

Graphical Abstract
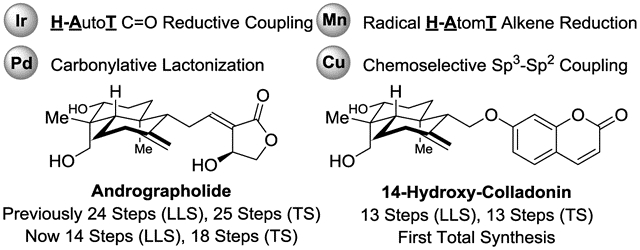
King of Bitters. An enantioselective total synthesis of the labdane diterpenoid andrographolide, the bitter principle of the medicinal herb Andrographis paniculata (known as “King of Bitters”), is accomplished in 14 steps (LLS) - 10 steps shorter than the prior total synthesis. Key transformations include iridium-catalyzed carbonyl reductive coupling to form the quaternary carbon stereocenter at C4, diastereoselective alkene reduction to establish the trans-decalin ring, and carbonylative lactonization to install the α-alkylidene-γ-butyrolactone.
Keywords: Enantioselective, Iridium, Labdane Diterpenoid, Hydrogen Transfer
Terpenoid natural products comprise a broad and structurally diverse class of secondary metabolites with applications spanning the fields of human medicine, agrochemistry and flavor/fragrance science.[1] The challenges posed by de novo terpenoid construction have evoked numerous innovative synthetic methods and synthesis design concepts. The vast majority of approaches rely on polyolefin cascade cyclizations,[2,3] which often require lengthy syntheses of the requisite polycyclization precursors. As recently illustrated to great effect, convergent routes to terpene natural products have the potential to be exceptionally concise.[4] Exploiting a transfer hydrogenative coupling of primary alcohols with isoprene oxide developed in our laboratory, products of carbonyl addition bearing acyclic quaternary carbon stereocenters could be generated with excellent control of diastereo- and enantioselectivity (Figure 1).[5,6,7] These adducts embody a tert-(hydroxy)-prenyl substructure that is found in over 2000 terpenoid natural products. Using this method, convergent syntheses of the terpenoid natural products oridamycin A,[8,9] triptoquinones B and C,[10,11] and isoiresin[12,13] were accomplished in fewer steps than previously possible (Scheme 1).14 The former 3 natural products all derive from a common intermediate, Fragment A, that is diversified through Suzuki cross-coupling followed by Friedel-Crafts cyclization (Scheme 1).[8]
Figure 1.
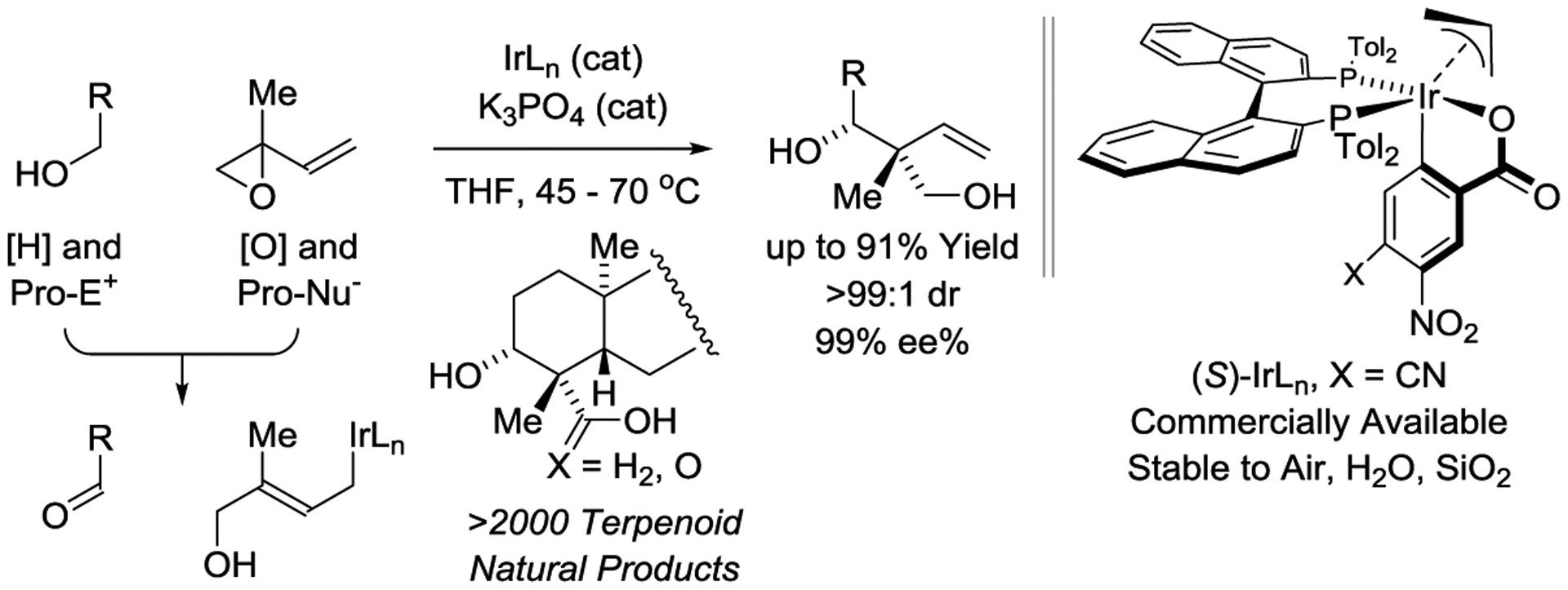
Entry to terpenoid natural products via enantioselective iridium-catalyzed carbonyl reductive coupling mediated by hydrogen auto-transfer.
Scheme 1.
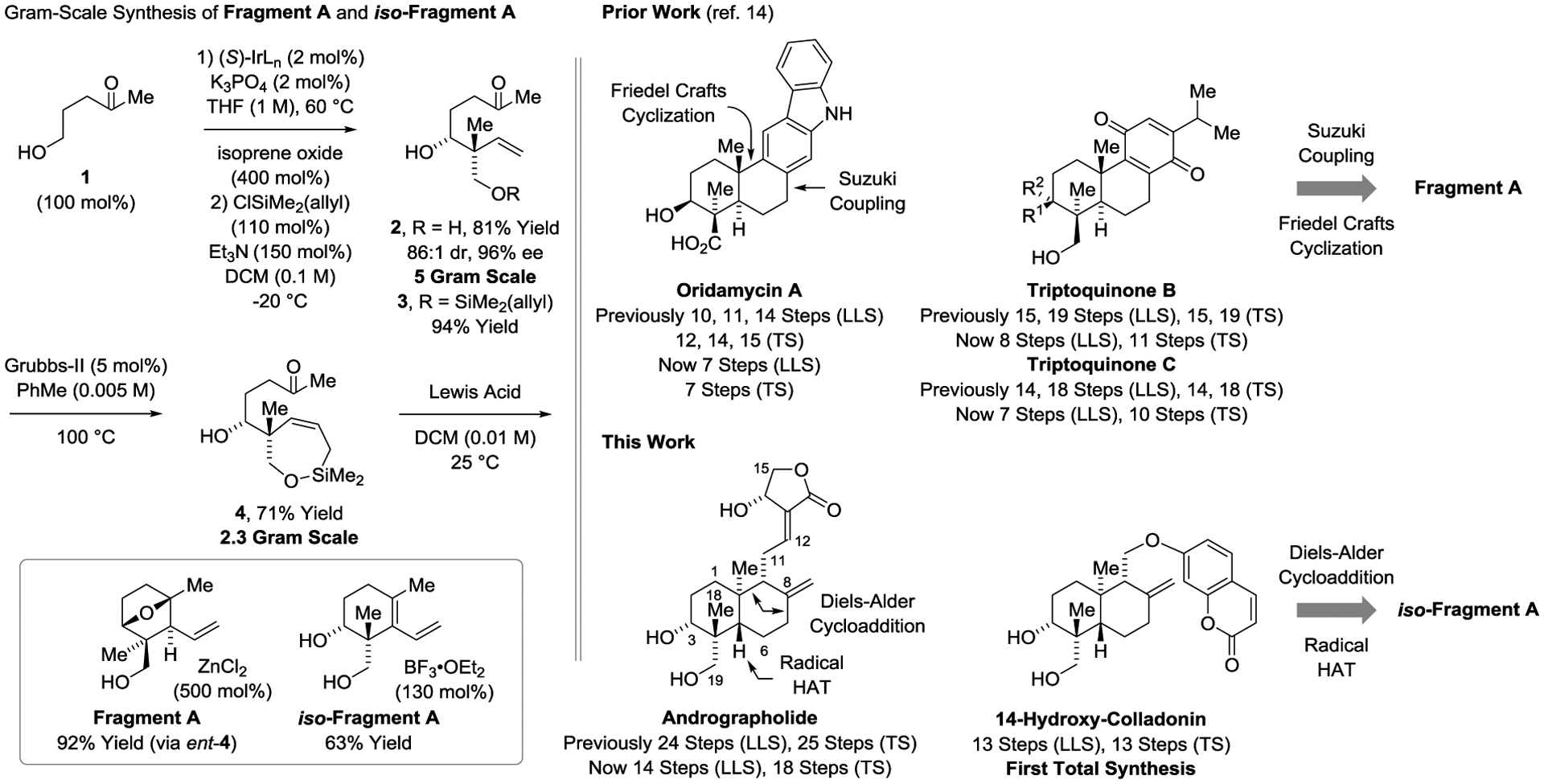
Linear divergent strategies for the total synthesis of terpenoid natural products via iridium-catalyzed carbonyl reductive coupling of 5-hydroxy-2-pentanone with isoprene oxide.
In the course of these studies, a divergent outcome in the intramolecular Sakurai allylation15 used to prepare Fragment A from compound 4 was observed (Scheme 1). Compound 4, which is prepared in 3 steps via isoprene oxide-mediated tert-(hydroxy)-prenylation[5] of commercially available 5-hydroxy-2-pentanone, exists in equilibrium with it’s 5-membered lactol. Using ZnCl2 as the Lewis acid, intramolecular Sakurai allylation[15] occurs by way of an endocyclic oxacarbenium ion to form the [2.2.1]oxabicycle Fragment A. However, using BF3•OEt2 as the Lewis acid, Fragment A ring opens and eliminates to deliver the conjugated diene iso-Fragment A. Access to iso-Fragment A raised the possibility of further broadening our convergent approach to terpenoid construction by opening linear divergent routes to other terpenoid natural products through successive Diels–Alder cycloaddition-olefin reduction to generate a trans-decalin motif, as initially communicated in our total synthesis of isoiresin. [12,13,14] In the present article, use of this strategy as a general point of entry to diverse terpenoids is demonstrated by the enantioselective total synthesis of the labdane diterpenoid andrographolide[16–19] in 14 steps (LLS) - 10 steps shorter than the prior synthesis[19] and the first total syntheses of the coumarin-containing sesquiterpene 14-hydroxy-colladonin[20] in 13 steps (LLS), which corroborates its structural and absolute stereochemical assignment (Figure 1).
The labdane diterpenoid andrographolide is the crystalline bitter principle of the annual herbaceous plant Andrographis paniculata of the family Acanthaceae, which, among many other names, is known as “King of Bitters”.[16] Native to India and Sri Lanka and used for centuries in Ayurvedic medicine, it has since entered the Chinese pharmacopeia and is now used throughout tropical regions of Asia and the West Indies. Andrographolide was first isolated in pure form in 1911,[16b] but it was not until 1963[16h] that the correct connectivity of andrographolide was proposed, and not until 1984 that it’s complete stereochemical assignment was determined by single crystal X-ray diffraction.[16j] Tinctures of Andrographis paniculata are used in traditional medicine to treat an unusually broad range of indications and, as documented in numerous reviews,[17] literally thousands of investigations into the biological properties of andrographolide have been conducted. As reported in a 2014 quantitative proteomics screen, the myriad phenotypic effects of andrographolide may derive from its extensive adduction of proteinaceous cysteine residues.[18a] Dehydroandrographolide inhibits TMEM16A expression,[18b] which is amplified in certain cancers,[18c] demonstrating derivatives of andrographolide can have translatable effects in human cells. Remarkably, despite the enormous attention andrographolide has received, only one total synthesis has been reported.[19]
Our approach to andrographolide begins with the transfer hydrogenative tert-(hydroxy)-prenylation[5] of commercially available 5-hydroxy-2-pentanone 1 (Scheme 1).[14] It was found that the more Lewis acidic (S)-Tol-BINAP modified π-allyliridium-C,O-benzoate derived from 3,4-dinitro-C,O-benzoic acid (IrLn, X = NO2) performed better than our standard catalyst (IrLn, X = CN). On 5 gram scale using low loadings of catalyst (2 mol%), the product of C–C coupling 2 could be obtained in 81% yield, 96% ee and 86:1 dr. Chemoselective allyldimethylsilylation of the primary alcohol followed by ring closing metathesis gives the cyclic allylsilane 4,[21] which upon BF3•OEt2 promoted intramolecular Sakurai allylation delivers iso-Fragment A. The Diels–Alder reaction of iso-Fragment A, which creates a quaternary carbon stereocenter, posed several challenges (Scheme 2).[22] iso-Fragment A itself could not be engaged in Diels–Alder cycloaddition, due to side-reactions associated with unprotected diol. Using the bis-pivalate derived from iso-Fragment A (compound 5), only highly activated dienophiles were effective partners for cycloaddition. Upon use of dimethyl acetylene dicarboxylate (DMAD), the desired cycloadduct 7 was formed as a 10:1 mixture of diastereomers. Cycloadditions of the corresponding bis-acetate and bis-isobutyrate gave 2.5:1 and 4.5:1 diastereomeric mixtures, respectively (not shown). It was posited that acetonide 6 might undergo cycloaddition with complete diastereofacial selectivity from the convex face of the bicycle. Indeed, cycloadduct 8 is formed as a single stereoisomer. Formation of the trans-decalin ring system of andrographolide was attempted next. Manganese-catalyzed hydrogen atom transfer (HAT) was selected for this purpose due to its ability to affect alkene hydrogenation with thermodynamically controlled diastereoselectivities, including formation of trans-decalins from precursors related in structure to cycloadduct 8.[23] In the event, HAT reduction of cycloadduct 8 occurs in a completely chemoselective manner, but with exclusive formation of the undesired cis-decalin 9, which was characterized by single crystal X-ray diffraction. This outcome was attributed to the conformational constraint imposed by the acetonide moiety, as HAT reduction of diol 10 delivers the trans-decalin 11 as a single diastereomer. The magnitude of the indicated 1H NMR coupling constants between the indicated C3 methine hydrogen and the adjacent C2 methylene moieties for acetonide 8 versus diol 10 suggests the acetonide inverts the preferred chair-like conformer such that the C3 methine hydrogen is equatorially disposed.24 Based on this insight, stereochemical models that account for the observed stereodivergence in the HAT reductions of compounds 8 and 10 were proposed (Scheme 2).
Scheme 2.
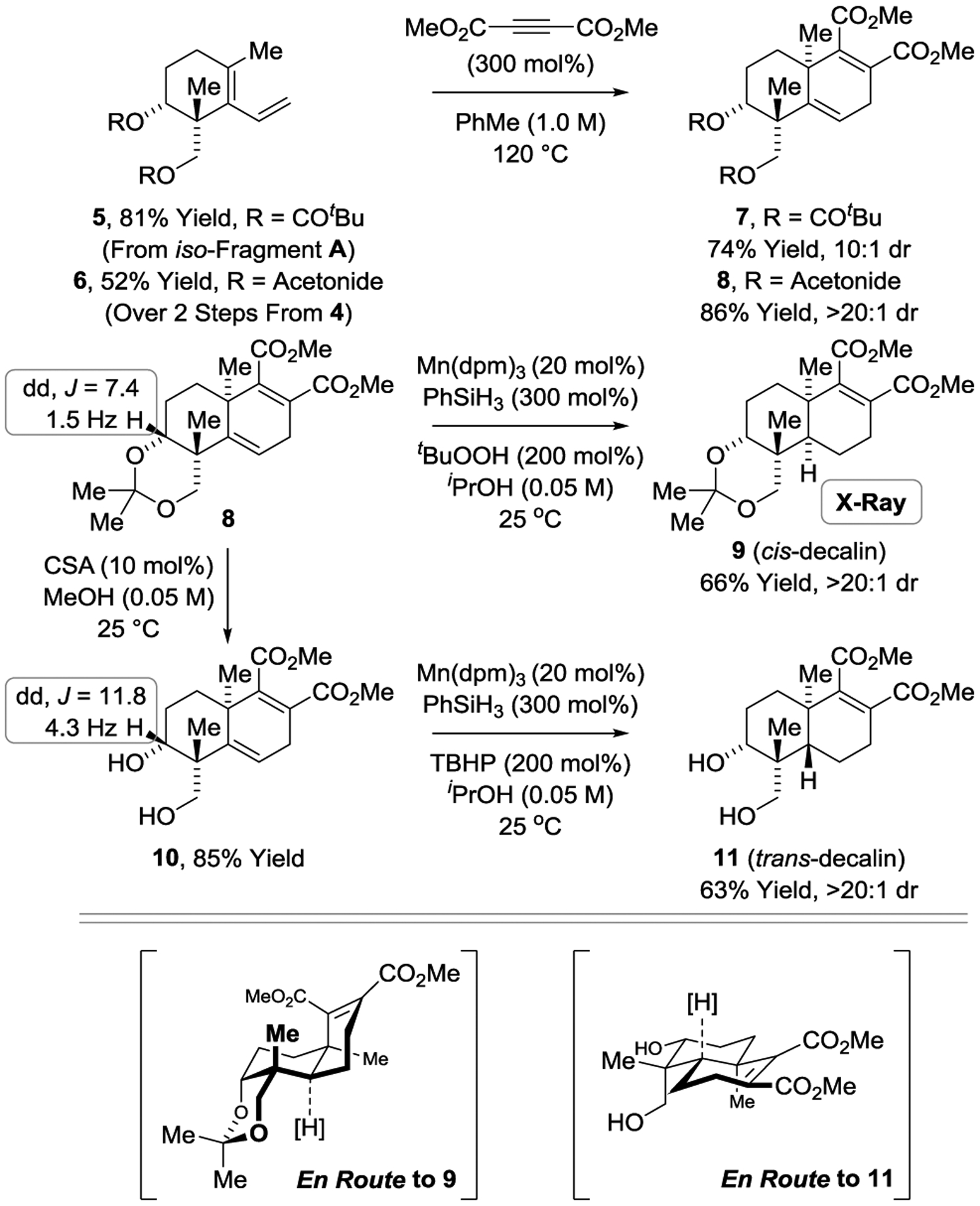
Stereodivergence in HAT reductions of Diels–Alder cycloadducts 8 and 10.
With this insight into the requirements for trans-decalin formation, the total synthesis of andrographolide was undertaken (Scheme 3). Diels–Alder cycloaddition of diene 6 with DMAD followed by hydrolysis of the acetonide in situ delivered cycloadduct 10 as a single diastereomer. Manganese-catalyzed HAT reduction[23] of 10 provides the trans-decalin 11, which upon successive treatment with triisopropylsilyl triflate (TIPSOTf) and diisobutyl aluminum hydride (DIBAL-H) furnishes the ene-diol 12. Reductive transposition of the less hindered allylic alcohol of 12 under Myers conditions[25] occurs in a completely chemoselective fashion to furnish homoallylic alcohol 13 as a 7:1 diastereomeric mixture at the newly formed C9 stereocenter. Using a modification of Appel’s method,[26] homoallylic alcohol 13 was converted to iodide 14, which was isolated as a single diastereomer.
Scheme 3.
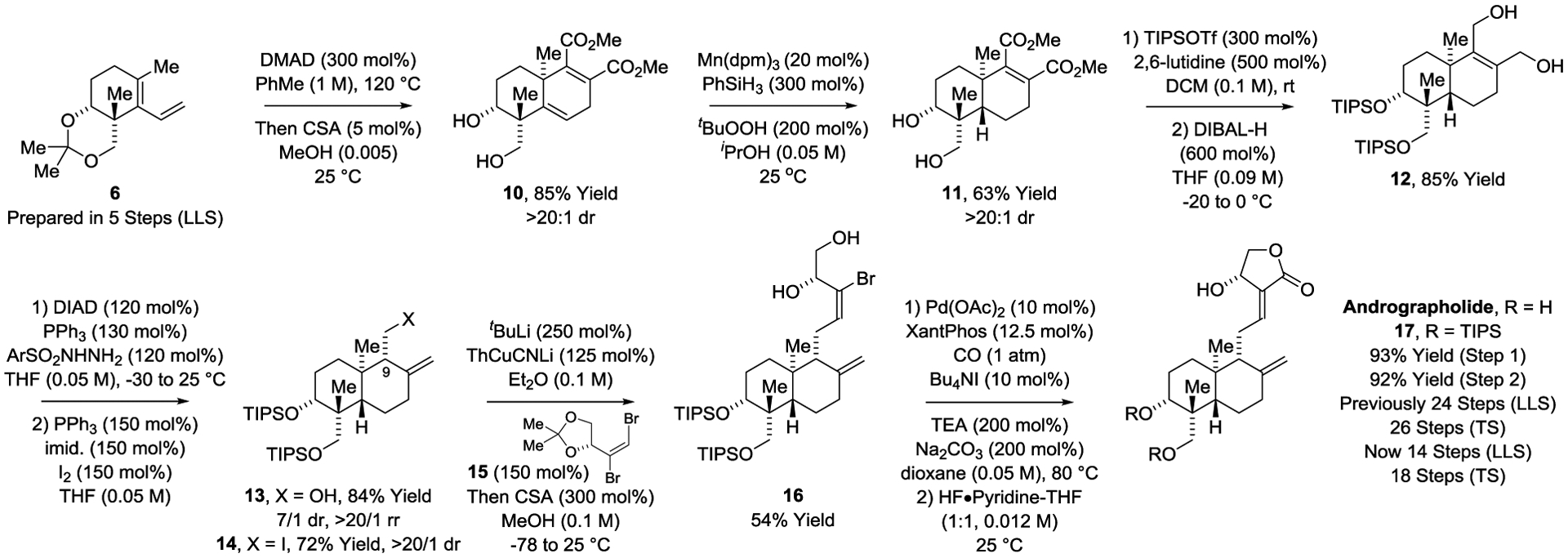
Total synthesis of the labdane diterpenoid andrographolide.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Installation of the α-methylene-γ-butyrolactone was especially challenging. Diverse catalytic methods for Sp3-Sp2 C–C coupling of iodide 14 with vinyl bromide 15[27] failed due to competing elimination to form diene byproducts or halide reduction. Consequently, Gillman-type C–C coupling of iodide 14 with vinyl bromide 15 was attempted.[28] To our delight, treatment of the organolithium derived from iodide 14 with 2-thienyl(cyano)copper lithium followed by exposure to vinyl bromide 15 resulted in completely chemoselective cross-coupling at the terminal vinyl bromide to provide (after hydrolysis of the acetonide in situ) diol 16 in 54% yield. Palladium- Xantphos-catalyzed carbonylative lactonization of bromoalcohol 16 in accordance with Buchwald’s protocol delivered the α-methylene-γ-butyrolactone 17 in excellent yield,[29,30] which upon removal of the triisopropylsilyl ethers afforded andrographolide. [16–19] Whereas the prior synthesis of andrographolide required 24 steps (LLS),[19] the present route is accomplished in 14 steps (LLS).
To illustrate how the elaboration of iso-Fragment A via Diels–Alder cycloaddition-HAT reduction might serve as a general, linear divergent conduit toward other terpenoid natural products, the total synthesis of the coumarin-containing sesquiterpene 14-hydroxy-colladonin[20] was undertaken (Scheme 4). Toward this end, diol 11 was exposed to methyl chloroformate, which resulted in exclusive acylation of the less hindered primary hydroxyl group to provide the allylic carbonate 18. Mitsunobu reaction of 18 with 7-hydroxycoumarin (also known as umbelliferone) proceeded smoothly to provide the product of substitution in 98% yield. Finally, Tsuji reduction[32] followed by fluoride-mediated removal of the silyl ethers delivered 14-hydroxy-colladonin[20] in a 13 steps (LLS). The spectroscopic characteristics of synthetic 14-hydroxy-colladonin were in excellent agreement with that of the natural material, corroborating it’s structural assignment.
Scheme 4.
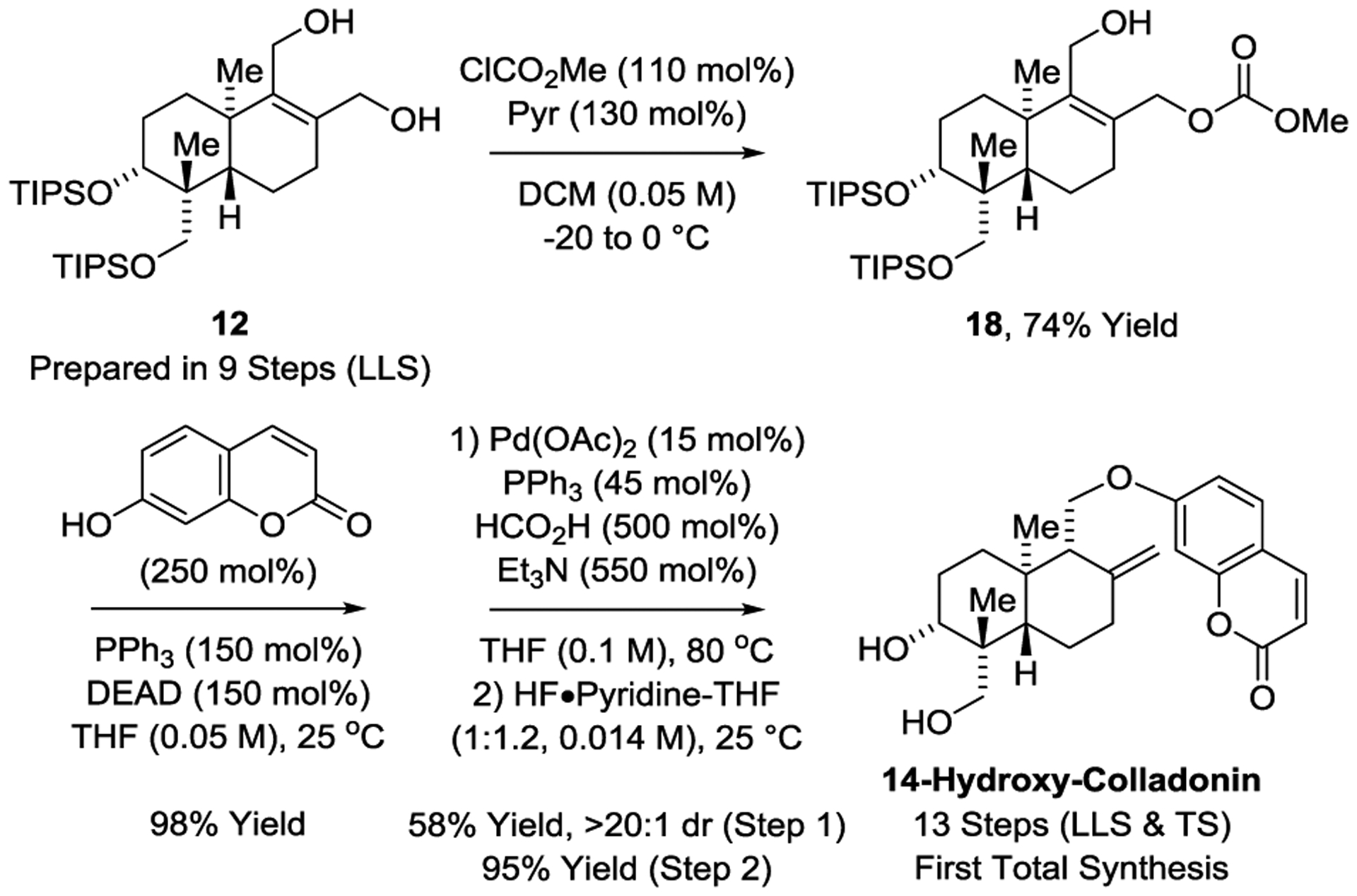
Total synthesis of the coumarin-containing sesquiterpene 14-hydroxy-colladonin.
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
In summary, the enantioselective total synthesis of the labdane diterpenoid andrographolide, the bitter principle of the herbaceous plant Andrographis paniculata (known as “King of Bitters”), was accomplished in 14 steps (LLS) - 10 steps shorter than the prior total synthesis - and the first total synthesis of the coumarin-containing sesquiterpene 14-hydroxy-colladonin was accomplished in 13 steps (LLS). Both natural products were prepared from a common intermediate, trans-decalin 11, which itself derives from the diene iso-Fragment A. Two hydrogen transfer reactions were uniquely enabling in the preparation of these compounds: the iridium-catalyzed carbonyl reductive coupling to form the quaternary carbon stereocenter at C4, and the manganese-catalyzed HAT reduction of the 1,4-cyclohexadiene 10 to form trans-decalin 11. Expansion of this approach to other medicinally relevant terpenoid natural products is currently underway.
Supplementary Material
Acknowledgments
The Welch Foundation (F-0038) and the NIH (RO1-GM093905) are acknowledged for partial support of this research. The Alexander von Humboldt Foundation is acknowledged for postdoctoral fellowship support under the aegis of the Feodor-Lynen program (T.W.). Dr. Jiajie Feng is acknowledged for preliminary synthetic studies toward andrographolide.
References
- [1].For selected reviews on terpenoid natural products, see:; a) Connolly JD, Hill RA, Dictionary of Terpenoids, Springer: Dordrecht, 1991; [Google Scholar]; b) Breitmaier E, Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, Wiley-VCH: Weinheim, Germany, 2006; [Google Scholar]; c) Harrewijn P, van Oosten AM, Piron PGM, Natural Terpenoids as Messengers, Springer: Dordrecht, 2000. [Google Scholar]
- [2].For selected reviews on terpenoid biosynthesis, see:; a) Davis EM, Croteau R, Top. Curr. Chem 2000, 209, 53–95; [Google Scholar]; b) Christianson DW, Chem. Rev 2006, 106, 3412–3442; [DOI] [PubMed] [Google Scholar]; c) Dewick PM, Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; Wiley & Sons: Chichester, 2009, Chapter 5, pp 187–306; [Google Scholar]; d) Tantillo DJ, Nat. Prod. Rep 2011, 28, 1035–1053. [DOI] [PubMed] [Google Scholar]
- [3].For selected reviews on de novo terpenoid synthesis, see:; a) Yoder RA, Johnston JN, Chem. Rev 2005, 105, 4730–4756; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Maimone TJ, Baran PS, Nat. Chem. Biol 2007, 3, 396–407; [DOI] [PubMed] [Google Scholar]; c) Urabe D, Asaba T, Inoue M, Chem. Rev 2015, 115, 9207–9231. [DOI] [PubMed] [Google Scholar]
- [4].For recent selected examples of concise, convergent terpenoid total synthesis, see:; a) Lu H, Martinez M, Shenvi R, Nature Chem. 2015, 7, 604–607; [DOI] [PubMed] [Google Scholar]; b) Ohtawa M, Krambis MJ, Cerne R, Schkeryantz J, Witkin JM, Shenvi RA, J. Am. Chem. Soc 2017, 139, 9637–9644; [DOI] [PubMed] [Google Scholar]; c) Hu X, Xu S, Maimone TJ, Angew. Chem. Int. Ed 2017, 56, 1624–1628; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Turlik A, Chen Y, Scruse AC, Newhouse TR, J. Am. Chem. Soc 2019, 141, 8088–8092; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Schuppe AW, Zhao Y, Liu Y, Newhouse TR, J. Am. Chem. Soc 2019, 141, 9191–9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Feng J, Garza VJ, Krische MJ, J. Am. Chem. Soc 2014, 136, 8911–8914; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guo Y-A, Liang T, Kim SW, Xiao H, Krische MJ, J. Am. Chem. Soc 2017, 139, 6847–6850; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guo Y-A, Lee W, Krische MJ, Chem. Eur. J 2017, 23, 2557–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For selected reviews on carbonyl reductive coupling via hydrogenation, transfer hydrogenation and hydrogen auto-transfer, see:; a) Hassan A, Krische MJ, Org. Proc. Res. Devel 2011, 15, 1236–1242; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354, 300 (aah5133); [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371–2380; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Doerksen RS, Meyer CC, Krische MJ, Angew. Chem. Int. Ed 2019, 58, 14055–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For selected reviews on enantioselective metal-catalyzed formation of acyclic quaternary carbon stereocenters, see:; a) Trost BM, Jiang C, Synthesis 2006, 369–396; [Google Scholar]; b) Marek I, Sklute G, Chem. Commun 2007, 1683–1691; [DOI] [PubMed] [Google Scholar]; c) Das JP, Marek I, Chem. Commun 2011, 47, 4593–4623; [DOI] [PubMed] [Google Scholar]; d) Quasdorf KW, Overman LE, Nature 2014, 516, 181–191; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Feng J, Holmes M, Krische MJ, Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For the isolation and structure determination of oridamycin A and B, and the stereoisomeric natural product xiamycin, see:; a) Takada K, Kajiwara H, Imamura N, J. Nat. Prod 2010, 73, 698–701; [DOI] [PubMed] [Google Scholar]; b) Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH, Hertweck C, Bioorg. Med. Chem. Lett 2010, 20, 6685–6687; [DOI] [PubMed] [Google Scholar]; c) Ding L, Maier A, Fiebig HH, Lin WH, Hertweck C, Org. Biomol. Chem 2011, 9, 4029–4031; [DOI] [PubMed] [Google Scholar]; d) Zhang Q, Mandi A, Li SM, Chen Y, Zhang W, Tian XP, Zhang H, Li HX, Zhang W, Zhang S, Ju JH, Kurtan T, Zhang C, Eur. J. Org. Chem 2012, 5256–5262; [Google Scholar]; e) Kim S-H, Ha T-K-Q, Oh WK, Shin J, Oh D-C, J. Nat. Prod 2016, 79, 51–58. [DOI] [PubMed] [Google Scholar]
- [9].For total syntheses of oridamycin A and B, and the closely related xiamycins, see:; a) Rosen BR, Werner EW, O’Brien AG, Baran PS, J. Am. Chem. Soc 2014, 136, 5571–5574; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meng Z, Yu H, Li L, Tao W, Chen H, Wan M, Yang P, Edmonds DJ, Zhong J, Li A, Nat. Commun 2015, 6, 6096–6104; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Trotta AH, Org. Lett 2015, 17, 3358–3361; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pfaffenbach M, Bakanas I, O’Connor NR, Herrick JL, Sarpong R, Angew. Chem. Int. Ed 2019, 58, 15304–15308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For the isolation and structure determination of triptoquinones B and C, see:; a) Takaishi Y, Shishido K, Wariishi N, Shibuya M, Goto K, Kido M, Ono Y, Tetrahedron Lett 1992, 33, 7177–7180; [Google Scholar]; b) Shishido K, Nakano K, Wariishi N, Tateishi H, Omodani T, Shibuya M, Goto K, Ono Y, Takaishi Y, Phytochemistry 1994, 35, 731–737; [Google Scholar]; c) Morota T, Qin W-Z, Takagi K, Xu L-H, Maruno M, Yang B-H, Phytochemistry 1995, 40, 865–870; [Google Scholar]; d) Duan HQ, Takaishi Y, Momota H, Ohmoto Y, Taki T, Jia YF, Li D, J. Nat. Prod 1999, 62, 1522–1525; [DOI] [PubMed] [Google Scholar]; e) Tanaka N, Ooba N, Duan H, Takaishi Y, Nakanishi Y, Bastow K, Lee K-H, Phytochemistry 2004, 65, 2071–2076; [DOI] [PubMed] [Google Scholar]; f) Wang X-D, Jia W, Gao W-Y, Zhang R, Zhang Y-W, Zhang J, Takaishi Y, Duan H-Q, J. Asian Nat. Prod. Res 2005, 7, 755–759; [DOI] [PubMed] [Google Scholar]; g) Zhang Y, Fan Y, Wang X, Gao W, Duan H, Zhong Cao Yao 2007, 38, 493–496; [Google Scholar]; h) Wang Y, Huang R, Yang J, Zhong Cao Yao 2010, 41, 1252–1254. [Google Scholar]
- [11].For total syntheses of triptoquinones B and C, see:; a) Shishido K, Goto K, Tsuda A, Takaishi Y, Shibuya M, J. Chem. Soc., Chem. Commun 1993, 793–794; [Google Scholar]; b) Yamamura I, Fujiwara Y, Yamato T, Irie O, Shishido K, Tetrahedron Lett. 1997, 38, 4121–4124. [Google Scholar]
- [12].For the isolation and structure determination of iresin and isoiresin, see:; a) Djerassi C, Sengupta P, Herran J, Walls F, J. Am. Chem. Soc 1954, 76, 2966–2968; [Google Scholar]; b) Crabbé P, Burstein S, Djerassi C, Bull. Soc. Chim. Belg 1958, 67, 632–641; [Google Scholar]; c) Bratoeff E, Perez-Amador MC, Ramirez E, Phyton 1996, 58, 119–123; [Google Scholar]; d) Structure Elucidation: Djerassi C, Rittel W, Nussbaum AL, Donovan FW, Herran J, J. Am. Chem. Soc 1954, 76, 6410–6411; [Google Scholar]; e) Djerassi C, Rittel W, J. Am. Chem. Soc 1957, 79, 3528–3534; [Google Scholar]; f) Djerassi C, Donovan FW, Burstein S, Mauli R, J. Am. Chem. Soc 1958, 80, 1972–1977; [Google Scholar]; g) Rossmann MG, Lipscomb WN, J. Am. Chem. Soc 1958, 80, 2592–2593; [Google Scholar]; h) Djerassi C, Burstein S, J. Am. Chem. Soc 1958, 80, 2593; [Google Scholar]; i) Djerassi C, Burstein S, Tetrahedron 1959, 7, 37–46; [Google Scholar]; j) X-ray structure: Rossmann MG, Lipscomb WN, Tetrahedron 1958, 4, 275–293; [Google Scholar]; k) NMR Assignments: Rios MY, Berber LA, Magn. Reson. Chem 2005, 43, 339–342. [DOI] [PubMed] [Google Scholar]
- [13].For total syntheses of iresin and isoiresin, see:; a) Pelletier SW, Prabhakar S, J. Am. Chem. Soc 1968, 90, 5318–5320; [Google Scholar]; b) Wang B-L, Gao H-T, Li W-DZ, J. Org. Chem 2015, 80, 5296–5301. [DOI] [PubMed] [Google Scholar]
- [14].Feng J, Noack F, Krische MJ, J. Am. Chem. Soc 2016, 138, 12364–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].For intramolecular Sakurai allylation of a 5-membered lactol, see:; Noguchi N, Nakada M, Org. Lett 2006, 8, 2039–2042. [DOI] [PubMed] [Google Scholar]
- [16].For studies pertaining to the isolation and structure determination of andrographolide, see:; a) Boorsma WA, Med.’s Lands Plant 1896, 18, 63–72; [Google Scholar]; b) Gorter MK, Rec. Trav. Chim 1911, 30, 151–160; [Google Scholar]; c) Gorter MK, Rec. Trav. Chim 1914, 33, 239–243; [Google Scholar]; d) Bhadun K, Amer. J. Pharm 1914, 84, 349–354; [Google Scholar]; e) Moktader A, GuhaSircar SS, J. Ind. Chem. Soc 1939, 16, 333–338; [Google Scholar]; f) Schwyzer R, Biswas HG, Karrer P, Helv. Chim. Acta 1951, 34, 652–677; [Google Scholar]; g) Kleipool RJC, Nature 1952, 169, 33–34;14910682 [Google Scholar]; h) Cava MP, Chan WR, Haynes LJ, Johnson LF, Weinstein B, Tetrahedron 1962, 18, 397–403; [Google Scholar]; i) Cava MP, Weinstein B, Chan WR, Haynes LJ, Johnson LF, Chem. Ind 1963, 167; [Google Scholar]; j) Cava MP, Chan WR, Stein RP, Willis CR, Tetrahedron 1965, 21, 2617–2632; [Google Scholar]; k) Fujita T, Fujitani R, Takeda Y, Takaishi Y, Yamada T, Kido M, Miura I, Chem. Pharm. Bull 1984, 32, 2117–2125. [Google Scholar]
- [17].For selected reviews on the wide-ranging biological properties of andrographolide, see:; a) Mishra SK, Sangwan NS, Sangwan RS, Pharmacognosy Rev 2007, 1, 283–298; [Google Scholar]; b) Lim JCW, Chan TK, Ng DSW, Sagineedu SR, Stanslas J, Wong WSF, Clin. Exp. Pharm. Phys 2012, 39, 300–310; [DOI] [PubMed] [Google Scholar]; c) Gupta S, Mishra KP, Ganju L, Arch. Virol 2017, 162, 611–623; [DOI] [PubMed] [Google Scholar]; d) Tan WSD, Liao W, Zhou S, Wong WSF, Biochem. Pharm 2017, 139, 71–81; [DOI] [PubMed] [Google Scholar]; e) Islam MT, Ali ES, Uddin, Md. SJ Islam A, Shaw S, Khan IN, Saravi SSS, Ahmad S, Rehman S, Gupta VK, Gaman M-A, Gaman AM, Yele S, Das AK, de Castro e Sousa J. Marcelo, Dantas SMMM, Rolim HML, Melo-Cavalcante AAC, Mubarak MS, Yarla NS, Shilpi JA, Mishra SK, Atanasov AG, Kamal MA, Cancer Lett. 2018, 420, 129–145; [DOI] [PubMed] [Google Scholar]; f) Lu J, Ma Y, Wu J, Huang H, Wang X, Chen Z, Chen J, He H, Huang C, Biomed. Pharm 2019, 117, 109078–109088; [DOI] [PubMed] [Google Scholar]; g) Zhang L, Bao M, Liu B, Zhao H, Zhang Y, Ji X, Zhao N, Zhang C, He X, Yi J, Tan Y, Li L, Lu C, Pharmacology 2020, 105, 123–134. [DOI] [PubMed] [Google Scholar]
- [18].a) Wang J, Tan XF, Nguyen VS, Yang P, Zhou J, Gao M, Li Z, Lim TK, He Y, Ong CS, Lay Y, Zhang J, Zhu G, Lai S-L, Ghosh D, Mok YK, Shen H-M, Lin Q, Mol. Cell. Proteomics 2014, 13, 876–866; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sui Y, Wu F, Lv J, Li H, Li X, Du Z, Sun M, Zheng Y, Yang L, Zhong L, Zhang X, Zhang G, PLoS One 2015, 10, e0144715/1–e0144715/18; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Crottès D, Jan LY, Cell Calcium 2019, 82, 102050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].For the total synthesis of andrographolide, see:; Gao H-T, Wang B-L, Li W-DZ, Tetrahedron 2014, 70, 9436–9448. [Google Scholar]
- [20].For the isolation and structure determination of 14-hydroxy-colladonin, see:; Appendino G, Ozen HC, Tagliapietra S, Cisero M, Phytochemistry 1992, 31, 3211–3213. [Google Scholar]
- [21].For a related cross-metathesis, see:; Taylor RE, Engelhardt FC, Schmitt MJ, Yuan H, J. Am. Chem. Soc 2001, 123, 2964–2969. [Google Scholar]
- [22].For Diels–Alder reactions of structurally related dienes, see:; a) Hollinshead DM, Howell SC, Ley SV, Mahon M, Ratcliffe NM, J. Chem. Soc. Perkin Trans. I 1983, 1579–1589; [Google Scholar]; b) Wang J, Ma D, Angew. Chem. Int. Ed 2019, 58, 15731–15735; [DOI] [PubMed] [Google Scholar]; c) Pitsinos EN, Mavridis I, Tzouma E, Vidali VP, Eur. J. Org. Chem 2020, 4730–4742. [Google Scholar]
- [23].Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi R, J. Am. Chem. Soc 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bifulco G, Dambruoso P, Gomez-Paloma L, Riccio R, Chem. Rev 2007, 107, 3744–3779. [DOI] [PubMed] [Google Scholar]
- [25].Myers AG, Zheng B, Tetrahedron Lett. 1996, 37, 4841–4844. [Google Scholar]
- [26].Lange GL, Gottardo C, Synth. Comm 1990, 20, 1473–1479. [Google Scholar]
- [27].Vinyl bromide 15 is a novel compound that was prepared via Ohira-Bestmann reaction-dibromination of (R)-glyceraldehyde acetonide. See Supporting Information for further details.
- [28].Lipshutz BH, Koerner M, Parker DA, Tetrahedron Lett. 1987, 28, 945–948. [Google Scholar]
- [29].Martinelli JR, Watson JR, Freckmann DMM, Barder TE, Buchwald SL, J. Org. Chem 2008, 73, 7102–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].For a review of carbonylative cyclizations in natural product total synthesis, see:; Ma K, Martin BS, Yin X, Dai M, Nat. Prod. Rep 2019, 36, 174–219. [DOI] [PubMed] [Google Scholar]
- [31].For a review, see:; Mitsunobu O, Synthesis 1981, 1–28. [Google Scholar]
- [32].a) Tsuji J, Minami I, Shimizu I, Synlett 1986, 623–627; [Google Scholar]; b) Mandai T, Matsumoto T, Kawada M, Tsuji J, Tetrahedron 1994, 50, 475–486; [Google Scholar]; c) Choi H.-w., Fang FG, Fang H, Kim D-S, Mathieu SR, Yu RT, Org. Lett 2017, 19, 6092–6095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.