

Abstract
Unlike other variants of transition-metal-catalyzed cross-coupling reactions, those based on organosilicon donors have not been used extensively in natural product synthesis. However, recent advances such as: 1) the development of mild reaction conditions, 2) the expansion of substrate scope, 3) the development of methods to stereoselectively and efficiently introduce the silicon-containing moiety, 4) the development of a large number of sequential processes, and 5) the advent of bifunctional bis(silyl) linchpin reagents, signify the coming of age of silicon-based cross-coupling reactions. The following case studies illustrate how silicon-based cross-coupling reactions play a strategic role in constructing carbon–carbon bonds in selected target molecules.
Keywords: cross-coupling, organopalladium, organosilicon reagents, silanols, tandem reactions
1. Introduction
Carbon–carbon bond-forming reactions are of central importance in organic chemistry. In the total synthesis of natural products, the efficiency of a synthetic plan often depends on a rapid and selective assembly of the carbon skeleton. Among the most powerful carbon–carbon bond-forming reactions, transition-metal-catalyzed cross-coupling reactions have, in recent decades, been extensively utilized in the total synthesis of a multitude of target molecules.[1] The most popular variants of these cross-coupling processes have achieved name reaction status, such as the Negishi, Suzuki–Miyaura, and Stille–Migita–Kosugi reactions. However, silicon-based cross-coupling reactions (despite a similarly prestigious appellation, the Hiyama Coupling)[2] has not received as much attention and is not as widely employed. Nevertheless, since the early days of their development in this context, the unique advantages of organosilane reagents, including their high stability, low toxicity, and ease of introduction into various substrates have been well recognized and have facilitated the development of silicon-based cross-coupling reactions. In addition, the long-standing problem of the incompatibility of many functional groups with the fluoride promoter has been solved by developments from our research group with the introduction of Brønsted base activation of organosilanols. This novel and general fluoride-free reaction protocol, complementary to the more conventional fluoride-promoted process, greatly increases the versatility of silicon-based cross-coupling reactions.[2b,c] This breakthrough becomes very important in total syntheses because of the ubiquitous use of silyl protecting groups.
Although hydrolytically sensitive, halosilanes were the first widely utilized organosilane cross-coupling reagents.[3] More stable variants, including triallylsilanes,[4] alkoxysilanes,[5] and [(2-hydroxymethyl)phenyl]dimethylsilanes,[6] were soon developed. Very significantly, dimethylsilanols, which can be employed under both fluoride-promoted[7] and fluoride-free conditions,[8] have been pioneered by our research group. In addition, “masked” silanol equivalents have been developed, such as alkoxydimethylsilanes,[9] benzyldimethylsilanes,[10] phenyldimethylsilanes,[11] 2-thienyldimethylsilanes,[10d,12] and 2-pyridyldimethylsilanes.[13] These “masking” groups are cleaved in the presence of fluoride and water to reveal the silanol, and thereby offer even higher stability and tolerance to harsh reaction conditions. Under fluoride-free conditions, the preferred reaction protocol is the stoichiometric conversion of silanols into their alkali metal salts.[14] The major advantage of preformed silanolates is that they are self activating and require no additional base to effect the coupling. Moreover, they possess additional advantages compared to their counterparts that are in situ generated such as enhanced reactivity, ease of handling (most arylsilanolates are free-flowing solids while most silanols are viscous liquids), and the stability for long-term storage.
A well-known feature of organosilicon chemistry is the wide variety of methods available for the introduction of different silyl groups.[15] These reactions have been parlayed with cross-coupling reactions to effect highly efficient constructions. The simplest example of this kind of transformation is the sequential intermolecular hydrosilylation/silicon-based cross-coupling developed independently by us[16] and others.[10c,e] The intramolecular versions of this reaction have also been developed to control the site and geometrical selectivity in the introduction of the organosilicon unit. For example, intramolecular syn-[5j, 17] and anti-hydrosilylation[18] of homopropargyloxyhydrosilanes and subsequent cross-coupling give highly substituted (E)- and (Z)-homoallylic alcohols, respectively. In addition, homopropargyloxysilanes also undergo intramolecular silylformylation/cross-coupling[19] or sequential silylcyanation/cross-coupling[20] to furnish β-disubstituted δ-hydroxy-(Z)-α,β-unsaturated aldehydes or nitriles with exclusive Z configuration. Other sequential processes include sequential ring-closing metathesis/cross-coupling that provides (Z)-homoallylic alcohols,[21] sequential silylcarbocyclization/cross-coupling that affords (Z)-alkylidene cyclopentanes,[10a] as well as sequential enyne coupling/allylic cyclization/cross-coupling that results in a 2-vinyl-4-(E)-benzylidenetetrahydropyran.[10b] In all the above processes, the cross-coupling step proceeds with fluoride-activation of the intermediate organosilane species. On the other hand, a recently reported tandem process that furnishes 2,3-disubstituted indoles combines a Larock indole synthesis with a fluoride-free cross-coupling reaction.[22] Finally, the complementarity of the fluoride-promoted and the fluoride-free modes of activation allows for the selective coupling of unsymmetrical 1,4-bis(silyl)diene reagents in which one terminus is substituted with a silanol whereas the other terminus is substituted with a “masked” silanol equivalent. Thus, when subjected sequentially to the fluoride-free cross-coupling and fluoride-promoted reaction conditions, these 1,4-bis(silyl)diene reagents act as a linchpin to unite two different cross-coupling partners to afford a large range of unsymmetrical dienes and polyenes.
The stability and versatility of organosilicon reagents provide a solid foundation for the strategic application of silicon-based cross-coupling reaction in the synthesis of natural products. The repertoire of tandem processes that allows rapid entry to complex structures makes silicon-based cross-coupling reaction more favorable compared to other variants of cross-coupling. In the following discussion, emphasis will be placed on the different methods by which the critical carbon–silicon bond is created as well as the diverse roles silicon-based cross-coupling plays in each total synthesis.
2. Survey of Target Molecules
2.1. NK-104[23]
In one of the first illustrations of the use of silicon-based cross-coupling reactions in synthesis, Hiyama and co-workers employed a hydrosilylation/cross-coupling sequence to unite the two main fragments with the biaryl core the synthesis of an artificial HMG-CoA reductase inhibitor, NK-104 (Scheme 1). To maximize the yield and selectivity of the process, the authors conducted a model study with alkyne 1 and halobenzenes to identify the optimal silylating reagent and platinum catalyst for the hydrosilylation. They discovered that the choice of the hydrosilylating agent also affected the results of the subsequent cross-coupling reaction (Table 1).
Scheme 1.
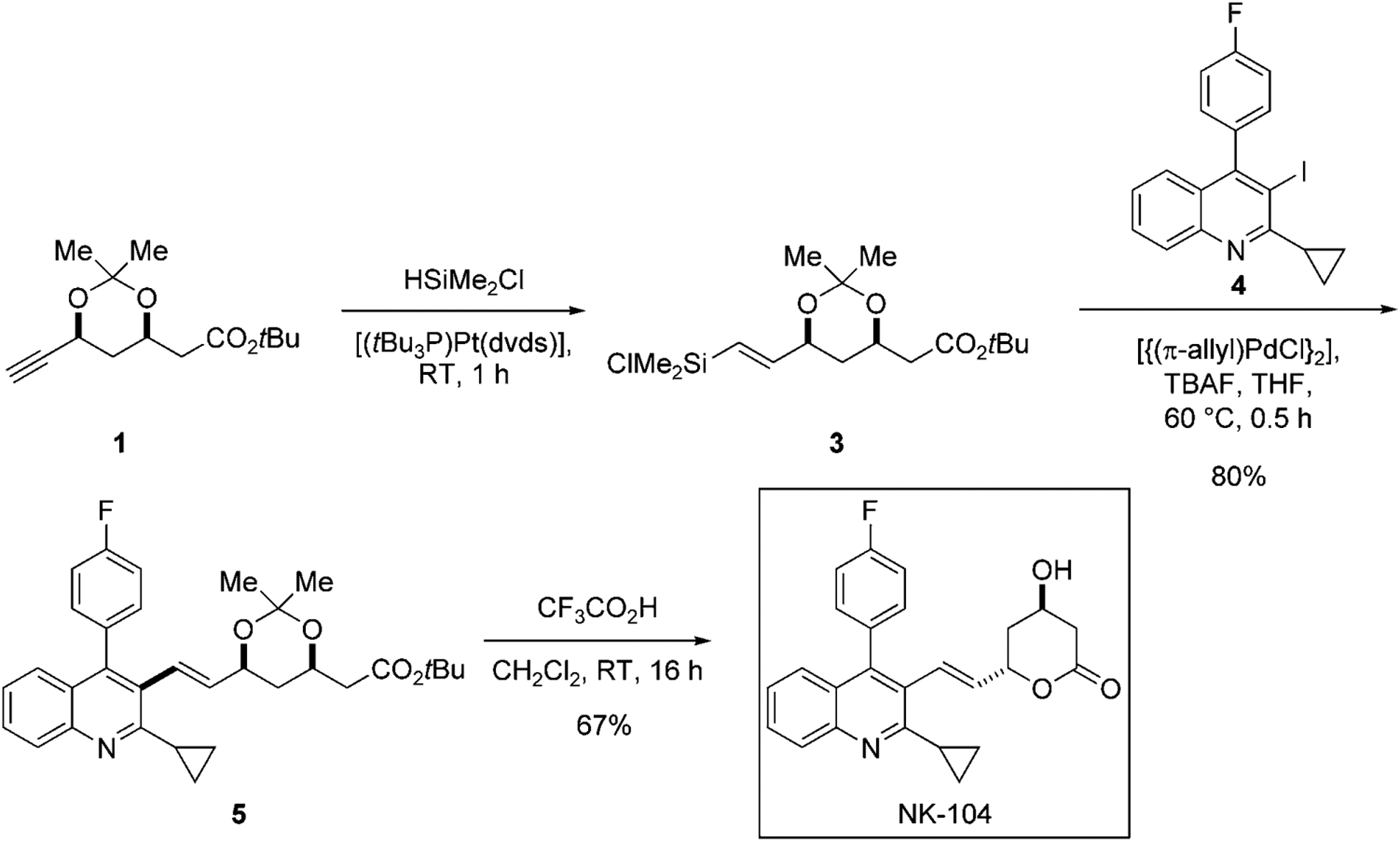
Table 1:
 | |||||
|---|---|---|---|---|---|
| Entry | HSiR3 | Pt cat. | PhX | 2a/2b/2c | Combined yield [%] |
| 1[b] | HSiMe(OEt)2 | H2PtCl6·H2O | Phl | 70:28:2 | 91 |
| 2[b] | HSiMe(OiPr)2 | H2PtCl6·H2O | Phl | 72:18:10 | 77 |
| 3[b] | HSiMe(OEt)2 | [(tBu3P)Pt(dvds)] | Phl | 89:1:10 | 80 |
| 4[c] | HSiMe2Cl | [(tBu3P)Pt(dvds)] | Phl | 96:4:0 | 78 |
| 5[c] | HSiMe2Cl | [(tBu3P)Pt(dvds)] | PhBr | 95:5:0 | 82 |
Reaction conditions: 1. HSiR3 (1.2 equiv), Pt cat. (5 mol%), RT, 1 h; 2. PhX (1.1 equiv), [{(π-allyl)PdCl}2] (2.5 mol%), TBAF (1.5 equiv), THF, 60°C, 1.5 h, unless otherwise specified.
(EtO)3P (5 mol%) used with [{(π-allyl)PdCl}2].
2.0 equivalents of TBAF employed. dvds=1,3-divinyl-1,1,3,3-tetramethyldisiloxane, TBAF=tetra-n-butylammonium fluoride, THF=tetrahydrofuran.
As part of the optimization, alkyne 1 was subjected to the hydrosilylation reaction conditions and after 1 hour the crude hydrosilylation product was employed directly in the cross-coupling reaction with iodobenzene in the presence of TBAF and [{(π-allyl)PdCl}2]. When HSiMe(OEt)2 was used in the hydrosilylation under catalysis by H2PtCl6·H2O, two constitutional isomers, 2a and 2b, as well as the protodesilylation product, 2c, were produced in a 70/28/2 ratio (entry 1). Efforts to improve the product ratio, including the use of a more sterically hindered silylating agent, HSiMe(OiPr)2 (entry 2) as well as a more bulky catalyst [(tBu3P)Pt(dvds)] (entry 3), resulted in only a moderate improvement but with lower overall yields. However, the combination of chlorodimethylsilane and [(tBu3P)Pt(dvds)] proved superior in both the hydrosilylation step (constitutional selectivity) and in the cross-coupling step (resistance towards protodesilylation; entry 4). Interestingly, the same conditions could be extended to the cross-coupling of aryl bromides. When bromobenzene was employed, both the product ratio and yield remained virtually unchanged (entry 5). Under optimal reaction conditions described in entry 4, the sequential hydrosilylation/cross-coupling of 1 proceeded smoothly. In the final route, the crude alkenylchlorosilane 3 was subjected to cross-coupling with 4 to afford 5 in 80% yield, which was converted into NK-104 by a simple acid treatment (Scheme 1).
2.2. Formal Synthesis of Nitidine[24]
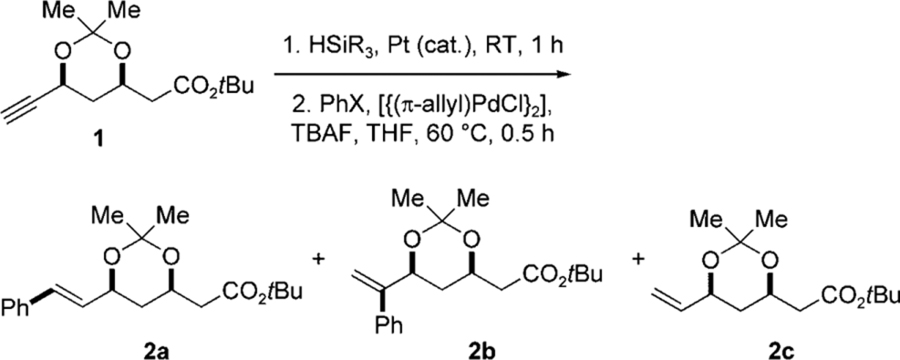
In the formal total synthesis of the antileukemic compound nitidine, Hanaoka and co-workers employed an interesting Heck/cross-coupling reaction sequence to unite the two oxygenated benzene subunits in stilbene 9 (Scheme 2). The commercially available building block ethoxydimethylvinylsilane serves as a linchpin reagent to assemble the carbon skeleton of the target molecule 10, thus intercepting a previous synthesis of nitidine.[25] In the first step of this sequential process, aryl iodide 6 undergoes a Heck reaction with ethoxydimethylvinylsilane catalyzed by [PdCl2(PPh3)2] to afford ethoxydimethylstyrylsilane 7.[26] Without purification, 7 is combined with aryl iodide 8, and the mixture is treated with TBAF in the presence of [{(π-allyl)PdCl}2],[5j] to afford the cross-coupling product 10 in 76% yield.
Scheme 2.
The Heck reaction/cross-coupling sequence in the formal synthesis of nitidine.[24]
2.3. Brasilenyne[27]
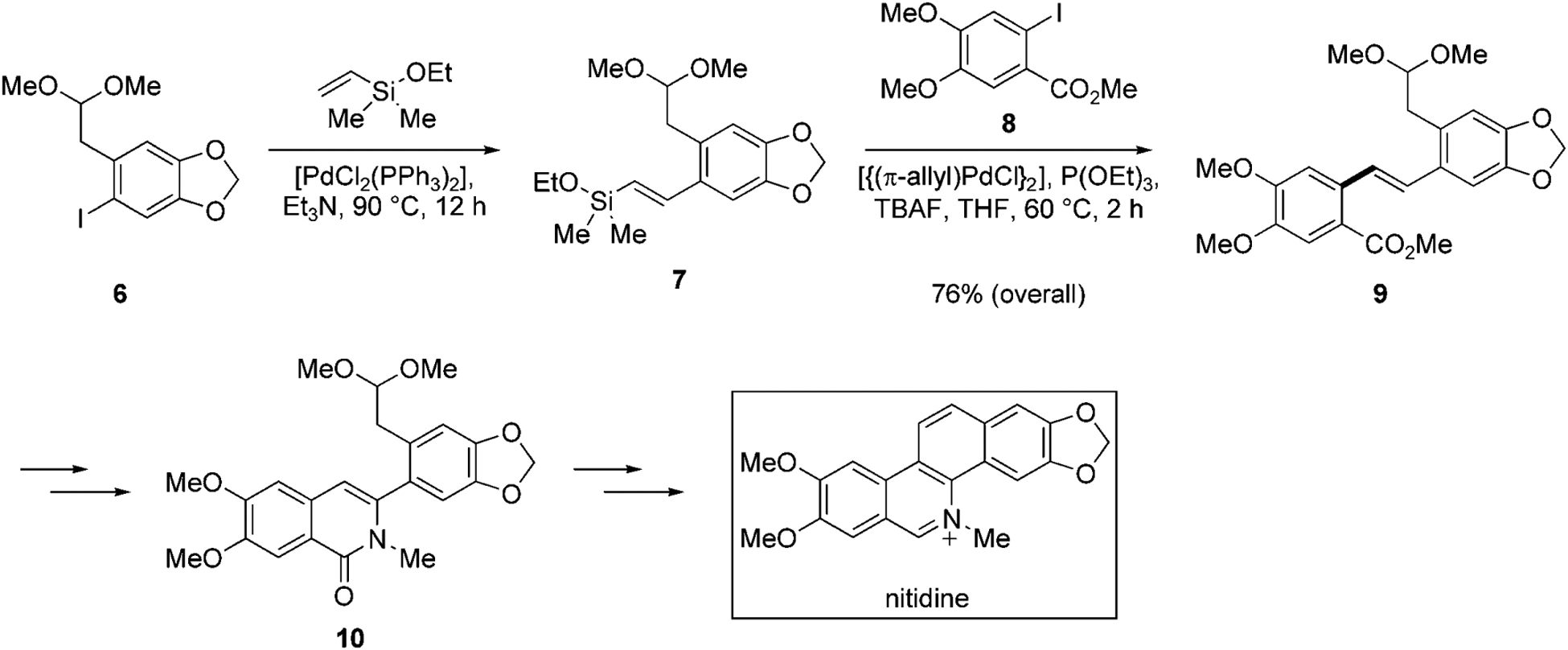
The amalgamation of the powerful ring-closing metathesis (RCM) process with silicon-based cross-coupling reaction allows for a general synthesis of medium-sized rings containing a 1,3-cis,cis-diene unit.[28] This sequence, illustrated in Scheme 3, is highly versatile because the size of the ring and the positioning of the latent functionality can be controlled by the lengths of the individual components in the starting material. In the example shown in Scheme 3, vinyl silyl ether 11 contains two different alkene termini extending from the hydroxy bearing carbon atom. The length of these chains will dictate the size of the medium-size ring and the positioning of the revealed hydroxy group. In the tandem process, the vinylsilane unit first undergoes a ring-closing metathesis reaction[29] in the presence of Schrock’s catalyst.[30] The resulting cyclic alkenylsilyl ether 12, is then subjected to an intramolecular cross-coupling reaction to effect a ring closure. Despite the unfavorable entropic and enthalpic factors,[31] ring systems containing as many as twelve atoms (such as 13) can be produced as single isomers.
Scheme 3.
Medium-sized ring formation using sequential RCM/intramolecular cross-coupling reaction.[28]
The power of this sequential process is illustrated in the total synthesis of the marine antifeedant brasilenyne reported recently by our research group (Scheme 4).[27] The most prominent structural feature of brasilenyne is the nine-membered cyclic ether bearing a 1,3-cis,cis-diene unit. The key precursor 14, containing all of the carbon atoms and the stereocenters needed for brasilenyne, is first subjected to a highly efficient RCM reaction using Schrock’s catalyst to afford six-membered cyclic alkenylsilyl ether 15. In the subsequent cross-coupling reaction, the six-membered siloxane ring is transformed by the combination of TBAF and [{(π-allyl)PdCl}2] to the nine-membered ring ether through the formation of the C4–C5 bond. The stereospecificity of the cross-coupling process assures the specific generation of the Z,Z-conjugated diene at the desired position in the nine-membered ether 16. Brasilyene could then be completed by a straightforward introduction of a chlorine atom at C8 and elaboration of the enyne side chain by following a literature precedent.
Scheme 4.
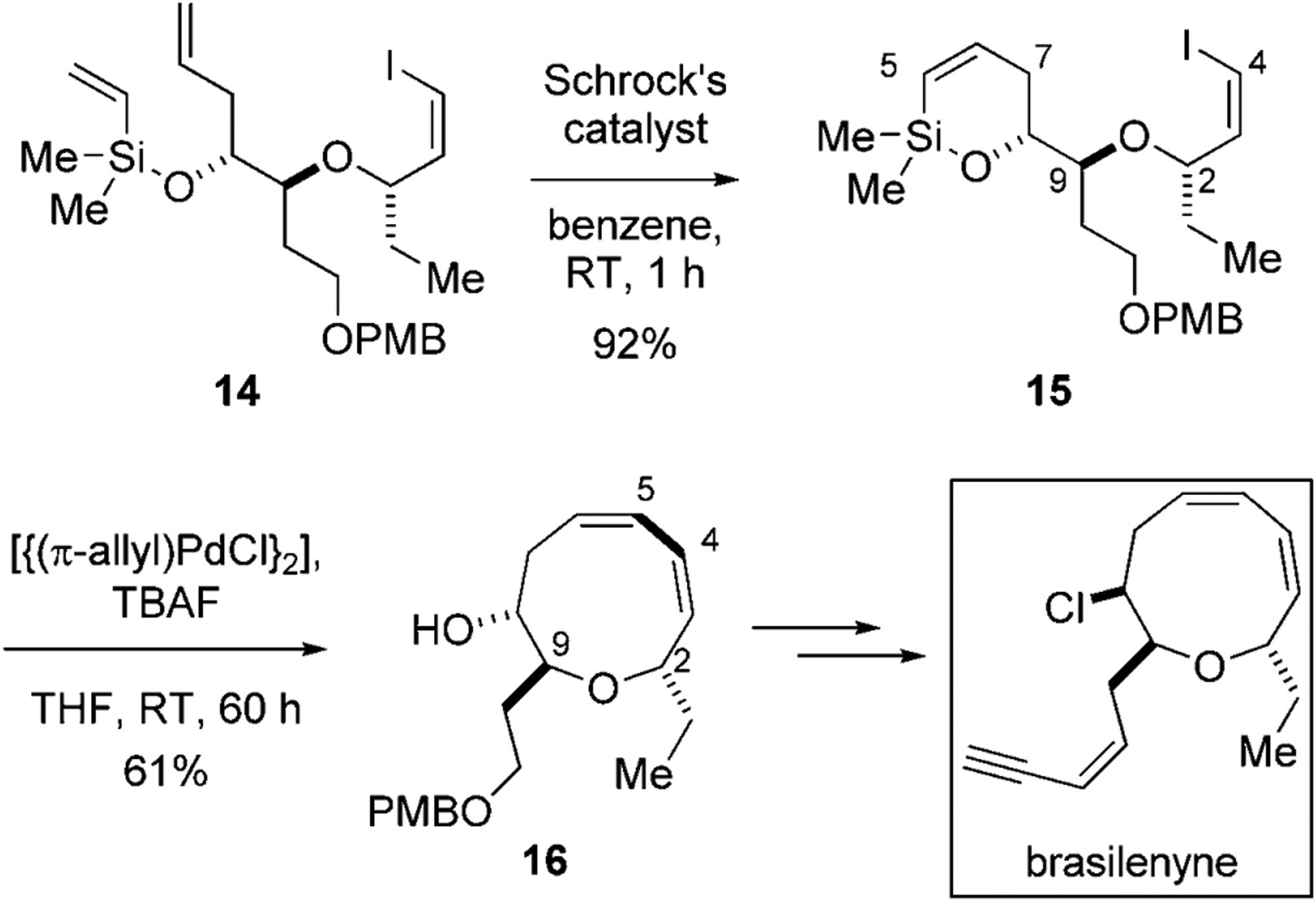
The key ring-closure steps in the total synthesis of brasilenyne.[27] PMB=para-methoxybenzyl.
2.4. Herboxidiene/GEX 1A[32]
An important advantage of silicon-based cross-coupling reactions for complex molecule synthesis is the stability of organosilicon groups to many different reaction conditions. As a result of this feature, the silyl group can be introduced early in the synthetic route (if the strategy calls for it) and it can be carried through until the key bond-forming process is called into service. The recent total synthesis of phytotoxic antitumor compound herboxidiene/GEX 1A, by Huang and Panek, highlights this empowering feature of silicon-based cross-coupling reactions. In the early stage of the synthesis, allylsilane 17 and (E)-3-benzyldimethylsilylmethacrolein are combined in a Lewis acid promoted [4+2] annulation reaction to prepare 19, a dihydropyran bearing an alkenylbenzyldimethylsilyl group (Scheme 5).[33] Alkenylbenzyldimethylsilane 18, a “masked” equivalent of an alkenylsilanol, is carried through four steps that involve an aggressive reducing agent such as lithium aluminum hydride, a basic nucleophile such as 4-dimethylaminopyridine, and elevated temperature (DMF at reflux), to arrive at the cross-coupling substrate 19. The treatment of benzylsilane 19 with TBAFand [{(π-allyl)PdCl}2] promotes the cross-coupling with a fourteen carbon fragment, alkenyl iodide 20, to afford the advanced intermediate 21,[10] which possesses the complete carbon skeleton of herboxidiene/GEX 1A (Scheme 5).
Scheme 5.
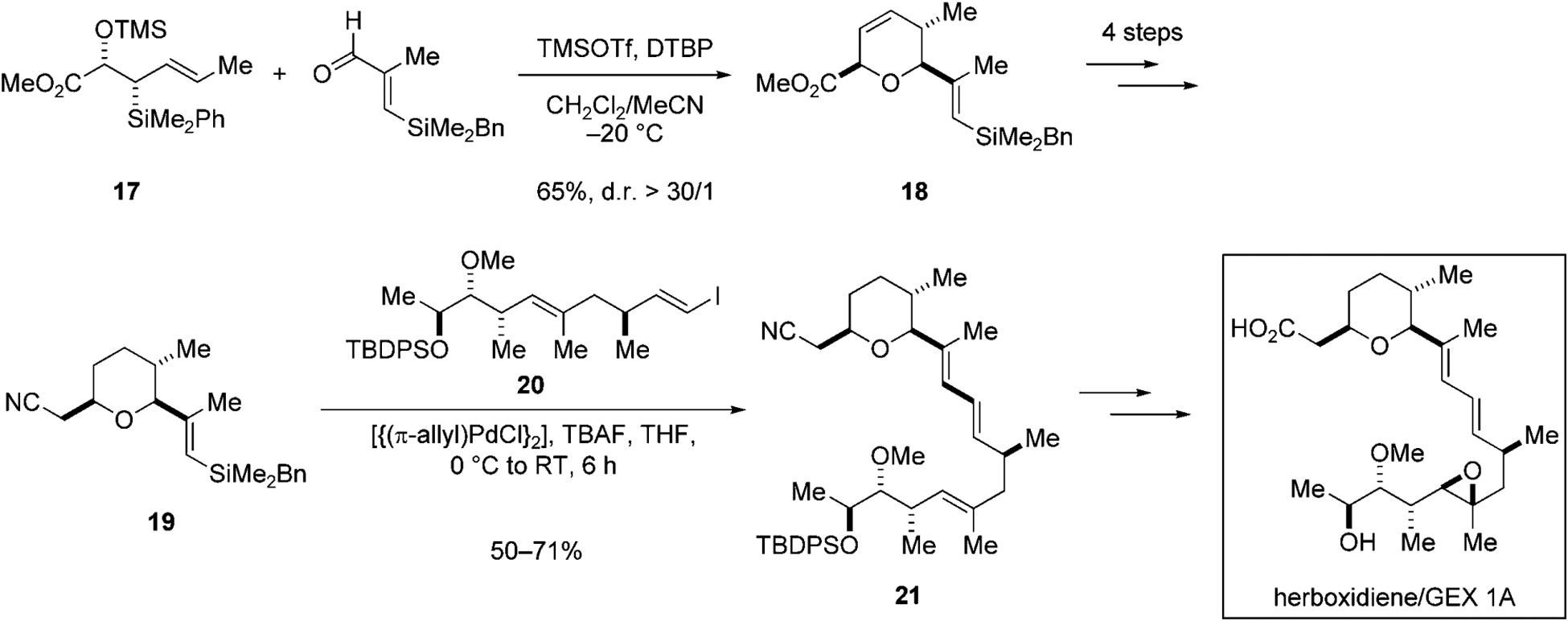
Key carbon–carbon bond-forming reactions in the total synthesis of herboxidiene/GEX 1A.[32] Bn=benzyl, DTBP=2,6-di-tert-butyl-pyridine, TBDPS=tert-butyldiphenylsilyl, Tf=trifluoromethanesulfonyl, TMS=trimethylsilyl.
2.5. RK-397[34]
In addition to being synthetically useful, the silicon-based cross-coupling reaction is also mechanistically unique in that two different modes of transmetalation are possible.[35] This mechanistic duality also has significant preparative utility because the different mechanistic pathways can be accessed under very different reaction conditions. The feasibility of using both modes of activation in a single reagent was established by our research group through the studies on sequential Brønsted base/fluoride-promoted cross-coupling reaction using the linchpin reagent (E,E)-[(4-benzyldimethylsilyl)-1,3-butadienyl]dimethylsilanol (22; Scheme 6).[10d] This bifunctional reagent can combine with two electrophiles under complementary conditions for the construction of unsymmetrical polyenes. In the first cross-coupling reaction, 22 is treated with the Brønsted base potassium trimethylsilanolate (TMSOK) in the presence of an aryl iodide and [Pd(dba)2] at room temperature. The silanolate generated in situ undergoes a direct cross-coupling reaction to afford the (1-aryl-1,3-butadienyl)benzylsilane (23). The terminus substituted with a benzylsilyl group is inert under these reaction conditions. Subsequently, when 23 is treated with TBAF and under otherwise similar conditions as the first coupling process, the second cross-coupling reaction proceeds smoothly to afford the unsymmetrical 1,4-diaryl-1,3-butadiene 24 (Scheme 6).
Scheme 6.
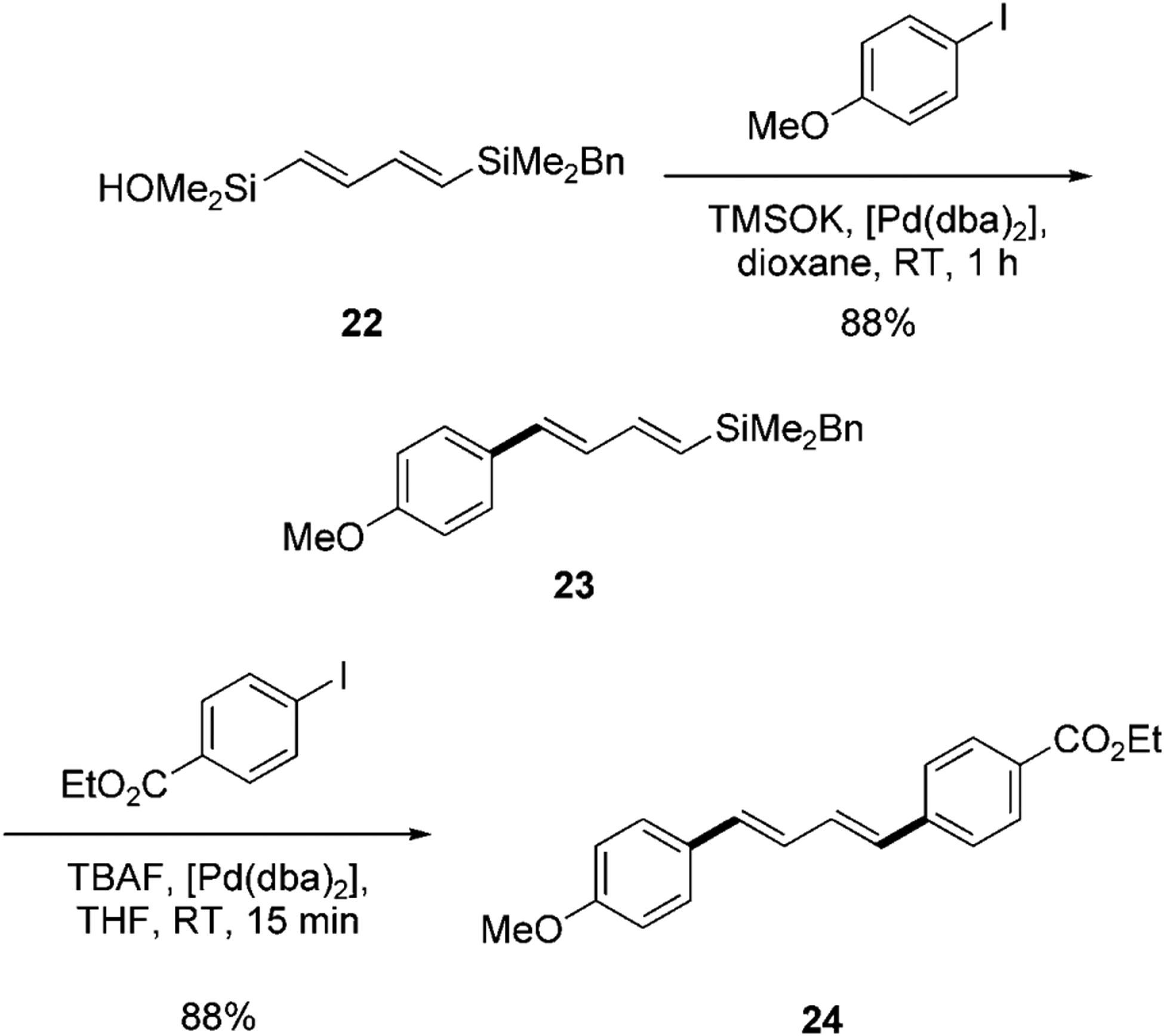
Sequential cross-coupling of 1,4-bis(silyl)diene 22.[10d] dba=trans,trans-dibenzylideneacetone.
The total synthesis of the polyene-polyol antifungal agent RK-397 aptly demonstrates the power of complementary modes of activation for silicon-based cross-coupling reaction (Scheme 7). Whereas in the above synthetic study, both electrophiles are aryl iodides for the construction of the polyene fragment of RK-397 however, the cross-coupling reaction with two alkenyl iodides is required. This extension is challenging because alkenyl iodides are known to be less reactive.[7] Thus, for the cross-coupling of 22 with alkenyl iodide 25, NaH is employed instead of TMSOK as the Brønsted base promoter. The stoichiometric generation of the silanolate using a strong base such as NaH provides heightened reactivity.[14] The silanolate undergoes the cross-coupling reaction smoothly at this stage. The resulting triene 26 is then combined with ethyl (E)-3-iodopropiolate under fluoride-promoted cross-coupling conditions to afford tetraene 27. This key fragment was then incorporated onto the polyol fragment, to complete the total synthesis of RK-397.
Scheme 7.
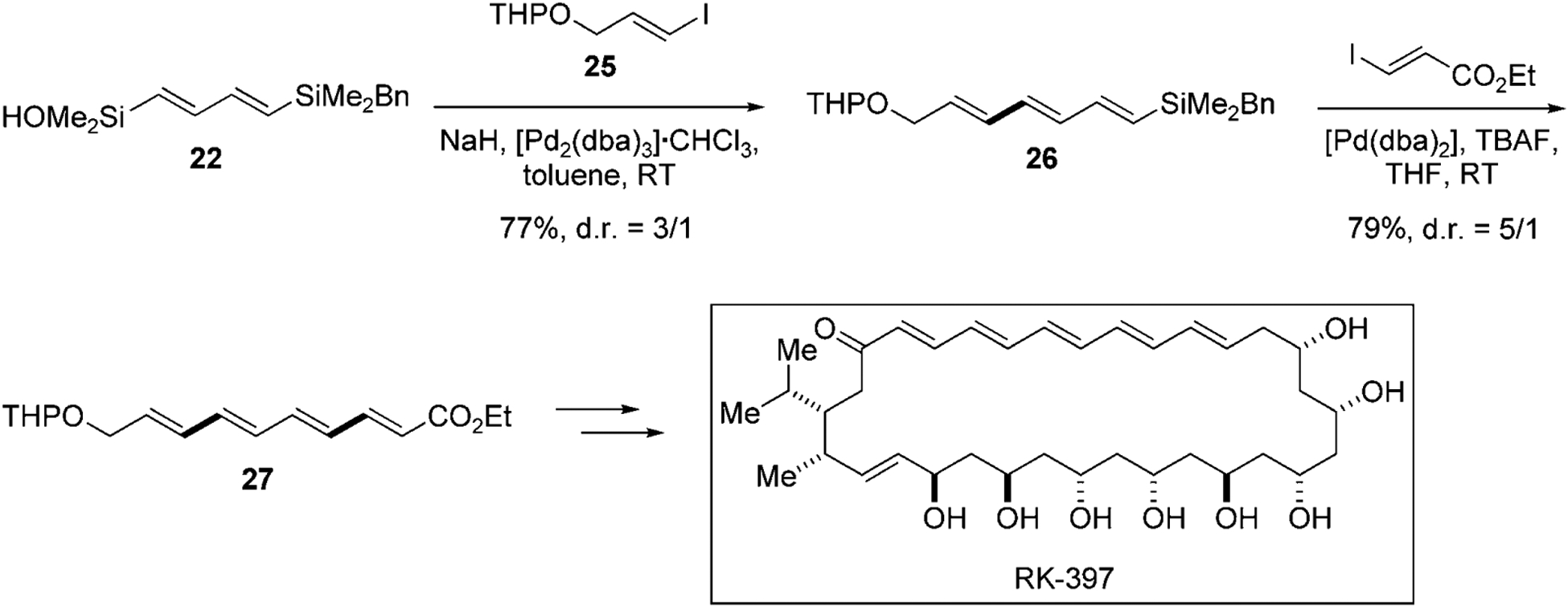
Preparation of the unsymmetrical polyene fragment of RK-397 using 22.[34] THP=tetrahydropyran.
2.6. Papulacandin D[36]
One of the most important aspects of the ability to promote silicon-based cross-coupling reactions without fluoride ions is the compatibility with silicon-based protecting groups and substrates that are prone to protodesilylation with fluoride. Both of these features were critical to the successful synthesis of the C-aryl glycoside antibiotic, papulacandin D, recently disclosed by our research group.[36] The key strategic disconnection required the cross-coupling of a 2-pyranylsilanol with an aryl halide. A similar transformation had been investigated in the early stage of the development of the cross-coupling utilizing silanols (Scheme 8).[7d] Dihydropyranylsilanol 28 (prepared by the lithiation of dihydropyran and subsequent quenching with hexamethyltrisiloxane) is a competent reagent for cross-coupling under fluoride activation. When 28 and an aryl iodide are combined with TBAF and a substoichiometric amount of [{(π-allyl)PdCl}2], the cross-coupling proceeds rapidly to afford a good yield of the 2-aryldihydropyran product 29.
Scheme 8.
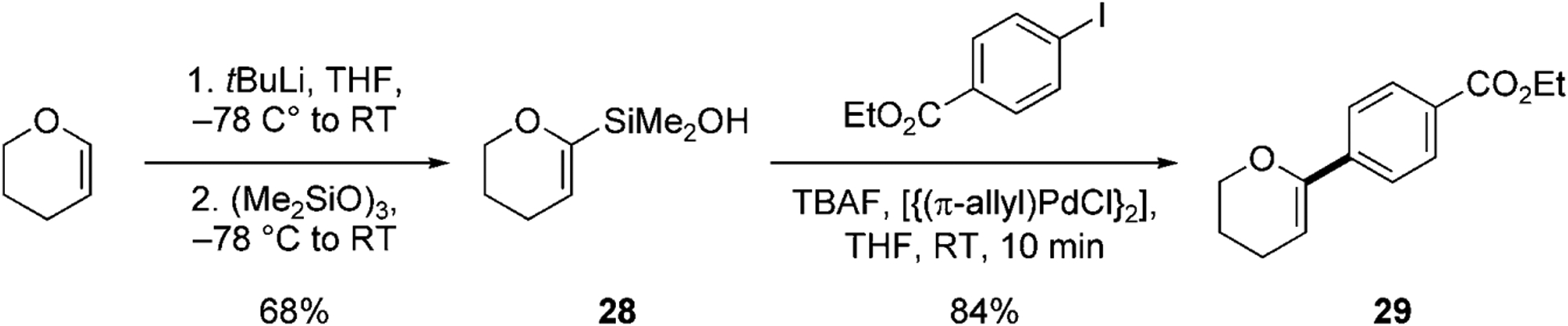
The preparation and the cross-coupling of dihydropyranylsilanol 28.[7d]
Although the above examples showed that, in principle, α-oxyalkenylsilanols are competent substrates for cross-coupling, the fluoride-containing reaction conditions are clearly incompatible with the silyl ether protecting groups planned in the total synthesis of papulacandin D. Therefore, fluoride-free conditions for the cross-coupling had to be developed.
The actual synthesis required the cross-coupling of the silyl-protected glycal 32 with the protected iodoresorcinol derivative 33 (Scheme 9). To prepare 32, silyl-protected glycal 30 is lithiated at C1 followed by capture with chlorodimethylsilane. The resulting hydrosilane 31 is subjected to a ruthenium-catalyzed, oxidative hydrolysis to afford the base-sensitive silanol, 32. The key cross-coupling reaction of 32 was quite challenging, because aryl iodide 33 is not only sterically encumbered (ortho-disubstituted) but also electron-rich. Even non-fluoride activators caused a significant amount of protodesilylation at C1. Ultimately, this critical transformation could be achieved efficiently using sodium tert-butoxide as the Brønsted base activator and [Pd2(dba)3]·CHCl3 as the catalyst to provide C-arylglycal 34 in good yield. Glycal 34 contains the entire carbon framework of the sugar fragment of papulacandin D.
Scheme 9.
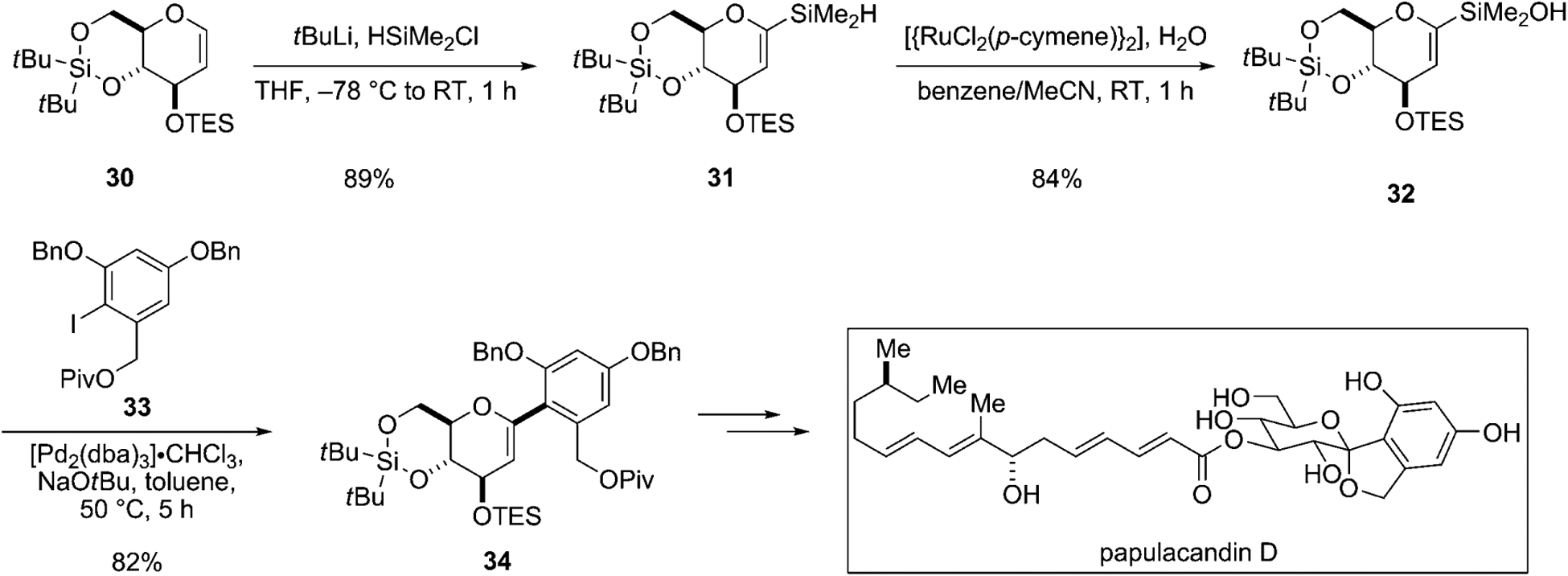
The assembly of the C-aryl glycoside of papulacandin D.[36] Piv=pivaloyl, TES=triethylsilyl.
2.7. Isodomoic Acids G and H[37]
Whereas most of the target molecules described in the foregoing sections are highly oxygenated natural products, the neuroactive marine natural products, isodomoic acids G and H, are the first alkaloids synthesized through silicon-based cross-coupling. The initial strategic connection of these targets was inspired by the recently published sequential silylcarbocyclization/cross-coupling reactions from our research group,[10a] involving a silicon-based donor related to 35 and a 5-iodopentenoate acceptor related to 37. Surprisingly, after an extensive survey of reaction conditions, none of the key cross-coupling product could be detected. The failure to effect this coupling led to a reversal in the roles of the donor and the acceptor.
Accordingly, treatment of alkenylphenyldimethylsilane 35 with iodine monochloride effects an iododesilylation that proceeds with a complete inversion of double bond configuration, presumably through an anchimeric participation of the neighboring carbonyl group (Scheme 10).[38] In the key cross-coupling reaction of 36, the fluoride hydration level plays a critical role. When the TBAF was tri-, tetra-, or hexahydrated, the conversion is modest and the reaction stalls within 2 hours. However, the reaction rate improved dramatically by employing TBAF·8H2O. With this activator, the coupling with silanol 37 proceeds rapidly to afford the protected isodomoic acid H, 38, in 92% yield. The synthesis is completed by the saponification of the three methyl ester groups of 38 using LiOH, and subsequent detosylation using sodium amalgam[39] to afford isodomoic acid H.
Scheme 10.
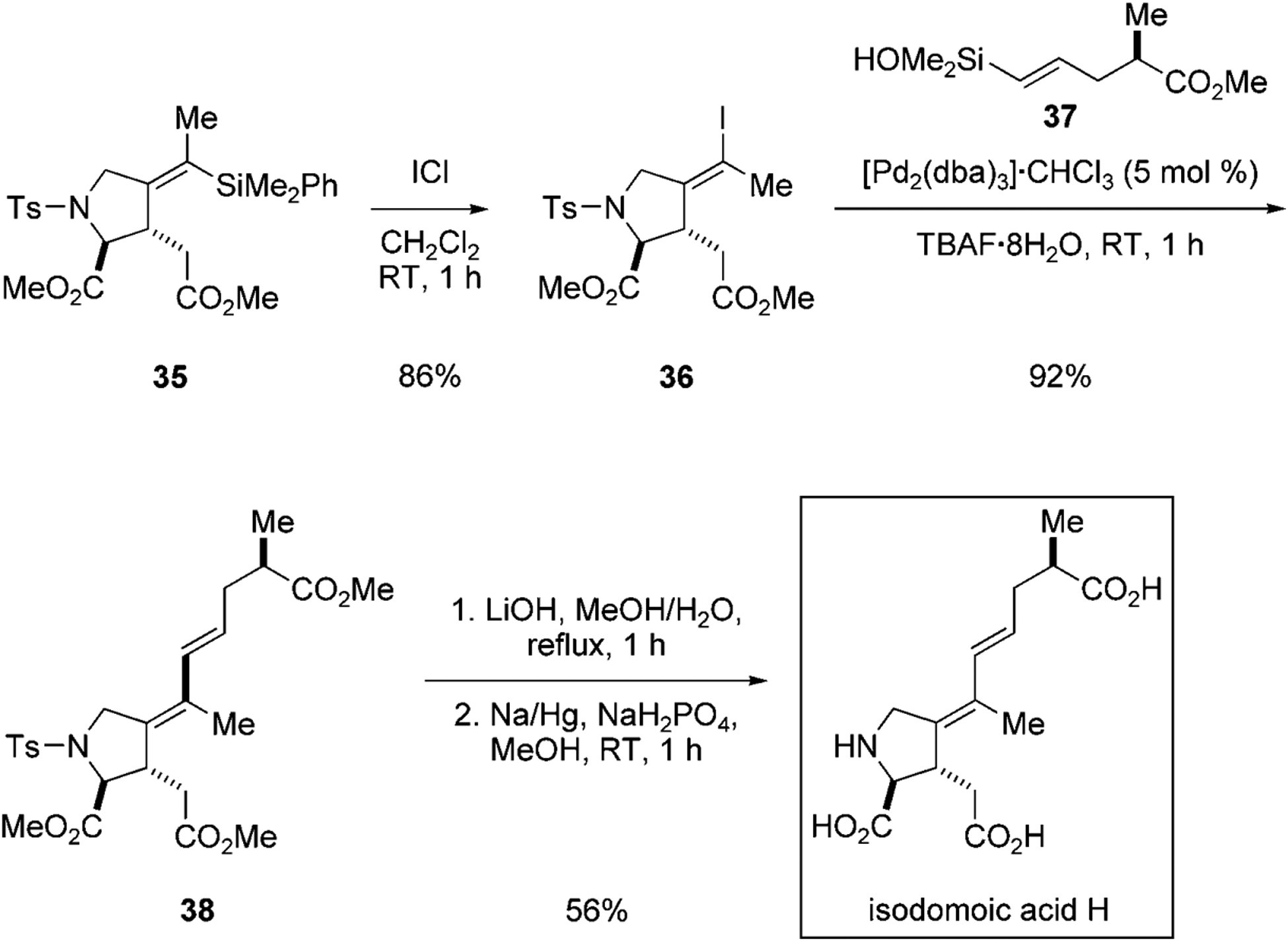
Key steps leading to the synthesis of isodomoic acid H.[37] Ts=4-toluenesulfonyl.
The invertive iododesilylation pathway could be suppressed by employing 39 in which the triisopropylsilyl ether inhibits the anchimeric participation of the ether oxygen atom, thus enabling the synthesis of isodomoic acid G (Scheme 11). The treatment of 39 with iodine monochloride proceeds with exclusive retention of double bond configuration, to give (E)-alkenyl iodide 40 in 73% yield. (E)-Alkenyl iodide 41 is subjected to the same sequence of cross-coupling and deprotection as described above, to achieve the total synthesis isodomoic acid G.
Scheme 11.
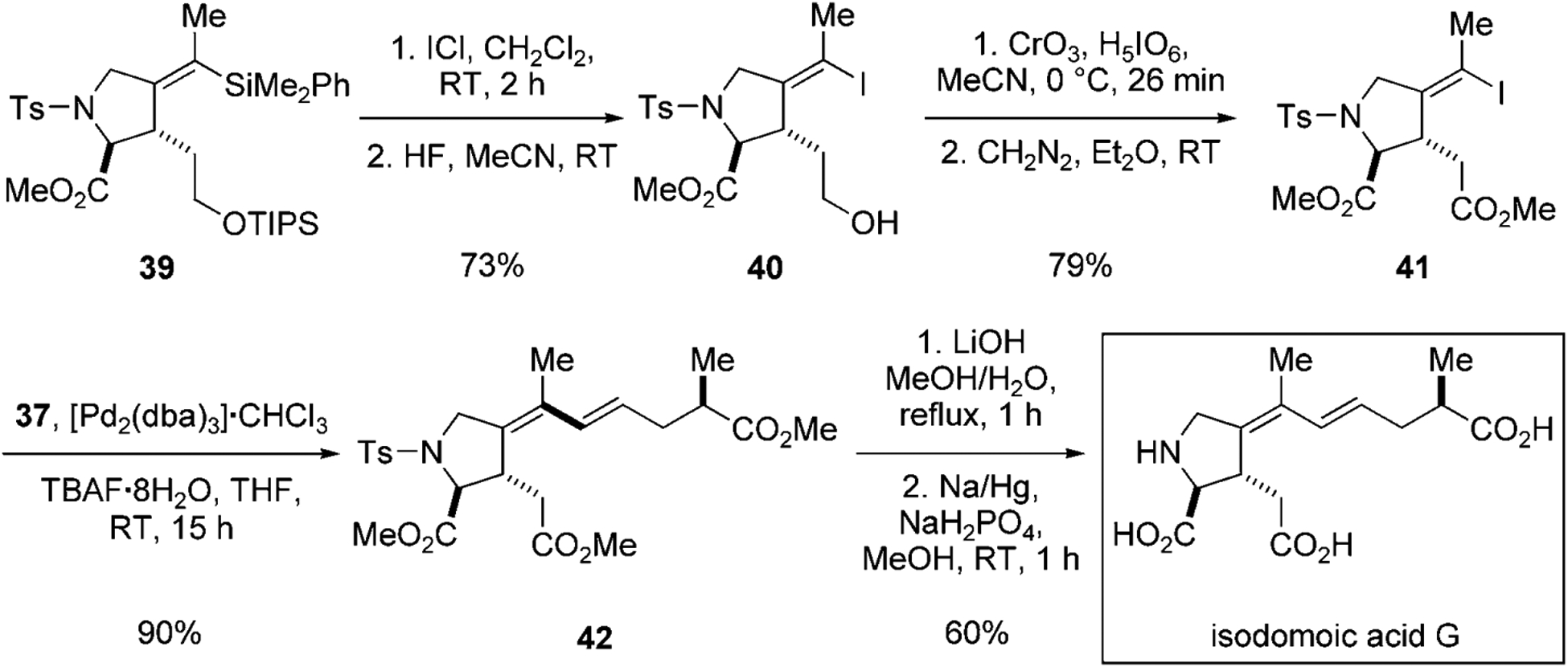
Conclusion of the synthesis of isodomoic acid G.[37] TIPS=triisopropylsilyl.
3. Summary and Outlook
The structural diversity of the natural and non-natural synthesis targets described in this Minireview illustrates the versatility of both the fluoride-promoted and Brønsted base promoted, silicon-based, cross-coupling reactions. In particular, in the case of RK-397, both modes of activation were employed with the same substrate sequentially for the synthesis of an unsymmetrical polyene. A broad range of reactions have been developed to selectively introduce the silicon-containing moiety, ranging from conventional lithiation and subsequent trapping with a silylating agent (papulacandin D), to hydrosilylation (NK-104), to the less commonly seen [4+2] annulation (herboxidiene/GEX 1A), to Heck reaction (nitidine), to ring-closing metathesis (brasilenyne), and to carbonylative silylcarbocyclization (isodomoic acids G and H). When parlayed with a mild cross-coupling process, complex molecules can be constructed expediently. Furthermore, the advent of “masked” silanol equivalents allows the silicon-containing intermediates to survive harsh reaction conditions, as in the case of herboxidiene/GEX 1A synthesis.
It should be pointed out that, presently the repertoire of conditions developed for silicon-based cross-coupling is far from being exhausted in total syntheses, since only alkenyl-alkenyl and alkenyl-aryl cross-couplings have been utilized in all the syntheses reviewed above. A promising new direction for applying silicon-based cross-coupling reactions to total synthesis is in the preparation of heteroaromatic natural products. The cross-coupling of a large number of heteroaromatic silanols, such as indolyl-, pyrrolyl-, furanyl-, thienyl-,[8b,14b,d] isoxazolyl-,[8a] and benzofuranylsilanols.[14a] has been extensively investigated by our research group. Moreover, the recent development of a sequential Larock indole synthesis/cross-coupling allows for a straightforward preparation of 2,3-disubstituted indoles, a common motif of many therapeutic agents.[22]
With all the empowering features discussed in this Minireview, it is foreseeable that more natural and non-natural compounds will be synthesized through silicon-based cross-coupling reactions. We hope that the strategies described above will encourage chemists to take advantage of this underutilized and yet valuable carbon–carbon bond-forming reaction. Ideally, we would also hope that new, sequential processes will be developed that expand the range of structure that can be accessed by their amalgamation with silicon-based cross-coupling reactions. The impact of organosilicon chemistry on the practice of organic synthesis over the past four decades has been spectacular and we anticipate that its impact on the era of cross-coupling will be no less impressive.
Acknowledgments
We are grateful to the National Institutes of Health for generous financial support (GM63167).
Biographies

Scott E. Denmark was born in Lynbrook, New York in 1953. He obtained an SB degree from MIT in 1975 (under Richard H. Holm and Daniel S. Kemp) and his DSc Tech. (under Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois. He was promoted to associate professor in 1986 and to full professor in 1987, and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry, and the origin of stereocontrol in fundamental carbon-carbon bond-forming processes. Prof. Denmark was selected as an ACS Fellow in the inaugural year.

Jack Hung-Chang Liu was born in Taipei, Taiwan in 1979. He obtained a BSc (Hon.) degree at University of Toronto in 2002 (working with Robert A. Batey and Mark Lautens). He then joined the research group of Scott E. Denmark at the University of Illinois at Urbana-Champaign, focusing on the development and application of silicon-based cross-coupling reactions. After obtaining his PhD in 2009, he moved to the University of California, Berkeley to begin postdoctoral research under F. Dean Toste.
References
- [1].Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem 2005, 117, 4516 – 4563; Angew. Chem. Int. Ed. 2005, 44, 4442 – 4489. [DOI] [PubMed] [Google Scholar]
- [2].For reviews on silicon-based cross-coupling reactions, see:; a) Denmark SE, J. Org. Chem 2009, 74, 2915 – 2927; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Denmark SE, Regens CS, Acc. Chem. Res 2008, 41, 1486 – 1499; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Denmark SE, Baird JD, Chem. Eur. J 2006, 12, 4954 – 4963; [DOI] [PubMed] [Google Scholar]; d) Nakao Y, Sahoo AK, Imanaka H, Yada A, Hiyama T, Pure Appl. Chem 2006, 78, 435 – 440; [Google Scholar]; e) Tsuji J, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, Wiley, Wessex, England, 2004, pp. 339 – 348; [Google Scholar]; f) Denmark SE, Sweis RF in Metal-Catalyzed Cross-Coupling Reactions: Second, Completely Revised and Enlarged Edition, (Eds.: de Meijere A, Diederich F), Wiley-VCH, Weinheim, 2004, Chapter 4; [Google Scholar]; g) Denmark SE, Ober MH, Aldrichimica Acta 2003, 36, 75 – 85; [Google Scholar]; h) Denmark SE, Sweis RF, Acc. Chem. Res 2002, 35, 835 – 846; [DOI] [PubMed] [Google Scholar]; i) Denmark SE, Sweis RF, Chem. Pharm. Bull 2002, 50, 1531 – 1541; [DOI] [PubMed] [Google Scholar]; j) Hiyama T, Shirakawa E, Top. Curr. Chem 2002, 219, 61 – 85; [Google Scholar]; k) DeShong P, Handy CJ, Mowery ME, Pure Appl. Chem 2000, 72, 1655 – 1658; [Google Scholar]; l) Hiyama T in Metal-Catalyzed Cross-Coupling Reactions (Eds.: Diederich F, Stang PJ), Weinheim, Wiley-VCH, 1998, Chapter 10; [Google Scholar]; m) Hiyama T, Hatanaka Y, Pure. Appl. Chem 1994, 66, 1471 – 1478; [Google Scholar]; n) Hatanaka Y, Hiyama T, Synlett 1991, 845 – 853; [Google Scholar]; o) Hatanaka Y, Hiyama T, J. Synth. Org. Chem. Jpn 1990, 48, 834 – 843. [Google Scholar]
- [3].a) Strotman NA, Sommer S, Fu GC, Angew. Chem 2007, 119, 3626 – 3628; Angew. Chem. Int. Ed. 2007, 46, 3556 – 3558; [DOI] [PubMed] [Google Scholar]; b) Homsi F, Hosoi K, Nozaki K, Hiyama T, J. Organomet. Chem 2001, 624, 208 – 216; [Google Scholar]; c) Homsi F, Nozaki K, Hiyama T, Tetrahedron Lett. 2000, 41, 5869 – 5872; [Google Scholar]; d) Kanie K, Mizuno K, Kuroboshi M, Takehara S, Hiyama T, Bull. Chem. Soc. Jpn 1999, 72, 2523 – 2535; [Google Scholar]; e) Hagiwara E, Gouda K.-i., Hatanaka Y, Hiyama T, Tetrahedron Lett. 1997, 38, 439 – 442; [Google Scholar]; f) Matsuhashi H, Asai S, Hirabayashi K, Hatanaka Y, Mori A, Hiyama T, Bull. Chem. Soc. Jpn 1997, 70, 1943 – 1952; [Google Scholar]; g) Matsuhashi H, Asai S, Hirabayashi K, Hatanaka Y, Mori A, Hiyama T, Bull. Chem. Soc. Jpn 1997, 70, 437 – 444; [Google Scholar]; h) Gouda K.-i., Hagiwara E, Hatanaka Y, Hiyama T, J. Org. Chem 1996, 61, 7232 – 7233; [DOI] [PubMed] [Google Scholar]; i) Matsuhashi H, Hatanaka Y, Kuroboshi M, Hiyama T, Heterocycles 1996, 42, 375 – 384; [Google Scholar]; j) Takahashi K, Minami T, Ohara Y, Hiyama T, Bull. Chem. Soc. Jpn 1995, 68, 2649 – 2656; [Google Scholar]; k) Matsuhashi H, Kuroboshi Y, Hatanaka Y, Hiyama T, Tetrahedron Lett. 1994, 35, 6507 – 6510; [Google Scholar]; l) Hatanaka Y, Goda K, Okahara T, Hiyama T, Tetrahedron 1994, 50, 8301 – 8316; [Google Scholar]; m) Takahashi K, Minami T, Ohara Y, Hiyama T, Tetrahedron Lett. 1993, 34, 8263 – 8266; [Google Scholar]; n) Hatanaka Y, Fukushima S, Hiyama T, Tetrahedron 1992, 48, 2113 – 2126; [Google Scholar]; o) Hatanaka Y, Fukushima S, Hiyama T, Heterocycles 1990, 30, 303 – 306; [Google Scholar]; p) Hatanaka Y, Fukushima S, Hiyama T, Chem. Lett 1989, 1711 – 1714; [Google Scholar]; q) Hatanaka Y, Hiyama Y, J. Org. Chem 1989, 54, 268 – 270. [Google Scholar]
- [4].a) Sahoo AK, Nakao Y, Hiyama T, Chem. Lett 2004, 33, 632 – 633; [Google Scholar]; b) Sahoo AK, Oda T, Nakao Y, Hiyama T, Adv. Synth. Catal 2004, 346, 1715 – 1727; [Google Scholar]; c) Nakao Y, Oda T, Sahoo AK, Hiyama T, J. Organomet. Chem 2003, 687, 570 – 573. [Google Scholar]
- [5].a) Dai X, Strotman NA, Fu GC, J. Am. Chem. Soc 2008, 130, 3302 – 3303; [DOI] [PubMed] [Google Scholar]; b) Lee J-Y, Fu GC, J. Am. Chem. Soc 2003, 125, 5616 – 5617; [DOI] [PubMed] [Google Scholar]; c) Lee HM, Nolan SP, Org. Lett 2000, 2, 2053 – 2055; [DOI] [PubMed] [Google Scholar]; d) Shibata K, Miyazawa K, Goto Y, Chem. Commun 1997, 1309 – 1310; [Google Scholar]; e) Seganish WM, DeShong P, J. Org. Chem 2004, 69, 1137 – 1143; [DOI] [PubMed] [Google Scholar]; f) McElroy WT, DeShong P, Org. Lett 2003, 5, 4779 – 4782; [DOI] [PubMed] [Google Scholar]; g) Mowery ME, DeShong P, Org. Lett 1999, 1, 2137 – 2140; [DOI] [PubMed] [Google Scholar]; h) Mowery ME, DeShong P, J. Org. Chem 1999, 64, 3266 – 3270; [DOI] [PubMed] [Google Scholar]; i) Mowery ME, DeShong P, J. Org. Chem 1999, 64, 1684 – 1688; [DOI] [PubMed] [Google Scholar]; j) Tamao K, Kobayashi K, Ito Y, Tetrahedron Lett. 1989, 30, 6051 – 6054. [Google Scholar]
- [6].a) Nakao Y, Ebata S, Chen J, Imanaka H, Hiyama T, Chem. Lett 2007, 36, 606 – 607; [Google Scholar]; b) Nakao Y, Imanaka H, Chen J, Yada A, Hiyama T, J. Organomet. Chem 2007, 692, 585 – 603; [Google Scholar]; c) Nakao Y, Chen J, Tanaka M, Hiyama T, J. Am. Chem. Soc 2007, 129, 11694 – 11695; [DOI] [PubMed] [Google Scholar]; d) Nakao Y, Sahoo AK, Yada A, Chen J, Hiyama T, Sci. Technol. Adv. Mater 2006, 7, 536 – 543; [Google Scholar]; e) Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T, J. Am. Chem. Soc 2005, 127, 6952 – 6953. [DOI] [PubMed] [Google Scholar]
- [7].a) Denmark SE, Pan W, J. Organomet. Chem 2002, 653, 98 – 104; [Google Scholar]; b) Denmark SE, Sweis RF, Org. Lett 2002, 4, 3771 – 3774; [DOI] [PubMed] [Google Scholar]; c) Chang S, Yang SH, Lee PH, Tetrahedron Lett. 2001, 42, 4833 – 4835; [Google Scholar]; d) Denmark SE, Neuville L, Org. Lett 2000, 2, 3221 – 3224; [DOI] [PubMed] [Google Scholar]; e) Denmark SE, Wehrli D, Org. Lett 2000, 2, 565 – 568. [DOI] [PubMed] [Google Scholar]
- [8].a) Denmark SE, Kallenmeyn JM, J. Org. Chem 2005, 70, 2839 – 2842; [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Baird JD, Org. Lett 2004, 6, 3649 – 3652; [DOI] [PubMed] [Google Scholar]; c) Denmark SE, Ober MH, Adv. Synth. Catal 2004, 346, 1703 – 1714; [Google Scholar]; d) Denmark SE, Tymonko SA, J. Org. Chem 2003, 68, 9151 – 9154; [DOI] [PubMed] [Google Scholar]; e) Denmark SE, Ober MH, Org. Lett 2003, 5, 1357 – 1360; [DOI] [PubMed] [Google Scholar]; f) Denmark SE, Sweis RF, J. Am. Chem. Soc 2001, 123, 6439 – 6440; [DOI] [PubMed] [Google Scholar]; g) Hirabayashi K, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T, J. Org. Chem 2000, 65, 5342 – 5349; [DOI] [PubMed] [Google Scholar]; h) Hirabayashi K, Kawashima J, Nishihara Y, Mori A, Hiyama T, Org. Lett 1999, 1, 299 – 302. [Google Scholar]
- [9].a) Ito H, Sensui H.-o., Arimoto K, Miura K, Hosomi A, Chem. Lett 1997, 639 – 640; [Google Scholar]; b) Tamao K, Kobayashi K, Ito Y, Tetrahedron Lett. 1989, 30, 6051 – 6054. [Google Scholar]
- [10].a) Denmark SE, Liu JH-C, J. Am. Chem. Soc 2007, 129, 3737 – 3744; [DOI] [PubMed] [Google Scholar]; b) Trost BM, Machacek MR, Faulk BD, J. Am. Chem. Soc 2006, 128, 6745 – 6754; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Trost BM, Ball ZT, J. Am. Chem. Soc 2005, 127, 17644 – 17655; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Denmark SE, Tymonko SA, J. Am. Chem. Soc 2005, 127, 8004 – 8005; [DOI] [PubMed] [Google Scholar]; e) Trost BM, Machacek MR, Ball ZT, Org. Lett 2003, 5, 1895 – 1898. [DOI] [PubMed] [Google Scholar]
- [11].Anderson JC, Anguille S, Bailey R, Chem. Commun 2002, 2018 – 2019. [DOI] [PubMed] [Google Scholar]
- [12].Hosoi K, Nozaki K, Hiyama T, Chem. Lett 2002, 138 – 139. [Google Scholar]
- [13].Itami K, Nokami T, Ishimura Y, Mitsudo K, Kamei T, Yoshida J.-i., J. Am. Chem. Soc 2001, 123, 11577 – 11585. [DOI] [PubMed] [Google Scholar]
- [14].a) Denmark SE, Smith RC, Chang W-TT, Muhuhi JM, J. Am. Chem. Soc 2009, 131, 3104 – 3118; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Denmark SE, Baird JD, Regens CS, J. Org. Chem 2008, 73, 1440 – 1445; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Denmark SE, Kallenmeyn JM, J. Am. Chem. Soc 2006, 128, 15958 – 15959; [DOI] [PubMed] [Google Scholar]; d) Denmark SE, Baird JD, Org. Lett 2006, 8, 793 – 795. [DOI] [PubMed] [Google Scholar]
- [15].Brook MA, Silicon in Organic, Organometallic and Polymer Chemistry, Wiley, New York, 2000. [Google Scholar]
- [16].Denmark SE, Wang Z, Org. Lett 2001, 3, 1073 – 1076. [DOI] [PubMed] [Google Scholar]
- [17].Denmark SE, Pan W, Org. Lett 2001, 3, 61 – 64. [DOI] [PubMed] [Google Scholar]
- [18].Denmark SE, Pan W, Org. Lett 2002, 4, 4163 – 4166. [DOI] [PubMed] [Google Scholar]
- [19].Denmark SE, Kobayashi T, J. Org. Chem 2003, 68, 5153 – 5159. [DOI] [PubMed] [Google Scholar]
- [20].Suginome M, Kinugasa H, Ito Y, Tetrahedron Lett. 1994, 35, 8635 – 8638. [Google Scholar]
- [21].Denmark SE, Yang S-M, Org. Lett 2001, 3, 1749 – 1752. [DOI] [PubMed] [Google Scholar]
- [22].Denmark SE, Baird JD, Tetrahedron 2009, 65, 3120 – 3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Hiyama T, Pure Appl. Chem 1996, 68, 609 – 612; [Google Scholar]; b) Takahashi K, Minami T, Ohara Y, Hiyama T, Bull. Chem. Soc. Jpn 1995, 68, 2649 – 2656; [Google Scholar]; c) Takahishi K, Minami T, Ohara Y, Hiyama T, Tetrahedron Lett. 1993, 34, 8263 – 8266; [Google Scholar]; d) Minami T, Takahashi K, Hiyama T, Tetrahedron Lett. 1993, 34, 513 – 516. [Google Scholar]
- [24].Minami T, Nishimoto A, Hanaoka M, Tetrahedron Lett. 1995, 36, 9505 – 9508. [Google Scholar]
- [25].Hanaoka M, Yamagishi H, Marutani M, Mukai C, Chem. Pharm. Bull 1987, 35, 2348 – 2354. [Google Scholar]
- [26].Yamashita H, Roan BL, Tanaka M, Chem. Lett 1990, 2175 – 2176. [Google Scholar]
- [27].a) Denmark SE, Yang S-M in Strategy and Tactics in Organic Synthesis, Vol. 6 (Ed.: Harmata MA), Elsevier, Amsterdam, 2005, pp. 100 – 136; [Google Scholar]; b) Denmark SE, Yang S-M, J. Am. Chem. Soc 2004, 126, 12432 – 12440; [DOI] [PubMed] [Google Scholar]; c) Denmark SE, Yang S-M, J. Am. Chem. Soc 2002, 124, 15196 – 15197. [DOI] [PubMed] [Google Scholar]
- [28].a) Denmark SE, Yang S-M, Tetrahedron 2004, 60, 9695 – 9708; [Google Scholar]; b) Denmark SE, Yang S-M, J. Am. Chem. Soc 2002, 124, 2102 – 2103. [DOI] [PubMed] [Google Scholar]
- [29].For recent reviews, see:; a) Monfette S, Fogg DE, Chem. Rev 2009, 109, 3783 – 3816; [DOI] [PubMed] [Google Scholar]; b) Hoveyda AH, Zhugralin AR, Nature 2007, 450, 243 – 251; [DOI] [PubMed] [Google Scholar]; c) Majumdar KC, Rahaman H, Roy B, Curr. Org. Chem 2007, 11, 1339 – 1365; [Google Scholar]; d) Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem 2005, 117, 4564 – 4601; Angew. Chem. Int. Ed. 2005, 44, 4490 – 4527; K. C. Nicolaou, P. G. Bulger, D. Sarlah, Angew. Chem. 2005, 117, 4564 – 4601; [DOI] [PubMed] [Google Scholar]; e) Deiters A, Martin SF, Chem. Rev 2004, 104, 2199 – 2238; [DOI] [PubMed] [Google Scholar]; f) McReynolds MD, Dougherty JM, Hanson PR, Chem. Rev 2004, 104, 2239 – 2258; [DOI] [PubMed] [Google Scholar]; g) Yeol S-Y, Chang S, In Handbook of Metathesis, Vol. II (Ed.: Grubbs RH), Wiley-VCH, Weinheim, 2004; [Google Scholar]; h) Trnka TM, Acc. Chem. Res 2001, 34, 18 – 29; [DOI] [PubMed] [Google Scholar]; i) Füstner A, Angew. Chem 2000, 112, 3140 – 3172; Angew. Chem. Int. Ed. 2000, 39, 3012 – 3043; [Google Scholar]; j) Grubbs RH, Chang S, Tetrahedron 1998, 54, 4413 – 4450. [Google Scholar]
- [30].Schrock RR, Murdzek JS, Bazan GC, Robbins J, DiMare M, O’Regan M, J. Am. Chem. Soc 1990, 112, 3875 – 3886; [Google Scholar]; for reviews on Schrock’s catalysts, see:; a) Schrock RR, Chem. Rev 2009, 109, 3211 – 3226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schrock RR, J. Mol. Catal. A 2004, 213, 21 – 30; [Google Scholar]; c) Schrock RR in Handbook of Metathesis, Vol. I (Ed.: Grubbs RH), Wiley-VCH, Weinheim, 2004, pp. 8 – 32; [Google Scholar]; d) Schrock RR, Hoveyda AH, Angew. Chem 2003, 115, 4740 – 4782; Angew. Chem. Int. Ed. 2003, 42, 4592 – 4633; [Google Scholar]; e) Schrock RR, Tetrahedron 1999, 55, 8141 – 8153; [Google Scholar]; f) Schrock RR, Top. Organomet. Chem 1999, 1, 1 – 36. [Google Scholar]
- [31].a) Illuminati G, Mandolini L, Acc. Chem. Res 1981, 14, 95 – 102; [Google Scholar]; b) Liebman JF, Greenberg A, Chem. Rev 1976, 76, 311 – 365. [Google Scholar]
- [32].Zhang Y, Panek JS, Org. Lett 2007, 9, 3141 – 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang H, Panek JS, J. Am. Chem. Soc 2000, 122, 9836 – 9837. [Google Scholar]
- [34].Denmark SE, Fujimori S, J. Am. Chem. Soc 2005, 127, 8971 – 8973. [DOI] [PubMed] [Google Scholar]
- [35].a) Denmark SE, Sweis RF, Wehrli D, J. Am. Chem. Soc 2004, 126, 4865 – 4875; [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Sweis RF, J. Am. Chem. Soc 2004, 126, 4876 – 4882. [DOI] [PubMed] [Google Scholar]
- [36].a) Kobayashi T, Regens CS, Denmark SE, J. Synth. Org. Chem. Jpn 2008, 66, 616 – 628; [Google Scholar]; b) Denmark SE, Regens CS, Kobayashi T, J. Am. Chem. Soc 2007, 129, 2774 – 2776. [DOI] [PubMed] [Google Scholar]
- [37].Denmark SE, Liu JH-C, Muhuhi JM, J. Am. Chem. Soc 2009, 131, 14188 – 14189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stamos DP, Taylor AG, Kishi Y, Tetrahedron Lett. 1996, 37, 8647 – 8650. [Google Scholar]
- [39].a) Yamada H, Sodeoka M, Shibasaki M, J. Org. Chem 1991, 56, 4569 – 4574; [Google Scholar]; b) Pyne SG, Hensel MJ, Fuchs PL, J. Am. Chem. Soc 1982, 104, 5719 – 5728; [Google Scholar]; c) Pyne SG, Hensel MJ, Byrn SR, McKenzie AT, Fuchs PL, J. Am. Chem. Soc 1980, 102, 5960 – 5962. [Google Scholar]