

Abstract
This work describes a quantitative high-throughput analytical method for the simultaneous measurement of small aliphatic nitrogenous biomarkers, i.e., 1,6-hexamethylenediamine (HDA), isophoronediamine (IPDA), ß-methylamino-L-alanine (BMAA), and trimethylamine N-oxide (TMAO), in human urine. Urinary aliphatic diamines, HDA and IPDA, are potential biomarkers of environmental exposure to their corresponding diisocyanates. Urinary BMAA forms as a result of human exposure to blue-green algae contaminated food. And, TMAO is excreted in urine due to the consumption of carnitine- and choline-rich diets. These urinary biomarkers represent classes of small aliphatic nitrogen-containing compounds (N-compounds) that have a high aqueous solubility, low logP, and/or high basic pKa. Because of the highly polar characteristics, analysis of these compounds in complex sample matrices is often challenging. We report on the development of ion-pairing chemistry based ultra-performance liquid chromatography–electrospray ionization–tandem mass spectrometry (UPLC–ESI–MS/MS) method for the simultaneous measurement of these biomarkers in human urine. Chromatographic separation was optimized using heptafluorobutyric acid- (HFBA-) based mobile phase and a reversed-phase C18 column. All four analytes were baseline separated within 2.6 minutes with an overall run time of five minutes per sample injection. Sample preparation involved four hours of acid hydrolysis followed by automated solid phase extraction (SPE) performed using strong cation exchange sorbent bed with 7 N ammonia solution in methanol as eluent. Limits of detection ranged from 0.05 ng/mL to 1.60 ng/mL. The inter-day and intra-day accuracy were within 10%, and reproducibility within 15%. The method is accurate, fast, and well-suited for biomonitoring studies within targeted groups, as well as larger population-based studies such as the U. S. National Health and Nutrition Examination Survey (NHANES).
Keywords: Diisocyanate exposure, urinary aliphatic diamine, UPLC, mass spectrometry, hexamethylenediamine, isophoronediamine, trimethylamine N-oxide, ß-methylamino-L-alanine
Graphical Abstract
INTRODUCTION
Aliphatic diamines and other nitrogen-containing compounds (N-compounds) in human urine form as a result of exposure to a broad range of sources and could serve as biomarkers of toxicant exposure. Urinary aliphatic diamines can be formed from exposure to their corresponding diisocyanates, which are used in polyurethane-based paints and coatings[1, 2]. These aliphatic diisocyanates may enter the environment through the discharge of industrial waste, application of paint spray, and degradation of consumer products; exposure may subsequently occur through dermal contact, ingestion, and inhalation. The most common aliphatic diisocyanates found in paints and coatings are hexamethylenediisocyanate (HDI) and isophoronediisocyanate (IPDI). Exposed human subjects metabolize these diisocyanates to hexamethylenediamine (HDA) and isophoronediamine (IPDA), respectively. Health effects of diisocyanate exposure include asthma, dermatitis, gastrointestinal irritation, chemical bronchitis, and pneumonitis[3].
Two other health-relevant biomarkers can be analyzed together with HDA and IPDA: ß-methylamino-L-alanine (BMAA) and trimethylamine N-oxide (TMAO). Exposure to BMAA, a non-proteinogenic amino acid toxin produced by cyanobacteria, occurs through consumption of contaminated seafood and water and is associated with increased risk of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Parkinson’s disease, and Alzheimer’s disease[4–6]. TMAO, a toxicant formed from metabolism of phosphatidylcholine- and L-carnitine-rich foods (e.g., dairy, red meat, fish, and certain dietary supplements), is associated with increased risk of cardiovascular disease[7–9]. TMAO affects cholesterol and sterol metabolism and is mechanistically involved in the development of atherosclerosis and cardiovascular disease[8].
Several mass spectrometry-based analytical methods have been developed previously for the measurement of these compounds individually, or simultaneously with a similar class of compounds in urine. HDA and IPDA have been widely analyzed as their perfluoroanhydride derivatives using GC-MS[10–14] and in a few cases using LC-MS[12, 15, 16]. Samples were acid- or base-hydrolyzed, extracted in a non-aqueous solvent (i.e., liquid-liquid extraction), and reacted with a derivatizing reagent prior to lengthy instrumental analysis (~30 minutes). Similarly, several GC-MS and LC-MS methods have been reported for BMAA measurements in neurological tissues, cyanobacterial samples, seafood, and staple foods, and in some cases in human and primate urine[4, 5, 17–23]. However, the chemical specificity has been a concern due to the lack of standard analytical technique for BMAA measurement as well as co-eluting isobaric compounds for GC- and LC-MS method [4, 19]. TMAO has been historically measured using GC-MS [24] and very recently using LC-MS [25–28]. However, the methods reported are based on protein precipitation and direct “dilute-and-shoot,” which lack proper sample clean-up to avoid potential chemical interferences. We developed a simple, fast, sensitive, rugged, and high-throughput ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) method to simultaneously quantify different classes of small aliphatic N-compounds in human urine using ion-pairing chemistry. The analytical challenges regarding the simultaneous measurement of these four analytes using liquid chromatography include their high aqueous solubility, low logP, and/or high basic pKa values. This paper reports the first analytical method that can simultaneously measure all four targeted urinary biomarkers of interest.
EXPERIMENTAL
Materials.
HPLC-grade heptafluorobutyric acid (HFBA), LCMS Optima™-grade methanol, AcroSeal® 7N ammonia in methanol, and synthetic urine were purchased from Fisher Scientific (Suwanee, GA). Reagents 1N sodium hydroxide (NaOH) and 6N hydrochloric acid (HCl) were purchased from Sigma-Aldrich (St. Louis, MO). Trifluoroacetic acid (TFA), pentafluoropropionic anhydride (PFPA), and analytical standard-grade HDA, IPDA, BMAA and TMAO were purchased from Sigma-Aldrich (St. Louis, MO). IPDA-[13C, 15N2], BMAA-[13C, 15N2], and TMAO-[13C3] were purchased from IsoSciences (Kings of Prussia, PA), and HDA-[2H12] from Toronto Research Chemicals (Ontario, Canada). Human urine pool was collected anonymously following a protocol approved by the CDC Institutional Review Board.
Calibration Solutions.
Individual master stocks of neat standards and internal standards (1 μg/mL) were prepared in methanol/water (50:50 v/v). Master stocks were diluted in water to make a set of working stocks of seven different concentrations. All calibration solutions wereprepared by diluting the working stocks by a factor of 10 in 0.1% HFBA (v/v) in water. The concentration range of calibration solutions is shown in Table 6. Internal standard concentrations ranged from 1 – 18 ng/mL and were assumed constant for the quantitation. All stock solutions were stored at −70 °C prior to use.
Table 6.
Limits of detection (LODs) in synthetic urine samples and range in the non-urine aqueous matrix. LODs were calculated at the concentration corresponding to 3S0, where S0 represents standard deviation at zero concentration obtained by extrapolation of standard deviation versus concentration curve (n=5)
| Analyte | Method LOD (ng/mL) | Range (ng/mL) |
|---|---|---|
| HDA | 0.15 | 0.032 – 32.0 |
| IPDA | 0.05 | 0.016 – 16.0 |
| BMAA | 0.15 | 0.063 – 63.0 |
| TMAO† | 1.60 | 1.600 – 1600 |
TMAO instrumental parameters were detuned to avoid detector saturation.
Sample Preparation.
The sample preparation workflow involved acid hydrolysis followed by automated solid phase extraction (SPE). For acid hydrolysis, 1000 μL sample was prepared by mixing 250 μL of urine followed by 100 μL of 6 N HCl (final concentration of acid is 0.6 N), 50 μL of the internal standard mixture, and 600 μL of water. The sample solution was placed in a heating block (VWR, Radnor, PA) set at 80 °C for 4 hours. The solution was allowed to cool to room temperature and was subsequently adjusted to pH ~1.0 using 500 μL of 1 N NaOH prior to performing SPE. Strata XC (30mg, 3mL) mixed-mode strong cation 96-well plates from Phenomenex (Torrance, CA) were used for sample clean-up. The SPE plates were conditioned using 1 mL of methanol followed by equilibration using 1 mL of HPLC grade water. The hydrolyzed samples were then loaded and washed with 1 mL of 0.1 N HCl acid followed by 2 mL of methanol. Finally, analytes were eluted twice with 500 μL of 7N NH3 in methanol solution (i.e., SPE elution solvent). The SPE elution solvent bottle was chilled at 4 °C using Thermal Lab Beads™ and a cooling tray (TecaLAB™, Chicago, IL). SPE was performed using a Biotage® Extrahera™ sample preparation system (Charlotte, NC). Following SPE, eluents were evaporated to dryness in an SPE Dry evaporation system from Biotage® (Charlotte, NC) under nitrogen gas at 60 °C. Prior to UPLC-MS/MS analysis, dried samples were reconstituted in 1 mL of water/methanol (95:5 v/v) with 0.1% of HFBA using an automated vortex mixer for 10 minutes (VWR, Radnor, PA).
UPLC-ESI-MS/MS Analysis.
The analytical run was performed using an Acquity I-Class UPLC system (Waters Corporation, Milford, MA) equipped with 2.1 × 100 mm, 2 μm ACE Excel2 SuperC18 Column (Mac-Mod Analytical, Chadds Ford, PA). The UPLC system was coupled to a 5500 triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source (Sciex, Framingham, MA). Chemical separation was performed using the solvent gradient of 0.1% aqueous HFBA (v/v, mobile phase A) and 0.1% HFBA in methanol (v/v, mobile phase B) as shown in Table 1. Column and sample manager temperatures were set to 50 °C and 10 °C, respectively. Injection volume was 2 μL using the full loop injection mode. The mass spectrometer was operated in positive ion ESI scheduled multiple reaction monitoring (sMRM) mode with a target scan time of 0.3 seconds for 30 seconds MRM detection window. Optimized ion source parameters included the following: ESI voltage, 2.5 kV; CAD gas, 7 psi; curtain gas flow, 30 psi; nebulizing gas (GS1) flow, 45 psi; heating gas (GS2) flow, 55 psi; and heater temperature, 650 °C. The chemical structures and mass-to-charge ratios of the precursor ions monitored are shown in Figure 1. Other compound-dependent parameters are shown in Table 2.
Table 1.
Gradient elution table for chromatographic separation. Mobile phases A and B are 0.1% (v/v) HFBA (aq.) and 0.1% HFBA (v/v) in methanol, respectively.
| Time (min) | Flow Rate (mL/min) | % Mobile Phase B (by volume) |
|---|---|---|
| Initial | 0.2 | 5 |
| 0.75 | 0.5 | 5 |
| 1.75 | 0.5 | 50 |
| 3.25 | 0.3 | 95 |
| 3.5 | 0.3 | 5 |
| 5.0 | 0.2 | 5 |
Figure 1.
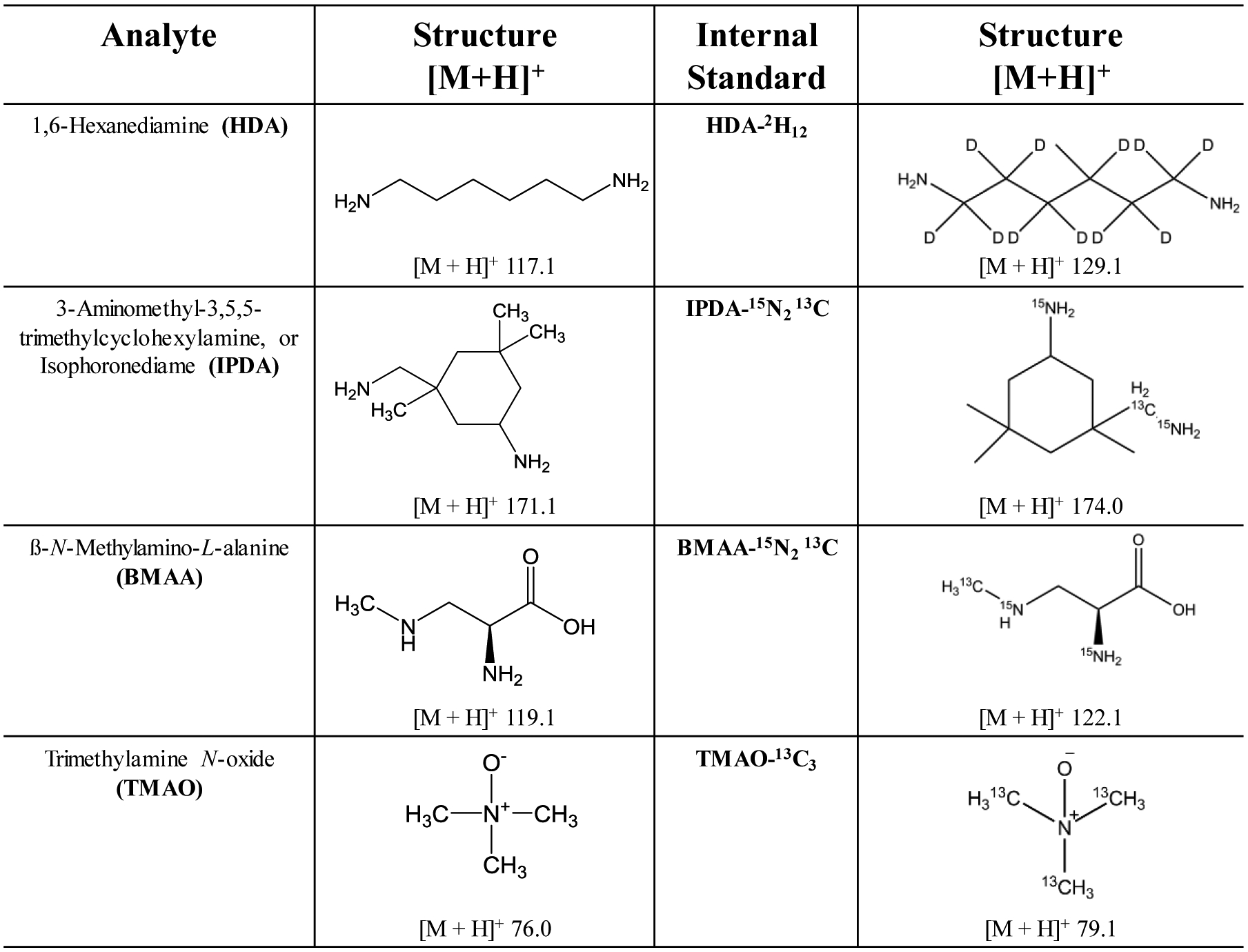
Chemical structures of compounds of interest and corresponding stable isotope labelled internal standards with their nominal proton adduct mass.
Table 2.
Compound specific mass spectrometry parameters.* The tolerance for the response ratio between quantitation and confirmation was ±25%.
| Analyte | Ion-transition (m/z) | DP | EP | CE | CXP | |
|---|---|---|---|---|---|---|
| Quantitation | Confirmation | |||||
| HDA | 117.1→100.1 | - | 13 | |||
| - | 117.1→55 | 40 | 5 | 25 | 10 | |
| HDA-2H12 | 129.1→112 | 13 | ||||
| IPDA | 171.1→154.0 | - | 18 | |||
| - | 171.1→95.0 | 50 | 10 | 30 | 9 | |
| IPDA-15N213C | 174.0→156.0 | 18 | ||||
| BMAA | 119.1→102.1 | - | 13 | |||
| - | 119.1→88.1 | 40 | 4 | 15 | 6 | |
| BMAA-15N213C | 122.1→104.1 | 13 | ||||
| TMAO | 76.0→43.0 | - | 25 | |||
| - | 76.0→60.0 | 50 | 8 | 25 | 8 | |
| TMAO-13C3 | 79.1→62.0 | 25 | ||||
DP = Declustering Potential; EP = Entrance Potential; CE = Collision Energy; CXP= Cell Exit Potential
TMAO data was detuned to avoid detector saturation. Instrument detuned using non-optimal but specific ion-transition and collision energy for TMAO
Data Analysis.
All LC-MS/MS data were generated in Analyst 1.6.2 (Sciex, Framingham, MA) and processed in MultiQuant 3.0.2 (Sciex, Framingham, MA).
RESULTS AND DISCUSSION
Hydrolysis.
Diisocyanate metabolites and BMAA are mostly covalently bonded with macromolecules, such as albumin and hemoglobin in biospecimens[4, 6, 11, 13, 14]. Studies have shown that conjugated diamines can be hydrolyzed using acid or base [11, 13, 14]. Acid hydrolysis is preferentially used over base hydrolysis when total concentration of urinary diamines is required. Acid hydrolyzes conjugated forms into free diamines, as well as converts mono- and di-acetylated forms into free diamines[14]. BMAA, a lysine analog, is typically hydrolyzed using hydrochloric acid. However, no systematic study on BMAA hydrolysis has been reported in the literature[4]. During the development of this assay, BMAA-exposed urine samples were not available, and hence it was not feasible to generate a temporal hydrolysis profile to fill this research gap. In addition, the hydrolysis of urinary TMAO has yet to be reported. Therefore, in the case of TMAO, the effect of acid hydrolysis on the calculated concentration of free TMAO is investigated here. This work was performed using anonymously-collected urine pool samples laden with endogenous TMAO. A set of samples was hydrolyzed in acid, and the other samples were processed without hydrolysis. The percentage differences between the calculated concentrations of hydrolyzed and non-hydrolyzed samples were within 3%, which indicates that there is no conjugated TMAO in urine. Therefore, hydrolysis is may not be an essential step for urinary TMAO analysis. However, acid hydrolysis is still a key step for the analysis of total diamines and BMAA concentrations in human urine as evident from our aromatic diamine study[29], and other past studies[6, 13, 14]. Therefore, based on our recent findings on the hydrolysis of urinary diamines[29], this work was performed in 0.6 N HCl at 80 °C for four hours unless stated otherwise.
In addition, ruggedness of the method associated with different hydrolysis time was tested. Urine samples were spiked with a known concentration of analyte mix and hydrolyzed separately for 3, 4, and 6 hours. Regardless of the hydrolysis time, the calculated concentration of all analytes was within 6% thereby providing the sufficient stability of spiked analytes for the 4 hour hydrolysis time chosen for the method. The ruggedness testing of the hydrolysis of conjugated compounds was not feasible because of the unavailability of the exposed urine specimens and off-the-shelf conjugated products.
Solid Phase Extraction.
Following acid hydrolysis, sample pH was adjusted to ~1. Based on pKa values[30] (Table 3), all analytes have a net positive charge at pH = 1 and therefore can be retained by a strong cation exchange SPE sorbent. However, elution chemistry was challenging because of the broad range of pKa values for the analytes. Shown in Table 4 are percent spiked recoveries at different concentrations of ammonia in methanol. Increased concentrations of ammonia in methanol had a notable effect on the spiked recovery for high pKa analytes, HDA and IPDA (i.e., pKa ~10.5), compared to low pKa TMAO (i.e., pKa ~4.66). These results suggest that a higher concentration ammonia solution is required to neutralize and, hence, elute aliphatic diamines relative to TMAO and BMAA. Both 5 N and 7 N ammonia in methanol resulted in comparable percent recoveries, while recoveries were lower at the 1 N and 3 N ammonia concentrations, with the exception of BMAA. For BMAA, the average percent spiked recovery was 81 ± 14% when using a 3 N solution and decreased to 65 ± 16% using a higher ammonia concentration of 7 N. This effect warrants further experimentation to fully comprehend the influence of eluent pH on strong cation exchange SPE for BMAA and similar amino acids. Nevertheless, the highest concentration of ammonia, 7 N, was chosen for this method because ammonia is volatile and, regardless of the use of chilling trays, there is a potential for evaporative loss during SPE. When the concentration of ammonia in methanol is 3 N or less, the spiked recovery for HDA can be reduced to 33% or lower.
Table 3.
Physicochemical properties of analytes of interest.30
| Analytes | Strongest basic pKa | Strongest acidic pKa | logP | logS† (at pH =1.7) |
|---|---|---|---|---|
| HDA | 10.51 | - | 0.04 | 2.16 |
| IPDA | 10.54 | - | 0.96 | 1.05 |
| BMAA | 9.86 | 1.96 | −3.77 | 2.70 |
| TMAO | 4.66 | - | −0.93 | 2.58 |
S = Aqueous solubility measured in mol/L. LogS value for pH =1.7 is reported because it is the closest pH reported in reference 30 that is equivalent to the pH of the mobile phase (pH = 2).
Table 4.
Percent spiked recovery for analytes at different concentrations of ammonia in methanol eluent
| Analytes | Percentage spiked recovery at various concentrations of NH3 in methanol | |||
|---|---|---|---|---|
| 1 N | 3 N | 5 N | 7 N | |
| HDA | NA | 33 ± 3% | 82 ± 6% | 89 ± 2% |
| IPDA | 65 ± 5% | 88 ± 5% | 91 ± 5% | 91 ± 4% |
| BMAA | 60 ± 5% | 81 ± 14% | 78 ± 22% | 65 ± 18% |
| TMAO | 92 ± 4% | 95 ± 8% | 96 ± 7% | 97 ± 6% |
UPLC-ESI-MS/MS.
The effect of various ion-pairing reagents, i.e., 0.1% (v/v) TFA, PFPA, and HFBA, on the chromatographic separation and mass spectrometric detection, was evaluated. All three ion-pairing reagents assessed in this study substantially improved analytical separation as well as mass spectrometric sensitivity compared to the traditional formic acid additive. With 0.1% formic acid additive in both mobile phases, all four analytes eluted close to the void volume with peak resolutions < 1. Substituting it with volatile ion-pairing reagents, chromatographic peak resolutions were improved to ≥ 4 using either HFBA or PFPA, compared to a resolution of 1.9 using TFA. In addition, mass spectrometric signal intensities increased in the following order: HFBA>PFPA>TFA. Considering the combined effect of optimal chromatographic peak resolution and enhanced signal intensity, HFBA was the most suitable ion-pairing reagent for the method. Figure 2 shows the extracted-ion chromatograms for the four analytes generated using 0.1% HFBA in both mobile phases, A and B. As opposed to higher aqueous solubility in their native state (Table 3, logS values), the analytes eluted according to their corresponding logP values when eluted using an ion-pairing reagent on a reversed-phase column. BMAA with the lowest logP eluted first, while IPDA with the highest logP eluted last.
Figure 2.
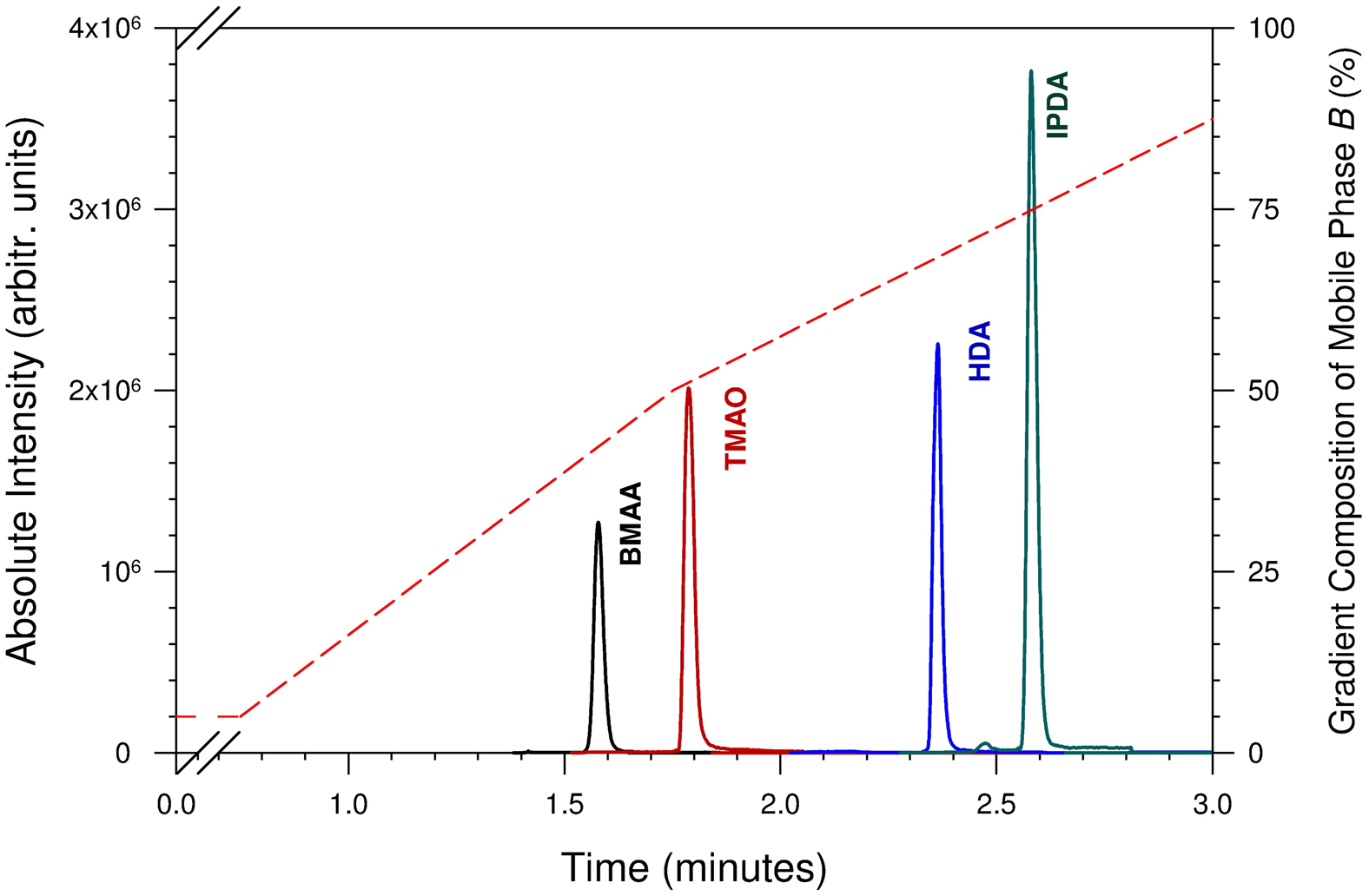
Representative extracted-ion chromatograms of BMAA, TMAO, HDA, and IPDA prepared in 0.1% (v/v) HFBA in water at arbitrary concentrations (non-equimolar) using HFBA and a reversed-phase column. The dotted line represents percentage composition of mobile phase B (right y-axis) against retention time (x-axis). The gradient profile does not illustrate the change in flow rate shown in Table 1.
Quantitative Measurement and Analytical Figures of Merit.
Quantitative studies were performed using a stable isotope dilution method. The calibration curves spanned over three orders of magnitude in concentration (Table 6) with coefficients of determination exceeding 0.99 for all curves fitted using linear regression on 1/x weighted data.
A matrix validation experiment on the use of non-urine-based calibrators (i.e., in mobile phase A) for quantitation was performed. The slopes of the calibration curves obtained using 0.1% HFBA (aq.), synthetic urine, and pooled human urine as matrices for calibrators were compared. The pooled urine and synthetic urine samples were processed through the entire sample preparation process, including hydrolysis and SPE. However, samples prepared in 0.1% HFBA (aq.) were directly analyzed using the dilute-and-shoot approach. Experiments were repeated three times for each matrix. Table 5 shows an overview of slopes and absolute percent differences of the slopes in various matrices. The percent errors in the slopes from the urine or synthetic urine matrices relative to the 0.1% HFBA (aq.) matrix were within 5%. The calibration slope for TMAO created using the urine pool matrix was not reported because TMAO-free human urine pool was not available.
Table 5.
Comparison of calibration curve slopes for different types of matrices. The linear calibration range used for all matrices was identical (please refer Table 6 for the range)
| Analyte | 0.1% HFBA (aq.) Matrix | Urine Pool Matrix | Synthetic Urine Matrix | ||
|---|---|---|---|---|---|
| Slope | Slope | Difference (%)* | Slope | Difference (%)* | |
| HDA | 0.244 | 0.238 | 2.49 | 0.238 | 2.49 |
| IPDA | 1.818 | 1.877 | 3.19 | 1.741 | 4.33 |
| BMAA | 0.302 | 0.292 | 3.37 | 0.303 | 0.33 |
| TMAO | 0.060 | NR† | NR† | 0.059 | 1.68 |
Absolute percent differences relative to 0.1% HFBA (aq.) matrix
NR = Not reported because of high endogenous TMAO content in a urine pool
Limits of detection (LODs) and limits of quantitation (LOQs) were calculated based on 3S0 and 10S0, respectively, where S0 is the standard deviation at zero concentration from the standard deviation versus concentration plot.[31] The calculated LODs in synthetic urine matrix are 0.15 ng/mL for HDA, 0.05 ng/mL for IPDA, 0.15 ng/mL for BMAA, and 1.6 ng/mL for TMAO (Table 6). The relatively high LOD calculated for TMAO results from using detuned instrumental parameters (i.e., less sensitive but distinct ion-transition and collision energy), which was necessary to shift the quantitation range to a suitable concentration based on the urine pool characterization, which was above 100 ng/mL. The concentrations of urinary HDA in an occupationally exposed urine were reported between 0.36 ng and 15 ng/mL [2, 13]. No clear reference value for the urinary IPDA concentration in exposed urine sample was available. However, the reported LODs were between 0.5 and 2.5 ng/mL [2, 11, 16]. The reference value for the urinary BMAA in exposed subjects, as well as method LODs related to the human urine specimens, has not been reported yet. Literature values reported were mostly for tissue, the staple food, and other environmental matrices [19–21]. The urinary TMAO concentration has been reported between μg/mL to mg/mL range with the method LODs of 600 ng/mL [8, 32]. These literature values are higher than the LODs calculated using our method. Hence, the method reported has sufficient sensitivity to provide an effective measurement of the analytes of interest.
Inter-day accuracy and precision of the method were evaluated by analyzing synthetic urine spiked at three different concentrations for 19 nonconsecutive days over several months. Similarly, intra-day accuracy and precision were evaluated from four replicates of synthetic urine spiked at three different concentrations. The intra- and inter-day absolute analytical accuracy ranged from 0.1 – 10.0% and 0.5 – 9.4%, respectively, and RSDs were 0.7 – 7.2% and 3.5 – 14.6%, respectively (Table 7). The carryover effect studied using the highest calibrator. The carryover following a highest calibrator was below LOD on the next injection of a blank solvent and in instances where samples follow a level higher than the highest standard, the following sample was flagged for repeat analysis with an adequate dilution.
Table 7.
Intra-day and inter-day accuracy and precision of spiked samples in synthetic urine at three different concentrations.
| Analyte | Actual Concentration (ng/mL) | Intra-day (n=4) | Inter-day (n=19) | ||||
|---|---|---|---|---|---|---|---|
| Calculated Concentration (ng/mL) | Accuracy (%) | RSD (%) | Calculated Concentration (ng/mL) | Accuracy (%) | RSD (%) | ||
| HDA | 0.316 | 0.294 | −7.0 | 1.6 | 0.303 | −4.1 | 14.6 |
| 1.000 | 0.900 | −10.0 | 3.9 | 0.906 | −9.4 | 7.8 | |
| 3.162 | 3.054 | −3.4 | 5.3 | 3.002 | −5.1 | 7.3 | |
| IPDA | 0.158 | 0.160 | 1.3 | 3.4 | 0.156 | −1.3 | 14.9 |
| 0.500 | 0.522 | 4.4 | 5.1 | 0.525 | 5.0 | 7.6 | |
| 1.581 | 1.725 | 9.1 | 7.2 | 1.716 | 8.5 | 6.0 | |
| BMAA | 0.632 | 0.601 | −4.9 | 4.9 | 0.599 | −5.2 | 5.3 |
| 2.000 | 1.976 | −1.2 | 4.0 | 1.973 | −1.4 | 3.8 | |
| 6.325 | 6.142 | −2.9 | 0.7 | 6.127 | −3.1 | 3.5 | |
| TMAO | 15.81 | 15.61 | −1.3 | 5.8 | 15.67 | −0.9 | 11.0 |
| 50.00 | 50.05 | 0.1 | 3.8 | 50.26 | 0.5 | 4.6 | |
| 158.1 | 159.1 | 0.6 | 2.1 | 159.0 | 0.6 | 4.7 | |
Stability.
Temporal stability at three temperatures and freeze-thaw stability for seven cycles were examined to determine optimal handling and storage conditions. For short-term temporal stability, synthetic urine was spiked, mixed, aliquoted, and then stored for time intervals of 0, 4, 8, and 16 hr, and 1 through 7 days at either room temperature, 4 °C, or −20 °C. After short-term storage at specified conditions, samples were transferred to a −70 °C storage freezer and analyzed later on the same run. At 4 °C and −20 °C, percentage loss for all analytes was within 10%. At room temperature, the percentage loss for TMAO was within 10%, while percentage losses for HDA, IPDA, and BMAA were 15, 20, and 30%, respectively. The percentage losses over the short term storage conditions were calculated between zero hour and 7th day samples. Furthermore, long-term stability at −70 °C was evaluated in a spiked synthetic urine. Experiments were performed at 1, 3, 6 and 9 months following sample storage at −70 °C. Concentrations were within 10% of those measured on the first month.
Freeze-thaw stability was assessed for spiked urine samples stored at −70 °C for seven freeze-thaw cycles. The average concentration of all analytes was within 10% between the 1st and the 7th freeze-thaw cycles.
Application to Human Specimens.
The method accuracy was tested in spiked human urine. Anonymous urine specimens were pooled and tested for baseline concentration prior to spiking neat standards. Except for TMAO, no other analytes were detected in the pooled urine. It is not uncommon to have non-detectable urinary HDA, IPDA, and BMAA because the urine pools were collected a non-occupational exposure environment. This aligns well with the fact that the urinary concentrations reported elsewhere are mainly from the occupational exposure of corresponding diisocyanates (for HDA & IPDA) and from dietary intake of BMAA contained staples. Following screening urine pool, the specimens were spiked with various known concentrations of standard analytes, and blind coded. For quantitative analysis, 25 spiked blind-coded urine samples were bracketed with a set of seven calibration solutions and both low- and high-concentration quality control samples. The calculated concentrations for human urine samples spiked with HDA, IPDA, and BMAA were within 13.5, 7.0, and 15.0% of the spiked amount (Table 8). The accuracy of TMAO-spiked urine was not assessed because of the existence of a high ng/mL concentration of endogenous TMAO in the pooled urine. To mitigate the quantitation issue associated with the high endogenous content of TMAO, urine specimens were required to be diluted by at least a factor of 10 for the accurate measurement using the method reported here.
Table 8.
Method accuracy for spiked human urine at different concentrations.
| Sample Number | HDA | IPDA | BMAA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Spiked (ng/mL) | Calculated (ng/mL) | Accuracy (%) | Spiked (ng/mL) | Calculated (ng/mL) | Accuracy (%) | Spiked (ng/mL) | Calculated (ng/mL) | Accuracy (%) | |
| 01 | 0.62 | 0.60 | −3.2 | 0.63 | 0.59 | −6.3 | 1.23 | 1.36 | 10.6 |
| 02 | 0.87 | 0.82 | −5.7 | 0.88 | 0.82 | −6.8 | 1.72 | 1.85 | 7.6 |
| 03 | 1.11 | 1.08 | −2.7 | 1.13 | 1.11 | −1.8 | 2.21 | 2.05 | −7.2 |
| 04 | 1.36 | 1.22 | −10.3 | 1.38 | 1.32 | −4.3 | 2.70 | 2.86 | 5.9 |
| 05 | 1.61 | 1.47 | −8.7 | 1.63 | 1.62 | −0.6 | 3.20 | 3.06 | −4.4 |
| 06 | 1.86 | 1.63 | −12.4 | 1.88 | 1.90 | 1.1 | 3.69 | 4.21 | 14.1 |
| 07 | 2.11 | 1.91 | −9.5 | 2.13 | 2.17 | 1.9 | 4.18 | 3.81 | −8.9 |
| 08 | 2.35 | 2.17 | −7.7 | 2.38 | 2.44 | 2.5 | 4.67 | 3.98 | −14.8 |
| 09 | 2.60 | 2.25 | −13.5 | 2.63 | 2.59 | −1.5 | 5.16 | 5.11 | −1.0 |
| 10 | 2.85 | 2.79 | −2.1 | 2.88 | 2.96 | 2.8 | 5.65 | 5.83 | 3.2 |
| 11 | 3.10 | 2.93 | −5.5 | 3.13 | 3.22 | 2.9 | 6.14 | 6.09 | −0.8 |
| 12 | 3.34 | 3.08 | −7.8 | 3.38 | 3.42 | 1.2 | 6.64 | 7.07 | 6.5 |
| 13 | 3.59 | 3.28 | −8.6 | 3.63 | 3.70 | 1.9 | 7.13 | 6.82 | −4.3 |
| 14 | 3.84 | 3.43 | −10.7 | 3.88 | 3.96 | 2.1 | 7.62 | 7.50 | −1.6 |
| 15 | 4.09 | 3.72 | −9.0 | 4.13 | 4.15 | 0.5 | 8.11 | 8.11 | 0.0 |
| 16 | 4.34 | 4.04 | −6.9 | 4.38 | 4.51 | 3.0 | 8.60 | 8.89 | 3.4 |
| 17 | 4.58 | 4.29 | −−6.3 | 4.63 | 4.69 | 1.3 | 9.09 | 10.2 | 12.2 |
| 18 | 4.83 | 4.45 | −7.9 | 4.88 | 5.03 | 3.1 | 9.59 | 9.61 | 0.2 |
| 19 | 5.08 | 4.96 | −2.4 | 5.13 | 5.21 | 1.6 | 10.1 | 10.7 | 5.9 |
| 20 | 5.33 | 5.27 | −1.1 | 5.38 | 5.63 | 4.6 | 10.6 | 11.8 | 11.3 |
| 21 | 5.57 | 5.37 | −3.6 | 5.63 | 5.64 | 0.2 | 11.1 | 11.6 | 4.5 |
| 22 | 5.82 | 5.58 | −4.1 | 5.88 | 5.87 | −0.2 | 11.6 | 12.3 | 6.0 |
| 23 | 6.07 | 5.79 | −4.6 | 6.13 | 6.35 | 3.6 | 12.0 | 12.3 | 2.5 |
| 24 | 6.32 | 6.15 | −2.7 | 6.38 | 6.55 | 2.7 | 12.5 | 13.0 | 4.0 |
| 25 | 6.57 | 6.21 | −5.5 | 6.63 | 6.86 | 3.5 | 13.0 | 13.5 | 3.8 |
Sample Throughput.
In our current method, all analytes elute within 2.6 minutes. However, the total run time was extended to five minutes to fully equilibrate the column for high-throughput sample analyses. Thus, the time required to analyze 96 samples is eight hours. In addition, with fast hydrolysis (4 hours) followed by automated SPE (1.5 hours) without any derivatization steps, the sample preparation per 96 samples is under 8 hours with an option to multiplex. At its current stage, this method can generate 384 analytical results in 16 hours including the entire sample preparation and analytical run time. The method is advantageous over the use of non-simultaneous methods reported in the literature for the analysis of low molecular weight polar N-compounds currently being analyzed in clinical and research laboratories.
CONCLUSIONS
The use of HFBA as an ion-pairing reagent for reversed-phase LC combined with mass spectrometric analysis provides low ng/mL sensitivity, linearity over three orders of magnitude, precision and accuracy within 15%, and baseline chromatographic separation for the quantitative analysis of four small (amu ≤ 170) aliphatic N-compound-based biomarkers of exposure in urine. In addition, because this method permits simultaneous measurement of HDA, IPDA, BMAA, and TMAO in urine within five minutes per sample injection, it is well suited for high-throughput analysis in which to examine environmental and dietary exposures associated with these biomarkers. As a result, this method can be implemented to assess human exposure to HDI, IPDI, BMAA, and carnitine/choline in population-based studies, such as the National Health and Nutritional Examination Survey (NHANES)[33] as well as other clinical studies that desire non-invasive urine sampling.
Footnotes
Publisher's Disclaimer: Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
- [1].Cocker J, Biological monitoring for isocyanates, Occup. Med, 57 (2007) 391–393. [DOI] [PubMed] [Google Scholar]
- [2].Skarping G, Dalene MD, Tinnerberg H, Biological monitoring of hexamethylene- and isophorone-diisocyanate by the determination of hexamethylene- and isophorone-diamine in hydrolysed urine using liquid chromatography and mass spectrometry, Analyst, 119 (1994) 2051–2055. [DOI] [PubMed] [Google Scholar]
- [3].Lockey JE, Redlich CA, Streicher R, Pfahles-Hutchens A, Hakkinen PJ, Ellison GL, Harber P, Utell M, Holland J, Comai A, White M, Isocyanates and human health: Multistakeholder information needs and research priorities, J. Occup. Environ. Med, 57 (2015) 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cohen SA, Analytical techniques for the detection of α-amino-β- methylaminopropionic acid, Analyst, 137 (2012) 1991–2005. [DOI] [PubMed] [Google Scholar]
- [5].Levine TD, Miller RG, Bradley WG, Moore DH, Saperstein DS, Flynn LE, Katz JS, Forshew DA, Metcalf JS, Banack SA, Cox PA, Phase I clinical trial of safety of L-serine for ALS patients, Amyotroph. La. Scl. Fr, 18 (2017) 107–111. [DOI] [PubMed] [Google Scholar]
- [6].Faassen EJ, Antoniou MG, Beekman-Lukassen W, Blahova L, Chernova E, Christophoridis C, Combes A, Edwards C, Fastner J, Harmsen J, Hiskia A, Ilag LL, Kaloudis T, Lopicic S, Lürling M, Mazur-Marzec H, Meriluoto J, Porojan C, Viner-Mozzini Y, Zguna N, A collaborative evaluation of LC-MS/MS based methods for BMAA analysis: Soluble bound BMAA found to be an important fraction, Mar. Drugs, 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WHW, Didonato JA, Lusis AJ, Hazen SL, Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease, Nature, 472 (2011) 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, Didonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ, Hazen SL, Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis, Nat. Med, 19 (2013) 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Senthong V, Wang Z, Fan Y, Wu Y, Hazen SL, Tang WHW, Trimethylamine N-oxide and mortality risk in patients with peripheral artery disease, J. Am. Heart Assoc, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rosenberg C, Savolainen H, Determination in urine of diisocyanate-derived amines from occupational exposure by gas chromatography - Mass fragmentography, Analyst, 111 (1986) 1069–1071. [DOI] [PubMed] [Google Scholar]
- [11].Budnik LT, Nowak D, Merget R, Lemiere C, Baur X, Elimination kinetics of diisocyanates after specific inhalative challenges in humans: Mass spectrometry analysis, as a basis for biomonitoring strategies, J. Occup. Med. Toxicol, 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tinnerberg H, Skarping G, Dalene M, Hagmar L, Test chamber exposure of humans to 1,6-hexamethylene diisocyanate and isophorone diisocyanate, Int. Arch. Occ. Env. Hea, 67 (1995) 367–374. [DOI] [PubMed] [Google Scholar]
- [13].Flack SL, Ball LM, Nylander-French LA, Occupational exposure to HDI: Progress and challenges in biomarker analysis, J. Chromatogr. B, 878 (2010) 2635–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gaines LGT, Fent KW, Flack SL, Thomasen JM, Ball LM, Richardson DB, Ding K, Whittaker SG, Nylander-French LA, Urine 1,6-hexamethylene diamine (HDA) levels among workers exposed to 1,6-hexamethylene diisocyanate (HDI), Ann. Occup. Hyg, 54 (2010) 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marand Å, Karlsson D, Dalene M, Skarping G, Determination of amines as pentafluoropropionic acid anhydride derivatives in biological samples using liquid chromatography and tandem mass spectrometry, Analyst, 129 (2004) 522–528. [DOI] [PubMed] [Google Scholar]
- [16].Dalene M, Skarping G, Tinnerberg H, Thermospray mass spectrometry of aliphatic diamines derivatized with trifluoroethyl chloroformate, with special reference to the biological monitoring of hexamethylenediisocyanate (HDI) and isophoronediisocyanate (IPDI), Chromatographia, 38 (1994) 776–780. [Google Scholar]
- [17].Duncan MW, Kopin IJ, Crowley JS, Jones SM, Markey SP, Quantification of the putative neurotoxin 2-amino-3-(methylamino)propanoic acid (BMAA) in cycadales: Analysis of the seeds of some members of the family Cycadaceae, J. Anal. Toxicol, 13 (1989). [DOI] [PubMed] [Google Scholar]
- [18].Oh CH, Brownson DM, Mabry TJ, Screening for non-protein amino acids in seeds of the Guam cycad, Cycas circinalis, by an improved GC-MS method, Planta Med, 61 (1995) 66–70. [DOI] [PubMed] [Google Scholar]
- [19].Combes A, El Abdellaoui S, Sarazin C, Vial J, Mejean A, Ploux O, Pichon V, Validation of the analytical procedure for the determination of the neurotoxin β-N-methylamino-l-alanine in complex environmental samples, Anal. Chim. Acta, 771 (2013) 42–49. [DOI] [PubMed] [Google Scholar]
- [20].Banack SA, Metcalf JS, Bradley WG, Cox PA, Detection of cyanobacterial neurotoxin β-N-methylamino-l-alanine within shellfish in the diet of an ALS patient in Florida, Toxicon, 90 (2014) 167–173. [DOI] [PubMed] [Google Scholar]
- [21].Combes A, El Abdellaoui S, Vial J, Lagrange E, Pichon V, Development of an analytical procedure for quantifying the underivatized neurotoxin β-N-methylamino-l-alanine in brain tissues, Anal. Bioanal. Chem, 406 (2014) 4627–4636. [DOI] [PubMed] [Google Scholar]
- [22].Glover WB, Baker TC, Murch SJ, Brown PN, Determination of β-N-methylamino-L-alanine, N-(2-aminoethyl)glycine, and 2,4-diaminobutyric acid in food products containing cyanobacteria by ultra-performance liquid chromatography and tandem mass spectrometry: Single-laboratory validation, J. AOAC Int, 98 (2015) 1559–1565. [DOI] [PubMed] [Google Scholar]
- [23].Duncan MW, Markey SP, Weick BG, Pearson PG, Ziffer H, Hu Y, Kopin IJ, 2-Amino-3-(methylamino)propanoic acid (BMAA) bioavailability in the primate, Neurobiol. Aging, 13 (1992) 333–337. [DOI] [PubMed] [Google Scholar]
- [24].daCosta KA, Vrbanac JJ, Zeisel SH, The measurement of dimethylamine, trimethylamine, and trimethylamine N-oxide using capillary gas chromatography-mass spectrometry, Anal. Biochem, 187 (1990) 234–239. [DOI] [PubMed] [Google Scholar]
- [25].Wang Z, Levison BS, Hazen JE, Donahue L, Li XM, Hazen SL, Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry, Anal. Biochem, 455 (2014) 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heaney LM, Jones DJL, Mbasu RJ, Ng LL, Suzuki T, High mass accuracy assay for trimethylamine N-oxide using stable-isotope dilution with liquid chromatography coupled to orthogonal acceleration time of flight mass spectrometry with multiple reaction monitoring, Anal. Bioanal. Chem, 408 (2016) 797–804. [DOI] [PubMed] [Google Scholar]
- [27].Grinberga S, Dambrova M, Latkovskis G, Strele I, Konrade I, Hartmane D, Sevostjanovs E, Liepinsh E, Pugovics O, Determination of trimethylamine-N-oxide in combination with l-carnitine and γ-butyrobetaine in human plasma by UPLC/MS/MS, Biomed. Chromatogr, 29 (2015) 1670–1674. [DOI] [PubMed] [Google Scholar]
- [28].Awwad HM, Geisel J, Obeid R, Determination of trimethylamine, trimethylamine N-oxide, and taurine in human plasma and urine by UHPLC–MS/MS technique, J. Chromatogr. B, 1038 (2016) 12–18. [DOI] [PubMed] [Google Scholar]
- [29].Bhandari D, Ruhl J, Murphy A, McGahee E, Chambers D, Blount BC, Isotope Dilution UPLC-APCI-MS/MS Method for the Quantitative Measurement of Aromatic Diamines in Human Urine: Biomarkers of Diisocyanate Exposure, Anal. Chem, 88 (2016) 10687–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. http://www.chemicalize.com.
- [31].Taylor J, Quality Assurance of Chemical Measurements, Lewis Publishers, New York, 1987. [Google Scholar]
- [32].Awwad HM, Geisel J, Obeid R, Determination of trimethylamine, trimethylamine N-oxide, and taurine in human plasma and urine by UHPLC–MS/MS technique, J Chromatogr B Analyt Technol Biomed Life Sci, 1038 (2016) 12–18. [DOI] [PubMed] [Google Scholar]
- [33]. https://www.cdc.gov/nchs/nhanes/.