

Abstract
Background
Identification of patients who may become hypercapnic, or develop acidotic hypercapnic respiratory failure (AHRF), is important in chronic obstructive pulmonary disease (COPD) to avoid hospital admission and select patients for use of home NIV. This study aimed to identify factors associated with presence and development of hypercapnia.
Methods
1224 patients, 637 with COPD and 587 with alpha 1 antitrypsin deficiency (AATD), from 4 previously established patient cohorts, were included in cross-sectional analyses of hypercapnia (PaCO2 ≥ 6.5 kPa or 48.8 mmHg), focusing on phenotypic features of COPD and mortality. Longitudinal associations of rising PaCO2 were also assessed. A second cohort of 160 COPD patients underwent sleep studies and 1-year follow-up, analysing in a similar way, incorporating additional information from their sleep studies if appropriate.
Results
Hypercapnia was 15 times more common in usual COPD than AATD (p < 0.01) after adjustment for baseline differences by regression. Independent predictors of hypercapnia in COPD included FEV1 and current use of oxygen; these variables, together with lack of emphysema, explained 11% of variance in CO2. Increasing PaCO2 also associated with higher risk of death (p=0.03). 44/160 patients exhibited sleep disordered breathing. The sleep study cohort also showed an association of low FEV1 with hypercapnia. Prior hospital admission for AHRF was also clinically significant, being a feature of almost double the number of hypercapnic patients in both test and sleep study COPD cohorts.
Conclusion
Lower FEV1 and prior AHRF are the main associations of hypercapnia in COPD, which carries a poor prognosis, particularly worsening over time.
1. Introduction
The most important function of the respiratory system is to maintain blood oxygenation and exhale carbon dioxide, when impaired respiratory failure defined as significant hypoxia with or without hypercapnia can develop. In chronic obstructive pulmonary disease (COPD), both hypoxia and hypercapnia may occur; in over 12 000 patients reviewed as part of the European COPD audit, the median PaCO2 was 5.8 kPa (43.5 mmHg) and PaO2 was 7 kPa (52.5 mmHg) on hospital admission [1]. In the UK, 35% of all COPD admissions have respiratory failure, and 44% have an elevated PaCO2, suggesting that acidotic hypercapnic respiratory failure (AHRF) is relatively frequent [2].
AHRF is a well-defined complication of COPD, and noninvasive ventilation (NIV) is an effective treatment in such patients [3]. NIV has also been used nightly at home to manage chronic respiratory failure in COPD [4] and is now a commonly used treatment in severe disease. An episode of AHRF in the context of COPD is a poor prognostic marker, with only 50% of patients alive one year later [5]. Early research suggested that stable-state hypercapnia was also a key prognostic factor in patients with COPD [6] with a hazard ratio of 2.90 for death at 1 year when PaCO2 ≥ 6 kPa (45 mmHg) measured on admission for an acute exacerbation of COPD (AECOPD) [7]. Furthermore, in 5-year follow-up data comparing those with type 1 respiratory failure, those who had acute type 2 respiratory failure that resolved (type 2.1) had a better long-term survival compared to those where it persisted (type 2.2) [8]. Consistent with this epidemiological evidence, the largest treatment benefit of home NIV is seen in patients with persistent hypercapnia [9, 10].
A number of factors in COPD have been reported to contribute to hypercapnia, including breathing pattern [11], inspiratory muscle weakness [11], hyperinflation [12], continued smoking [12], and FEV1 below 36% [13]. However, in each of these studies, the overall proportion of variance in carbon dioxide level explicable by the reported variables was low. Identification of patients who are likely to become hypercapnic, or to develop AHRF, is important in COPD if we are to avoid the deleterious consequences of hospital admission and select patients for use of home NIV in a timely manner. We hypothesized that inadequate characterisation might be the reason that previous studies had been unable to construct an adequate predictive model for hypercapnia. In particular, we felt that more data on emphysema, comorbidity, and sleep parameters would be important missing variables. The current study aimed to identify COPD patients predisposed to hypercapnia through a combined approach within COPD cohorts, including a large group with detailed COPD phenotyping, enriched for those at risk of hypercapnia by including those with prior AHRF or severe disease (defined by current LTOT use), and a newly recruited group who also underwent detailed sleep studies.
2. Methods
2.1. COPD Phenotype Cohort
There were four sources of patients, three with usual COPD and one with COPD related to AATD. The first with usual COPD was the Birmingham Heartlands Hospital NIV cohort, a population that had required NIV for AHRF described elsewhere [14]; patients with alternative pathologies (e.g., obesity-related hyperventilation) were excluded. Patients were recruited at admission and had blood gas analyses approximately 3 months after discharge. The second usual COPD cohort included all patients from the West Midlands COPD cohort, described previously [15]. The third usual COPD cohort was those in a long-term oxygen (LTOT) clinic [16]. In these cohorts, the presence of emphysema was determined by the appearance on computed tomography (CT) scan. If present, its location was defined as upper and lower zone, or both; quantitative analysis was not possible due to differences in scanners and scan protocol.
The fourth cohort was patients with AATD known to the Birmingham AATD registry who had at least one stable-state blood gas; procedures for assessment are described elsewhere [17]. Rate of decline per year in blood gas parameters was calculated in those that had at least 4 values over 3 years, using methods described in our previous work pertaining to FEV1 decline [18]. The presence and zone of emphysema was determined by appearance and densitometry (where possible), as described previously [19]. In the combined cohort, survival was determined at the end of 2014, using medical/GP records. Local ethical approval was given for all research cohorts, and patients gave informed consent.
2.2. Sleep Study Cohort
Patients with clinically diagnosed COPD, confirmed by postbronchodilator spirometry, and with at least 1 hospitalisation for COPD in the last 12 months were recruited prospectively from University Hospitals Birmingham NHS Foundation Trust between August 2014 and 2015; the study was ethically approved, and all patients gave informed consent. We enriched the cohort for those at risk of hypercapnia by approaching all patients with a previous admission for AHRF. Patients on home NIV or CPAP were excluded. All patients were then assessed in the stable state with a full medical history, Epworth sleepiness score (ESS) and CAT questionnaires, spirometry, blood gases, and limited channel polysomnography (ResMed Embletta Gold) at baseline, and this process is repeated, without the sleep study, at follow-up 12 months later. Where chest CT scans were available, these were reviewed in the same manner as the previous cohorts. All sleep studies were analysed only if there was >240 minutes of sleep recorded and were of good quality; those that did not meet these criteria were repeated. Studies were reported according to AASM scoring manual 2.2 by a single researcher (CD) and an independent sleep technician.
2.3. Statistical Analysis
Data were analysed in SPSS version 24.0, and results were considered statistically significant when p < 0.05. Data normality was determined visually and by the Kolmogorov–Smirnov test; normally and nonnormally distributed variables are expressed as mean (standard error) or median (interquartile range), respectively. Of 21 studies of home NIV vs usual care in our systematic review, 12 studies defined hypercapnia as PCO2 >6.5 kPa (48.8 mmHg) [4]; hence, this was taken to define hypercapnia in our study. In the COPD phenotype cohort, univariate analyses were conducted and variables were then taken forward, in a stepwise manner if p < 0.05 to multiple logistic regression models assessing CO2, presence of hypercapnia, or rising CO2 during follow-up. Cox-regression analysis was used to assess the impact of hypercapnia and rising CO2 on mortality, using a stepwise approach to determine covariates. Those variables that appeared to relate to outcome were then tested in the sleep study cohort, in whom independent multivariable analyses of hypercapnia, rising CO2, and mortality were also conducted, which incorporated additional information from their sleep studies if appropriate. Linear regression was used to assess CO2 as a quantitative outcome, both to align to prior research assessing it in this way and give an indication of what proportion of variance was explicable by included variables. Independent associations of obstructive sleep apnoea (OSA) were also sought using similar techniques.
3. Results
3.1. COPD Phenotype Cohort
1224 patients, 637 with COPD and 587 with AATD, were included. Characteristics of the COPD patients are shown in Table 1, stratified by presence of AHRF in COPD. This was not relevant in AATD since no patients had reported this event previously. Characteristics of the AATD group are shown in Supplementary Table 1. The prevalence of hypercapnia was much higher in usual COPD than AATD (p < 0.01), with usual COPD patients 14.82 (95% CI: 6.67–32.92, p < 0.01) times more likely to be hypercapnic after adjustment for baseline differences by regression. Consequently, we decided to analyse the AATD and COPD groups separately and present the AATD results only in the supplement allowing to focus on usual COPD in the main manuscript. In usual COPD, there were significant associations between hypercapnia and reduced FEV1 (but not with DLco or volumatic measures of gas trapping), use of LTOT, lower PO2, higher HCO3, and base excess (Table 1). Independent predictors of hypercapnia in COPD in the multivariate model included FEV1 and current use of LTOT (Table 2); these variables explained 11% of variance in CO2. In multivariate analyses, the risk of hypercapnia was doubled per 1 kPa (7.5 mmHg) lower PaO2 (odds ratio (OR): 2.05, 95% confidence interval: 1.23–3.33, p < 0.01) and current smoking status conferred an OR for hypercapnia of 9.69 (95% CI: 2.85–33.08, p < 0.01); together these accounted for 28% of variance in CO2. Emphysema zone did not relate to hypercapnia in either group, whether examined visually or (in AATD) by densitometry (458 scans; p > 0.1).
Table 1.
Characteristics of the COPD cohort.
| All COPD N = 637 | Comparison based on prior AHRF in COPD | |||
|---|---|---|---|---|
| No AHRF N = 323 | AHRF N = 314 | p | ||
| Age (years) | 74 (66–80) | 75 (68–82) | 65 (73–78) | <0.01 |
| Sex (male) | 278 (43.6) | 148 (45.8) | 140 (44.59) | 0.75 |
| Pack years | 40 (33–56) | 40 (32–55) | 40 (33–56) | 0.59 |
| Current smokers | 126 (19.8) | 49 (15.2) | 77 (24.5) | 0.02 |
| Hypercapnia | 233 (36.6) | 73 (22.6) | 145 (46.18) | <0.01 |
| BMI | 25.5 (213–32.5) | 24.6 (21.3–32.0) | 26·7 (21.4–33.6) | 0.28 |
| FEV1 | 0.9 (0.65–1.26) | 0.98 (0.72–1.43) | 0·82 (0.59–1.12) | <0.01 |
| FEV1pp | 41 (41–55) | 47 (34–60) | 36 (28–49) | <0.01 |
| FVCpp | 73 (57–90) | 78 (63–94) | 69 (51–85) | <0.01 |
| DLCOpp | 48.9 (38.9–73.3) | 60.9 (55.1–66.7) | 43.6 (36.2–64.9) | <0.01 |
| RVpp | 104.35 (89.5–123.4) | 100 (88.03–108.93) | 112 (93.4–134.25) | 0.09 |
| PaO2 | 7.9 (7.1–8.7) | 7.9 (7.2–8.7) | 7.8 (7.0–8.6) | 0.08 |
| PaCO2 | 5.9 (4.9–6.9) | 5.5 (4.7–6.4) | 6.4 (5.4–7.5) | <0.01 |
| HCO3 | 28.4 (26.2–31.5) | 27.5 (25.6–30.1) | 29 (26.7–32.8) | <0.01 |
| BE | 3.7 (1.5–6.5) | 3.4 (1.2–5.7) | 4.3 (2.3–7.2) | <0.01 |
| On LTOT | 304 (47.7) | 146 (45.2) | 158 (50.3) | 0.2 |
| Emphysema | 284 (44.6) | 177 (54.8) | 142 (45.2) | 0.04 |
| Upper lobe emphysema | 113 (39.8) | 58 (51.3) | 55 (48.7) | 0.21 |
| Lower lobe emphysema | 14 (4.9) | 12 (85.7) | 2 (14.3) | 0.12 |
| No zonal predominance | 157 (55.3) | 89 (56.69) | 68 (43.3) | 0.79 |
| LZVI-UZVI | — | — | — | — |
| Bronchiectasis | 101 (15.9) | 50 (15.5) | 51 (16.2) | 0·79 |
| Change in FEV1/ml/year | −54 (−160-23.5) | −65.5 (−172.8-8) | −49 (−143.5−32.5) | 0·33 |
| Follow-up (years) | 1.33 (0.31–2.65) | 1.30 (0.31–2.56) | 0.31 (1.34–2.87) | 0·62 |
| Death | 415 (65.1) | 192 (59.4) | 223 (71.02) | <0.01 |
Data are shown as mean (SE) or median (IQR) for quantitative measures, and n (%) for binary outcomes, which are further denoted by italic text.
Table 2.
Multiple regression analysis assessing hypercapnia.
| OR | p value | |
|---|---|---|
| FEV1 pp | 0.98 (0.97–0.99) | <0·01 |
| Presence of emphysema | 0.72 (0.50–1.05) | 0·09 |
| Use of LTOT | 1.74 (1.19–2.53) | <0·01 |
| DLCOpp | 0.92 (0.89–0.94) | <0.01 |
| FRCpp | 1.17 (1.09–1.25) | <0.01 |
The ORs shown represent the effect of increasing FEV1 by 1% of predicted, and DLCO, by 1% predicted, having emphysema v not, using LTOT v not, increasing age by 1 year, and current smoking v not. p = percent predicted.
Follow-up blood gases were not carried out often in the COPD groups; thus, the number of patients in whom deteriorating CO2 could be assessed was relatively small, and no clinically significant associations were seen in univariate models. Although serial blood gases were available in all AATD patients, few developed hypercapnia and no meaningful regression analysis were possible due to small numbers (Supplementary Table 2).
Survival curves for COPD and AATD populations, stratified by the presence of hypercapnia or rising PaCO2, are shown in Figure 1. Hypercapnia did not influence mortality within the usual COPD group (OR: 0.93 (95% CI: 0.70–1.25)). However, the presence of an increasing stable-state PaCO2 was a predictor of mortality; patients were 1.65 (95% CI: 1.06–2.56, p=0.03) times more likely to die if they had an increasing PaCO2.
Figure 1.
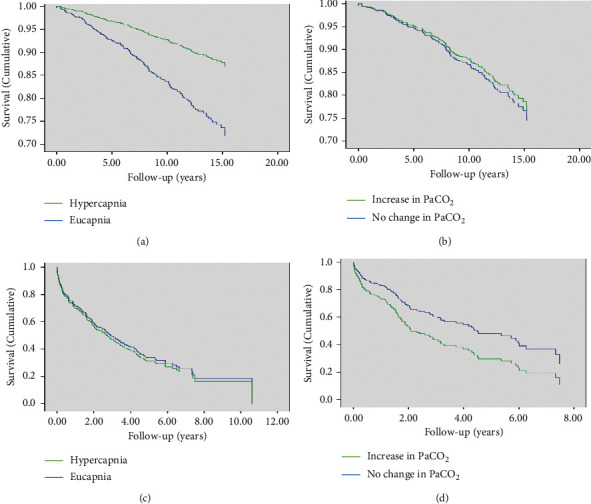
Cox regression for mortality in AATD and COPD. (a) AATD, hypercapnia and survival, (b) AATD, increasing CO2 and survival, (c) COPD, hypercapnia and survival, (d) COPD, increasing CO2 and survival.
3.2. Sleep Study Cohort
176 patients were recruited, of whom 16 were excluded, 8 due to spirometry not being diagnostic of COPD, 3 due to current CPAP use, 2 due to home NIV use, and 3 due to inability to obtain an adequate sleep study after 2 attempts. Of the 160 remaining, 94 had never had AHRF and 66 had required NIV for AHRF on a previous hospital admission; their clinical characteristics are shown in Table 3 stratified by presence of prior AHRF. Thirty-four (21%) were current smokers, and the remainder all were ex-smokers. Comorbidities were common; 66 (41%) had hypertension, 33 (20%) had documented vascular disease (ischaemic heart disease, atrial fibrillation, and cerebral or peripheral vascular disease), and 16 (10%) had osteoporosis. 108/160 patients (68%) had a CT scan available for analysis; no patient had lower zone dominant disease. Of the variables independently associated with hypercapnia in the phenotype cohort, this was replicated only for FEV1 (p < 0.05). Notably, hypercapnia was generally less common in this cohort and LTOT use less frequently (expected due to nature of recruitment of test cohort).
Table 3.
Characteristics of the sleep study cohort at baseline and 1 year.
| No AHRF (n = 94) | AHRF (n = 66) | p value | |
|---|---|---|---|
| Age (years) | 65.4 (63.4–67.5) | 67.6 (65.4–69.8) | 0.16 |
| Sex (male) | 50 (53.2) | 34 (51.5) | 0.83 |
| Weight (kg) | 71.0 (60.1–84.4) | 76.8 (71.2–82.3) | 0.47 |
| BMI | 26.6 (23.2–31.7) | 29.1 (27.1–31.1) | 0.31 |
| pH | 7.4 (7.4–7.4) | 7.4 (7.4–7.4) | 0.04 |
| PaO2 | 9.2 (8.9–9.7) | 8.5 (8.1–8.8) | <0.01 |
| PaCO2 | 5.0 (4.5–5.6) | 5.7 (5.5–5.9) | <0.01 |
| HCO3− | 25.2 (24.0–26.5) | 27.4 (26.2–28.7) | <0.01 |
| BE | 0.6 (−1.1-2.5) | 2.35 (1.2–4.9) | <0.01 |
| Hypercapnia | 7 (7.4) | 13 (19.7) | 0.02 |
| CAT | 23.5 (21.8–25.2) | 22.7 (20.5–25.0) | 0.5 |
| Epworth sleep score | 9.0 (5.0–14.0) | 8.00 (4.0–12.0) | 0.2 |
| Bronchiectasis (% of those with CT) | 13/68 (19.1) | 5/40 (12.5) | 0.37 |
| Emphysema | 56/68 (82.35) | 30/40 (75) | 0.59 |
| LTOT | 10/94 (10.6) | 10/66 (15.2) | 0.40 |
| Pack years | 45.0 (30.0–60.0) | 48.5 (38.0–64.3) | 0.12 |
| FEV1 | 1.4 (0.9–1.8) | 0.9 (0.7–1.5) | <0.01 |
| FEV1 % predicted | 56.5 (52.2–60.9) | 43.77 (39.55–47.99) | <0.01 |
| FVC | 2.7 (2.6–2.9) | 2.3 (2.1–2.5) | <0.01 |
| FVC % predicted | 86.5 (72.0–100.0) | 76.1 (71.2–81.0) | <0.01 |
| FEV1/FVC | 0.5 (0.5–0.6) | 0.5 (0.4–0.5) | 0.02 |
| OSA on sleep study | 14 (14.9) | 29 (43.9) | <0.01 |
| AHI | 2.4 (0.6–4.6) | 3.7 (0.5–8.8) | 0.09 |
| ODI | 5.6 (3.3–10.1) | 7.5 (4.5–14.1) | 0.08 |
| Mean oxygen sats during sleep study | 90.4 (87.7–92.5) | 89.6 (85.6–92.6) | 0.26 |
| Time under 90% | 12.0 (1.6–52.3) | 23.1 (3.5–96.1) | 0.04 |
| Death at 12 months | 5/94 (5.3) | 10/66 (15.2) | 0.35 |
| Follow-up PaCO2 | 4.9 (4.5–5.6) | 4.9 (4.5–5.4) | 0.66 |
Data are shown as mean (SE) or median (IQR) for quantitative measures, and n (%) for binary outcomes, which are further denoted by italic text. CAT = COPD assessment test, LTOT = long-term oxygen therapy, AHI = apnoea hyponoea index, and ODI = oxygen desaturation index.
Forty-four patients exhibited sleep disordered breathing, 43 having OSA and 1 having central apnoeic events; amongst these, 17 had ESS>10 (i.e., obstructive sleep apnoea hypopnoea syndrome, OSAHS), and all of whom were then offered CPAP. The only independent association of OSA in this group was baseline CO2 (Supplementary Tables 3 and 4). Few differences in the most recent NIV admission were seen; only a small increase was seen in time on NIV and length of hospital stay, although neither was significant in multivariate analysis (Supplementary Tables 5 and 6). Nocturnal hypoventilation also appeared to differ between those who had or had not had AHRF, as shown by time during which O2 saturations were<90%. Addition of OSA to the multivariate model for hypercapnia did not improve its positive predictive value which may reflect few OSA cases in hypercapnic patients (n = 4, Supplementary Table 2).
No patient was lost to follow-up, although 15 deaths occurred, 5 in those who had never had AHRF, and 10 in those with prior AHRF. Although the average CO2 level did not appear markedly different at follow-up, there were a higher number of patients with hypercapnia in both those who had AHRF and those who had not. Baseline hypercapnia also significantly associated with death at 1 year (11/15 deaths if hypercapnic, 10/145 who were not; p < 0.01, OR: 1.67 (1.45–2.89), Supplementary Figure 1).
4. Discussion
Our study has shown that patients with AATD are at low risk of hypercapnia compared to usual COPD patients. The main novel findings of our work are that longitudinal change in CO2 may be more important than hypercapnia itself when considering risk of death and that hyperinflation (irrespective of its cause) could be more relevant to development of hypercapnia than emphysema itself.
4.1. Which COPD Patients Become Hypercapnic?
We chose to study defined hypercapnia, rather than just PaCO2 variance, as we felt this would be more applicable to clinical decision-making. The results confirmed previous work assessing PaCO2 [12], concurring with previous evidence that poor lung function is associated with hypercapnia. In the usual COPD cohort, a number of markers of severe disease (lower FEV1 or DLCOpp, higher FRCpp, and current LTOT prescription) were associated with hypercapnia. Historically, spirometry has been poorly associated with blood gas parameters, but PaCO2 does rise more sharply after FEV1 falls below 40% predicted [13]. Almost half our hypercapnic patients were under this threshold compared to only 33% of eucapnic patients. This suggests that overall alveolar ventilation is adequate up to a threshold, beyond which it becomes difficult to maintain.
Hypercapnia tended to be less likely if there was emphysema on CT. This was to some extent unexpected as previous studies linked hyperinflation, a measure of air trapping, and potentially therefore an indirect measure of emphysema, to higher PaCO2 [6, 20]. Conversely, the classical cachectic “pink puffer” with emphysema has long been thought more likely to have type 1 not type 2 respiratory failure [21]. Studies have suggested that the location of emphysema could have a direct physiological effect because of differences in the mechanical forces present, as lower zone emphysema directly impairs diaphragmatic motion [22]; however, in our data, emphysema location had no impact on hypercapnia, and moreover, in AATD (where most patients have lower zone dominant disease), hypercapnia was markedly less common. Although visual CT scan inspection might be considered a crude method to quantify the extent of the emphysema, even densitometry, which was assessed for many of the AATD patients, did not reveal an association of emphysema zone or severity with hypercapnia, suggesting that any impact on the diaphragm alone is insufficient to cause alveolar hypoventilation. Direct measurement of diaphragmatic function might have strengthened the evidence supporting this hypothesis, but this was not possible in our study.
Whilst emphysema did not associate positively with the presence of hypercapnia, we acknowledge that a quantitative CT analysis could not be done in the COPD cohort; thus, it may be relevant that other surrogate markers of air trapping (lower DLCOpp, increasing hyperinflation as measured by FRCpp) were associated with hypercapnia. Such air trapping could occur with airways disease or emphysema. It has been well documented that NIV in stable hypercapnic COPD patients reduces hyperinflation amongst other parameters [23], which in turn increases daytime PaO2 and reduces daytime PaCO2. This suggests that amount of hyperinflation independent of presence or amount of emphysema may be more of an influence on PaCO2. Other potential unmeasured influences relevant to the relationship between emphysema and hypercapnia include the degree of VQ matching, which could differ by affected zone, and contributions from dynamic rather than static hyperinflation.
4.2. What Is the Prognosis with Hypercapnia?
Although hypercapnic patients have a short-term mortality risk reported by others [8] and confirmed in our data (albeit not as high a risk as earlier studies have found) if survival is present beyond 2 years and certainly over 10 years, there does not seem to be any obvious difference compared to nonhypercapnic COPD patients [24]. A more recent study showed that 5.12% of a eucapnic COPD cohort progressed to develop hypercapnia (PaCO2>6 kPa or 45 mmHg) during long-term follow-up [25] and 13.3% of these had reversible hypercapnia following discharge from hospital. Thus, eucapnic COPD may actually represent a subphenotype and not just simply an “early stage” of COPD that will eventually develop hypercapnia. In the same cohort of Chinese patients, median survival was longer in the eucapnic vs hypercapnic groups (6.5 vs 5 years, p=0.02) [25]. With the advent of home NIV as a treatment proven to reduce mortality [9] and hospital admissions [10] in COPD, it is possible that prognosis will continue to improve, but only if these high-risk patients are identified early for treatment.
4.3. Which COPD Patients Should Have a Sleep Study?
OSA was more likely in patients with a history of AHRF, but this did not increase the likelihood of OSAHS, and thus such patients would not necessarily present to medical services with symptoms of sleepiness. Furthermore, even though mean PaCO2 was slightly higher in the OSAHS group (5.68 vs 5.4), it did not reach statistical significance; this implies that identifying OSAHS in COPD patients would require widespread screening, since the CO2 would be an insufficiently sensitive distinguishing feature to adopt as a case finding approach. Such patients also appeared to have delayed weaning from acute NIV, although this was also not significant in multivariate analysis due to the small differences in ventilation duration driven by strict protocols for weaning in our unit [26] which may have limited our ability to detect any relationship. The value of routine sleep studies in the non-AHRF population was questionable given that significant problems occurred in only 15% of patients.
The PaCO2 is a well-established physiological difference in patients with COPD-OSA overlap and COPD alone. Two hundred and thirteen consecutive patients compared in one study had a mean PaCO2 difference of 5.94 kPa (44.5 mmHg) vs 5.28 kPa (39.6 mmHg) [27] although why this occurred remains unclear. Regression analysis within the same cohort could only partially explain the occurrence of hypercapnia by the combination of reduced respiratory function and being overweight, which suggests that there is clearly a multifactorial element to this phenomenon. In our cohort, no associations with anthropometric features were seen, but it is notable that few of our patients exhibited very high BMI.
Overnight hypoxia in overlap syndrome has been shown to be more severe compared to COPD alone as both COPD and OSA cause desaturations [28]. However, in a large cohort of nonobese, severe COPD patients, the number of patients needing supplemental oxygen did not differ between OSA and overlap syndrome patients [29]. Other studies have found mean oxygen saturations at similar levels between COPD and overlap groups [30]. Our data showed that time spent under 90% saturations during the sleep study was significantly more in the OSA patients. However, there was no difference in mean saturations, which suggests that there are more extreme hypoxia events when the COPD patients seem to be more at risk of having an apnoeic or hypopnoeic episode. Whilst simple overnight oximetry to screen for OSA within COPD appeared attractive based on this feature, it was not statistically significant in the regression model seeking independent predictors of OSA, so time spent under 90% saturations is unlikely to be a useful screening tool. We, therefore, propose an algorithm to detect COPD-OSA overlap whereby any patient who has AHRF and remains hypercapnic at discharge should have repeat blood gases at 4 weeks, and if they remain hypercapnic, a limited channel sleep study should be done in order to formally calculate AHI.
4.4. What Do Our Data Tell Us about Natural History of COPD?
The two cohorts studied comprised one with a very broad range of disease severity (the AATD group) and some that were more homogeneous (COPD groups); in part, this is related to the way that cohorts were recruited, in which all COPD patients presented routinely to respiratory services with symptoms whereas some AATD patients could have been diagnosed whilst asymptomatic as a result of family screening. In this broader cohort, we observed less hypercapnia and even over long periods of follow-up and little progression in terms of blood CO2 level. However, in the more homogeneous, generally more severe COPD cohort deterioration in this parameter was notable and related to survival. Taken together with our previous results, we suggest that hyperinflation develops in severe disease and contributes to development of hypercapnia only in advanced disease. Once it has developed, further deterioration is indicative of end-stage disease with a high risk of death, which is why rising CO2 in this group was associated with death. These people might be the group most relevant for home NIV and active palliative care.
4.5. Strengths and Limitations
The large cohorts studied, assessing a wider range of phenotypic features and outcomes, and robustly assessing COPD-OSA overlap in the replication cohort are strengths. Although we focussed on patients at high risk of hypercapnia (LTOT use and previous episodes of AHRF), there was good representation of disease severities; a fifth of hypercapnic patients had an FEV1 >50% predicted. Furthermore, the inclusion of the AATD population allowed inclusion of those with mild disease, yet at risk of rapid progression. However, there were clear limitations; the measure of “increasing PaCO2” is not validated, and we did not use a comorbidity score (e.g., Charlson) to adjust for this in our multivariable analyses. Finally, the AATD cohort may be prone to acquisition bias, since those with hypercapnia may have poorer survival, or less ability to travel for assessment; nevertheless, the large number of very severe COPD patients seen suggests that this is not a major factor.
5. Conclusions
Lower FEV1 and prior AHRF are the main associations of stable-state hypercapnia in COPD, which carries a poor prognosis, particularly if it worsens over time in the stable state. Whilst sleep disordered breathing does occur and is clinically important, conducting a sleep study is likely only to be valuable in patients who are persistently hypercapnic in the stable state.
Acknowledgments
The authors would like to thank members of the Respiratory Physiology Department at Heartlands Hospital who aided sleep study conduct and reporting, and with whom informative discussions about the implications of results occurred. CD was funded during the study by Heartlands Hospital, equipment for sleep studies was provided by ResMed, and funding for review of AATD patients in the PAST was provided by Grifols biotherapeutics.
Data Availability
Data are available upon reasonable request.
Conflicts of Interest
Prof. Turner reports nonfinancial support from ResMed, during the conduct of the study, grants from NIHR, grants, personal fees, and nonfinancial support from AstraZeneca, grants and nonfinancial support from Chiesi, grants from Health Foundation, grants from Alpha 1 Foundation, grants from ATS Foundation, and personal fees and nonfinancial support from Boehringer Ingelheim, outside the submitted work. Dr. Mukherjee reports personal fees and nonfinancial support from Pfizer, personal fees from Boehringer Ingelheim, and personal fees from ResMed, outside the submitted work. Prof. Stockley has nor commercial activity or current or past grants that pertain to the current submission but is a member of the COLD scientific committee and contributes annually to the update of the strategy document although does not participate in discussions about publications on which he is an author. Otherwise, the authors declare that there are no conflicts of interest regarding the publication of this article.
Authors' Contributions
CD collected and analysed all data. AMT conceived the study and drafted the manuscript. SW, RM, BMF, and RAS reviewed and edited the manuscript.
Supplementary Materials
Supplementary Table 1: characteristics of AATD cohort. Supplementary Table 2: characteristics of patients whose arterial CO2 worsened over time. Supplementary Table 3: characteristics of COPD patients with or without OSA. Supplementary Table 4: independent associations of COPD-OSA overlap. Supplementary Table 5: impact of undiagnosed OSA on prior hospital admission with AHRF. Supplementary Table 6: independent associations of undiagnosed OSA. Supplementary Figure 1: hypercapnia and survival in the sleep cohort.
References
- 1.Lopez-Campos J. L., Hartl S., Pozo-Rodriguez F., Roberts C. M., European C. A. Antibiotic prescription for COPD exacerbations admitted to hospital: European COPD audit. PLoS One. 2015;10(4) doi: 10.1371/journal.pone.0124374.e0124374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts C. M., Stone R. A., Buckingham R. J., Pursey N. A., Lowe D. Acidosis, non-invasive ventilation and mortality in hospitalised COPD exacerbations. Thorax. 2011;66(1):43–48. doi: 10.1136/thx.2010.153114. [DOI] [PubMed] [Google Scholar]
- 3.Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA. 2002;288(8):932–935. doi: 10.1001/jama.288.8.932. [DOI] [PubMed] [Google Scholar]
- 4.Dretzke J., Blissett D., Dave C., et al. The cost-effectiveness of domiciliary non-invasive ventilation in patients with end-stage chronic obstructive pulmonary disease: a systematic review and economic evaluation. Health Technology Assessment. 2015;19(81):1–246. doi: 10.3310/hta19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu C. M., Chan V. L., Lin A. W., Wong I. W., Leung W. S., Lai C. K. Readmission rates and life threatening events in COPD survivors treated with non-invasive ventilation for acute hypercapnic respiratory failure. Thorax. 2004;59(12):1020–1025. doi: 10.1136/thx.2004.024307. Epub 2004/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrows B., Earle R. H. Chronic obstructive lung disease. New England Journal of Medicine. 1969;280(21):1183–1184. doi: 10.1056/nejm196902202800801. [DOI] [PubMed] [Google Scholar]
- 7.Slenter R. H. J., Sprooten R. T. M., Kotz D., Wesseling G., Wouters E. F. M., Rohde G. G. U. Predictors of 1-year mortality at hospital admission for acute exacerbations of chronic obstructive pulmonary disease. Respiration. 2013;85(1):15–26. doi: 10.1159/000342036. [DOI] [PubMed] [Google Scholar]
- 8.Costello R., Deegan P., Fitzpatrick M., McNicholas W. T. Reversible hypercapnia in chronic obstructive pulmonary disease: a distinct pattern of respiratory failure with a favorable prognosis. The American Journal of Medicine. 1997;102(3):239–244. doi: 10.1016/s0002-9343(97)00017-x. [DOI] [PubMed] [Google Scholar]
- 9.Kohnlein T., Windisch W., Kohler D., et al. Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: a prospective, multicentre, randomised, controlled clinical trial. The Lancet Respiratory Medicine. 2014;2 doi: 10.1016/S2213-2600(14)70153-5. [DOI] [PubMed] [Google Scholar]
- 10.Murphy P. B., Rehal S., Arbane G., et al. Effect of home noninvasive ventilation with oxygen therapy vs oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation. JAMA. 2017;317(21):2177–2186. doi: 10.1001/jama.2017.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorini M., Misuri G., Corrado A., et al. Breathing pattern and carbon dioxide retention in severe chronic obstructive pulmonary disease. Thorax. 1996;51(7):677–683. doi: 10.1136/thx.51.7.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saure E. W., Eagan T. M. L., Jensen R. L., et al. Explained variance for blood gases in a population with COPD. The Clinical Respiratory Journal. 2012;6(2):72–80. doi: 10.1111/j.1752-699x.2011.00248.x. [DOI] [PubMed] [Google Scholar]
- 13.Rodríguez D. A., Jover L., Drakulovic M. B., et al. Below what FEV1 should arterial blood be routinely taken to detect chronic respiratory failure in COPD? Archivos de Bronconeumología (English Edition) 2011;47(7):325–329. doi: 10.1016/j.arbr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Dave C., Turner A., Thomas A., et al. Utility of respiratory ward-based NIV in acidotic hypercapnic respiratory failure. Respirology. 2014;19(8):1241–1247. doi: 10.1111/resp.12366. [DOI] [PubMed] [Google Scholar]
- 15.Wood A. M., de Pablo P., Buckley C. D., Ahmad A., Stockley R. A. Smoke exposure as a determinant of autoantibody titre in 1-antitrypsin deficiency and COPD. European Respiratory Journal. 2011;37(1):32–38. doi: 10.1183/09031936.00033710. [DOI] [PubMed] [Google Scholar]
- 16.Turner A. M., Sen S., Steeley C., et al. Evaluation of oxygen prescription in relation to hospital admission rate in patients with chronic obstructive pulmonary disease. BMC Pulmonary Medicine. 2014;14:p. 127. doi: 10.1186/1471-2466-14-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pillai A. P., Turner A. M., Stockley R. A. Global initiative for chronic obstructive lung disease 2011 symptom/risk assessment in α 1 -antitrypsin deficiency. Chest. 2013;144(4):1152–1162. doi: 10.1378/chest.13-0161. [DOI] [PubMed] [Google Scholar]
- 18.Wood A. M., Harrison R. M., Semple S., Ayres J. G., Stockley R. A. Outdoor air pollution is associated with rapid decline of lung function in -1-antitrypsin deficiency. Occupational and Environmental Medicine. 2010;67(8):556–561. doi: 10.1136/oem.2009.047589. [DOI] [PubMed] [Google Scholar]
- 19.Green C. E., Parr D. G., Edgar R. G., Stockley R. A., Turner A. M. Lung density associates with survival in alpha 1 antitrypsin deficient patients. Respiratory Medicine. 2016;112:81–87. doi: 10.1016/j.rmed.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Rochester D. F., Braun N. M. Determinants of maximal inspiratory pressure in chronic obstructive pulmonary disease. The American Review of Respiratory Disease. 1985;132(1):42–47. doi: 10.1164/arrd.1985.132.1.42. [DOI] [PubMed] [Google Scholar]
- 21.Miravitlles M., Soler-Cataluña J. J., Calle M., Soriano J. B. Treatment of COPD by clinical phenotypes: putting old evidence into clinical practice. European Respiratory Journal. 2013;41(6):1252–1256. doi: 10.1183/09031936.00118912. [DOI] [PubMed] [Google Scholar]
- 22.Kang H. W., Kim T. O., Lee B. R., et al. Influence of diaphragmatic mobility on hypercapnia in patients with chronic obstructive pulmonary disease. Journal of Korean Medical Science. 2011;26(9):1209–1213. doi: 10.3346/jkms.2011.26.9.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallet R. H., Diaz J. V. The physiologic effects of noninvasive ventilation. Respiratory Care. 2009;54(1):102–115. [PubMed] [Google Scholar]
- 24.Saryal S., Celik G., Karabiyikoğlu G. Distinctive features and long-term survival of reversible and chronic hypercapnic patients with COPD. Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace. 1999;54(3):212–216. [PubMed] [Google Scholar]
- 25.Yang H., Xiang P., Zhang E., et al. Is hypercapnia associated with poor prognosis in chronic obstructive pulmonary disease? A long-term follow-up cohort study. BMJ Open. 2015;5(12) doi: 10.1136/bmjopen-2015-008909.e008909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trethewey S. P., Edgar R. G., Morlet J., Mukherjee R., Turner A. M. Late presentation of acute hypercapnic respiratory failure carries a high mortality risk in COPD patients treated with ward-based NIV. Respiratory Medicine. 2019;151:128–132. doi: 10.1016/j.rmed.2019.04.013. [DOI] [PubMed] [Google Scholar]
- 27.Resta O., Foschino Barbaro M. P., Brindicci C., Nocerino M. C., Caratozzolo G., Carbonara M. Hypercapnia in overlap syndrome: possible determinant factors. Sleep & Breathing = Schlaf & Atmung. 2002;6(1):11–18. doi: 10.1007/s11325-002-0011-6. [DOI] [PubMed] [Google Scholar]
- 28.Chaouat A., Weitzenblum E., Krieger J., Ifoundza T., Oswald M., Kessler R. Association of chronic obstructive pulmonary disease and sleep apnea syndrome. American Journal of Respiratory and Critical Care Medicine. 1995;151(1):82–86. doi: 10.1164/ajrccm.151.1.7812577. [DOI] [PubMed] [Google Scholar]
- 29.Soler X., Gaio E., Powell F. L., et al. High prevalence of obstructive sleep apnea in patients with moderate to severe chronic obstructive pulmonary disease. Annals of the American Thoracic Society. 2015;12(8):1219–1225. doi: 10.1513/annalsats.201506-379le. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He B.-T., Lu G., Xiao S.-C., et al. Coexistence of OSA may compensate for sleep related reduction in neural respiratory drive in patients with COPD. Thorax. 2017;72(3):256–262. doi: 10.1136/thoraxjnl-2016-208467. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: characteristics of AATD cohort. Supplementary Table 2: characteristics of patients whose arterial CO2 worsened over time. Supplementary Table 3: characteristics of COPD patients with or without OSA. Supplementary Table 4: independent associations of COPD-OSA overlap. Supplementary Table 5: impact of undiagnosed OSA on prior hospital admission with AHRF. Supplementary Table 6: independent associations of undiagnosed OSA. Supplementary Figure 1: hypercapnia and survival in the sleep cohort.
Data Availability Statement
Data are available upon reasonable request.