermal gene expression and the loss of epidermal cell identity in Eed; Ring1a/b 3KO EpSCs most likely stem from indirect mechanisms.
Figure 4.

Indirect PRC1 and PRC2 functions preserve EpSC identity. (A) Fold change in epidermal gene expression in P0 Eed cKO, Ring1a/b 2KO, and Eed; Ring1a/b 3KO epidermis. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (B,C) IF staining of the epidermal markers p63 (B) and SATB1 (C) in P0 control and Eed; Ring1a/b 3KO epidermis. The basement membrane is labeled by Integrin β4 (ITGB4; green). Scale bars, 50 µm. (D) IGV browser view of Polycomb marks and subunits for PRC1/2 down-regulated genes. Arrows indicate transcription start sites (TSSs) and transcription direction.
PRC1 and PRC2 act redundantly in transcriptional repression
We next investigated whether the pronounced transcriptional changes observed in Eed; Ring1a/b 3KO EpSCs could be direct consequences of the loss of functional redundancy between PRC1 and PRC2 in transcriptional repression. Integration between RNA-seq and ChIP-seq data showed that >60% of the up-regulated genes were direct targets of PRC1 and/or PRC2, with ∼65% of direct targets comarked by both complexes (Fig. 5A). Among PRC1/2 demarcated genes up-regulated in Eed; Ring1a/b 3KO EpSCs, >35% remained silent in both Eed cKO and Ring1a/b 2KO EpSCs (Fig. 5B), indicating that those genes were redundantly repressed by PRC1 and PRC2. Further analysis also showed a higher level of gene derepression in Eed; Ring1a/b 3KO EpSCs compared with Eed cKO or Ring1a/b 2KO EpSCs (Fig. 5C,D). Among PRC1/2 comarked genes, we observed enrichment for nonepidermal TFs that are normally silent in control EpSCs (Fig. 5E). ChIP-seq and ChIP-qPCR analysis showed that these TF genomic loci maintained high levels of H2AK119ub in Eed cKO and high levels of H3K27me3 in Ring1a/b 2KO EpSCs (Supplemental Fig. S5A–C). Consistent with the ChIP data, loss of both PRC1 and PRC2 resulted in stronger up-regulation of those TFs in Eed; Ring1a/b 3KO EpSCs than in single complex KOs (Fig. 5F). Taken together, these data indicate that PRC1 and PRC2 function redundantly to repress their cotargeted genes and that loss of both PRC1 and PRC2 leads to a massive derepression of nonlineage TFs in EpSCs.
Figure 5.

PRC1 and PRC2 redundantly repress nonlineage transcription factors in EpSCs. (A) Distribution of H3K27me3 and H2AK119ub marks on the up-regulated genes in Eed; Ring1a/b 3KO. (B) Distribution of PRC1 and PRC2 up-regulated direct target genes among P0 Eed cKO, Ring1a/b 2KO, and Eed; Ring1a/b 3KO EpSCs. (C) Expression levels of PRC1 and PRC2 direct target genes in P0 control, Eed cKO, and Eed; Ring1a/b 3KO EpSCs. (D) Expression levels of PRC1 and PRC2 direct target genes in P0 control, Ring1a/b 2KO, and Eed; Ring1a/b 3KO EpSCs. (E) Proportion of up-regulated transcription factor (TF) genes marked by PRC1, PRC2, or PRC1/2. (F) Relative expression levels of nonepidermal TFs in P0 Eed cKO, Ring1a/b 2KO, and Eed; Ring1a/b 3KO EpSCs. (G) Relative expression levels of epidermal genes in keratinocytes ectopically expressing combinations of nonepidermal TFs. Combination 1 included NR4A2, GATA2, FOXA1, TBX3, NKX2-9, and TBX5. Combination 2 included PITX2, SIX2, HOXD13, SIX3, OTX2, and SATB2.
Expression of nonlineage TFs suppresses epidermal cell identity
Probing deeper into the molecular mechanisms of Polycomb functional redundancy, we tested whether derepression of nonlineage TFs led to the changes in epidermal identity phenotype observed in Eed; Ring1a/b 3KO EpSCs. Many genes bound by PRC1 and PRC2 are central regulators of development, believed to be capable of modifying cell fates on their own (Mattick et al. 2010). To select the most prominent nonlineage TFs among a list of ∼300 TFs significantly up-regulated in Eed; Ring1a/b 3KO EpSCs, we performed motif enrichment analysis on the promoter and DNA regulatory regions with ATAC-seq and H3K27ac peaks present near epidermal genes that were down-regulated in Eed; Ring1a/b 3KO EpSCs (Supplemental Fig. S5D,E). We used the HOMER software (Heinz et al. 2010) to find enriched transcription factor binding motifs, and from the results, we further selected a list of 19 transcription factors that were up-regulated in Eed; Ring1a/b 3KO EpSCs (Supplemental Fig. S5F; Supplemental Table S6). This analysis revealed enrichment for TFs of the T-box (TBX) gene family, the sine oculis homeobox (SIX) gene family, and several additional TFs with known roles in the neural, cardiac, or mesodermal lineages (Supplemental Fig. S5F; Oliver et al. 1995; Bruneau 2002; Evseenko et al. 2010). To test the role of these nonepidermal TFs, we ectopically expressed two different combinations of six TFs in wild-type EpSCs and tested their effect on the expression of epidermal genes. The combined expression of these nonepidermal TFs was sufficient to suppress epidermal genes (Fig. 5G), similar to the suppressed epidermal identity observed in Eed; Ring1a/b 3KO epidermis (Figs. 3E, 4A). Thus, functional redundancy between PRC1 and PRC2 in the repression of unwanted TFs preserves epidermal transcriptional identity (Fig. 6).
Figure 6.
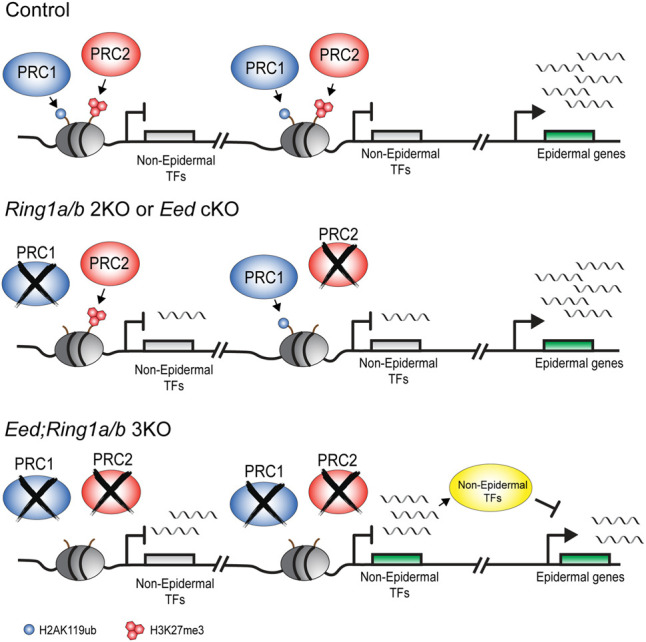
Redundant repressive functions of PRC1 and PRC2 promote epidermal cell fate in EpSCs. (Top) In control EpSCs, PRC1 and PRC2 repressive marks are present on the promoters of nonepidermal genes. Epidermal genes are expressed. (Middle) In Eed cKO or Ring1a/b 2KO EpSCs, loss of either PRC complex results in mild derepression of nonepidermal genes. Epidermal gene expression is not suppressed. (Bottom) In Eed; Ring1a/b 3KO EpSCs, loss of both complexes results in a robust derepression of nonepidermal transcription factor (TF) genes, highlighting that PRC1 and PRC2 redundantly repress these genes. Epidermal genes are suppressed in Eed; Ring1a/b 3KO EpSCs, emphasizing the physiological importance of PRC1 and PRC2 functional redundancy in cell identity maintenance.
Discussion
Maintenance of specific gene expression patterns has a central role in the establishment of cell differentiation programs during development. In this study, we provide evidence for the importance of functional redundancy between repressive functions of PRC1 and PRC2 in the maintenance of somatic stem cell identity, differentiation, and tissue development. We show that genetic ablation of both PRC1 and PRC2 in EpSCs impairs epidermal stratification and tissue development, a phenotype that is not observed upon ablation of individual complexes. Analysis of gene expression indicates a loss of epidermal identity in Eed; Ring1a/b 3KO EpSCs that is coupled with a strong up-regulation of nonlineage TFs that is redundantly repressed by both Polycomb complexes. Expression of these nonlineage TFs drives loss of epidermal identity, highlighting the importance of functional redundancy between repressive functions of PRC1 and PRC2 in preserving cellular identity in vivo (Fig. 6).
Genome-wide studies of Polycomb occupancy have shown that PRC1 and PRC2 binding largely overlaps (Boyer et al. 2006; Tolhuis et al. 2006; Ku et al. 2008). In line with these observations, loss-of-function studies in Drosophila have shown similar homeotic transformations in PRC1 and PRC2 mutants, leading to a model of interdependent function of PRC1 and PRC2, both genetically and biochemically (Simon et al. 1992; Cao et al. 2002; Min et al. 2003; Wang et al. 2004b). In contrast, studies in mammalian tissues revealed surprising independent and even opposing functions of PRC1 and PRC2 in multiple tissues, including the skin, the tongue, the intestine, and the hematopoietic system. In many of these studies, the phenotype of PRC1 loss was often more severe than that of PRC2 loss (Lessard et al. 1999; Majewski et al. 2010; Chiacchiera et al. 2016a,b; Koppens et al. 2016; Loubiere et al. 2016; Cohen et al. 2018, 2019; Bar et al. 2019; Fursova et al. 2019), suggesting a dominant role for PRC1 in gene repression. Overall, mammalian Polycomb loss-of-function studies indicate that Polycomb regulation of gene expression is more complex than originally suggested by the Drosophila studies.
Studies in mESCs started to explore functional redundancy between PRC1 and PRC2 in gene repression. Indeed, mESCs in which Ring1b and Eed were ablated failed to differentiate into the three germ layers, a phenotype not observed in the single Ring1b or Eed knockouts (Leeb et al. 2010). Moreover, developmental genes were strongly derepressed in the double knockout cells compared with single knockouts, where they largely remained repressed (Leeb et al. 2010). Similarly, a later study revisiting the functional redundancy between Polycomb complexes has shown that deletion of both complexes results in a stronger up-regulation compared with individual complex ablation (King et al. 2018). However, the significance of functional redundancy between Polycomb complexes in vivo remains to be explored. Our studies in the epidermis show that ablation of a single Polycomb complex (either PRC1 or PRC2) results in the retention of the other complex's histone mark. Also, ablation of single Polycomb complexes results in mild gene derepression of PRC1/2 corepressed genes, and robust derepression is achieved only when both Polycomb complexes are ablated, in line with the mESCs studies (Leeb et al. 2010; King et al. 2018). Furthermore, loss of both PRC1/2 complexes resulted in a severely impaired epidermal stratification, a phenotype not observed in single knockouts lacking either PRC1 or PRC2 activity. To the best of our knowledge, our results provide the first in vivo evidence of functional redundancy between PRC1 and PRC2 in transcriptional repression, which is required for proper development of the epidermis, a tissue essential for our survival.
The observed functional redundancy between PRC1 and PRC2 transcriptional repression in the epidermis raises questions about the physiological significance of this coregulation. Eed; Ring1a/b 3KO EpSCs displayed down-regulation in expression of key epidermal lineage identity genes such as the TF Trp63 (Mills et al. 1999; Koster et al. 2004) and the chromatin regulator Satb1 (Fessing et al. 2011), as well as the signaling pathway component Fgfr2, which also plays a role in mechanosensory Merkel cell formation (Nguyen et al. 2018, 2019). Interestingly, these genes are not direct targets of Polycomb complexes, indicating that the changes in epidermal gene expression in Eed; Ring1a/b 3KO EpSCs are likely indirect. We observed a strong up-regulation of nonepidermal genes in Eed; Ring1a/b 3KO EpSCs compared with single knockouts or control cells, including many developmental master regulator TFs that are direct targets of PRC1 and PRC2 in EpSCs. Remarkably, overexpression of small combinations of these nonepidermal TFs resulted in significant down-regulation of epidermal lineage genes, highlighting the role of redundant PRC1 and PRC2 repression in preserving lineage identity. Studies of Polycomb complexes in multiple systems have shown that loss of either PRC1 or PRC2 leads to up-regulation of nonlineage genes (Calés et al. 2008; Chiacchiera et al. 2016a; Ikawa et al. 2016; von Schimmelmann et al. 2016; Yakushiji-Kaminatsui et al. 2016; Vidal and Starowicz 2017; Jadhav et al. 2019). It would be intriguing to investigate whether functional redundancy is similarly critical for preserving the identity of those tissues. Notably, coablation of PRC1 and PRC2 in the tongue epithelium did not result in exacerbation of the phenotype of the PRC1-null tongue epithelium as seen in the skin epidermis (Bar et al. 2019). This is somewhat in line with the observation made in mESCs, where nucleosome occupancy is controlled predominantly by PRC1 (King et al. 2018). These variabilities suggest that the importance of functional redundancy between PRC1 and PRC2 may be tissue specific or context dependent or both.
The discovery of functional redundancy between PRC1 and PRC2 in vivo has multiple translational implications. First, proteins of both Polycomb complexes, in particular EZH2 and BMI1, are often overexpressed in epithelial cancers, including skin cancers (Balasubramanian et al. 2008; Eckert et al. 2011; Adhikary et al. 2015; Ferretti et al. 2016; Fisher et al. 2016). These studies suggest specific roles of Polycomb subunits in tumorigenesis; however, how their overexpression affects the function of PRC1 and PRC2 repression in cancer cells remains unclear. The therapeutic use of EZH2 inhibitors has been clinically studied (Comet et al. 2016), with one inhibitor recently approved for human use (Mullard 2020). Understanding whether functional redundancy between PRC1 and PRC2 exists in cancer cells may help improve cancer therapy by simultaneous targeting of both complexes. Second, functional redundancy between PRC1 and PRC2 in the repression of developmental genes may also be relevant for reprogramming. Pluripotent cells can be isolated either from the inner cell mass of the blastocyst or from the epiblast of a post-implantation embryo (Marks et al. 2012; Gafni et al. 2013; Weinberger et al. 2016). One molecular difference between these two pluripotent cell types is deposition of H3K27me3 on lineage genes in the epiblast cells (Weinberger et al. 2016). The physiological significance of PRC1 and PRC2 functional redundancy raises the question whether loss of both the PRC1 and PRC2 complexes can facilitate the reprogramming of somatic cells to a pluripotent state. Here we show that functional redundancy between PRC1 and PRC2 has physiological significance in the protection of cellular identity, which is essential for proper tissue development, regeneration, and organ function.
Materials and methods
Mice
All mice used in this study were housed at the Center of Comparative Medicine and Surgery (CCMS), Icahn School of Medicine at Mount Sinai (ISMMS), according to the Institutional Animal Care and Use Committee (IACUC) guidelines (protocol no. LA11-0020). Eedflox/flox mice were kindly provided by Dr. Weipeng Mu and Dr. Terry Magnuson (Mu et al. 2014). Ring1a−/− and Ring1bflox/flox mice were previously described (del Mar Lorente et al. 2000), and were kindly provided by Dr. Miguel Vidal. Both male and female mice were used in this study. Primer sequences used for genotyping are available in Supplemental Table S7.
Cell cultures
HEK293T cells were cultured in DMEM, supplemented with 10% heat-inactivated fetal bovine serum (R&D Systems) and 1% pen/strep solution (Life Technologies). Primary epidermal progenitors were isolated from newborn mouse epidermis as previously described (Cohen et al. 2018) and cultured in E-media (Rheinwald and Green 1977) supplemented with 15% serum and 0.05 mM CaCl2.
Ectopic expression of nonepidermal TFs in primary epidermal progenitors
HEK293T cells were transfected with the 20 µg of lentiviral construct (Horizon Discovery lentiviral library), 2.5 µg of packing plasmids (pPAX2, pMD2.G, AddGene plasmids 12260 and 12259, a kind gift from Didier Trono), 100 mM CaCl2, and 1× HBS (2× HBS stock solution: 280 mM NaCl, 10 mM KCl, 1.5 mM Na2HPO4, 12 mM dextrose, 50 mM HEPES) in a total volume of 1 mL, diluted into 7 mL of DMEM media supplemented with 10% heat-inactivated fetal bovine serum (R&D Systems) and 1% pen/strep solution (Life Technologies). Media were changed 8 h post transfection, and cells were cultured for an additional 48 h to produce lentiviral particles. Lentivirus-containing media were collected, filtered with a 0.45-µm strainer, and concentrated with Amicon ultracentrifuge concentration units (Sigma-Millipore), producing total of 2.5 mL of high-titer virus.
Fifty-thousand cells were plated for lentiviral combinations (six viruses per combination). Plasmids used in this study include pLVX304-Blast-LacZ (control) and TF-expressing vectors, all are pLVX304-Blast, each expressing one TF. Combo 1 consisted of pLX304-NR4A2, GATA2, FOXA1, TBX3, TBX5, and NKX2-9. Combo 2 consists of PLVX304-SIX2, HOXD13, SIX3, OTX2, PITX2, and SATB2. Cells were infected twice with 100 µL of each virus in a total volume of 3 mL of E-media and 5 µg/mL polybrene and spun for 30 min at 32°C. Media were changed 8 h after each infection, and cells were cultured for an additional 48 h in E media and were then were selected with 0.4 µg/mL blasticidin for 48 h. After selection, cells were cultured until confluent in E-media and collected in RLT plus buffer (Qiagen) for RNA extraction.
Fluorescence-activated cell sorting
Isolation of epidermal progenitors from control, Eed cKO, Ring1a/b 2KO, and Eed; Ring1a/b 3KO mice was done as previously described (Cohen et al. 2019). P0 back skins were collected and incubated for 4–6 h in 1.26 U/mL dispase (Invitrogen) at 4°C. The epidermis was gently peeled from the underlying dermis, followed by 0.25% trypsin treatment for 15 min at 37°C. The cell suspension was washed twice with 1× PBS; stained with 1:200 Sca1-PerCP-Cy5.5 (Biolegend), 1:100 α6-integrin-FITC (eBiosciences), and 1:200 EpCAM-APC (Biolegend) for 30 min on ice; and washed twice with 1× HBSS before cell sorting. Interfollicular epidermis, enriched for epidermal progenitors, was sorted as EpCAM(+), Sca1(+), and α6-integrin(high). For ChIP and ChIP-seq analyses, cell suspensions were stained for cell viability and cross-linked before staining and FACS sorting as described above. All cell isolations were performed on a FACS Influx instrument (BD) in the Flow Cytometry Core Facility at Icahn School of Medicine at Mount Sinai.
Chromatin immunoprecipitation, ChIP-qPCR, and library preparation
Chromatin immunoprecipitation (ChIP) was performed on FACS-sorted populations using materials and methods as previously described (Bar et al. 2020). A total of 0.4 × 106 cells was used for each histone mark per ChIP. Before cell sorting, cells were stained for viability using Zombie violet (Biolegend) and then cross-linked using fresh solution with a final concentration of 1% formaldehyde (Thermo Fisher Scientific) for 10 min at room temperature. Cross-linking was stopped by the addition of glycine (final concentration 125 mM) for 5 min of incubation at room temperature, followed by two washes with 1× PBS. Cells were incubated in lysis buffer 1 (50 mM HEPES at pH 7.5, 140 mM Nacl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, protease inhibitor cocktail [Roche]) for 10 min on ice and then incubated for 10 min with lysis buffer 2 (10 mM Tris-HCl at pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA). Before ChIP, cells were resuspended in lysis buffer 3 (10 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, 0.5% N-laurylsarcosine, 1% Triton X-100) and sonicated using a Bioruptor sonicator (Diagenode UCD-200) according to a 25× regimen of 30 sec of sonication followed by 90 sec of rest at 2.7°C. Chromatin was incubated overnight at 4°C with antibodies as indicated in Supplemental Table S7. To detect changes in H2AK119ub and H3K27me3 levels between control, Eed cKO, and Ring1a/b 2KO samples, we used Drosophila spike-in chromatin and antibody (Active Motif) as normalization controls, as previously described (Egan et al. 2016). DYNAL protein G magnetic beads (Invitrogen) were added the next day and incubated for 4 h. The beads were sequentially washed with low-salt, high-salt, LiCl, and Tris-EDTA buffers for 10 min each at 4°C. Bound chromatin was eluted, and cross-linking was reversed by overnight incubation at 65°C, followed by RNase A (Sigma-Aldrich) and proteinase K (Roche Diagnostics) treatments. Samples were purified using a ChIP DNA Clean and Concentrator kit (Zymo Research). Samples were analyzed by qPCR using LightCycler 480 SYBR Green I master mix (Roche Diagnostics) on a LightCycler 480 instrument (Roche). Primer sequences are available in Supplemental Table S7. For high-throughput ChIP sequencing, libraries were constructed from 3 ng of purified DNA using the DNA SMART ChIP-seq kit (Clontech) according to the manufacturer's instructions. Constructed ChIP-seq libraries were sequenced on the Illumina HiSeq 4000 or the Illumina NextSeq 500 platform, and two biological replicates were used.
ATAC-seq and library preparation
Skin was harvested as previously described in “Fluorescence-Activated Cell Sorting.” Sorted cells were collected in PBS+10%FBS. ATAC-seq was performed as previously described (Buenrostro et al. 2015). Briefly, cells were precipitated by centrifugation and resuspended in lysis buffer (10 mM Tris-HCl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Cell lysate was precipitated, and pellets were incubated with a Tn5 transposase kit (Nextra) for 30 min at 37°C. The transposition reaction was stopped by addition of ATAC clean-up buffer (900 mM NaCl, 300 mM EDTA) and cleaned using Qiagen MinElute kit. Transposed DNA fragments were amplified using a NEBNext high-fidelity library construction kit (New England Biolabs). The ATAC-seq library was prepared as previously described (Buenrostro et al. 2015) with the following modifications: Transposed DNA was amplified by PCR with a common forward primer and barcoded reverse primer, using a NEBNext high fidelity kit, for 10 cycles. The number of cycles required for final amplification (N) was determined by a qPCR side reaction. Final amplification was performed with N cycles, using 20 µL of each sample. The amplified library was cleaned using AMPure XP beads according to the manufacturer's instructions.
RNA purification, RT-qPCR, and library preparation
FACS-purified cells were collected directly into RLT Plus buffer (Qiagen), and total RNA was isolated with the RNeasy Plus micro kit (Qiagen). Complimentary DNA was reverse-transcribed from total RNA using qScript cDNA SuperMix (Quanta Biosciences) according to the manufacturer's instructions. Samples were analyzed by RT-qPCR using LightCycler 480 SYBR Green I master mix (Roche Diagnostics) on a LightCycler 480 instrument (Roche). Results were normalized to Ppib mRNA levels. Primer sequences are available in Supplemental Table S7. Before library construction, the sample quality was measured using an Agilent Bioanalyzer, and samples with RNA integrity numbers greater than eight were used. Ten nanograms of total RNA was reverse transcribed and amplified using the Ovation RNA-seq system V2 (Nugen). Libraries were constructed from 100 ng of sonicated cDNA (Covaris) using the Ovation ultralow DR multiplex system (Nugen). The concentration and quality of the libraries were determined using Qubit (Invitrogen) and Bioanalyzer (Agilent). Constructed RNA-seq libraries were sequenced at GENEWIZ on the Illumina HiSeq platform, obtaining 150-nt paired-end reads.
Immunofluorescence staining and microscopy
Back skin tissues were collected from newborn mice, embedded in OCT compound (Tissue-Tek) and subsequently cut into 7-µm sections using a Leica cryostat. Slides were then prefixed in 4% PFA for 10 min at room temperature and blocked overnight at 4°C in blocking solution (1× PBS supplemented with 0.1% Triton X-100, 1% BSA, 0.25% normal donkey serum, 0.01% gelatin). Primary antibodies were diluted in blocking solution and incubated for 1 h, followed by 1-h incubation with secondary antibodies. Slides were counterstained with DAPI to visualize nuclei. For FGFR2 staining, slides were incubated with 100% methanol for 15 min after PFA fixation for tissue permeabilization. TUNEL apoptosis detection assay was performed using the in situ cell death detection kit, fluorescein (Roche). All antibodies and dilutions are available in Supplemental Table S7. Slides were imaged using a Leica DM5500 slide microscope using 10× or 20× objectives. For each immunofluorescence assay, at least three animals from at least two independent litters were analyzed per mutant genotype.
ChIP-seq analysis and data visualization
The generation, data processing, and peak calling of the H3K27me3, H2AK119ub, H3K4me3, H3K27ac, SUZ12, and RYBP ChIP-seq data were described previously (Cohen et al. 2018; Zhao and Zheng 2018). Those data were used to cluster the ChIP-seq peaks shared between H3K27m3 and H2AK119ub by randomly sampling 15 million reads from each sample and using the software seqMINER (v1.3.4) (Ye et al. 2011). The new H3K27me3 and H2AK119ub ChIP-seq data were analyzed by the same method, but the reads were also aligned to the fly genome, which was used as a spike-in control (Active Motif) to adjust for potential read-depth differences and other ChIP factors in different samples as previously described (Egan et al. 2016). The H3K27me3 or H2AK119ub peaks called in the CTL samples were sorted by peak sizes and then used by the seqMINER to generate the read density heat maps and aggregated profiles in Figure 1. For visualization, we also used the Integrative Genomics Viewer (IGV; http://software.broadinstitute.org/software/igv) and TDF files from the igvtools (v2.3.57; https://software.broadinstitute.org/software/igv/igvtools).
RNA-seq data analysis
The RNA-seq reads were aligned to the mouse transcriptomes corresponding to the Gencode annotation (vM20) (Harrow et al. 2012) using the software Kallisto (v0.42.5) (Bray et al. 2016). The genes with an average TPM (transcripts per million reads mapped) > 1 in either control or 3KO were used for differentially gene expression analysis with the software DESeq2 (Love et al. 2014) at the significance level of adjust P-value < 0.05 and more than twofold changes. To compare the gene expression changes across different genotypes and their controls (Eed cKO vs. CTL, Ring1a/b 2KO vs. CTL, and Eed;Ring1a/b 3KO vs. CTL), we used ComBat (v3.36.0) (Johnson et al. 2007) to remove batch effects.
ATAC-seq data analysis and data visualization
The ATAC-seq reads were aligned to the mouse genome (mm10), and peaks were called by the software MACS2 (Feng et al. 2012), otherwise analyzed the same as ChIP-seq data.
Gene ontology enrichment analysis
Identification of significantly overrepresented functional categories was done using the DAVID tool with default setting (Huang da et al. 2009). For PRC1/2-marked clusters, the combined gene list of clusters 1 + 2 and the entire gene list for clusters 3 and 4 were functionally annotated for biological processes. Selected GO terms were considered significant with P < 0.05 and are shown in Supplemental Figure S1E and Supplemental Table S2. Genes significantly down-regulated or up-regulated in Eed; Ring1a/b 3KO versus control epidermal progenitors were annotated using the same significance parameters, and selected GO terms are shown in Figure 3, C and E, and Supplemental Table S5.
Cell proliferation analysis quantification
Ki67(+) cells in the basal layer of the epidermis were quantified using the Leica LAS AF software. Nuclear DAPI staining was used to quantify the total number of cells in the basal layer, and the data are shown as the percentage of Ki67(+) cells in the basal layer. At least 50 random epidermal regions were measured for each animal group, from three animals (n = 3) of two independent litters. Comparisons and statistics were performed between matching knockout and control littermates.
Quantification of KRT8(+) Merkel cells
Merkel cells were quantified by the number of KRT8(+) cells per millimeter of skin. Sections had a typical skin length ranging between 7 and 14 mm. At least 100 mm of total skin length was analyzed per condition from at least three animals (n ≥ 3) of two independent litters. Comparisons and statistics were performed between matching knockout and control littermates.
Motif enrichment analysis
To perform motif enrichment analysis, the DNA sequences within the ±1-kb promoter region, ATAC-seq, or H3K27ac peaks associated with differentially expressed genes that were down-regulated in Eed; Ring1a/b 3KO versus control EpSCs were extracted, and the HOMER software (v4.7, default parameters) was used to find enriched transcription factor binding motifs. From the results, 19 transcription factors were selected whose expression was up-regulated in Eed; Ring1a/b 3KO EpSCs, and HOMER was rerun to obtain the enrichment scores shown in Supplemental Figure S5F.
Statistics
To determine the statistical significance between two groups, a two-sided t-test was performed. To determine the statistical significance between more than two groups, comparisons were made using one-way ANOVA with the Tukey post-hoc test correction for multiple comparisons. For box-and-whisker box plots in Supplemental Figure S1, K and M and violin plots in Supplemental Figures S1D, S3, and S4D and Figure 5, C and D, the midline represents the median. Box limits and colored dashed lines in violin plots represent the 25th percentile (lower quartile) and 75th percentile (upper quartile). The upper whisker represents the 75th–95th percentiles, and the lower whisker represents the 5th–25th percentiles. All data in bar graphs are presented as mean ± SEM. The number of biological replicates used for comparison is indicated in each figure. For each comparison, at least three animals for each group from two independent litters were used. Significance levels were defined as P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), and not significant (NS). For statistical analyses, GraphPad Prism 8 was used.
No randomization or blinding was performed in this study. Sample size is indicated in figure legends, and statistical methods are indicated in the quantification and statistical analysis paragraph.
Data availability
The accession number for the sequencing data reported here is GSE158324.
Competing interest statement
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
We thank Sergei Ezhkov, Venu Pothula, and all Ezhkova laboratory members for help and critical suggestions. We thank Miguel Vidal for the Ring1a−/− and Ring1bflox/flox mice and Weipeng Mu and Terry Magnuson for the Eedflox/flox mice. C.B. is a Merksamer Fund scholar. Research reported here was supported by the National Institutes of Health (NIH)/National Institute of Arthritis and Musculoskeletal and Skin Diseases under award numbers R01AR069078 and R01AR063724 to E.E., NIH/National Heart, Lung, and Blood Institute grants R01HL148128 and R01HL153920 to D. Zheng, and the Tisch Cancer Institute P30 Cancer Support Grant to E.E.
Author contributions: I.C., C.B., and E.E. conceived and designed the experiments. I.C., C.B., and V.J.V. performed the experiments. J.M.S provided the Horizon Discovery lentiviral library. H.L., P.M.G., D. Zhao, and D. Zheng performed the bioinformatic analyses. H.K. provided the Ring1a−/− and Ring1bflox/flox mice. I.C., C.B., D. Zhao, H.L., P.M.G. Jr., J.M.S, D. Zheng, and E.E. analyzed the data. I.C., C.B., and E.E. wrote the manuscript with input from all of the other authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.345363.120.
References
- Adhikary G, Grun D, Balasubramanian S, Kerr C, Huang JM, Eckert RL. 2015. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 36: 800–810. 10.1093/carcin/bgv064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nat Rev Genet 17: 487–500. 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- Aloia L, Di Stefano B, Di Croce L. 2013. Polycomb complexes in stem cells and embryonic development. Development 140: 2525–2534. 10.1242/dev.091553 [DOI] [PubMed] [Google Scholar]
- Atlasi Y, Stunnenberg HG. 2017. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 18: 643–658. 10.1038/nrg.2017.57 [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Lee K, Adhikary G, Gopalakrishnan R, Rorke EA, Eckert RL. 2008. The Bmi-1 polycomb group gene in skin cancer: regulation of function by (-)-epigallocatechin-3-gallate. Nutr Rev 66: S65–S68. 10.1111/j.1753-4887.2008.00071.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar C, Cohen I, Zhao D, Pothula V, Litskevitch A, Koseki H, Zheng D, Ezhkova E. 2019. Polycomb repressive complex 1 controls maintenance of fungiform papillae by repressing sonic hedgehog expression. Cell Rep 28: 257–266.e5. 10.1016/j.celrep.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar C, Valdes VJ, Ezhkova E. 2020. Chromatin immunoprecipitation of low number of FACS-purified epidermal cells. Methods Mol Biol 2154: 197–215. 10.1007/978-1-0716-0648-3_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardot ES, Valdes VJ, Zhang J, Perdigoto CN, Nicolis S, Hearn SA, Silva JM, Ezhkova E. 2013. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J 32: 1990–2000. 10.1038/emboj.2013.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, et al. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157: 1445–1459. 10.1016/j.cell.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Fursova NA, Kelley JR, Huseyin MK, Feldmann A, Klose RJ. 2020. PRC1 catalytic activity Is central to Polycomb system function. Mol Cell 77: 857–874.e9. 10.1016/j.molcel.2019.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. 10.1038/nature04733 [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Bruneau BG. 2002. Transcriptional regulation of vertebrate cardiac morphogenesis. Circ Res 90: 509–519. 10.1161/01.RES.0000013072.51957.B7 [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109: 21 29 21–21 29 29. 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calés C, Román-Trufero M, Pavón L, Serrano I, Melgar T, Endoh M, Pérez C, Koseki H, Vidal M. 2008. Inactivation of the polycomb group protein Ring1B unveils an antiproliferative role in hematopoietic cell expansion and cooperation with tumorigenesis associated with Ink4a deletion. Mol Cell Biol 28: 1018–1028. 10.1128/MCB.01136-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang LJ, Wang HB, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Chiacchiera F, Rossi A, Jammula S, Piunti A, Scelfo A, Ordóñez-Morán P, Huelsken J, Koseki H, Pasini D. 2016a. Polycomb complex PRC1 preserves intestinal stem cell identity by sustaining Wnt/β-catenin transcriptional activity. Cell Stem Cell 18: 91–103. 10.1016/j.stem.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Chiacchiera F, Rossi A, Jammula S, Zanotti M, Pasini D. 2016b. PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J 35: 2301–2314. 10.15252/embj.201694550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Zhao D, Bar C, Valdes VJ, Dauber-Decker KL, Nguyen MB, Nakayama M, Rendl M, Bickmore WA, Koseki H, et al. 2018. PRC1 fine-tunes gene repression and activation to safeguard skin development and stem cell specification. Cell Stem Cell 22: 726–739.e7. 10.1016/j.stem.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Zhao D, Menon G, Nakayama M, Koseki H, Zheng D, Ezhkova E. 2019. PRC1 preserves epidermal tissue integrity independently of PRC2. Genes Dev 33: 55–60. 10.1101/gad.319939.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Bar C, Ezhkova E. 2020. Activity of PRC1 and histone H2AK119 monoubiquitination: revising popular misconceptions. Bioessays 42: e1900192. 10.1002/bies.201900192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, Helin K. 2016. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer 16: 803–810. 10.1038/nrc.2016.83 [DOI] [PubMed] [Google Scholar]
- Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, et al. 2014. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7: 1456–1470. 10.1016/j.celrep.2014.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196. 10.1016/S0092-8674(02)00975-3 [DOI] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. 2000. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127: 4775–4785. [DOI] [PubMed] [Google Scholar]
- Dauber KL, Perdigoto CN, Valdes VJ, Santoriello FJ, Cohen I, Ezhkova E. 2016. Dissecting the roles of Polycomb repressive complex 2 subunits in the control of skin development. J Invest Dermatol 136: 1647–1655. 10.1016/j.jid.2016.02.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Lorente M, Marcos-Gutierrez C, Perez C, Schoorlemmer J, Ramirez A, Magin T, Vidal M. 2000. Loss- and gain-of-function mutations show a polycomb group function for Ring1A in mice. Development 127: 5093–5100. [DOI] [PubMed] [Google Scholar]
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, et al. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 7: 663–676. 10.1016/j.devcel.2004.10.005 [DOI] [PubMed] [Google Scholar]
- Eckert RL, Adhikary G, Rorke EA, Chew YC, Balasubramanian S. 2011. Polycomb group proteins are key regulators of keratinocyte function. J Invest Dermatol 131: 295–301. 10.1038/jid.2010.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, Maile TM, Busby J, Henry C, Kelly TK, et al. 2016. An alternative approach to ChIP-seq normalization enables detection of genome-wide changes in histone H3 lysine 27 trimethylation upon EZH2 inhibition. PLoS One 11: e0166438. 10.1371/journal.pone.0166438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M, Endo TA, Endoh T, Isono K, Sharif J, Ohara O, Toyoda T, Ito T, Eskeland R, Bickmore WA, et al. 2012. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet 8: e1002774. 10.1371/journal.pgen.1002774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evseenko D, Zhu Y, Schenke-Layland K, Kuo J, Latour B, Ge S, Scholes J, Dravid G, Li X, MacLellan WR, et al. 2010. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci 107: 13742–13747. 10.1073/pnas.1002077107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. 2012. Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. 10.1038/nprot.2012.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti R, Bhutkar A, McNamara MC, Lees JA. 2016. BMI1 induces an invasive signature in melanoma that promotes metastasis and chemoresistance. Genes Dev 30: 18–33. 10.1101/gad.267757.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessing MY, Mardaryev AN, Gdula MR, Sharov AA, Sharova TY, Rapisarda V, Gordon KB, Smorodchenko AD, Poterlowicz K, Ferone G, et al. 2011. P63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J Cell Biol 194: 825–839. 10.1083/jcb.201101148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881. 10.1101/gad.1110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher ML, Adhikary G, Grun D, Kaetzel DM, Eckert RL. 2016. The Ezh2 polycomb group protein drives an aggressive phenotype in melanoma cancer stem cells and is a target of diet derived sulforaphane. Mol Carcinog 55: 2024–2036. 10.1002/mc.22448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangini A, Sjöberg M, Roman-Trufero M, Dharmalingam G, Haberle V, Bartke T, Lenhard B, Malumbres M, Vidal M, Dillon N. 2013. The aurora B kinase and the polycomb protein ring1B combine to regulate active promoters in quiescent lymphocytes. Mol Cell 51: 647–661. 10.1016/j.molcel.2013.08.022 [DOI] [PubMed] [Google Scholar]
- Fursova NA, Blackledge NP, Nakayama M, Ito S, Koseki Y, Farcas AM, King HW, Koseki H, Klose RJ. 2019. Synergy between variant PRC1 complexes defines Polycomb-mediated gene repression. Mol Cell 74: 1020–1036.e8. 10.1016/j.molcel.2019.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni O, Weinberger L, Mansour AA, Manor YS, Chomsky E, Ben-Yosef D, Kalma Y, Viukov S, Maza I, Zviran A, et al. 2013. Derivation of novel human ground state naive pluripotent stem cells. Nature 504: 282–286. 10.1038/nature12745 [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. 2012. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45: 344–356. 10.1016/j.molcel.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, Reinberg D. 2014. An AUTS2–Polycomb complex activates gene expression in the CNS. Nature 516: 349–354. 10.1038/nature13921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Fuchs E. 2018. Stretching the limits: from homeostasis to stem cell plasticity in wound healing and cancer. Nat Rev Genet 19: 311–325. 10.1038/nrg.2018.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res 22: 1760–1774. 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy E, Mucha M, Glancy E, Fitzpatrick DJ, Conway E, Neikes HK, Monger C, Van Mierlo G, Baltissen MP, Koseki Y, et al. 2019. PRC2.1 and PRC2.2 synergize to coordinate H3K27 trimethylation. Mol Cell 76: 437–452.e6. 10.1016/j.molcel.2019.08.012 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ikawa T, Masuda K, Endo TA, Endo M, Isono K, Koseki Y, Nakagawa R, Kometani K, Takano J, Agata Y, et al. 2016. Conversion of T cells to B cells by inactivation of polycomb-mediated epigenetic suppression of the B-lineage program. Genes Dev 30: 2475–2485. 10.1101/gad.290593.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Cavazza A, Banerjee KK, Xie H, O'Neill NK, Saenz-Vash V, Herbert Z, Madha S, Orkin SH, Zhai H, et al. 2019. Extensive recovery of embryonic enhancer and gene memory stored in hypomethylated enhancer DNA. Mol Cell 74: 542–554.e5. 10.1016/j.molcel.2019.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127. 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- King HW, Fursova NA, Blackledge NP, Klose RJ. 2018. Polycomb repressive complex 1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res 28: 1494–1507. 10.1101/gr.237180.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppens MA, Bounova G, Gargiulo G, Tanger E, Janssen H, Cornelissen-Steijger P, Blom M, Song JY, Wessels LF, van Lohuizen M. 2016. Deletion of polycomb repressive complex 2 from mouse intestine causes loss of stem cells. Gastroenterology 151: 684–697.e12. 10.1053/j.gastro.2016.06.020 [DOI] [PubMed] [Google Scholar]
- Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. 2004. P63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 18: 126–131. 10.1101/gad.1165104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242. 10.1371/journal.pgen.1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905. 10.1101/gad.1035902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. 2010. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev 24: 265–276. 10.1101/gad.544410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Schumacher A, Thorsteinsdottir U, van Lohuizen M, Magnuson T, Sauvageau G. 1999. Functional antagonism of the Polycomb-Group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev 13: 2691–2703. 10.1101/gad.13.20.2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loubiere V, Delest A, Thomas A, Bonev B, Schuettengruber B, Sati S, Martinez AM, Cavalli G. 2016. Coordinate redeployment of PRC1 proteins suppresses tumor formation during Drosophila development. Nat Genet 48: 1436–1442. 10.1038/ng.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, Busslinger M, Koseki H, Hu Y, Smyth GK, et al. 2010. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood 116: 731–739. 10.1182/blood-2009-12-260760 [DOI] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. 2012. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149: 590–604. 10.1016/j.cell.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS, Taft RJ, Faulkner GJ. 2010. A global view of genomic information—moving beyond the gene and the master regulator. Trends Genet 26: 21–28. 10.1016/j.tig.2009.11.002 [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. 1999. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398: 708–713. 10.1038/19531 [DOI] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM. 2003. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17: 1823–1828. 10.1101/gad.269603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu W, Starmer J, Fedoriw AM, Yee D, Magnuson T. 2014. Repression of the soma-specific transcriptome by Polycomb-repressive complex 2 promotes male germ cell development. Genes Dev 28: 2056–2069. 10.1101/gad.246124.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. 2020. FDA approves an inhibitor of a novel ‘epigenetic writer.’ Nat Rev Drug Discov 19: 156. [DOI] [PubMed] [Google Scholar]
- Nguyen MB, Cohen I, Kumar V, Xu Z, Bar C, Dauber-Decker KL, Tsai PC, Marangoni P, Klein OD, Hsu YC, et al. 2018. FGF signalling controls the specification of hair placode-derived SOX9 positive progenitors to Merkel cells. Nat Commun 9: 2333. 10.1038/s41467-018-04399-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MB, Valdes VJ, Cohen I, Pothula V, Zhao D, Zheng D, Ezhkova E. 2019. Dissection of Merkel cell formation in hairy and glabrous skin reveals a common requirement for FGFR2-mediated signalling. Exp Dermatol 28: 374–382. 10.1111/exd.13901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver G, Mailhos A, Wehr R, Copeland NG, Jenkins NA, Gruss P. 1995. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 121: 4045–4055. [DOI] [PubMed] [Google Scholar]
- Petiot A, Conti FJ, Grose R, Revest JM, Hodivala-Dilke KM, Dickson C. 2003. A crucial role for Fgfr2-IIIb signalling in epidermal development and hair follicle patterning. Development 130: 5493–5501. 10.1242/dev.00788 [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. 1977. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265: 421–424. 10.1038/265421a0 [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. 2017. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell 171: 34–57. 10.1016/j.cell.2017.08.002 [DOI] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. 2009. Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10: 697–708. 10.1038/nrm2763 [DOI] [PubMed] [Google Scholar]
- Simon J, Chiang A, Bender W. 1992. Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114: 493–505. [DOI] [PubMed] [Google Scholar]
- Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, van Lohuizen M. 2006. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet 38: 694–699. 10.1038/ng1792 [DOI] [PubMed] [Google Scholar]
- Vidal M, Starowicz K. 2017. Polycomb complexes PRC1 and their function in hematopoiesis. Exp Hematol 48: 12–31. 10.1016/j.exphem.2016.12.006 [DOI] [PubMed] [Google Scholar]
- von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK, Wang Z, Lachmann A, Dewell S, Ma'ayan A, et al. 2016. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat Neurosci 19: 1321–1330. 10.1038/nn.4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. 2004a. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431: 873–878. 10.1038/nature02985 [DOI] [PubMed] [Google Scholar]
- Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. 2004b. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell 14: 637–646. 10.1016/j.molcel.2004.05.009 [DOI] [PubMed] [Google Scholar]
- Weinberger L, Ayyash M, Novershtern N, Hanna JH. 2016. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat Rev Mol Cell Biol 17: 155–169. 10.1038/nrm.2015.28 [DOI] [PubMed] [Google Scholar]
- Yakushiji-Kaminatsui N, Kondo T, Endo TA, Koseki Y, Kondo K, Ohara O, Vidal M, Koseki H. 2016. RING1 proteins contribute to early proximal-distal specification of the forelimb bud by restricting Meis2 expression. Development 143: 276–285. 10.1242/dev.127506 [DOI] [PubMed] [Google Scholar]
- Yang J, Meyer M, Müller AK, Böhm F, Grose R, Dauwalder T, Verrey F, Kopf M, Partanen J, Bloch W, et al. 2010. Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J Cell Biol 188: 935–952. 10.1083/jcb.200910126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L. 2011. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res 39: e35. 10.1093/nar/gkq1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Zheng D. 2018. SMARTcleaner: identify and clean off-target signals in SMART ChIP-seq analysis. BMC Bioinformatics 19: 544. 10.1186/s12859-018-2577-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. 2011. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12: 7–18. 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported here is GSE158324.