

Abstract
More than a year after its emergence, COVID-19, the disease caused by SARS-CoV-2, continues to plague the world and dominate our daily lives. Even with the development of effective vaccines, this coronavirus pandemic continues to cause a fervor with the identification of major new variants hailing from the United Kingdom, South Africa, Brazil, and California. Coupled with worries over a distinct mink strain that has caused human infections and potential for further mutations, SARS-CoV-2 variants bring concerns for increased spread and escape from both vaccine and natural infection immunity. Here, we outline factors driving SARS-CoV-2 variant evolution, explore the potential impact of specific mutations, examine the risk of further mutations, and consider the experimental studies needed to understand the threat these variants pose.
In this review, Plante et al. examine SARS-CoV-2 variants including B.1.1.7 (UK), B.1.351 (RSA), P.1 (Brazil), and B.1.429 (California). They focus on what factors contribute to variant emergence, mutations in and outside the spike protein, and studies needed to understand the impact of variants on infection, transmission, and vaccine efficacy.
Keywords: coronavirus, 2019-nCoV, SARS-CoV-2, COVID-19, furin cleavage, spike
Introduction
Over the past year, the emergence and continued spread of SARS-CoV-2 has reshaped the world in which we live. The outbreak has tested public health infrastructures, spurred economic turmoil, and led to millions of infections and deaths worldwide (Hu et al., 2021). With the recent rollout of vaccines, the identification of major SARS-CoV-2 variants has caused a new fervor with concerns for increased spread and escape from immunity (Eurosurveillance editorial team, 2021). To understand SARS-CoV-2 variants and the risk that they pose, it is important to consider the factors driving their selection, define important mutations, and outline experiments needed to understand their impact. In this review, we examine these areas and provide a nuanced perspective on the threat posed by the current and future SARS-CoV-2 variants.
What drives variant emergence?
Like other RNA viruses, coronaviruses (CoVs) rely on an error-prone RNA-dependent RNA polymerases (RdRps) to facilitate virus infection, replication, and adaptation (Smith and Denison, 2013). However, CoVs also encode a 3′-to-5′ exoribonuclease (nsp14-ExoN) that provides a proofreading function and reduces its error rate 100–1,000-fold compared with other RNA viruses. This proofreading capacity likely allows for the large size and stability of the coronavirus genome relative to other RNA viruses, which might otherwise be susceptible to error catastrophe (Domingo et al., 2005). However, errors still occur in the CoV genome at a higher rate than eukaryotic cells and, coupled with robust replication rates, allow for the accumulation of mutations in the viral genome (Smith and Denison, 2013). Many of these changes are synonymous mutations impacting only the RNA sequence and not the protein; in contrast, other mutations can result in amino acid changes, truncations, or loss of viral proteins with implications on infection and spread. Over time, these mutations can be positively selected for if they confer a fitness advantage.
For SARS-CoV-2, multiple selection parameters may have led to the development of novel variants in the human population. The most likely selective pressure is for changes that improve intrinsic fitness, either via direct replication of the virus within a host or in host-to-host transmission. As SARS-CoV-2 has evolved in humans over the past year, the virus has had ample opportunity to explore the sequence space, resulting in altered replication and transmission. One notable example is the D614G substitution in the virus spike protein that engages the host receptor, ACE2, for cell entry. D614G has been shown to improve SARS-CoV-2 infection in the upper airways, allowing the mutant to out-compete the original wild-type virus in vivo (Plante et al., 2020; Hou et al., 2020a). This advantage for D614G in SARS-CoV-2 has allowed it to quickly become the dominant variant in the world (Korber et al., 2020). Because SARS-CoV-2 appears to be new to human populations, it may still be adapting to its human host with the selection of variants capable of improved replication and spread. Similar adaption has been observed for both SARS-CoV and influenza strains in the past (Song et al., 2005; Su et al., 2015).
Alternatively or in addition, a SARS-CoV-2 variant may incorporate mutations that alter interactions with key host components. SARS-CoV-2 proteins have been shown to interact with over 300 host proteins (Gordon et al., 2020) and variant changes may augment or ablate these interactions. For example, substitutions in key viral proteins may alter control of the host translational machinery, disrupt membrane/vesicle trafficking, or influence epigenetic regulation in the host cell (Gordon et al., 2020). The variant mutations may also impact known viral antagonists of host defenses, such as NSP1, NSP6, or ORF6, changing the dynamics of the host immune response (Xia and Shi, 2020). While the spike protein is known to affect replication and spread, it has limited interactions with host factors following infection; as such, substitutions on the spike surface are more likely to impact binding and entry mechanisms.
A third selective pressure can produce a mutation that permits escape from adaptive immune responses. The spike protein is a primary target for host immunity after natural infection and current approved vaccines. Antibodies that target the spike can prevent binding/entry and disrupt infection. Changes in key spike residues may also reduce or ablate antibody binding/neutralization, reducing the efficacy of vaccine- and natural-infection-derived antibodies. However, with a naive initial population, SARS-CoV-2 encountered minimal adaptive immunity to select for escape mutations early on during the pandemic. Even now, with few exceptions, prevalence of SARS-CoV-2-specific immune memory at the population level is likely not sufficient to generate variants driven primarily by escape from antibodies. Even in areas like Manaus, Brazil, predicted to have already experienced high infection and immunity rates (Buss et al., 2021), identified variant mutations are similar to those that evolved in independent lineages in regions with lower exposure. These results suggest that many of the current mutations confer a fitness advantage based on infection, replication, and/or transmission efficiency, although potential escape from antibodies may be a byproduct of these mutations. However, as herd immunity is achieved through natural infection and vaccination, the increased selective pressure for escape variants may become much more prominent, further challenging vaccine development.
Finally, variant mutations may result in no significant advantage or disadvantage for infection or transmission. Such mutations may result from genetic drift including genetic hitchhiking—the fixation of a mutation that occurs with an advantageous mutation—or founder effects, when a lineage is introduced into a new geographic region, carrying chance mutations that are rarely beneficial. These changes may have been incorporated early on during SARS-CoV-2 spread and were subsequently maintained through the resulting daughter lineages. Alternatively, heterogeneity can also be influenced by super-spreading and impact genetic drift on variant frequencies (Gómez-Carballa et al., 2020). Yet, variant mutations may have minimal impact on the overall infection and spread. For example, the D614G substitution was accompanied by mutations in ORF1a/b (P4715L) that encodes a polyprotein that is processed into several nonstructural proteins and nucleocapsid or N (G204R) proteins (Korber et al., 2020). Based on competition and transmission studies, D614G appears to be the major target of selection, suggesting that the other changes may have been due to genetic drift (Zhou et al., 2020a; Plante et al., 2020; Hou et al., 2020a). However, further studies on these individual mutations are required to decipher their individual phenotypes and impact.
Key non-spike variant mutations
As SARS-CoV-2 variants have emerged, the major focus has been on changes in the spike protein, most notably in the receptor-binding domain (RBD) and N-terminal domain (NTD) (Figure 1 A). However, the B.1.1.7 (originally identified in the United Kingdom), B.1.351 (Republic of South Africa; RSA), P1 (Brazil), and B.429 (California, USA) variants also incorporate a number of mutations outside the spike protein that may impact SARS-CoV-2 infection (Table 1 ). The vast majority of mutations occur in only a single variant, suggesting that they resulted from genetic drift. However, several common amino acid changes suggest convergent evolution, potentially resulting from strong positive selection. For example, a substitution in nonstructural protein 12 or NSP12 (P323L), a component of the viral RdRp, is observed in all four human variants as well as the Danish mink strain, B.1.298, that has been linked to various human infections. This amino acid change corresponds to the interaction site with another RdRp component, NSP8, in the replication complex and suggests a potential role in its formation (Kirchdoerfer and Ward, 2019). Similarly, a deletion in NSP6 (del106-108) is observed in three of the four human variants and may alter interferon (IFN) antagonism (Xia et al., 2020). The T85I substitution in NSP2 is observed in both B.1.351 (RSA) and B.429 (Cal) variants and may slightly destabilize the protein (Wang et al., 2020). Finally, changes in the N protein at R203K and G204R are found in B.1.1.7 (UK), P.1 (Brazil), and B.1.298 (Mink), while the B.1.351 (RSA) and B.1.429 (Cal) variants incorporate a T205I change in the adjacent residue; these convergent or similar changes also suggest that they enhance SARS-CoV-2 fitness. This region of the nucleocapsid represents the edge of the serine/arginine (SR)-rich domain, thought to play a role in viral assembly (Nikolakaki and Giannakouros, 2020). It also governs N protein interactions with host (GSK-3, SR Kinase 1 and 2) and viral (NSP3) kinases (Cong et al., 2020) that facilitate virus replication. Further studies with individual mutations are required to confirm their effects on fitness and to determine their specific impacts.
Figure 1.

Variant substitutions in the conserved and NTD portions of the spike
(A) Diagram of the coronavirus spike protein divided into four domains: N-terminal domain of S1 (NTD), RBD, C-terminal domain of S1 (CTS1), and S2 subunit (S2). Sequence identities were extracted from alignments constructed from representative 2B CoVs using alignment data paired with neighbor-joining phylogenetic trees built using Geneious (V.9.1.5) using accession numbers: QHU79204 (SARS-CoV-2 WA1), QHR63300.2 (RATG13), QND76034.1 (HKU3), and AYV99817.1(SARS-CoV Urbani). Heatmaps constructed using EvolView (ww.evolgenius.info/) with SARS-CoV-2 WA1 as the reference sequence.
(B) SARS-CoV trimer (PDB: 6VSB ) defining the NTD (orange), RBD (red), CTS1 (pink), and S2 (gray) of spike. RBD noted in the up or down position. (C–D) Spike substitutions found in each variant of concern defined in (C) table and on (D) corresponding ribbon structure (PDB: 6VSB) by color. (E) NTD surface-exposed substitutions found on the SARS-CoV-2 trimer (PDB: 7DF3). (F) Reported NTD substitutions in each variant of concern, identified escape mutations, and proximity to glycosylation sites. Hashed boxes represent amino acid sites that are adjacent to escape or glycosylated residues.
Table 1.
Summary of coding changes in SARS-CoV-2 variants of concern
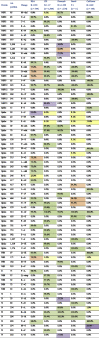 |
Data gathered from GISAID (https://doi.org/10.6084/m9.figshare.14110265) (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017). Lineages B.1.351, B.1.1.7, and B.1.1.298 reflect sequences deposited on or before 06 January 2021. Lineage P.1 and B.1.429 reflect sequences gathered 16 Januray 2021. Datasets selected by country and lineage. All relevant sequences included except for UK B.1.1.7, from which 2,500 of 7,785 were randomly selected. Sequence alignments based on hCoV-19/Wuhan/WIV04/2019. For reference, USA_WA1/2020 contains only a single coding change (NS8 L84S) in comparison to the reference sequence. Green indicates ≥75%, orange indicates 50%–74.9%, purple 25%–49.9% yellow indicates 1%–24.9%.
Analyzing spike variant mutations
With its critical role in binding, entry, and immunity, attention has been focused on variant substitutions in the SARS-CoV-2 spike protein (Table 1). However, the location within different domains of the spike protein ultimately impacts the degree and mechanism by which each substitution affects viral characteristics. The spike protein of coronaviruses is traditionally divided into the S1 and S2 portions, but here we further divide them into a head and stalk portion (Figure 1B). The NTD (orange) and the RBD (red) make up the globular head of the spike trimer, the most diverse region of the spike protein (Figure 1A). While most attention is paid to the RBD due to its interaction with the host receptor, ACE2, the NTD is heavily glycosylated and may play a role in attachment, as seen with related coronaviruses MERS-CoV and Human CoV OC43 (Tortorici et al., 2019). In contrast to the globular head, the stalk portion comprising the C-terminal domain of S1 (CTS1, pink) and the S2 domain (gray) are more conserved across the SARS-like strains found in group 2B CoVs (Figures 1A and 1B). The CTS1 contains the S1 cleavage site that is typically processed by a host protease and is required in the first step of virus activation and entry. The S2 portion maintains a second cleavage site and the highly conserved fusion machinery. Together, each of the domains plays a key role in CoV infection and variant substitutions within these domains may have a major impact on infectivity and immunity.
CTS1 and S2 variant mutations
Examining the most conserved domains, it is difficult to predict how substitutions in the CTS1 and S2 will alter infection. The D614G substitution that was first detected during early 2020 is found in all of the discussed variants, but initially, its potential impact was not easily predicted based on its location. Examination of the structure suggests that D614G increases the ability to shift the RBD into the up position required for ACE2 receptor interaction (Yurkovetskiy et al., 2020). This structural prediction is consistent with augmented infection and transmission observed for the D614G substitution relative to the original SARS-CoV-2 strains (Plante et al., 2020; Zhou et al., 2020a; Hou et al., 2020a). Among the other substitutions in these conserved domains, none is found in more than one of the variants (Table 1). Several of the changes occur in sites exposed on the spike surface adjacent to the S1/S2 cleavage site (H655Y, A701V, and T716I) (Figures 1C and 1D). Other substitutions are located in more internal alpha helices in the spike protein (T1027I, D1118H). Two changes (A570D, S982A), while internal to the spike protein, are adjacent to the RBD and are more exposed when the RBD is in the up conformation (Figures 1C and 1D). In terms of functional consequences, V1176F is located in the second heptad repeat of the S2 domain—part of the fusion core necessary for entry (Huang et al., 2020). Similarly, P681H is adjacent to the SARS-CoV-2 furin cleavage site; absent in other group 2B viruses, substitutions at this site may impact infection and pathogenesis (Johnson et al., 2021). Together, these CTS1 and S2 variant substitutions do not imply a clear mechanism of action but require further examination to reveal their potential roles in SARS-CoV-2 spread and escape from immunity.
NTD variant mutations
In contrast to the S2 and CTS1, the NTD of the SARS-CoV-2 spike protein is the least conserved domain, likely due to its prominent location on the globular head of the spike (Figures 1A and 1B). The antigenically exposed location and lack of NTD conservation across the group 2B family suggests minimal functional impact for this portion of the spike. However, for several CoV family members including MERS-CoV and Human CoV OC43, the NTD is responsible for attachment to host cells via diverse polysaccharide moieties (Tortorici et al., 2019). While not described for SARS-CoV-2, the NTD may play a similar role in attachment or in escaping antibody binding, thus influencing substitutions in the variants. Several substitutions and deletions are found in the NTD and the majority of these changes involve amino acid positions with external surface exposure (Figures 1E and 1F). In addition, several of these substitutions are in positions adjacent to glycosylation sites (L18F, P26S, D138Y, del145, R246I), making them more likely targets for antibody recognition (Figure 1E) (Watanabe et al., 2020; McCarthy et al., 2021).
In exploring the variants, several of the NTD substitutions appear in independent lineages. For example, a small portion of the B.1.351 (RSA, 25.8%) and P.1. (Brazil, 9.1%) variants encode the L18F substitution, which evolved independently; this change occurs in a NTD surface residue adjacent to a glycosylation site (Figures 1E and 1F) (Watanabe et al., 2020). Notably, the mutation has been associated with escape from several NTD-targeted antibodies (McCallum et al., 2021), leaving the possibility for immune-pressure-based selection. However, while the Brazilian variant has been attributed to high numbers of infections in the Manaus region (Buss et al., 2021), the presence of this change in the B.1.351 (RSA) variant occurred in regions without high rates of immunity, suggesting that other factor drove the selection of L18F. Similarly, del69-70 has been observed in both B.1.1.7 (UK) and B.1.298 (Danish mink) variants. This deletion, found in a prominent exterior loop of the spike, has not been associated with escape from NTD antibodies (Figures 1E and 1F). Early studies suggest that del69-70 had only a marginal impact on plaque reduction neutralization (PRNT) titer by 50% (PRNT50) of the values of vaccine sera (Xie et al., 2021). For both L18F and del69-70, further studies are required to determine their overall impact on SARS-CoV-2 infection, spread, and immune escape.
The other NTD substitutions are lineage specific within these variants of concern (Figure 1E). Among these, D80A and R246I, found in the B.1.351 (RSA) variant, have both been identified as predicted escape mutations in the identical locations (McCallum et al., 2021). D138Y and del145, found in P.1. (Brazil) and B.1.1.7 (UK) variants, respectively, are adjacent to both glycosylation sites and residues associated with escape (Watanabe et al., 2020). Del 242–244, the only non-surface-exposed change found in B.1.351 (RSA), is also adjacent to an escape residue and is notable for its proximity to the R246I change (McCarthy et al., 2021). Substitutions at T20N, P26S, and D215G occur on external loops that may be impacted by immune selection. Finally, R190S, found in the P.1. (Brazil) variant, has minimal surface exposure but is buried relatively deeply in the NTD; it has not been associated with adjacent glyocyslation sites or any escape mutants to date (Watanabe et al., 2020). Overall, the substitutions in the NTD mostly correspond to surface-exposed, antigenically significant loops and may be driven by both antibody escape and other fitness advantages.
RBD variant changes
As new variants have emerged over the past few months, the major focus has been on the substitutions in the RBD and their impact on infection. RBD changes are key to receptor binding and are also potent sites for antibody neutralization. They are also easily tested in a variety of experimental systems and numerous reagents are available to explore changes across variants (Widge et al., 2021). While the RBD interface with ACE2 has some pliability based on changes across the group 2B CoV family, substitutions at only eight RBD positions have been identified in the variants being examined here (Table 1). Among them, the most prominent is N501Y found in the B.1.1.7 (UK), B.1.351 (RSA), and P.1. (Brazil) variants (Figures 2A and 2B). The N501Y substitution renders a robust change in the RBD with the tyrosine substitution allowing additional interaction with human ACE2 at residue 353, possibly improving binding affinity (Figure 2C). Notably, initial tests with the N501Y substitution found only marginal changes in PRNT50 values, suggesting greater implications for receptor binding rather than antibody escape (Xie et al., 2021). In addition, adaptation studies have found that the N501Y substitution confers the capacity for replication in mice, which are typically resistant to SARS-CoV-2 infection as their Ace2 does not readily engage the spike protein. Thus, N501Y may improve binding to mouse Ace2 (Gu et al., 2020). Overall, the N501Y substitutions in three of the four human variants argue for its importance in infection and spread of SARS-CoV-2.
Figure 2.

RBD substitutions found across the variants of concern
(A) Reported RBD substitutions in each variant of concern, identified escape mutations, and mouse-adapted SARS-CoV-2 strains.
(B) Receptor-binding domain interaction with human ACE2 (PDB: 6M0J) highlighting substitutions in SARS-CoV-2 variants.
(C–E) Predicted amino acid structure of substitutions: (C) N501 (red) to Y501 (blue), (D) K417 (green) to N417 (red) or T417 (blue); and (E) E484 (blue) to K484 (red).
In addition to N501Y, substitutions at K417N/T and E484K have been identified as key changes that evolved in multiple variant lineages (Figures 2A and 2B). Both B.351 (RSA) and P.1 (Brazil) have a substitution at K417 to N and T, respectively, replacing the positively charged amino acid with a neutral residue (Table 1). The change from K417 results in loss of the polar interaction with residue D30 on human ACE2 (Figure 2D). The same substitution, K417N, has been observed in a virulent mouse-adapted strain of SARS-CoV-2, suggesting an improvement in receptor binding(Sun et al., 2020). In addition, K417E is a predicted escape substitution in the same position described by a pseudotyping study and would likely also disrupt interaction with D30 (Starr et al., 2021). As with 417N/T, E484K has been observed in both the RSA and the Brazilian variants (Figure 2E). Located in the RBD binding cleft, this substitution extends the amino acid side chain and changes the charge from negative to positive (Figure 2D). This change may correspond with improved binding to ACE2 but has also been associated with immune escape from both monoclonal antibodies and polyclonal sera (Starr et al., 2021; Andreano et al., 2020; Weisblum et al., 2020). Examination of E484K in concert with D614G revealed a modest reduction in vaccine neutralization titers (Xie et al., 2021). Overall, the accumulation of both K417N/T and E484K in independent variant lineages suggests strong selective pressure driving their emergences.
While N501Y, K417N/T, and E484K have garnered the most attention, several other substitutions in the RBD are worthy of surveillance and further diligence (Figures 2A and 2B). Among these, Y453F, found in the B.1.298 (Danish/Mink) variant, is located in the center of the RBD and has been associated with predicted escape from monoclonal antibody neutralization (Starr et al., 2021). Similarly, the adjacent L452R has been identified in a newer California variant, B.1.429 (Figures 2A and 2B). This change is within the RBD but may not have a robust impact on the binding interface with human ACE2. For N439K and S477N, these residues are found on the far edges of the binding domain, making them exposed and relatively pliable for substitutions. Both positions have been associated with predicted escape mutations, and human sequence data have revealed sporadic occurrence of these substitutions in deposited sequences on GSIAD(Liu et al., 2020; McCallum et al., 2021). Finally, Q493K has been implicated in a number of escape mutants and in mouse-adaptation studies (Weisblum et al., 2020; Starr et al., 2021; Leist et al., 2020). However, the mouse-adapted SARS-CoV-2 viruses show a loss of fitness in primary human airway cultures that may preclude their emergence in human populations (Leist et al., 2020). Overall, these other RBD substitutions have not yet emerged in the variants of concern, but with increased herd immunity due to vaccine deployment and further natural infections, these changes may be selected for by increasing immune pressure.
Analysis, testing, and interpretations
As the COVID-19 pandemic continues, research examining SARS-CoV-2 variants will require piecing together information from imperfect data and experiments. The identification of lineage B.1.17 (UK), the most examined and first variant of concern, was a product of extensive surveillance (2021). The robust sequencing infrastructure in the UK allowed for both its identification and for evidence of it becoming the dominant variant in that region(Kupferschmidt, 2021). Several genomic epidemiological analyses have demonstrated an increase in prevalence of B.1.1.7 over time and the results are consistent with enhanced transmissibility/fitness in humans (Volz et al., 2021). Subsequently, additional geographic variants have been identified, but their detection has been limited by less-established systems for sequencing SARS-CoV-2 strains and genomic epidemiology studies are still in progress (2021). As such, the lack of sequencing infrastructure limits the identification and tracking of new variants. This problem will continue until more extensive surveillance systems are established around the world.
While the sequencing data allow for variant identification and geographical tracking of variants, epidemiological data can provide insights into the disease and damage they cause (Monod et al., 2021). However, several factors complicate this analysis. Age, sex, co-morbidities, and other factors all impact disease outcomes in humans and complicate analysis with the need for large sample sizes for statistical power. Furthermore, due to the absence of a uniform system for data collection, the information is often incomplete and difficult to compile. In addition, the time between symptom onset and disease resolution can stretch from weeks to months, resulting in a range of data and requiring input from multiple providers. Together, the epidemiology data have great potential to illuminate the impact of individual SARS-CoV-2 variants, but the absence of uniform, aggregate data leaves an unclear picture.
In terms of basic experimental studies, a number of factors complicate analysis and interpretations. The majority of studies to date have focused on how variant mutations impact immunity following COVID-19 (2021, Cele et al., 2021, Wibmer et al., 2021, Xie et al., 2021). Many of these studies rely on either pseudotyped or live-virus infections to evaluate how given variant mutations may alter antibody-mediated protection. However, despite their utility, both methods have issues that complicate the analysis. For pseudotype approaches, the myriad of systems and laboratories running these assays leads to variability in the data and analysis (Widge et al., 2021). The lack of uniform standards can lead to contradictory results and murky conclusions. While live-virus-neutralization platforms have more uniformity, the availability and condition of the isolates used can alter outcomes (Widge et al., 2021). Availability of the variant isolates is dependent on their location in the world, local capacity to generate material for distribution, and the relationships in place to foster their transfer. In addition, cell-culture adaption and amplification approaches can impact mutations in variant isolates. For example, deletion of the furin cleavage site shifts neutralization values 2–3-fold (Johnson et al., 2021) and has already been reported in certain isolates (Cele et al., 2021). Overall, the neutralization data will need to be evaluated across several platforms to confirm the impact of mutations on immune protection.
Beyond the impact on immunity, basic science studies should also explore changes in fitness for replication and transmission using in vitro and in vivo models. For cell culture studies, the variants should be examined in a variety of cell types including standard IFN-deficient cell lines (VeroE6), respiratory cell lines (A549, Calu3), and primary human airway cultures. In addition to traditional kinetic and replication studies, competition experiments with the original SARS-CoV-2 strains will give insights into any advantages the variant may possess. Similarly, in vivo studies should take advantage of the mice, hamster, and ferret models of SARS-CoV-2 disease(Lakdawala and Menachery, 2020). While mouse ACE2 is not compatible with wild-type SARS-CoV-2 (Zhou et al., 2020b), a N501Y substitution does appear in a SARS-CoV-2-adapted strain that permits mouse infection of BALB/C, similar to changes at Q498Y/P499T in another mouse-adapted strain (Gu et al., 2020; Dinnon et al., 2020). The hamster has been established as a robust model for SARS-CoV-2 and has been used for both in vivo competition and transmission studies (Hou et al., 2020a; Plante et al., 2020). Finally, studies with ferrets provide robust evidence that the D614G substitution facilitates aerosol transmission, and this model may have utility in studying the current variants (Zhou et al., 2020a). However, neither the in vitro models nor the in vivo models capture all elements of human disease or transmission dynamics, leaving some questions about data interpretation.
Moving forward, efforts will expand to examine the role of individual variant mutations. Early studies have focused on changes in the spike, most notably within the RBD (Xie et al., 2021). However, leveraging reverse genetic systems (Hou et al., 2020b; Xie et al., 2020) provides an opportunity to study the impact of changes in SARS-CoV-2, both individually and in combination. These studies will confirm which mutations drive phenotypic differences and allow mechanistic exploration. For example, individual substitutions at N501Y alone and in combination with del69-70 had only a marginal impact on SARS-CoV-2 neutralization by vaccine sera (Xie et al., 2021). Additional studies should examine how these and other changes impact neutralization, replication, and transmission as previously described for the D614G-mutant SARS-CoV-2(Zhou et al., 2020b; Plante et al., 2020; Hou et al., 2020a). However, for each variant, the mutations may work individually or in parallel to drive the phenotypic outcome, requiring significant efforts and time to decipher.
Conclusions
As SARS-CoV-2 circulates around the globe, variants will continue to emerge due to selective pressure and ongoing virus replication in the human population. Shaped by fitness for replication and transmission, and escape from antibody-mediated immunity, the current and new variants will impact the spread of the SARS-CoV-2 and the efficacy of vaccines. In deciphering a variant’s impact, mutations will need to be examined both individually and in combination to understand their contributions and determine their mechanisms, including possible epistatic interactions. Like so many other aspects of the COVID-19 outbreak, these studies will take time to complete and the findings might complicate efforts to mitigate the pandemic. Nonetheless, with the threat of SARS-CoV-2 mutations now clearly identified, we are increasingly equipped to respond and to anticipate challenges to come.
Acknowledgments
Research was supported by grants from NIA and NIAID of the NIH (AI153602 and AG049042 to V.D.M.; R24AI120942 [WRCEVA] to S.C.W.). Research was also supported by STARs Award provided by the University of Texas System to V.D.M. Sequence data derived from deposited sequences to GISAID and SARS-CoV-2 genome sources outlined in supplementary data sheets uploaded at: https://figshare.com/articles/dataset/The_Variant_Gambit_COVID_s_Next_Move/14110265 [https://doi.org/10.6084/m9.figshare.14110265].
Author contributions
Conceptualization, J.A.P. and V.D.M.; investigation, J.A.P., B.M.M., K.S.P., K.D., V.D.M.; resources, S.C.W. and V.D.M.; data curation, J.A.P., K.D., and V.D.M.; writing–original draft, V.D.M.; writing–review & editing, J.A.P., K.D., S.C.W., and V.D.M.; data visualization, J.A.P., K.D., and V.D.M.; supervision, K.D., S.C.W., and V.D.M.; funding acquisition, S.C.W., V.D.M.
Declaration of interests
V.D.M. has filed a US patent on the reverse genetic system and SARS-CoV-2 reporter. The other authors declare no competing interests.
References
- Andreano E., Piccini G., Licastro D., Casalino L., Johnson N.V., Paciello I., Monego S.D., Pantano E., Manganaro N., Manenti A., et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv. 2020 doi: 10.1101/2020.12.28.424451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss L.F., Prete C.A., Abrahim C.M.M., Mendrone A., Salomon T., de Almeida-Neto C., França R.F.O., Belotti M.C., Carvalho M.P.S.S., Costa A.G., et al. Three-quarters attack rate of SARS-CoV-2 in the Brazilian Amazon during a largely unmitigated epidemic. Science. 2021;371:288–292. doi: 10.1126/science.abe9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cele S., Gazy I., Jackson L., Hwa S.-H., Tegally H., Lustig G., Giandhari J., Pillay S., Wilkinson E., Naidoo Y., et al. Escape of SARS-CoV-2 501Y.V2 variants from neutralization by convalescent plasma. medRxiv. 2021 doi: 10.1038/s41586-021-03471-w. https://www.medrxiv.org/content/10.1101/2021.01.26.21250224v1#:∼:text=This%20observation%20indicates%20that%20501Y,SARS%2DCoV%2D2%20variants [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y., Ulasli M., Schepers H., Mauthe M., V'Kovski P., Kriegenburg F., Thiel V., De Haan C.A.M., Reggiori F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020;94 doi: 10.1128/JVI.01925-19. e01925–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinnon K.H., Leist S.R., Schäfer A., Edwards C.E., Martinez D.R., Montgomery S.A., West A., Yount B.L., Hou Y.J., Adams L.E., et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature. 2020;586:560–566. doi: 10.1038/s41586-020-2708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Escarmís C., Lázaro E., Manrubia S.C. Quasispecies dynamics and RNA virus extinction. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurosurveillance editorial team Updated rapid risk assessment from ECDC on the risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA - first update. EURO Surveill. 2021;26 doi: 10.2807/1560-7917.ES.2021.26.3.2101211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Carballa A., Bello X., Pardo-Seco J., Martinón-Torres F., Salas A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020;30:1434–1448. doi: 10.1101/gr.266221.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Chen Q., Yang G., He L., Fan H., Deng Y.Q., Wang Y., Teng Y., Zhao Z., Cui Y., et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science. 2020;369:1603–1607. doi: 10.1126/science.abc4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Chiba S., Halfmann P., Ehre C., Kuroda M., Dinnon K.H., 3rd, Leist S.R., Schäfer A., Nakajima N., Takahashi K., et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370:1464–1468. doi: 10.1126/science.abe8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M., et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182:429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Yang C., Xu X.F., Xu W., Liu S.W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B.A., Xie X., Bailey A.L., Kalveram B., Lokugamage K.G., Muruato A., Zou J., Zhang X., Juelich T., Smith J.K., et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021 doi: 10.1038/s41586-021-03237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R.N., Ward A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019;10:2342. doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt K. Fast-spreading U.K. virus variant raises alarms. Science. 2021;371:9–10. doi: 10.1126/science.371.6524.9. [DOI] [PubMed] [Google Scholar]
- Lakdawala S.S., Menachery V.D. The search for a COVID-19 animal model. Science. 2020;368:942–943. doi: 10.1126/science.abc6141. [DOI] [PubMed] [Google Scholar]
- Leist S.R., Dinnon K.H., 3rd, Schäfer A., Tse L.V., Okuda K., Hou Y.J., West A., Edwards C.E., Sanders W., Fritch E.J., et al. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard Laboratory Mice. Cell. 2020;183:1070–1085.e12. doi: 10.1016/j.cell.2020.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Vanblargan L.A., Bloyet L.M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., Ellebedy A.H., et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv. 2020 doi: 10.1016/j.chom.2021.01.014. https://www.biorxiv.org/content/10.1101/2020.11.06.372037v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccallum M., Marco A., Lempp F., Tortorici M.A., Pinto D., Walls A.C., Beltramello M., Chen A., Liu Z., Zatta F., et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. bioRxiv. 2021 doi: 10.1016/j.cell.2021.03.028. https://www.biorxiv.org/content/10.1101/2021.01.14.426475v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccarthy K.R., Rennick L.J., Nambulli S., Robinson-Mccarthy L.R., Bain W.G., Haidar G., Duprex W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science. 2021 doi: 10.1126/science.abf6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod M., Blenkinsop A., Xi X., Hebert D., Bershan S., Tietze S., Baguelin M., Bradley V.C., Chen Y., Coupland H., et al. Age groups that sustain resurging COVID-19 epidemics in the United States. Science. 2021 doi: 10.1126/science.abe8372. https://science.sciencemag.org/content/early/2021/02/01/science.abe8372#:∼:text=We%20estimate%20that%20as%20of,20%2D49%20in%20the%20US [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakaki E., Giannakouros T. SR/RS motifs as critical determinants of coronavirus life cycle. Front. Mol. Biosci. 2020;7:219. doi: 10.3389/fmolb.2020.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante J.A., Liu Y., Liu J., Xia H., Johnson B.A., Lokugamage K.G., Zhang X., Muruato A.E., Zou J., Fontes-Garfias C.R., et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020 doi: 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., Mccauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. EURO Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.C., Denison M.R. Coronaviruses as DNA wannabes: a new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013;9:e1003760. doi: 10.1371/journal.ppat.1003760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.-D., Tu C.-C., Zhang G.-W., Wang S.-Y., Zheng K., Lei L.-C., Chen Q.-X., Gao Y.-W., Zhou H.-Q., Xiang H., et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Addetia A., Hannon W.W., Choudhary M.C., Dingens A.S., Li J.Z., Bloom J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science. 2021;371:850–854. doi: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.C.F., Bahl J., Joseph U., Butt K.M., Peck H.A., Koay E.S.C., Oon L.L.E., Barr I.G., Vijaykrishna D., Smith G.J.D. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat. Commun. 2015;6:7952. doi: 10.1038/ncomms8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Gu H., Cao L., Chen Q., Yang G., Li R.-T., Fan H., Ye Q., Deng Y.-Q., Song X., et al. Characterization and structural basis of a lethal mouse-adapted SARS-CoV-2. bioRxiv. 2020 doi: 10.1038/s41467-021-25903-x. https://www.biorxiv.org/content/10.1101/2020.11.10.377333v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.A., Walls A.C., Lang Y., Wang C., Li Z., Koerhuis D., Boons G.J., Bosch B.J., Rey F.A., De Groot R.J., Veesler D. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019;26:481–489. doi: 10.1038/s41594-019-0233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz E., Mishra S., Chand M., Barrett J.C., Johnson R., Geidelberg L., Hinsley W.R., Laydon D.J., Dabrera G., O’Toole Á., et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: insights from linking epidemiological and genetic data. medRxiv. 2021 https://www.medrxiv.org/content/10.1101/2020.12.30.20249034v2 [Google Scholar]
- Wang R., Chen J., Gao K., Hozumi Y., Yin C., Wei G. Characterizing SARS-CoV-2 mutations in the United States. Res. Sq. 2020 doi: 10.21203/rs.3.rs-49671/v1. [DOI] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., Mclellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum Y., Schmidt F., Zhang F., Dasilva J., Poston D., Lorenzi J.C., Muecksch F., Rutkowska M., Hoffmann H.H., Michailidis E., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibmer C.K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Lambson B.E., Vermeulen M., Van Den Berg K., Rossouw T., Boswell M., et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. bioRxiv. 2021 doi: 10.1038/s41591-021-01285-x. https://www.biorxiv.org/content/10.1101/2021.01.18.427166v1 [DOI] [PubMed] [Google Scholar]
- Widge A.T., Rouphael N.G., Jackson L.A., Anderson E.J., Roberts P.C., Makhene M., Chappell J.D., Denison M.R., Stevens L.J., Pruijssers A.J., et al. Durability of responses after SARS-CoV-2 mRNA-1273 vaccination. N. Engl. J. Med. 2021;384:80–82. doi: 10.1056/NEJMc2032195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H., Cao Z., Xie X., Zhang X., Chen J.Y., Wang H., Menachery V.D., Rajsbaum R., Shi P.-Y. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33:108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H., Shi P.Y. Antagonism of type I interferon by severe acute respiratory syndrome coronavirus 2. J. Interferon Cytokine Res. 2020;40:543–548. doi: 10.1089/jir.2020.0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Muruato A., Lokugamage K.G., Narayanan K., Zhang X., Zou J., Liu J., Schindewolf C., Bopp N.E., Aguilar P.V., et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe. 2020;27:841–848.e3. doi: 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Liu Y., Liu J., Zhang X., Zou J., Fontes-Garfias C.R., Xia H., Swanson K.A., Cutler M., Cooper D., et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nature Medicine. 2021 doi: 10.1038/s41591-021-01270-4. [DOI] [PubMed] [Google Scholar]
- Yurkovetskiy L., Wang X., Pascal K.E., Tomkins-Tinch C., Nyalile T.P., Wang Y., Baum A., Diehl W.E., Dauphin A., Carbone C., et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183:739–751.e8. doi: 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Thao T.T.N., Hoffmann D., Taddeo A., Ebert N., Labroussaa F., Pohlmann A., King J., Portmann J., Halwe N.J., et al. SARS-CoV-2 spike D614G variant confers enhanced replication and transmissibility. bioRxiv. 2020 doi: 10.1101/2020.10.27.357558. [DOI] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]