

Abstract

The activation barriers ΔG⧧ for kcat/Km for the reactions of whole substrates catalyzed by 6-phosphogluconate dehydrogenase, glucose 6-phosphate dehydrogenase, and glucose 6-phosphate isomerase are reduced by 11–13 kcal/mol by interactions between the protein and the substrate phosphodianion. Between 4 and 6 kcal/mol of this dianion binding energy is expressed at the transition state for phosphite dianion activation of the respective enzyme-catalyzed reactions of truncated substrates d-xylonate or d-xylose. These and earlier results from studies on β-phosphoglucomutase, triosephosphate isomerase, and glycerol 3-phosphate dehydrogenase define a cluster of six enzymes that catalyze reactions in glycolysis or of glycolytic intermediates, and which utilize substrate dianion binding energy for enzyme activation. Dianion-driven conformational changes, which convert flexible open proteins to tight protein cages for the phosphorylated substrate, have been thoroughly documented for five of these six enzymes. The clustering of metabolic enzymes which couple phosphodianion-driven conformational changes to enzyme activation suggests that this catalytic motif has been widely propagated in the proteome.
A 12 kcal/mol intrinsic binding energy (IBE)1 is observed for the substrate phosphodianion in reactions catalyzed by triosephosphate isomerase (TIM)2−4 and glycerol 3-phosphate dehydrogenase (GPDH),5,6 where <50% of the 12 kcal/mol dianion binding energy is expressed at the Michaelis complex and >50% is specifically expressed at the transition state for activation of the reaction of a phosphodianion-truncated substrate by a phosphite dianion.6−8 β-Phosphoglucomutase (PGM) likewise utilizes the binding energy of the phosphite dianion to produce a 30 000-fold activation of the enzyme for transfer of a covalent phosphoryl reaction intermediate to the anomeric hydroxyl of β-d-xylopyranose.9
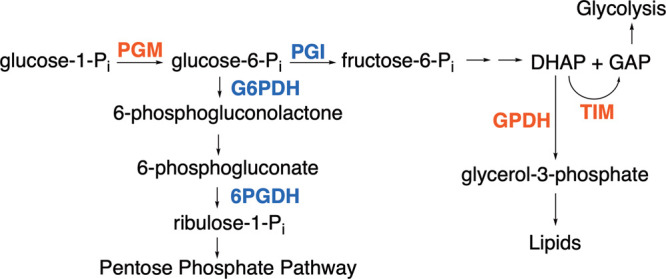
Glucose 6-phosphate (G6P) and 6-phosphogluconate (6PG) feature as substrates in the following reactions from Scheme 1: aldose–ketose isomerization catalyzed by glucose 6-phosphate isomerase (PGI),10 hydride transfer catalyzed by glucose 6-phosphate dehydrogenase (G6PDH),11 and oxidative decarboxylation catalyzed by 6-phosphogluconate dehydrogenase (6PGDH).12−14 We report that (i) d-xylose and d-xylonate are poor substrates for catalysis by PGI and G6PDH or by 6PGDH, respectively; (ii) the phosphodianion of whole G6P or 6-phosphogluconate substrates provides ≥11 kcal/mol stabilization of the respective enzymatic transition states; and (iii) between 30 and 50% of this total dianion binding energy is recovered as HPO32– activation of the enzyme-catalyzed reactions of phosphodianion-truncated substrates d-xylose (PGI and G6PDH) or d-xylonate (6PGDH). The utilization of dianion binding interactions in catalysis by this tight cluster of six enzymes (Scheme 1), which function at ancient metabolic pathways,15,16 provides evidence that large phosphodianion and phosphite binding energies are intrinsic to a catalytic motif that appeared early in evolution and which was propagated to glycolytic enzymes and enzymes that pivot intermediates of glycolysis toward the production of pentose phosphates or lipids (Scheme 1).
Scheme 1. Enzymes with Transition States Strongly Stabilized by Interactions to the Substrate Phosphodianion or Phosphite Dianion.
The sources for the chemicals and enzymes used in these studies are reported in the Supporting Information (SI). Ec6PGDH from Escherichia coli, LmG6PDH from Leuconostoc mesenteroides, and ScPGI from Saccharomyces cerevisiae were shown to each give a single major band by sodium dodecyl sulfate polyacrylamide gel electrophoresis (Figure S1). The experimental protocols for the following enzyme assays are described in the SI: Ec6PGDH-, LmG6PDH-, and ScPGI-catalyzed reactions of physiological substrates; the oxidative decarboxylation of d-xylonate catalyzed by Ec6PGDH; and LmG6PDH-catalyzed oxidation of d-xylose. The ScPGI-catalyzed isomerization of d-xylose (Scheme 2) was monitored by coupling formation of the product d-xylulose to oxidation of NADH catalyzed by sorbitol dehydrogenase.17
Scheme 2. Reactions of Whole [(kcat/Km)SPi] and Truncated [(kcat/Km)S] Substrates, and HPO32–-Activated Reactions of Truncated Substrates (kcat/KXylKHPi) Catalyzed by (A) Ec6PGDH, (B) LmG6PDH, and (C) ScPGI.

Figure S2A–C shows Michaelis–Menten plots of v/[E] against [6PG], [G6P], and [F6P], respectively, for the reactions catalyzed by Ec6PGDH, LmG6PDH, and ScPGI. These plots give the values of (kcat/Km)SPi for reactions of whole substrates reported in Table 1, where (kcat/Km)SPi for the ScPGI-catalyzed reaction of G6P was calculated from kcat/Km = 2.38 × 106 M–1 s–1 for isomerization of F6P to form G6P and Keq = 3.45 for this isomerization reaction.19Figure S3A–C shows plots of v/[E] against [d-xylonate] and [d-xylose] for reactions catalyzed by Ec6PGDH (d-xylonate), LmG6PDH (d-xylose), and ScPGI (d-xylose). The slopes of the linear correlations from Figure S3A,B are equal to (kcat/Km)S for the reaction of the truncated substrates. Values of (kcat)S = 0.0047 s–1 and (kcat/Km)S = 3.6 × 10–4 M–1 s–1 (Table 1) were determined from the fit of data from Figure S3C to the Michaelis–Menten equation. The contribution of dianion binding interactions to transition-state stabilization, (ΔG⧧)Pi = RT ln[(kcat/Km)SPi/(kcat/Km)S], are reported in Table 1.1
Table 1. Kinetic Parameters at pH 7.5 and 25 °C for Enzyme-Catalyzed Reactions of Whole and Phosphodianion Truncated Substrates (Scheme 2), the Total Phosphodianion Binding Energies, and the Dianion Binding Energy Utilized for Enzyme Activation.
| (kcat/Km)SPi | (kcat/Km)S | 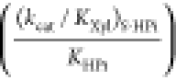 |
(ΔG⧧)Pi (kcal/mol),h,j | ΔGHPi⧧ (kcal/mol),i,j | ||
|---|---|---|---|---|---|---|
| enzyme | (M–1 s–1)e,f | (M–1 s–1)e,f | (M–2 s–1)f,g | [IBE]T | [IBE]HPi | [IBE]HPi/[IBE]T |
| ScPGIa | (6.9 ± 0.2) × 105 | (3.6 ± 0.2) × 10–4 | 0.52 ± 0.10 | 12.6 ± 0.1 | 4.3 ± 0.1 | 0.34 |
| kcat = 400 s–1 | ||||||
| LmG6PDHa | (2.0 ± 0.2) × 106 | (8.3 ± 0.1) × 10–3 | 53.0 ± 0.3 | 11.4 ± 0.1 | 5.2 ± 0.1 | 0.46 |
| kcat = 320 s–1 | ||||||
| Ec6PGDHb | (8.4 ± 0.4) × 105 | (9.9 ± 0.2) × 10–3 | 140 ± 1 | 10.8 ± 0.1 | 5.7 ± 0.1 | 0.53 |
| kcat = 12 s–1 | ||||||
| OMPDCc | 1.1 × 107 | 0.026 | 12000 | 11.7 ± 0.1 | 7.7 ± 0.1 | 0.66 |
| TIMd | 2.2 × 108 | 0.062 | 2700 | 13.0 ± 0.1 | 6.3 ± 0.1 | 0.48 |
| GPDHd | 4.6 × 106 | 0.050 | 16000 | 10.8 ± 0.1 | 7.5 ± 0.1 | 0.69 |
SPi = G6P; S = d-xylose.
SPi = 6-PG; S = d-xylonate.
Kinetic data for the catalyzed reactions of whole or truncated substrate (see SI).
The quoted uncertainty is the standard error obtained from the least-squares fit of experimental data to the appropriate kinetic equation.
Third-order rate constant for the phosphite dianion-activated reaction of truncated substrate (Scheme 3).
(ΔG⧧)Pi = RT ln[(kcat/Km)SPi/(kcat/Km)S].
The approximate uncertainties are calculated from the standard errors in the kinetic parameters.
The Ec6PGDH-, LmG6PDH-, and ScPGI-catalyzed reactions of dianion-truncated substrates are strongly activated by phosphite dianion. Note that only Ec6PGDH-catalyzed oxidation of d-xylonate by NADP was monitored. We have not determined if this enzyme catalyzes the subsequent decarboxylation reaction. We first fit plots of v/[E] against [HPO32–] to the full rate equation for Scheme 3 (not shown). However, the uncertainties in the kinetic parameters obtained from these fits (not shown) range from 25 to 100%, because the data do not clearly define the value for KXyl for weakly bound d-xylose or d-xylonate (Kxyl ≫ [Xyl], Scheme 3). We report these data (Figure 1A–C) as plots of (v – v0)/[E] against [HPO32–] for Ec6PGDH-, LmG6PDH-, and ScPGI-catalyzed reactions at different fixed concentrations of d-xylonate (1A) or d-xylose (1B and 1C), where (v – v0) is the difference in the initial velocity for reactions in the presence and absence of HPO32–. The HPO32–-activated reactions of Ec6PGDH and LmG6PDH were carried out at saturating [NADP] = 1 mM. Data from Figure 1 show that essentially the same enzyme activation is observed for reactions at 0.5 mM (open symbols) and 1.0 mM (solid symbols) [NADP]. The solid lines from Figure 1A–C show the fits of these kinetic data to eq 1, derived for Scheme 3, with the assumption that KXyl ≫ [Xyl] and using the derived values of (kcat/KHPi)obs (eq 2).
| 1 |
| 2 |
Scheme 3. Activation of the Catalyzed Reactions of d-Xylonate (Ec6PGDH) or d-Xylose (LmG6PDH and ScPGI) by HPO32–

The cofactor NADP is bound to LmG6PDH and Ec6PGDH and is reduced to NADPH for the enzyme-catalyzed reactions of d-xylose or d-xylonate.
Figure 1.

Effect of increasing [HPO32–] on (v – v0)/[E] for (A) Ec6PGDH-catalyzed reactions of d-xylonate and (B and C) LmG6PDH- and ScPGI-catalyzed reactions of d-xylose, respectively. (A) Reactions at the following [d-xylonate]: 4.1, 10, 12.4, 16.5, 20, and 24 mM. (B and C) Reactions at the following [d-xylose]: 10, 20, 30, 40, and 50 mM. The solid and open symbols for panels A (10 mM d-xylonate) and B (50 mM d-xylose) show data for reactions at 1.0 and 0.5 mM NADP, respectively.
Figure 2A shows plots of (kcat/KHPi)obs, determined for the corresponding plots from Figure 1, against [Xyl] for the reactions catalyzed by Ec6PGDH and LmG6PDH, while Figure 2B shows a similar plot for reactions catalyzed by ScPGI. The slopes, (kcat)S•HPi/KXylKHPi (eq 2), for these linear plots are reported in Table 1. The absence of detectable curvature for plots from Figure 2 shows that KXylKHPi ≫ KXyl[HPO32–] (eq 1), and that there is no significant accumulation of ternary [E·Xyl·HPO32–] complexes. By contrast, robust binding of phosphorylated substrates is observed (Km = 10–100 μM, SI) because of the entropic advantage to reactions of the whole substrates compared with the corresponding substrate pieces.18,20,21
Figure 2.

Effect of increasing [d-xylonate] or [d-xylose] on (kcat/KHPi)obs for dianion activation of the catalyzed reactions of truncated substrates (Scheme 3). (A) Ec6PGDH-catalyzed reactions of d-xylonate (●) and LmG6PDH-catalyzed oxidation of d-xylose by NADP (■). (B) ScPGI-catalyzed isomerization of d-xylose (●).
Table 1 also reports (1) the total transition-state stabilization from binding interactions with phosphite dianion (intrinsic binding energy [IBE]HPi), calculated from eq 3 derived for Scheme 4;
| 3 |
(2) the fraction ([IBE]HPi/[IBE]T) of the total intrinsic phosphodianion binding energy [IBE]T that is expressed at the transition state for the phosphite dianion-activated reaction of truncated substrate (Scheme 4); and (3) the corresponding kinetic parameters determined for isomerization, decarboxylation, and hydride-transfer reactions catalyzed by TIM, orotidine 5′-monophosphate decarboxylase (OMPDC), and GPDH, respectively.2,5,6,18ScPGI catalyzes both ring-opening of cyclic sugar phosphates and subsequent isomerization of the acyclic sugar,22 while Ec6PGDH catalyzes the coupled hydride-transfer and decarboxylation reactions of 6PG. We have not determined the transition-state stabilization for the individual enzymatic reaction steps from interactions with the substrate phosphodianion or phosphite dianion piece. We note earlier reports of phosphite dianion activation of both enzyme-catalyzed hydride transfer5,23,24 and decarboxylation reaction18 of phosphodianion-truncated substrates.
Scheme 4. Ground-State (KHPi) and Transition-State (KHPi⧧]) Binding of HPO32– to Ec6PGDH, LmG6PDH, and ScPGI.

The data from Table 1 and from an earlier study on catalysis by PGM9 define a cluster of six metabolic enzymes (Scheme 1) that show strong activation by HPO32– for the catalytic turnover of substrates, d-xylose, d-xylonate, or glycolaldehyde. Ec6PGDH, LmG6PDH, and ScPGI show smaller values of kcat/Km for the catalyzed reactions of both whole and truncated substrates, compared with the corresponding kinetic parameters for the TIM- and GPDH-catalyzed reactions of dihydroxyacetone phosphate and glycolaldehyde substrates. However, similar total intrinsic phosphodianion binding energies of 11–13 kcal/mol are observed for these five enzymes and for OMPDC (Table 1). Apparently, 13 kcal/mol, which corresponds to a >1010-fold rate acceleration, represents an operational limit for transition-state stabilization from catalytic protein–dianion interactions. A larger fraction of the total dianion binding energy, ≤70% compared with ≤50%, is recovered in the activation of reactions of glycolaldehyde compared with d-xylonate or d-xylose by HPO32–. This corresponds to a larger specificity in the utilization of HPO32– binding energy for the ground state relative to the transition state in enzymes that catalyze the reactions of 6-carbon versus 3-carbon substrates.7,25
We have proposed a model for enzyme activation in which unliganded catalysts exist in flexible, open, but inactive conformations, and protein–dianion interactions are utilized to stabilize a fully active, rigid, and closed enzyme.3,25 This model is supported by the results from experimental and computational studies on TIM,20,26,27 OMPDC,28,29 and GPDH.30,31 It provides a rationalization for dianion activation of reactions catalyzed by 6PGDH14 and PGI32 because substrate binding to these enzymes gives rise to sizable phosphodianion-driven conformational changes. It may hold for catalysis by G6PDH; however, we are not aware of X-ray crystal structures for G6PDH which show the bound cofactor and substrate positioned to undergo hydride transfer.
Evolutionary pressure to optimize energy production from nutrients has driven TIM to perfection in catalysis of the isomerization of triosephosphate.33 This catalytic perfection is presumably reflected in the structure for the iconic TIM barrel.34 We propose that enzyme catalysis, with utilization of phosphodianion binding energy to drive an enzyme-activating conformational change, appeared early in protein evolution and that this powerful catalytic motif has been replicated in the evolution of metabolic pathways (Scheme 1) and of enzymes that serve a host of cellular functions.
Glossary
Abbreviations
- IBE
intrinsic binding energy
- TIM
triosephosphate isomerase
- GPDH
glycerol 3-phosphate dehydrogenase
- PGM
β-phosphoglucomutase
- G6P
glucose 6-phosphate
- 6PG
6-phosphogluconate
- PGI
glucose 6-phosphate isomerase
- G6PDH
glucose 6- phosphate dehydrogenase
- 6PGDH
6-phosphogluconate dehydrogenase
- Ec6PGDH
6-phosphogluconate dehydrogenase from Escherichia coli
- LmG6PDH
glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides
- OMPDC
orotidine 5′-monophosphate decarboxylase
- ScPGI
glucose 6-phosphate isomerase from Saccharomyces cerevisiae
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c13423.
Experimental Section with (i) the sources of the enzymes and chemicals used in this work; (ii) experimental protocol for the determination of kinetic parameters for whole substrate and the substrate pieces; (iii) Figure S1 showing the SDS-PAGE gel of the commercial enzymes Ec6PGDH, LmG6PDH, ScPGI, and sorbitol dehydrogenase used in these studies; and (iv) figures with kinetic data for enzyme-catalyzed reactions of the whole substrate (Figure S2) and the phosphodianion-truncated substrate piece (Figure S3) (PDF)
The authors acknowledge the National Institutes of Health Grant GM134881 for support of this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Jencks W. P. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 2007, 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; O’Donoghue A. C.; Richard J. P. Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J. Am. Chem. Soc. 2001, 123, 11325–11326. 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 2008, 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Protein Flexibility and Stiffness Enable Efficient Enzymatic Catalysis. J. Am. Chem. Soc. 2019, 141, 3320–3331. 10.1021/jacs.8b10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 2017, 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J. Jr.; Long J. W. Thermodynamics and mechanism of the PO3 transfer process in the phosphoglucomutase reaction. Biochemistry 1976, 15, 3993–4006. 10.1021/bi00663a014. [DOI] [PubMed] [Google Scholar]
- Meng M.; Lin H.-Y.; Hsieh C.-J.; Chen Y.-T. Functions of the conserved anionic amino acids and those interacting with the substrate phosphate group of phosphoglucose isomerase. FEBS Lett. 2001, 499, 11–14. 10.1016/S0014-5793(01)02507-8. [DOI] [PubMed] [Google Scholar]
- Vought V.; Ciccone T.; Davino M. H.; Fairbairn L.; Lin Y.; Cosgrove M. S.; Adams M. J.; Levy H. R. Delineation of the Roles of Amino Acids Involved in the Catalytic Functions of Leuconostoc mesenteroides Glucose 6-Phosphate Dehydrogenase. Biochemistry 2000, 39, 15012–15021. 10.1021/bi0014610. [DOI] [PubMed] [Google Scholar]
- Rippa M.; Giovannini P. P.; Barrett M. P.; Dallocchio F.; Hanau S. 6-Phosphogluconate dehydrogenase: the mechanism of action investigated by a comparison of the enzyme from different species. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1998, 1429, 83–92. 10.1016/S0167-4838(98)00222-2. [DOI] [PubMed] [Google Scholar]
- Cervellati C.; Dallocchio F.; Bergamini C. M.; Cook P. F. Role of methionine-13 in the catalytic mechanism of 6-phosphogluconate dehydrogenase from sheep liver. Biochemistry 2005, 44, 2432–2440. 10.1021/bi0476679. [DOI] [PubMed] [Google Scholar]
- Chen Y.-Y.; Ko T.-P.; Chen W.-H.; Lo L.-P.; Lin C.-H.; Wang A. H. J. Conformational changes associated with cofactor/substrate binding of 6-phosphogluconate dehydrogenase from Escherichia coli and Klebsiella pneumoniae: Implications for enzyme mechanism. J. Struct. Biol. 2010, 169, 25–35. 10.1016/j.jsb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Fothergill-Gilmore L. A.; Michels P. A. M. Evolution of Glycolysis. Prog. Biophys. Mol. Biol. 1993, 59, 105–235. 10.1016/0079-6107(93)90001-Z. [DOI] [PubMed] [Google Scholar]
- Goldman A. D.; Beatty J. T.; Landweber L. F. The TIM Barrel Architecture Facilitated the Early Evolution of Protein-Mediated Metabolism. J. Mol. Evol. 2016, 82, 17–26. 10.1007/s00239-015-9722-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toteva M. M.; Silvaggi N. R.; Allen K. N.; Richard J. P. Binding Energy and Catalysis by D-Xylose Isomerase: Kinetic, Product, and X-ray Crystallographic Analysis of Enzyme-Catalyzed Isomerization of (R)-Glyceraldehyde. Biochemistry 2011, 50, 10170–10181. 10.1021/bi201378c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 2005, 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Tewari Y. B.; Steckler D. K.; Goldberg R. N. Thermodynamics of isomerization reactions involving sugar phosphates. J. Biol. Chem. 1988, 263, 3664–3669. 10.1016/S0021-9258(18)68976-8. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Enzyme Architecture: Remarkably Similar Transition States for Triosephosphate Isomerase-Catalyzed Reactions of the Whole Substrate and the Substrate in Pieces. J. Am. Chem. Soc. 2014, 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 4046–50. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schray K. J.; Benkovic S. J.; Benkovic P. A.; Rose I. A. Catalytic reactions of phosphoglucose isomerase with cyclic forms of glucose 6-phosphate and fructose 6-phoshate. J. Biol. Chem. 1973, 248, 2219–24. 10.1016/S0021-9258(19)44208-7. [DOI] [PubMed] [Google Scholar]
- Kholodar S. A.; Allen C. L.; Gulick A. M.; Murkin A. S. The Role of Phosphate in a Multistep Enzymatic Reaction: Reactions of the Substrate and Intermediate in Pieces. J. Am. Chem. Soc. 2015, 137, 2748–2756. 10.1021/ja512911f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodar S. A.; Murkin A. S. DXP Reductoisomerase: Reaction of the Substrate in Pieces Reveals a Catalytic Role for the Nonreacting Phosphodianion Group. Biochemistry 2013, 52, 2302–2308. 10.1021/bi400092n. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Specificity in transition state binding: The Pauling model revisited. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Byléhn F.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Role of Ligand-Driven Conformational Changes in Enzyme Catalysis: Modeling the Reactivity of the Catalytic Cage of Triosephosphate Isomerase. J. Am. Chem. Soc. 2018, 140, 3854–3857. 10.1021/jacs.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Role of Loop-Clamping Side Chains in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 2015, 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 2014, 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydy L. S.; Cristobal J. R.; Katigbak R. D.; Bauer P.; Reyes A. C.; Kamerlin S. C. L.; Richard J. P.; Gulick A. M. Human Glycerol 3-Phosphate Dehydrogenase: X-Ray Crystal Structures that Guide the Interpretation of Mutagenesis Studies. Biochemistry 2019, 58, 1061–1073. 10.1021/acs.biochem.8b01103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.; Reyes A. C.; Amyes T. L.; Richard J. P. Enzyme Architecture: The Role of a Flexible Loop in Activation of Glycerol-3-phosphate Dehydrogenase for Catalysis of Hydride Transfer. Biochemistry 2018, 57, 3227–3236. 10.1021/acs.biochem.7b01282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenieva D.; Jeffery C. J. Conformational Changes in Phosphoglucose Isomerase Induced by Ligand Binding. J. Mol. Biol. 2002, 323, 77–84. 10.1016/S0022-2836(02)00892-6. [DOI] [PubMed] [Google Scholar]
- Knowles J. R.; Albery W. J. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res. 1977, 10, 105–111. 10.1021/ar50112a001. [DOI] [Google Scholar]
- Sterner R.; Hocker B. Catalytic versatility, stability, and evolution of the (β/α)8-barrel enzyme fold. Chem. Rev. 2005, 105, 4038–4055. 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.