

Significance Statement
Regulation of endothelial cells is important in many biologic processes, including development, organ function, and disease. The kidney vasculature is highly sensitive to hypoxic injury and has a limited capacity for repair. AKI as a result of decreased blood flow is common, and there are no current therapies. MicroRNAs are small noncoding RNAs that inhibit expression of target genes. Endothelial-derived miR-17∼92 is a cluster of microRNAs critical for endothelial function and repair during AKI in mice. Furthermore, pharmacologic treatment with mimics of the cluster mitigates AKI, promoting angiogenesis. These microRNAs are the first potential therapeutic target for kidney endothelial damage after AKI, and mimics may be broadly applicable to disease processes that involve endothelial injury.
Keywords: microRNA, acute kidney injury, renal microvasculature
Abstract
Background
Damage to the renal microvasculature is a hallmark of renal ischemia-reperfusion injury (IRI)–mediated AKI. The miR-17∼92 miRNA cluster (encoding miR-17, -18a, -19a, -20a, -19b-1, and -92a-1) regulates angiogenesis in multiple settings, but no definitive role in renal endothelium during AKI pathogenesis has been established.
Methods
Antibodies bound to magnetic beads were utilized to selectively enrich for renal endothelial cells from mice. Endothelial-specific miR-17∼92 knockout (miR-17∼92endo−/−) mice were generated and given renal IRI. Mice were monitored for the development of AKI using serum chemistries and histology and for renal blood flow using magnetic resonance imaging (MRI) and laser Doppler imaging. Mice were treated with miRNA mimics during renal IRI, and therapeutic efficacies were evaluated.
Results
miR-17, -18a, -20a, -19b, and pri–miR-17∼92 are dynamically regulated in renal endothelial cells after renal IRI. miR-17∼92endo−/− exacerbates renal IRI in male and female mice. Specifically, miR-17∼92endo−/− promotes renal tubular injury, reduces renal blood flow, promotes microvascular rarefaction, increases renal oxidative stress, and promotes macrophage infiltration to injured kidneys. The potent antiangiogenic factor thrombospondin 1 (TSP1) is highly expressed in renal endothelium in miR-17∼92endo−/− after renal IRI and is a target of miR-18a and miR-19a/b. miR-17∼92 is critical in the angiogenic response after renal IRI, which treatment with miR-18a and miR-19b mimics can mitigate.
Conclusions
These data suggest that endothelial-derived miR-17∼92 stimulates a reparative response in damaged renal vasculature during renal IRI by regulating angiogenic pathways.
One of the hallmarks of ischemic AKI is damage to the renal microvasculature, including peritubular capillaries.1 This damage, mediated by vascular oxidative stress,2 inhibits endothelial function and increases leukocyte-endothelial interaction. Endothelial injury stimulates adhesion of platelets and inflammatory cells, resulting in thrombosis.3 This cascade of events results in reduced blood flow to the kidneys and activation of inflammation. This, in turn, leads to hypoxic and inflammatory injury in the renal parenchyma.4 Angiogenic response is key to the repair of injured endothelial cells, but the renal microvasculature is thought to have a limited capacity for repair.5 Thus, development of preventative therapies specifically targeting the renal microvasculature is important to combat ischemic AKI.
miRNAs are endogenous small noncoding RNAs that function by binding to their respective mRNA targets and causing them to undergo post-transcriptional repression.6,7 miR-17∼92 is a evolutionarily conserved cluster of polycistronic miRNAs.8 miR-17∼92 is known to be required for normal kidney development and is implicated in polycystic kidney disease.9–12 miR-17∼92 shows proangiogenic capacity13,14 through direct repression of an antiangiogenic factor, thrombospondin 1 (TSP1),13 among others. TSP1 has been found to promote vasoconstriction and ischemic AKI.15,16 Recent studies have implicated miR-17∼92 in recovery from various ischemic insults. Treatment with miR-19a/b mimics was protective against myocardial infarction,17,18 and loss of endothelial-derived miR-17∼92 resulted in increased arteriogenesis and increased blood flow recovery after limb ischemia.19 Important roles of miRNAs in AKI have recently been documented,20–24 including a beneficial role for proximal tubular–derived miR-17 and miR-17∼92 postrenal ischemia-reperfusion injury (IRI).25,26 It is not clear how the miR-17∼92 cluster functions in the renal vasculature after AKI.
Here, we show renal endothelium–specific regulation of miR-17∼92 miRNAs following renal IRI. Loss of miR-17∼92 in endothelial cells promotes renal dysfunction following renal IRI through dysfunction of renal microvasculature. We further demonstrate proof of concept data that cotreatment with miR-18a and miR-19b mimics protects against renal IRI, providing evidence for a potential novel therapeutic approach for the treatment of AKI (Supplemental Figures 1–11, Supplemental Material, Supplemental Tables 1–4).
Methods
C57BL/6J wild-type (WT; stock no. 000664), Tie2 Cre [FVB-Tg(Tek-cre)2352Rwng/J, stock no. 008537],27 R26-tdTomato Cre reporter [B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, stock no. 007914],28 and miR-17∼92 floxed (Mirc1tm1.1Tyj/J; stock no. 008458)29 mice were purchased from the Jackson Laboratories. Tie2 Cre; R26-tdTomato reporter mice, which harbor heterozygous Tie2 Cre and R26 tdTomato reporter alleles, were generated. Endothelial-specific miR-17∼92 knockout, which harbors heterozygous Tie2 Cre and homozygous floxed alleles of miR-17∼92 (miR-17–92endo−/−), was also generated. The offspring were genotyped by PCR according to the protocol from the Jackson Laboratories with the primer sequences in Supplemental Table 3.29,30 The primer pair F4/R5 (to amplify 255-bp miR-17–92 WT allele or the 289-bp miR-17–92 floxed allele fragment) and the primer pair F4/AV145 (to amplify the 441-bp miR-17–92 deleted allele fragment) were used.30,31 Tie2 Cre–negative sex- and age-matched littermates of miR-17∼92endo−/− mice were used as controls. The genetic backgrounds of miR-17∼92endo−/− and the control mice are C57BL/6J. For the miRNA mimics experiments, WT FVB/NJ (stock no. 001800) males were purchased from the Jackson Laboratories, 10- to 14-week-old mice were used. Male mice were used in the studies unless otherwise noted. The University of Pittsburgh Institutional Animal Care and Use Committee approved the animal experiments performed (approval no. 16088935).
CD31+ endothelial cells from mouse kidneys were isolated from a single-cell suspension of kidney cells obtained from 10- to 14-week-old male miR-17∼92endo−/− or Cre-negative littermate control mice using the Dynabeads Antibody Coupling Kit (14311D; Thermo Fisher Scientific) conjugated to CD31 antibody (Clone MEC 13.3, 553370; BD Biosciences) according to the manufacturer’s instruction.
To induce ischemic AKI, a renal IRI model was performed as previously described32 with modifications. Briefly, mice were anesthetized with the inhalant 2% isoflurane. Core body temperature of the mice was monitored with a rectal thermometer probe and was maintained at 36.8–37.2°C throughout the procedure with a water-heating circulation pump system (EZ-7150; BrainTree Scientific) and a heat lamp (Shat-R-shield). Buprenorphine (Par Pharmaceutical) was administered for pain control (0.1 mg/kg body wt, subcutaneous). With aseptic technique, a dorsal incision was made to expose the kidney. Renal ischemia was induced by unilateral clamping of the left kidney pedicle with a nontraumatic microvascular clamp (18055–04; Fine Science Tools). Length of ischemia was optimized as 22 minutes for WT C57BL/6J males for expression analysis of miR-17∼92 miRNAs (Figure 1), 22 minutes for males and 31 minutes for females of miR-17∼92endo−/− or Cre-negative littermate control, and 21 minutes for WT FVB/NJ males for the miRNA mimic experiments. The 21-minute ischemia time for WT FVB/NJ males was determined following a series of ischemia titration experiments (Supplemental Figure 7). Reperfusion was visually verified. Delayed contralateral nephrectomy of the right kidney was performed 1 day prior to harvesting samples. Mice were euthanized on day 7 to harvest blood and the remaining kidney. Sham procedure followed the same protocol except for the step of renal pedicle clamping. Serum was separated from the blood and analyzed by the Kansas State Veterinary Diagnostic Laboratory for the levels of creatinine, BUN, potassium, sodium, phosphorus, and bicarbonate.
Figure 1.

Time course analysis for expression of miR-17∼92 miRNAs and pri–miR-17∼92 in renal endothelial cells at days 1, 3, and 7 postrenal IRI demonstrates peak expression of mature miRs at day 3 postrenal IRI. Expression of primary and mature miRNAs in the miR-17∼92 cluster was analyzed by qRT-PCR using isolated CD31+ endothelial cells from WT C57BL/6J kidneys 1, 3, or 7 days after renal IRI or uninjured (Un) sham operation. Expression of miR-19a and miR-92 was not detected. Data normalized to Rps17 for pri–miR-17∼92 and U6 snRNA for miRNAs. n=3–11. Tukey multiple comparison. *P<0.05; **P<0.01; ***P<0.001; ****P<0.001.
All miRNA mimics were purchased from Dharmacon and formulated with Max-Suppressor in vivo RNALancerII, a lipid-based delivery reagent (341001; BIOO Scientific, Inc.), according to the manufacturer’s instructions and a previous report.18 Combined miRNA mimics miR-18a (C-310513–07–0050)/miR-19b (C-311179–00–0050) or control mimics (0.25 µg/kg body wt, intravenous) were administered via tail vein. Caenorhabditis elegans miRNA cel-miR-67–based miRNA mimic (CN-001000–01–50) was used for control. miRNA mimic or control was administered daily starting 24 hours prior to renal IRI for 8 days. This vehicle, dose, and daily treatment regimen were effective in a mouse cardiac infarction model.18
For the biodistribution analysis, 100 μl of 10% FITC-labeled RNA oligo (13750062, BLOCK-iT Fluorescent Oligo; Thermo Fisher) or PBS control was administered via tail vein injection. Kidneys were harvested 40 minutes postinjection and processed for cryosections.
Cortex areas of the left kidneys in mice were longitudinally analyzed by in vivo magnetic resonance imaging (MRI) noninvasively for anatomic and functional evaluation before and 72 hours postrenal IRI. T2-weighted MRI images were used for anatomic images. Rate of renal blood flow (RBF) was quantified by arterial spin labeling (ASL) MRI. ASL uses the endogenous water molecules in the blood as tracers to quantify RBF without needing to administer exogenous contrast agents. A continuous adiabatic pulse train in a 1-mm slab localized 9 mm proximal to the renal artery was applied to the abdominal aorta to spin tag all of the incoming proton spins through the abdominal aorta to reach the steady state. The spin attenuation by the steady-state labeling, labeling efficiency, blood-tissue partition coefficient, and T1obs were measured to calculate the RBF with the following equation:
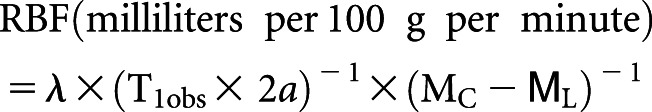 |
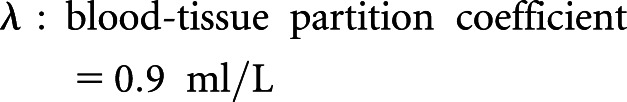 |
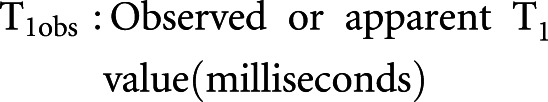 |
α was measured in the abdominal aorta 0.9 cm from the arterial spin inversion site.
MRI analysis was performed at the Animal Imaging Core at the Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center.33 Anatomic MRI images were referred to define cortex area of the kidneys in ASL MRI images (Supplemental Figure 6).
RBF was assessed 24 hours after renal IRI. Briefly, mice were anesthetized with 2.5% isoflurane and placed in a supine position on a heating pad. Core temperatures were maintained at 36°C and continuously monitored by rectal probe thermometer. Left kidneys were exposed with abdominal incisions, and real-time blood flow was measured using laser Doppler imaging (MoorLDI-2λ; Moor Instruments, Devon, United Kingdom).
The translating ribosome affinity purification microarray dataset for upregulated genes in renal endothelial cells 24 hours postrenal IRI was obtained from the National Center for Biotechnology Information Gene Expression Omnibus (accession no. GSE2004) originally published by Liu et al.34 TargetScan35 (http://www.targetscan.org/vert_72/) web server was used for miRNA target prediction. Ingenuity Pathway Analysis (Qiagen) was used for pathway analysis.
Kidneys were homogenized with a BeadBug homogenizer (Benchmark Scientific), and total RNA, including miRNA, was isolated using the miRNeasy Mini or Micro Kit (Qiagen). For miRNA analysis, cDNA was reverse transcribed from 10 ng of total RNA with the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher). For pri–miR-17∼92 analysis, cDNA was reverse transcribed from 20 ng of total RNA with the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). Quantitative RT-PCR (qRT-PCR) analysis was performed with TaqMan Small RNA Assay probes (Thermo Fisher), TaqMan Universal PCR Master Mix No AmpErase UNG (Thermo Fisher), and the CFX96 Touch Real-Time PCR Detection System with C1000 Thermal Cycler (Bio-Rad). Cycling conditions were 95°C for 10 minutes and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. As endogenous controls, U6 snRNA was used to normalize expression of miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, and miR-92a-1, whereas Rps17 was used for pri–miR-17∼92. Expression was analyzed using the 2−∆∆Ct method.36
For mRNA analysis, cDNA was reverse transcribed from 500 ng of total RNA with SuperScript First-Strand Synthesis System II (Thermo Fisher) with primers of Oligo dT and random hexamers. qRT-PCR analysis was performed with gene-specific primer oligos (Supplemental Table 2), SsoAdvanced SYBR Green Supermix (Bio-Rad), and the CFX96 Touch Real-Time PCR Detection System with C1000 Thermal Cycler. Cycling conditions were 95°C for 10 minutes and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Rn18S was used for endogenous control, which expression was normalized to, using the 2−∆∆Ct method.36
Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin or OCT. The paraffin-embedded tissues were sectioned at 4 μm. Hematoxylin and eosin staining was performed for histology evaluation. Semiquantitative scoring (zero to five) for tubular injury was performed in terms of tubular dilation, proteinaceous cast formation, and loss of brush border with microscopic fields at ×40 magnification that cover the entire renal cortex with an AX-500 microscope (Fisher Scientific), in a blinded fashion. The criteria are described in Supplemental Table 4. The representative images for the figures were obtained with a Leica DM2500 optical microscope and its DFC 7000T camera (Leica).
Immunolabeling was performed as described previously.37 Briefly, OCT-mounted frozen tissue sections at 10-μm thickness were used for TSP1 immunostaining. For all other immunostaining, all antigen retrieval of deparaffinized 4-μm sections was performed with 10.0 mM sodium citrate (pH 6) boiled with microwave for 15 minutes. Tissues were labeled with 1:200 rat anti-Endomucin (V.7C7; sc-65495; Santa Cruz), 1:50 rat anti-Neutrophil gelatinase–associated lipocalin (anti-NGAL; ab70287; Abcam), 1:50 mouse monoclonal anti-TSP1(A6.1) biotin (MA5–13395; Thermo Fisher), and 1:1000 rat anti-F4/80 (NB600–404; Novus Biologicals) with 1:200 secondary antibodies of donkey anti-rat IgG Alexa Fluor 594 (A-21209; Thermo Fisher), donkey anti-rat IgG Alexa Fluor 488 (A-21208; Thermo Fisher), or NeutrAvidin Dylight 594 (22842; Thermo Fisher); 1:100 FITC-conjugated Lotus Tetragonolobus Lectin (FL-1321; Vector Laboratories) was used to colabel with NGAL. F4/80-labeled tissues were visualized with the Bond Polymer Refine Detection System (DS9800; Leica). The representative images for the figures were obtained with a Leica DM2500 optical microscope and its DFC 7000T camera.
To quantify Endomucin+, F4/80+, or NGAL+ area in a tissue section, systems for tiling imaging (ECLIPSE 90i; Nikon or TissueFAXS PLUS; TissueGnostics) were used to capture the entire field of an immunolabeled kidney section with ×20 objectives. Immunostaining-labeled area in corticomedullary region was quantified with NIS Elements software (Nikon). To quantify microvasculature in kidneys, Endomucin+ glomeruli were manually excluded from the images. All tissue evaluation was performed in a blinded fashion.
The mouse kidneys were collected and immediately snap frozen after euthanasia. The protein homogenates derived from the whole-kidney tissues were immunoblotted with 1:200 goat anti-NGAL antibody (AF1757; R&D Systems). The immunoblot images for the figures were obtained with a ChemiDoc XRS+ System (Bio-Rad).
F2-isoprostanes (IsoPs) and isofurans were measured in the Vanderbilt University Eicosanoid Core Laboratory after lipid extraction from snap-frozen kidney samples using gas chromatography-mass spectrometry, as previously described.38
Data are presented as mean ± SEM. Prism 7.0C software (GraphPad) was used for statistical analysis. To determine whether sample data have been drawn from a normally distributed population, D’Agostino–Pearson Omnibus test or Shapiro–Wilk test was performed. For parametric data, t test was used to compare two different groups. Tukey multiple comparison test was used to compare multiple groups. For nonparametric data, Mann–Whitney U test was used. The threshold of P<0.05 was set to consider data statistically significant.
Results and Discussion
We first analyzed levels of all six miRNAs in isolated CD31+ renal endothelial cells from WT C57BL/6J mouse kidneys after renal IRI or sham operation (Figure 1). miR-17, miR-18a, miR-19b, and miR-20a expressions were downregulated or unchanged at day 1, upregulated and peaked at day 3, and downregulated at day 7 postrenal IRI. pri–miR-17∼92 expression was upregulated throughout the time course when compared with the sham controls. Furthermore, the expression pattern of pri–miR-17∼92 was inversely correlated to miR-17, -18, -19b, and -20a miRNAs, consistent with post-transcriptional regulation of the production of these mature miRNAs, as has been reported previously.
Because miR-17∼92 is a known proangiogenic factor,14 we hypothesized that mice with endothelial-specific loss of function experience exacerbated renal IRI. To test this, we generated a mouse model of Tie2 Cre-mediated, endothelial-specific miR-17∼92 knockout (hereafter referred to as miR-17–92endo−/−). We confirmed that Tie2 Cre was expressed in renal endothelium by colocalized expression of its R26-tdTomato reporter and an endothelial marker, endomucin, and that miR-17–92endo−/− mediated efficient knockout of miR-17∼92 in renal endothelium (Supplemental Figure 1). No difference was noted in renal function, renal vasculature, or the amount of F4/80+ resident macrophages of uninjured miR-17–92endo−/− mice compared with controls (Figures 2, A and B and 3, A, B, and D).
Figure 2.
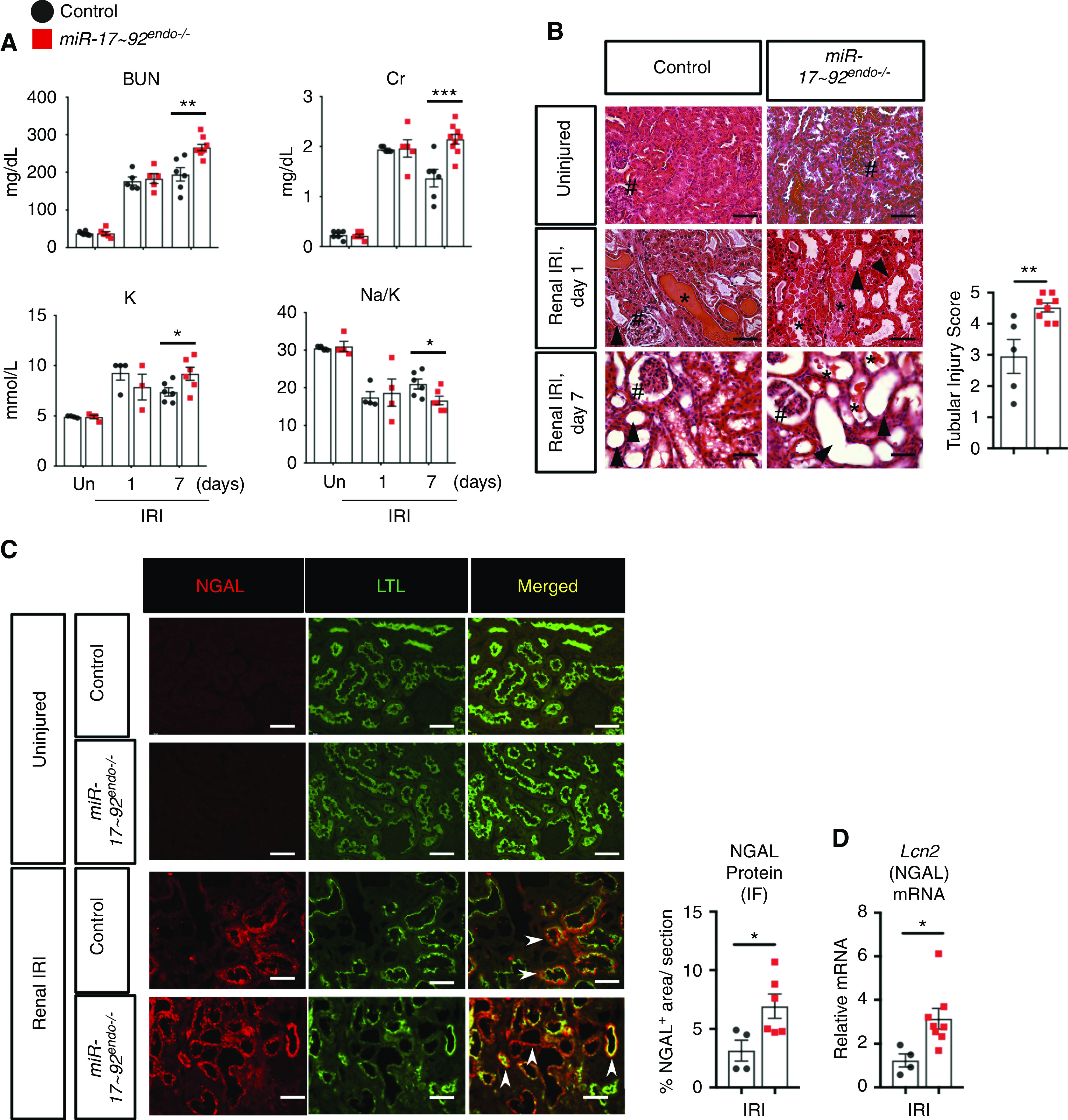
Endothelial-specific knockout of miR-17∼92 exacerbates renal IRI in male mice. (A) Serum analyses for BUN, creatinine (Cr), potassium (K), and sodium (Na)-K ratio indicate that the level of reduction of renal function is unchanged at 1 day but decreased in miR-17∼92endo−/− kidneys at 7 days postrenal IRI. n=4–6 for uninjured (Un); n=6–9 for renal IRI groups. (B) Hematoxylin and eosin–stained kidney tissues demonstrate that renal tubular injury is exacerbated in miR-17∼92endo−/− mutation kidneys 7 days postrenal IRI. Glomeruli (#), dilated tubules (arrowheads), and proteinaceous cast (*) are shown. n=5–8. Representative images and semiquantitative scoring for renal tubular damage at day 7 postrenal IRI are shown. (C and D) miR-17∼92endo−/− kidneys have increased mRNA (Lcn2) and protein levels of NGAL in whole kidneys 7 days after renal IRI. n=4–8. Arrowheads indicate NGAL+ (red)/Lotus Tetragonolobus Lectin+ (LTL+; green)–injured proximal tubules. IF, immunofluorescence (t test). Scale bars: 50 µm. *P<0.05; **P<0.01; ***P<0.001.
Figure 3.
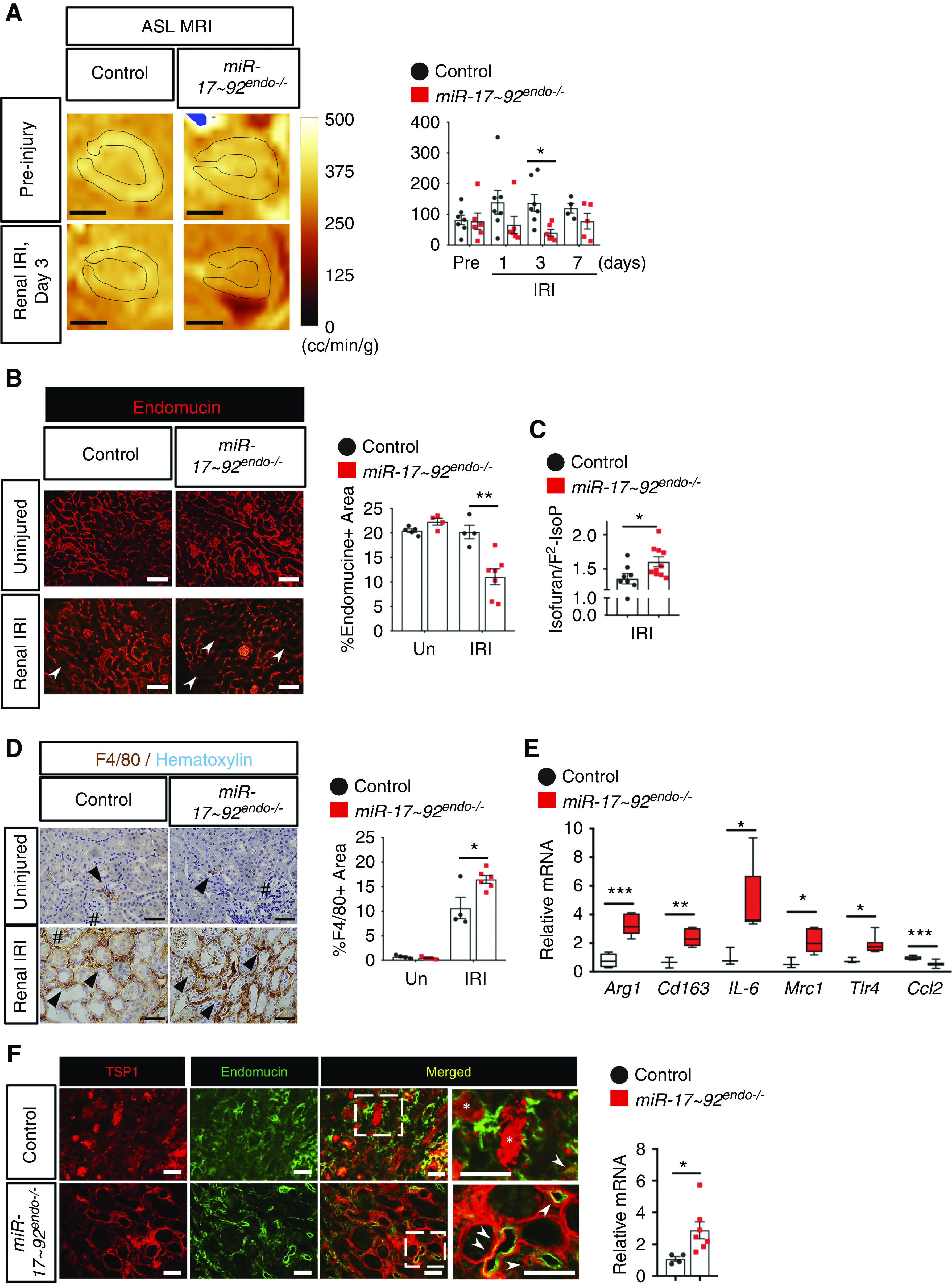
Endothelial-specific knockout of miR-17∼92 decreases microvascular function in kidneys after renal IRI. (A) ASL MRI analysis revealed that RBF is reduced in miR-17∼92endo−/− kidneys 3 days postrenal IRI (orange). Representative images for preinjury and day 3 after renal IRI are shown. MRI signals in renal cortex area with black lines were quantified. (B) Endomucin-labeled (red) renal microvasculature is decreased in miR-17∼92endo−/− kidneys 7 days postrenal IRI, whereas there is no change in uninjured (Un) kidneys. (C) The in vivo oxidative stress marker, isofuran–F2-IsoP ratio, is increased in miR-17∼92endo−/− kidneys 7 days postrenal IRI. (D) F4/80-labeled (brown) infiltrating macrophages are increased in miR-17∼92endo−/− kidneys 7 days postrenal IRI. (E) There is upregulation of the infiltrating macrophage markers in miR-17∼92endo−/− kidneys 7 days postrenal IRI using qRT-PCR. Data normalized to Rn18S, n=3–8 (F) miR-17∼92endo−/− kidneys have increased mRNA (Thbs1) levels of TSP1 7 days postrenal IRI, n=4–7. TSP1 is colocalized with renal microvasculature 7 days postrenal IRI (arrowheads). TSP1 is also expressed in renal tubular epithelium (*). The dotted box areas are magnified in the images on the right. t test. Endomucin+ and F4/80+ areas were quantified by tiling imaging methods. Glomeruli (#) and F4/80+ cells (arrowheads) are shown. Scale bars: 2.5 mm in A; 100 µm in B; 50 µm in D and F. *P<0.05; **P<0.01; ***P<0.001.
We next performed renal IRI on male miR-17–92endo−/− and Cre-negative littermate controls. miR-17–92endo−/− had increased renal injury 7 days postrenal IRI, assessed by increased serum levels of BUN, creatinine, and potassium, with a decrease in Na-K ratio (Figure 2A), as well as increased serum phosphorus and decreased serum bicarbonate (Supplemental Figure 3). Hematoxylin and eosin–stained renal tissue further confirmed the presence of increased renal tubular injury in miR-17–92endo−/− kidneys at day 7 (Figure 2B, arrowheads, Supplemental Figure 2). NGAL is an established injury marker for kidney tubules in mice and humans. It is expressed in a punctate cytoplasmic pattern in both proximal tubules and in multiple cell types in the distal segments.39,40 Levels of NGAL mRNA (Lcn2) and protein were increased in miR-17–92endo−/− kidneys 7 days postrenal IRI (Figure 2, C and D, Supplemental Figure 4). There were more widespread NGAL+ Lotus Tetragonolobus Lectin+ proximal tubular epithelial cells in miR-17–92endo−/− compared with controls (Figure 2C, arrowheads). Consistent results were obtained with the female mice (Supplemental Figure 5), suggesting that our results were independent of sex. The differences in kidney injury were not observed at 1 day postrenal IRI, suggesting that endothelial miR-17∼92 is important after this time point.
Ischemic AKI in mice is also characterized by reduction of RBF4 and microvascular rarefaction,41 which is often associated with incomplete endothelial repair after injury.5 We next hypothesized that miR-17–92endo−/− impairs the function of the renal vasculature. ASL MRI, which allows noninvasive/longitudinal evaluation of RBF with its excellent quantitative capacity,42 demonstrated that the cortex areas of miR-17–92endo−/− kidneys had reduced RBF 3 days postrenal IRI (Figure 3A). A consistent trend toward a decrease in RBF at day 1 postrenal IRI by ASL MRI was confirmed by laser Doppler analysis (Supplemental Figure 7). Consistent with this, endomucin-labeled renal microvasculature was decreased in miR-17–92endo−/− 7 days postrenal IRI when compared with controls (Figure 3B).
Endothelial damage by renal IRI increases infiltration of inflammatory leukocytes, including macrophages, to the kidneys and promotes AKI.4,43,44 F4/80+ macrophages are increased in miR-17–92endo−/− kidneys (Figure 3D). Furthermore, qRT-PCR analysis revealed that multiple macrophage markers of M1 and M2 types were increased in miR-17–92endo−/− kidneys 7 days postrenal IRI (Figure 3E). Interestingly, mRNA levels of a monocyte recruiting factor, Ccl2, were downregulated in miR-17–92endo−/− kidneys. These data may suggest that infiltrated monocytes are stuck in the kidney and are unable to exit from the miR-17–92endo−/− kidneys at day 7 due to enhanced microvascular damage. Oxidative stress induces endothelial senescence that limits angiogenesis, and miR-17∼92 is known to limit reactive oxygen species generation in lung cancer cells.45 Thus, we measured the isofuran–F2-IsoP ratio, a well-established in vivo oxidative stress marker.38 The isofuran–F2-IsoP ratio was increased in miR-17∼92endo−/− kidneys 7 days postrenal IRI (Figure 3C), suggesting that lack of endothelial-derived miR-17∼92 increases renal oxidative stress postrenal IRI. Together, these data suggest that miR-17–92endo−/− kidneys exacerbate functional impairment of renal vasculature after renal IRI.
Next, we explored putative downstream target genes for miR-17∼92 in the renal endothelium during renal IRI. By using a previously published translating ribosome affinity purification microarray dataset of upregulated genes in renal endothelial cells 24 hours postrenal IRI,34,46 we performed a computational prediction for miR-17∼92 targets using TargetScan 7.1.35 Focusing on the four miR-17∼92 members that were upregulated after renal IRI, we identified 407 predicted genes that are targeted by miR-17/20 (183), miR-18a (44), and/or miR-19a/b (180) (Supplemental Table 1). Pathway analysis for the predicted miR-17∼92 targets revealed that multiple angiogenesis-associated pathways, including extracellular signal–regulated kinase 5 (ERK5), p53, and HIF1α signaling, were suggested to be regulated by miR-17∼92 in renal endothelial cells after renal IRI (Supplemental Figure 8). ERK5 was a common downstream pathway for targets of miR-17/20, miR-18a, and miR-19a/b and is known to negatively regulate VEGF signaling.47 Furthermore, TSP1 is a potent antiangiogenic factor and a well-characterized miR-17∼92 target.13 Our computational prediction also identified TSP1 as a target for miR-18a and miR-19a/b (Supplemental Table 1, B and C). Consistent with this, Thbs1 (TSP1) mRNA was increased in miR-17–92endo−/− kidneys 7 days postrenal IRI (Figure 3F), and TSP1 protein was highly expressed in endomucin-labeled renal microvasculature (Figure 3F, arrowheads). These results suggest that miR-17–92 suppresses antiangiogenic factors and thus, increases the angiogenic capacity of renal endothelial cells in response to renal IRI.
Our data revealed that miR-18a and miR-19b were the most highly expressed miR-17∼92 family members in renal endothelial cells 3 days postrenal IRI (Figure 1). We hypothesized that pharmacologic cotreatment of miRNA mimics for miR-18a and miR-19b mitigates renal IRI. Biodistribution of FITC-labeled RNA oligos to the kidneys was confirmed (Supplemental Figures 9–11). We then demonstrated that daily cotreatment of miR-18a/miR-19b miRNA mimics for WT FVB/NJ male mice, starting 24 hours prior to renal IRI for 8 days, decreases serum levels of BUN and potassium, increases Na-K ratio, and reduces renal tubular injury (Figure 4, A–C). Endomucin-labeled renal microvasculature was increased in miR-18a/-19b–treated kidneys, suggesting that the mimics act on the renal microvasculature for its beneficial effects (Figure 4D).
Figure 4.
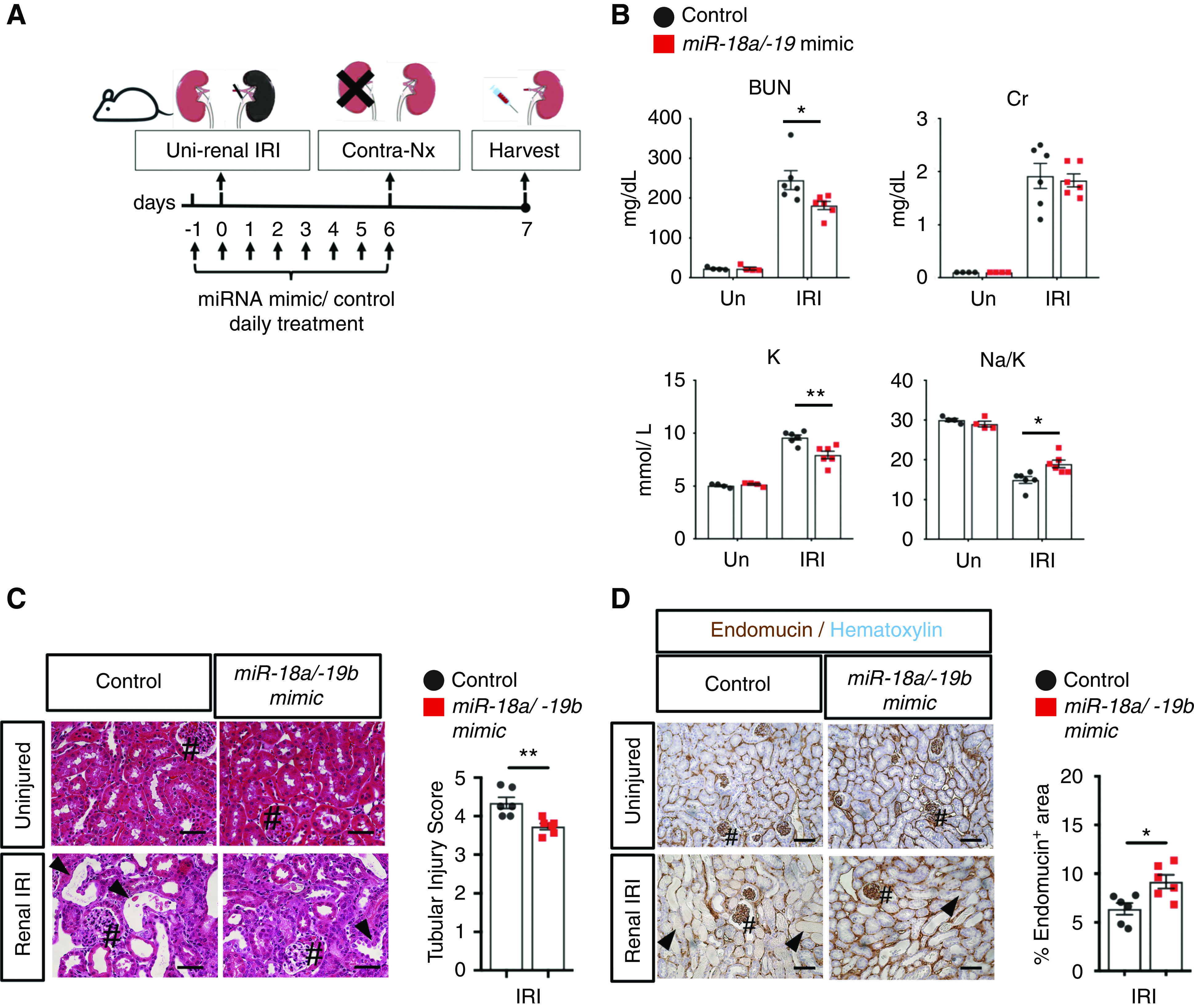
Pharmacologic cotreatment with miR-18a and miR-19b mimics mitigates renal IRI. (A) Schematic for renal IRI model with WT FVB/NJ male mice and mimic treatment regimen (details are in Methods). (B) Serum analyses for BUN, creatinine (Cr), potassium (K), and sodium (Na)-K ratio measured at 7 days postrenal IRI indicate that deterioration of renal function is mitigated by cotreatment with miR-18a/miR-19b mimics administered daily starting 24 hours prior to renal IRI for 8 days, whereas there is no change in uninjured (Un) kidneys. n=4–6. (C) Hematoxylin and eosin–stained kidney tissues demonstrate that renal tubular injury is mitigated by cotreatment of miR-18a/miR-19b mimics when evaluated at 7 days postrenal IRI. n=4–6. Glomeruli (#) and dilated tubules (arrowheads) are shown. Representative images and semiquantitative score for renal tubular injury score are shown. (D) Endomucin-labeled (brown) renal microvasculature is increased in the mimic-treated kidneys 7 days postrenal IRI. Glomeruli (#) and vascular rarefaction (arrowheads) are shown. t test. Scale bars: 50 µm. *P<0.05; **P<0.01.
This study revealed that renal endothelial cells dynamically regulate miR-17, miR-18a, miR-19b, and miR-20 in the miR-17∼92 cluster in response to renal IRI. These miRNAs were predicted to target antiangiogenesis pathways, including ERK5 signaling. Our genetic loss-of-function study revealed the critical role for endothelial-derived miR-17∼92 in the renal vasculature during renal IRI. Lack of endothelial-derived miR-17∼92 results in impaired endothelial function and stimulates expression of a potent antiangiogenic factor, TSP1, in the renal microvasculature. Consistent with the angiogenic role of miR-17∼92 known only in vitro or in cancer settings,13,14 our novel in vivo findings demonstrate the angiogenic capacity of mIR-17∼92 in the setting of renal IRI. Finally, we demonstrate that pharmacologic intervention by miR-18a/miR-19b mimics robustly protects against renal IRI. Oligonucleotide drugs are a promising therapeutic modality with their high target specificity and improved pharmacokinetic properties.48 This study provides the basis for developing delivery of miR-18a and miR-19b as a potential novel AKI therapy.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by Children’s Hospital of Pittsburgh Research Advisory Council Postdoctoral Fellowship (to D.M. Cerqueira); Nephrotic Syndrome Study Network (NEPTUNE) Career Development Award (to D.M. Cerqueira); American Heart Association postdoctoral fellowship 17POST33670685 (to T. Chiba); Richard K. Mellon Institute Award for Postdoctoral Trainees (to T. Chiba); National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants F31-DK116338 (to S.L. Hemker), and T32-DK061296 (to S.L. Hemker), R01-DK103776 (to J. Ho), and R01-DK125015 (to J. Ho and S. Sims-Lucas); University of Pittsburgh Medical Center Vascular Medicine Institute grant P3HVBVM (to J. Ho); and American Society of Nephrology Ben J. Lipps Research Fellowship Program (to Y.L. Phua).
Supplementary Material
Acknowledgments
The Center for Biologic Imaging provided expert microscopy support. We thank the UPMC Children’s Hospital of Pittsburgh Histology Core Laboratory and the Kansas State University Veterinary Diagnostic Laboratories for technical assistance. This work was previously presented in part at the annual meetings of the American Society for Nephrology in November 7–10, 2019 and October 19-25, 2020, and was published in abstract form.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020050717/-/DCSupplemental.
Supplemental Figure 1. miR-17∼92endo−/− shows endothelial-specific knockout of miR-17∼92 in kidneys.
Supplemental Figure 2. High– and low–power field images of miR-17∼92endo−/− or its controls postrenal IRI.
Supplemental Figure 3. Endothelial-specific knockout of miR-17∼92 increases serum phosphorus and decreases serum bicarbonate postrenal IRI.
Supplemental Figure 4. Endothelial-specific knockout of miR-17∼92 shows a trend to increase NGAL expression postrenal-IRI.
Supplemental Figure 5. Endothelial-specific knockout of miR-17∼92 exacerbates renal IRI in female mice.
Supplemental Figure 6. Cortex areas of left kidneys in ASL MRI images are demarcated by referring to each corresponding anatomical MRI image.
Supplemental Figure 7. Laser Doppler analysis reveals that endothelial-specific knockout of miR-17∼92 shows a trend to decrease of renal blood flow postrenal
IRI.
Supplemental Figure 8. Angiogenesis-associated pathways were predicted to be targeted by miR-17∼92 in renal endothelial cells after renal IRI.
Supplemental Figure 9. Renal IRI model with delayed contralateral nephrectomy induces AKI in an ischemia time-dependent manner at day 7 postinjury.
Supplemental Figure 10. Intravenously administered RNA oligo molecules are delivered to the kidneys.
Supplemental Figure 11. High– and low–power field images of miR-18a/-19b–treated or control-treated kidneys with or without renal IRI at day 7.
Supplemental Table 1. Putative miR-17∼92 targets that are upregulated in renal endothelial cells during ischemic AKI.
Supplemental Table 2. Primer sequences for mRNA qRT-PCR analysis.
Supplemental Table 3. Primer sequences for genotyping miR-17∼92endo−/− mice.
Supplemental Table 4. Criteria for renal tubular injury scoring.
Supplemental Material. Discussion and acknowledgments.
References
- 1.Le Dorze M, Legrand M, Payen D, Ince C: The role of the microcirculation in acute kidney injury. Curr Opin Crit Care 15: 503–508, 2009. 10.1097/MCC.0b013e328332f6cf [DOI] [PubMed] [Google Scholar]
- 2.Sena CM, Leandro A, Azul L, Seiça R, Perry G: Vascular oxidative stress: Impact and therapeutic approaches. Front Physiol 9: 1668, 2018. 10.3389/fphys.2018.01668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aksu K, Donmez A, Keser G: Inflammation-induced thrombosis: Mechanisms, disease associations and management. Curr Pharm Des 18: 1478–1493, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Bonventre JV, Yang L: Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011. 10.1172/JCI45161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basile DP, Donohoe D, Roethe K, Osborn JL: Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. 10.1152/ajprenal.2001.281.5.F887 [DOI] [PubMed] [Google Scholar]
- 6.Carthew RW, Sontheimer EJ: Origins and Mechanisms of miRNAs and siRNAs. Cell 136: 642–655, 2009. 10.1016/j.cell.2009.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebert MS, Sharp PA: Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524, 2012. 10.1016/j.cell.2012.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al.: A microRNA polycistron as a potential human oncogene. Nature 435: 828–833, 2005. 10.1038/nature03552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrone AK, Stolz DB, Bastacky SI, Kostka D, Bodnar AJ, Ho J: MicroRNA-17∼92 is required for nephrogenesis and renal function. J Am Soc Nephrol 25: 1440–1452, 2014. 10.1681/ASN.2013040390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel V, Williams D, Hajarnis S, Hunter R, Pontoglio M, Somlo S, et al.: miR-17∼92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A 110: 10765–10770, 2013. 10.1073/pnas.1301693110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, et al.: microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun 8: 14395, 2017. 10.1038/ncomms14395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yheskel M, Lakhia R, Cobo-Stark P, Flaten A, Patel V: Anti-microRNA screen uncovers miR-17 family within miR-17∼92 cluster as the primary driver of kidney cyst growth. Sci Rep 9: 1920, 2019. 10.1038/s41598-019-38566-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, et al.: Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38: 1060–1065, 2006. 10.1038/ng1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chamorro-Jorganes A, Lee MY, Araldi E, Landskroner-Eiger S, Fernández-Fuertes M, Sahraei M, et al.: VEGF-induced expression of miR-17-92 cluster in endothelial cells is mediated by ERK/ELK1 activation and regulates angiogenesis. Circ Res 118: 38–47, 2016. 10.1161/CIRCRESAHA.115.307408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thakar CV, Zahedi K, Revelo MP, Wang Z, Burnham CE, Barone S, et al.: Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J Clin Invest 115: 3451–3459, 2005. 10.1172/JCI25461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao M, Rogers NM, Csányi G, Rodriguez AI, Ross MA, St Croix C, et al.: Thrombospondin-1 activation of signal-regulatory protein-α stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J Am Soc Nephrol 25: 1171–1186, 2014. 10.1681/ASN.2013040433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al.: mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 112: 1557–1566, 2013. 10.1161/CIRCRESAHA.112.300658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao F, Kataoka M, Liu N, Liang T, Huang ZP, Gu F, et al.: Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat Commun 10: 1802, 2019. 10.1038/s41467-019-09530-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landskroner-Eiger S, Qiu C, Perrotta P, Siragusa M, Lee MY, Ulrich V, et al.: Endothelial miR-17∼92 cluster negatively regulates arteriogenesis via miRNA-19 repression of WNT signaling. Proc Natl Acad Sci U S A 112: 12812–12817, 2015. 10.1073/pnas.1507094112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandenburger T, Salgado Somoza A, Devaux Y, Lorenzen JM: Noncoding RNAs in acute kidney injury. Kidney Int 94: 870–881, 2018. 10.1016/j.kint.2018.06.033 [DOI] [PubMed] [Google Scholar]
- 21.Xu X, Kriegel AJ, Liu Y, Usa K, Mladinov D, Liu H, et al.: Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int 82: 1167–1175, 2012. 10.1038/ki.2012.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao J, Lou Q, Wei Q, Mei S, Li L, Wu G, et al.: MicroRNA-375 is induced in cisplatin nephrotoxicity to repress hepatocyte nuclear factor 1-β. J Biol Chem 292: 4571–4582, 2017. 10.1074/jbc.M116.754929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bijkerk R, van Solingen C, de Boer HC, de Vries DK, Monge M, van Oeveren-Rietdijk A, et al.: Silencing of miRNA-126 in kidney ischemia reperfusion is associated with elevated SDF-1 levels and mobilization of Sca-1+/Lin- progenitor cells. MicroRNA 3: 144–149, 2014. 10.2174/2211536604666150121000340 [DOI] [PubMed] [Google Scholar]
- 24.Bijkerk R, van Solingen C, de Boer HC, van der Pol P, Khairoun M, de Bruin RG, et al.: Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J Am Soc Nephrol 25: 1710–1722, 2014. 10.1681/ASN.2013060640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao J, Wei Q, Mei S, Li L, Su Y, Mei C, et al.: Induction of microRNA-17-5p by p53 protects against renal ischemia-reperfusion injury by targeting death receptor 6. Kidney Int 91: 106–118, 2017. 10.1016/j.kint.2016.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song T, Chen M, Rao Z, Qiu Y, Liu J, Jiang Y, et al.: miR-17-92 ameliorates renal ischemia reperfusion injury. Kaohsiung J Med Sci 34: 263–273, 2018. 10.1016/j.kjms.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Proctor JM, Zang K, Wang D, Wang R, Reichardt LF: Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci 25: 9940–9948, 2005. 10.1523/JNEUROSCI.3467-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al.: A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140, 2010. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, et al.: Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat Genet 43: 1026–1030, 2011. 10.1038/ng.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Xu B, Tian GG, Sun T, Wu J: Ablation of the MiR-17-92 microRNA cluster in germ cells causes subfertility in female mice. Cell Physiol Biochem 45: 491–504, 2018. 10.1159/000487028 [DOI] [PubMed] [Google Scholar]
- 31.Hurtado A, Real FM, Palomino R, Carmona FD, Burgos M, Jiménez R, et al.: Sertoli cell-specific ablation of miR-17-92 cluster significantly alters whole testis transcriptome without apparent phenotypic effects. PLoS One 13: e0197685, 2018. 10.1371/journal.pone.0197685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skrypnyk NI, Harris RC, de Caestecker MP: Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J Vis Exp 78: 50495, 2013. 10.3791/50495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu YL, Lo CW: Diverse application of MRI for mouse phenotyping. Birth Defects Res 109: 758–770, 2017. 10.1002/bdr2.1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Krautzberger AM, Sui SH, Hofmann OM, Chen Y, Baetscher M, et al.: Cell-specific translational profiling in acute kidney injury [published correction appears in J Clin Invest 124: 2288, 2014]. J Clin Invest 124: 1242–1254, 2014. 10.1172/JCI72126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agarwal V, Bell GW, Nam JW, Bartel DP: Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005, 2015. 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta C(T)) Method. Methods 25: 402–408, 2001. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 37.Mukherjee E, Maringer K, Papke E, Bushnell D, Schaefer C, Kramann R, et al.: Endothelial marker-expressing stromal cells are critical for kidney formation. Am J Physiol Renal Physiol 313: F611–F620, 2017. 10.1152/ajprenal.00136.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milne GL, Gao B, Terry ES, Zackert WE, Sanchez SC: Measurement of F2- isoprostanes and isofurans using gas chromatography-mass spectrometry. Free Radic Biol Med 59: 36–44, 2013. 10.1016/j.freeradbiomed.2012.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuwabara T, Mori K, Mukoyama M, Kasahara M, Yokoi H, Saito Y, et al.: Urinary neutrophil gelatinase-associated lipocalin levels reflect damage to glomeruli, proximal tubules, and distal nephrons. Kidney Int 75: 285–294, 2009. 10.1038/ki.2008.499 [DOI] [PubMed] [Google Scholar]
- 40.Langelueddecke C, Roussa E, Fenton RA, Wolff NA, Lee WK, Thévenod F: Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J Biol Chem 287: 159–169, 2012. 10.1074/jbc.M111.308296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kramann R, Tanaka M, Humphreys BD: Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J Am Soc Nephrol 25: 1924–1931, 2014. 10.1681/ASN.2013101121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tewes S, Gueler F, Chen R, Gutberlet M, Jang MS, Meier M, et al.: Functional MRI for characterization of renal perfusion impairment and edema formation due to acute kidney injury in different mouse strains. PLoS One 12: e0173248, 2017. 10.1371/journal.pone.0173248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huen SC, Cantley LG: Macrophages in renal injury and repair. Annu Rev Physiol 79: 449–469, 2017. 10.1146/annurev-physiol-022516-034219 [DOI] [PubMed] [Google Scholar]
- 44.Chiba T, Skrypnyk NI, Skvarca LB, Penchev R, Zhang KX, Rochon ER, et al.: Retinoic acid signaling coordinates macrophage-dependent injury and repair after AKI. J Am Soc Nephrol 27: 495–508, 2016. 10.1681/ASN.2014111108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ebi H, Sato T, Sugito N, Hosono Y, Yatabe Y, Matsuyama Y, et al.: Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 28: 3371–3379, 2009. 10.1038/onc.2009.201 [DOI] [PubMed] [Google Scholar]
- 46.Chiba T, Hukriede N, de Caestecker MP: Kidney regeneration: Lessons from development. Curr Pathobiol Rep 3: 67–79, 2015. 10.1007/s40139-015-0069-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sohn SJ, Sarvis BK, Cado D, Winoto A: ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem 277: 43344–43351, 2002. 10.1074/jbc.M207573200 [DOI] [PubMed] [Google Scholar]
- 48.Khvorova A, Watts JK: The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 35: 238–248, 2017. 10.1038/nbt.3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.