

Summary
Phagocytosis and autophagy play critical roles in immune defense. The human fungal pathogen Cryptococcus neoformans (Cn) subverts host autophagy-initiation complex (AIC)-related proteins, to promote its phagocytosis and intracellular parasitism of host cells. The mechanisms by which the pathogen engages host AIC-related proteins remain obscure. Here, we show that the recruitment of host AIC proteins to forming phagosomes is dependent upon the activity of CD44, a host cell surface receptor that engages fungal hyaluronic acid (HA). This interaction elevates intracellular Ca2+ concentrations and activates CaMKKβ and its downstream target AMPKα, which results in activation of ULK1 and the recruitment of AIC components. Moreover, we demonstrate that HA-coated beads efficiently recruit AIC components to phagosomes and CD44 interacts with AIC components. Taken together, these findings show that fungal HA plays a critical role in directing the internalization and productive intracellular membrane trafficking of a fungal pathogen of global importance.
Subject areas: Immunology, Mycology
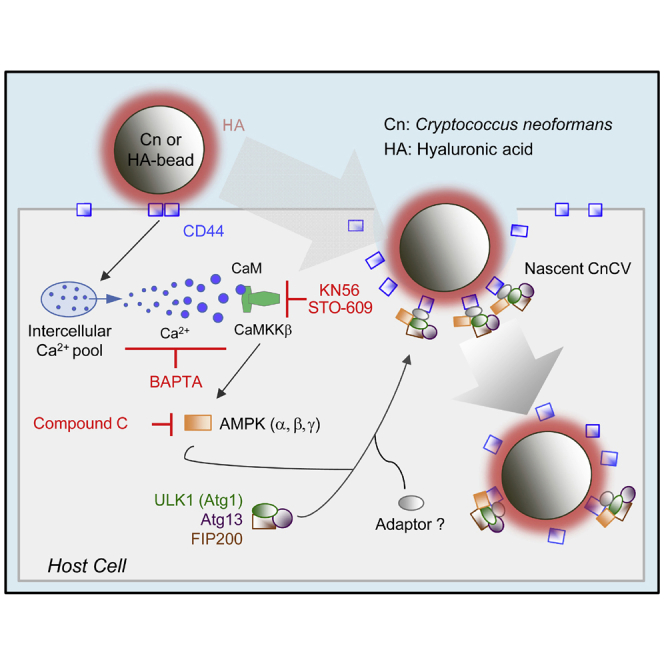
Graphical abstract
Highlights
-
•
Fungal HA drives non-canonical and ligand-induced autophagy in phagocytic cells
-
•
Cn recruits host CD44 to forming phagocytic cups to initiate fungal internalization
-
•
Fungal HA-CD44 interactions elevate intracellular Ca2+ levels and activate CaMKKβ
-
•
A Ca2+-CaMKKβ-AMPK-ULK1 signaling axis is involved in HA-CD44 induced autophagy
Immunology; Mycology
Introduction
Autophagy is an orderly “self-eating” process in cells that coordinates the degradation of cellular components. Various types of autophagy have been described, including macrophagy, microphagy, mitophagy, chaperone-mediated autophagy and xenophagy (Galluzzi et al., 2017; Khandia et al., 2019; Kirkin and Rogov, 2019). Some pathogens subvert autophagic machinery to promote their intracellular survival and replication (Case et al., 2016; de Figueiredo and Dickman, 2016; de Figueiredo et al., 2015; Dickman et al., 2017; Pandey et al., 2018). Several signaling pathways control the onset, duration and outcome of autophagy induction in mammalian cells (Abada and Elazar, 2014; Rubinsztein et al., 2012). Pathways that include components of the autophagy initiation complex (AIC), including AMPK, ULK1, ATG13, FIP200 and ATG9, play important roles in these processes (Ganley et al., 2009; Hosokawa et al., 2009; Jung et al., 2009). For example, AMPK or ULK1 signaling can regulate ATG9 recruitment to nascent phagosome or autophagosome membranes (Mack et al., 2012). This recruitment process is believed to contribute to the elongation of the autophagosomal membrane (Mack et al., 2012).
Cryptococcus neformans (Cn) is a pathogen of global consequence that causes fatal fungal meningoencephalitis worldwide (Kozubowski and Heitman, 2012; Olszewski et al., 2010; Sabiiti and May, 2012). Cn is particularly pernicious in immunocompromised individuals, where lethal infection constitutes a significant risk (Warkentien and Crum-Cianflone, 2010). Cn can survive, replicate and persist in both intracellular and extracellular environments within mammalian hosts (Garcia-Rodas and Zaragoza, 2012). However, the molecular mechanisms that control intracellular parasitism remain poorly understood (Evans et al., 2018; Zaragoza, 2019). Toward addressing this issue, we reported a functional analysis of host factors that regulate the infection, intracellular replication, and non-lytic release of Cn from host cells (Qin et al., 2011). We extended these findings by performing a phosphoproteomic analysis of the host response to Cn infection (Pandey et al., 2017). This analysis demonstrated that host AIC proteins, and upstream regulatory molecules, contribute to the internalization and intracellular replication of the pathogen in macrophages (Pandey et al., 2017). This work also raised questions about the cellular and molecular mechanisms by which these proteins contribute to this phenotype.
The internalization of Cn into host cells is regulated, in part, by interactions between fungal components and host associated CD44, a major receptor for hyaluronic acid (HA) in mammalian cells (Jong et al., 2008, 2012). Moreover, CD44 has been shown to control phagocytosis of the pathogen (Jong et al., 2008, 2012). Interestingly, mice deficient for CD44 display reduced susceptibility to infection (Jong et al., 2012). The deficiency in phagocytosis accounts for this phenotype, leaving open the question of the mechanism by which CD44 controls fungal internalization. Here, we show that Cn phagocytosis by macrophages occurs by a novel mechanism whereby AIC proteins, including ULK1, ATG9 and ATG13, as well as the key upstream signaling component AMPKα, are recruited to forming phagosomes to promote the phagocytosis of the pathogen in a CD44-dependent fashion. Interaction of fungal HA and host CD44 activates a Ca2+-CaMKKβ (calcium/calmodulin-dependent protein kinase kinase β subunit)-AMPK-ULK1 signaling axis that supports Cn internalization into host cells. Taken together, our findings uncover unexpected roles for HA-CD44 interactions in conferring susceptibility to fungal infection and open up new avenues for therapeutic intervention for a fungal pathogen of global importance.
Results
Cn recruits host AIC components to forming Cn-containing phagosomes
To test the hypothesis that Cn infection of macrophages promotes the formation of a physical complex that contains AIC components, we used Förster Resonance Energy Transfer (FRET) imaging microscopy, which detects close molecular associations (<10 nm) (Irving et al., 2014), to measure such interactions. The quenching of fluorescence in the donor fluorophore of an FRET pair accompanies the establishment of a close physical association between the pairs (Irving et al., 2014). We measured photon transfer between antibody labeled ATG13 and ULK1 or AMPKα and FIP200 in infected and uninfected RAW264.7 macrophages. We observed significant increases in the amount of FRET between these proteins in infected cells (Figures S1A–S1D). However, comparable FRET interactions were not observed in controls that were stained with a single label, or in uninfected samples (Figures S1A–S1D), thereby indicating the establishment of a close association between these proteins during infection.
Recruitment of AIC components to forming phagosomes containing Cn is galectin 8 independent
Previous studies have shown that galectin 8, a β-galactoside-binding lectin, monitors endosomal and lysosomal integrity by binding to host glycans on damaged pathogen-containing vacuoles. This binding drives the ubiquitin-dependent recruitment of autophagy adaptor proteins (e.g., NDP52) and microtubule associated light chain kinase 3 (LC3), which in turn, promotes the recruitment of autophagosome biogenesis proteins to damaged pathogen-containing vacuoles (Thurston et al., 2012). Galectin 8 mediated autophagosomal targeting is relevant to the observed recruitment of AIC components to phagocytic cups because Cn containing vacuoles (CnCVs) are permeabilized after phagocytosis by macrophages (Johnston and May, 2010). Moreover, nascent CnCVs in infected macrophages recruit host LC3 (Figure S1E) (Nicola et al., 2012; Qin et al., 2011), thereby implicating trafficking pathways that recruit LC3 to phagosomal membranes in controlling the intracellular lifestyle of Cn. With these ideas in mind, we tested the hypothesis that AIC recruitment to nascent CnCVs is associated with galectin 8 recruitment to these subcellular structures. We infected RAW264.7 macrophages that express a GFP-tagged variant of galectin 8 (GFP-Gal8) with Cn, and then used immunofluorescence microscopy (IFM) to determine whether GFP-Gal8 colocalized with AMPKα on nascent phagosomes containing Cn cells. We found that GFP-Gal8 did not display quantitative colocalization with either AMPKα or nascent phagosomes (Figure S1F). Consistent with these observations was the finding that UBEI-41, a potent and cell-permeable inhibitor of ubiquitin E1 activity (Yang et al., 2007), when added to macrophages at non-toxic doses (30 μM), did not diminish AMPKα recruitment to CnCVs (Figure S1G). Importantly, the inhibitor was washed out of the host cell culture media before Cn infection, thereby ensuring that the drug only targeted host cell components in these experiments. In addition, we found that nascent phagosomes decorated with the AIC component ULK1 colocalized with the endosomal marker EEA1 (Figure S1H). We also found that LC3 displayed minimal colocalization with AIC components on nascent CnCVs (Figure S1I). CnCVs in B6J2 macrophages expressing dominant negative variants of the AIC regulatory component AMPKα recruited less LC3 (Figure S1J) and AIC components (Figure S1K), thereby suggesting that AIC recruitment to nascent Cn-containing phagosomes and LC3 recruitment to phagosome membranes could be morphologically and genetically dissected. Taken together, these findings indicated that recruitment of AIC to the nascent phagosomes and the induction of autophagy were galectin 8-independent events.
A non-proteinaceous and/or non-capsular component controls AIC recruitment to nascent CnCVs
The observation that AIC and AIC regulatory components were recruited to forming phagosomes during Cn infection of macrophages encouraged us to determine the mechanism of recruitment. We infected host cells with live or heat-killed (HK) Cn and found that fungal viability was not required for AIC recruitment because nascent CnCVs containing the HK organism also efficiently recruited these proteins (Figure 1A). However, compared to host cells with internalized Cn cells, differential expression or fluorescence signal levels as well as differential localization of AIC and AIC regulatory components in host cells lacking Cn cells were observed (Figures 1A, S1F–S1G, S1J, and S2). These observations were consistent with the hypothesis that non-proteinaceous, Cn-associated molecules activate the host AIC pathway.
Figure 1.

Non-proteineous components on Cryptococcus neoformans (Cn) direct the recruitment of host AIC components to nascent phagosomes
(A and B) Colocalization of AMPKα and AIC components surrounding nascent CnCVs in host cells infected with live or heat-killed (HK) H99 (A) and opsonized or unopsonized acapsular cap59 strains (B) of Cn. RAW264.7 macrophages were infected with the indicated Cn cells at a multiplicity of infection (MOI) of 5. At 3 hr post infection (h.p.i.), the infected cells were fixed with 3.7% formaldehyde in 1×PBS for 1 to 2 hr and then subjected to immunofluorescence microscopy assays with the indicated antibodies. Host cells bounded with yellow or white dash lines indicate cells containing or lacking internalized Cn cells, respectively.
(C and D) Recruitment of AMPKα and ULK1 to nascent CnCVs in host cells incubated with HK-cap59 (5 MOI) at 3 h.p.i. (C) and the fluorescence intensity profile of AMPKα (green) and ULK1 (red) along the two crossed white lines (D).
(E and F) Activation of host cell AMPKα (E) and ULK1 (F) by acapsular Cn strain cap59. Representative blots and data from one of three independent experiments are shown in (E to H).
(G and H) Activation of host cell AMPKα (G) and ULK1 (H) by HK Cn strain H99 (HK-H99).
Cn is encased in a carbohydrate-enriched capsule that is essential for intracellular parasitism and virulence (O'Meara and Alspaugh, 2012). To test the hypothesis that capsular components direct the recruitment of AIC and AIC regulatory proteins to forming phagosomes, we infected host cells with an acapsular mutant of Cn (Cap59), which displays defects in extracellular trafficking of glucuronoxylomannan (GXM) (Garcia-Rivera et al., 2004), and analyzed AIC recruitment to phagosomes containing cap59 strains. We found that forming and formed phagosomes that contained the acapsular strain also efficiently recruited AIC proteins (Figures 1B–1D and S3A–S3C). AIC components showed close associations with internalized Cn cells as detected by indirect immunofluorescence with the monoclonal antibody 18B7 against GXM (Garcia-Rivera et al., 2004) (Figure S3D). Moreover, infection of host cells with live wild-type (WT) Cn resulted in the activation by phosphorylation of host cell ULK1 (Ser555), a key component of the AIC, and activation of the AIC regulatory protein AMPKα (Thr172) (Pandey et al., 2017). Similarly, the acapsular mutant and HK Cn also activated host AMPKα (Thr172) and/or ULK1 (Figures 1E–1H and S3E). However, when S. cerevisiae was incubated with host cells, similar AIC recruitment was not observed (Figure S3F), suggesting that phagocytosis of yeast cells by macrophages involves the participation of a distinct mechanism. Taken together, these findings suggested that Cn-specific, non-proteinaceous, non-capsular components activated the host AMPK-ULK1 signaling axis and promoted AIC recruitment to forming phagosomes.
CD44-deficient host cells fail to recruit AIC proteins to forming phagosomes
CD44 regulates fungal internalization (Jong et al., 2008, 2012). This observation raised the intriguing possibility that CD44 interactions with other host cell components may also control the recruitment of AIC components to nascent CnCVs. To test this hypothesis, we first used fluorescence microscopy to determine whether CD44 colocalized with Cn cells or was recruited to forming or formed nascent CnCVs. We also tested whether CD44 deficient macrophages recruited AIC components to the nascent pathogen-containing phagosomes. We found that forming or formed phagosomes containing both capsular or acapsular strains displayed strong colocalization with CD44 (Figure 2A), and that CD44 was enriched at sites of contact between the pathogen and the host cell surface during a time course of infection (Figure S3G). Next, we tested whether CD44 colocalized with AIC regulatory AMPK and AIC components on nascent CnCVs. We found that AIC components, including ATG9 and ULK1, colocalized with CD44 on nascent CnCVs (Figures 2B and 2C). Compared to CD44+/+ bone-marrow derived macrophage (BMDM) controls, AMPKα showed reduced colocalization in CD44−/− BMDMs (Figure 2D). As a result, Cn internalization in CD44-deficient cells was reduced (Figures 2E and 2F), indicating that host cell CD44 is required for Cn internalization, and suggesting that activation of the AMPK-ULK1 signaling axis and AIC recruitment is CD44-dependent.
Figure 2.

Host-associated CD44 is required for Cn host cell internalization
(A) Recruitment of host CD44 to nascent CnCVs that contained the wild-type (WT) strain H99 (leaf panel) or acapsular mutant strain cap59 (right panel) of Cn. At 1 h.p.i., host cells were fixed, permeabilized and processed for immunofluorescence microscopy using antibodies directed against the indicated host proteins. The antibody-stained samples were then subjected to confocal microscopy image analysis. Bars: 5 μm.
(B) Colocalization of CD44 with the indicated AIC components in the vicinity of nascent CnCVs in host cells infected with the indicated Cn strains. Bars: 5 μm.
(C) The fluorescence intensity profile of CD44 (red) and AIC components (ATG9 or ULK1) (green) along the two crossed white lines shown in the insets from (B, Merge 1).
(D) AMPK recruitment to nascent CnCVs in CD44 knockout (KO, CD44−/−) and WT (CD44+/+) bone marrow-derived macrophages (BMDMs) at 3 h.p.i.
(E and F) Cn internalization in CD44 WT and KO BMDMs infected by Cn H99 (1 or 10 MOI) assessed using image analysis approaches (E) and corresponding quantification (F) at 3 h.p.i. Data represent the means ± standard error of mean (SEM) from three independent experiments. ∗∗∗: significance at p < 0.001.
cps1 deletion mutants fail to recruit AIC components to nascent CnCVs
HA, a component of the Cn cell wall, is known to interact with CD44, an HA receptor on host cells, and to promote the internalization of the pathogen into human and murine brain microvascular endothelial cells (BMECs) (Jong et al., 2008, 2012). This observation raised the intriguing possibility that HA-CD44 interactions may promote the recruitment of AIC components to nascent CnCVs. To test this hypothesis, we first examined the internalization of Cn strains that harbor mutations in cps1, a gene that encodes hyaluronic synthase (Jong et al., 2007). Consistent with previous findings where Cn displayed reduced association with CD44-depleted murine BMECs compared to controls (Jong et al., 2012), we found that deletion of cps1 displayed reduced internalization of Cn into host macrophages (Figures 3A–3C and S4A). Next, we used fluorescence microscopy to determine whether cps1-deficient Cn strains recruited AIC components to nascent pathogen-containing phagosomes. We found the mutant strain displayed reduced recruitment compared to WT controls (Figure 3D). However, nascent CnCVs that contained strain C558 (cps1Δ::CPS1), in which the cps1 mutation was complemented with a WT copy of the gene (Jong et al., 2008), displayed higher levels of AIC recruitment than their cps1-deficient counterparts (Figure 3D, bottom panel). These data suggested that HA, the product of cps1 activity, contributed to directing the recruitment of AIC components to nascent CnCVs, and implicated a role for CD44, the dominant HA receptor on macrophages, in regulating this process.
Figure 3.

Fungal HA is required for recruitment of AMPK or AIC components to nascent phagosomes and Cn internalization
(A) Internalization of the wild-type, cps1Δ and complemented Cn strains via colony formation unit (CFU) assay. Re_Cn: relative Cn.
(B and C) Internalization of the indicated Cn strains in BMDMs (B) and quantification of intracellular Cn cells (C) at 3 h.p.i. via microscopy image analysis. BMDMs were infected with the indicated Cn strains at an MOI of 10.
(D) Confocal microscopy image analysis of recruitment of CD44 and the AIC component ULK1 by wild-type, cps1Δ, and complemented (C588) Cn strains (5 MOI for each strain) at 3 h.p.i.
(E and F) Recruitment of host CD44 and AMPKα (E), or CD44 and AIC component ULK1 (F) to nascent phagosomes or CnCVs by HA-coated beads or Cn (H99, 5 MOI), respectively. Bars: 5 μm.
(G and H) Quantification of CD44 and AMPKα positive beads (G) or CD44 and ULK1 positive beads based on confocal microscopy images as shown in (E, F). Data represent the means ± SEM from three independent experiments. ∗∗∗: significance at p < 0.001.
Pathogen-derived HA drives interactions between CD44 and AIC components
To test whether HA was sufficient to induce CD44-mediated recruitment of AIC components to nascent CnCVs, we determined whether HA-coated beads induced similar recruitment to forming phagosomes. For these experiments, we covalently coupled HA to polystyrene beads and then incubated the HA-coupled beads with RAW264.7 macrophages for various lengths of time. We also used antibodies directed against AIC components in immunofluorescence microscopy experiments to visualize the recruitment of AIC components to forming phagosomes that contained beads. We found robust recruitment of AMPKα and AIC component ULK1 to forming phagosomes containing HA-coated beads (Figures 3E–3H). We also incubated HA-coated beads with GFP-Gal8 expressing macrophages and found that co-recruitment of AMPKα and galectin 8 to the sites of bead internalization was not detected (Figure S4B, upper). However, co-localization of CD44 and AMPKα was observed to be colocalized with the HA-coated beads (Figure S4B, lower), Our observations therefore suggested that HA interactions with CD44 were necessary and sufficient to induce the formation of an AIC protein complex on forming phagosomes.
Interaction of fungal HA with host CD44 activates AMPK and AIC pathways
The observation that interactions between HA and host CD44 recruited AIC components to forming phagosomes raised questions about the mechanism of AIC recruitment by these components. HA induces Ca2+ elevation in cells and may increase the activity of Ca2+-associated signaling pathways (Singleton and Bourguignon, 2002). For example, an increase of cytoplasmic Ca2+ levels can induce autophagy through CaMKKβ and AMPK pathways (Feng et al., 2020; Green et al., 2011). To test whether HA and CD44 interactions elevate intracellular Ca2+ ([Ca2+]i) levels and induce autophagy by Ca2+-mediated activation of CaMKKβ and AMPK pathways, we incubated host cells with Cn cells, HA or HA-coated beads, and then visualized [Ca2+]i levels and activation of CaMKKβ-AMPK pathways in the treated cells. We observed that compared to the controls, cps1+-Cn induced Ca2+-fluxes and increased [Ca2+]i concentrations in a pulsed manner (Figures 4A–4C; Videos S1 and S2). Corresponding to the increase of [Ca2+]i levels, activation of CaMKKβ and AMPKα was observed in host cells incubated with Cn cells (Pandey et al., 2017), HA or HA-coated beads (Figures 4D and 4E). Importantly, phosphorylated AMPKα and ATG9 were co-immunoprecipitated (Co-IP) from host cells incubated with HA-coated, but not naked, beads (Figure 4F). Moreover, during Cn infection, host CD44 expression was induced (Figure 4G) and physical interactions of host CD44 and AIC components, including ATG13 and ATG9, were detected (Figure 4H).
Figure 4.

Cn infection activates CaMKKβ-AMPK-ULK1 signaling axis
(A and B) Cn infection results in intracellular Ca2+-flux in the infected host cells. Host cells incubated with cps1+ Cn (A) or PBS (B) and the intracellular Ca2+ level was detected by fluorescence emission between 500 and 550 nm. Red arrows: Intracellular Ca2+ increase in cps1+-Cn-infected host cells. Yellow arrows: Ca2+ releases from cells treated with ionomycin, a membrane permeable calcium ionophore used to increase intracellular calcium levels.
(C) Mean intracellular Ca2+ flux intensity of Cn-infected (upper panel) and control (lower panel) cells.
(D and E) Activation of host CaMKKβ (D) and AMPKα (E) in cells incubated with HA or HA-coated beads. Representative blots and data from one of three independent experiments are shown in (D to K).
(F) Co-immunoprecipitation (Co-IP) assays of host phosphorylated AMPKα and ATG9 trigged by HA-coated beads. Host cells were incubated with naked or HA-coated beads. At 3 h.p.i., the cells were lysed and immunoprecipitated using antibodies against CD44. The precipitated material was then visualized by Western blot analysis using antibodies directed against p-AMPKα or ATG9.
(G) Induction of CD44 protein (blue arrow) level by Cn infection. Host cells were infected with Cn H99 and at the indicated h.p.i., the infected cells were lysed and then subjected to immunoblotting assays using antibodies against CD44 and GAPDH.
(H) Co-IP assays show physical interactions between host CD44 and AIC components ATG13 or ATG9. BMDMs were infected with Cn H99 and at the indicated h.p.i., the infected cells were lysed and immunoprecipitated using antibodies against CD44. The precipitated material was then subjected to Western blot analysis using antibodies directed against CD44, ATG13 or ATG9. M: Prestained Protein Molecular Weight Marker.
(I and J) Chelation of intracellular Ca2+ by BAPTA-AM and inhibition of the activity of CaMKKβ by STO-609 reduce activation of AMPKα (I) and ULK1 (J). RAW264.7 macrophages were infected with Cn H99 in the absence or presence of BAPTA-AM (7.5 μM) or STO-609 (5 μM). At the indicated time points post infection, the infected cells were lysed for Western blot analysis using antibodies directed against p-AMPKα, p-ULK1, and pan-AMPKα or pan-ULK1.
(K) Depletion of AMPKα reduces activation of ULK1 during Cn internalization. Sta: starvation.
(L) Cn internalization in host cells treated by the indicated drug (BAPTA-AM: 7.5 μM, KN62: 5μM, STO-609: 5 μM) via CFU assays. Data represent the means ± SEM from three independent experiments. ∗, ∗∗, ∗∗∗: significance at p < 0.05, 0.01, and 0.001, respectively.
To test whether activation of AMPK resulted from the increase of [Ca2+]i, we treated host cells with or without supplementation of assorted inhibitors, including BAPTA-AM (a [Ca2+]i-chelator), KN62 (a specific CaMK inhibitor), and STO-609 (a specific CaMKKα/β inhibitor), during infection with Cn. We found that host cells treated with these compounds displayed reduced activation of AMPKα (Figure 4I); the activation of the downstream target ULK1 was also almost completely blocked (Figure 4J). During Cn infection, ULK1 is activated in a p-AMPKα-dependent fashion (Figures 4I–4K). Consequently, Cn internalization was reduced in cells in which [Ca2+]i was chelated, the activities of CaMK or CaMKKα/β were inhibited or depleted (Figures 4L and S5A–S5D), or AMPK or AIC components were depleted (Figures S5E and S5F). These findings suggest that interaction of HA with CD44 recruits AIC to forming phagosomes by increase of [Ca2+]i and activation of the CaMKKβ-AMPK-ULK1 signaling axis.
Discussion
The intracellular lifestyle of Cn is pivotal for pathogen colonization, dissemination and disease progression (Garcia-Rodas and Zaragoza, 2012; Johnston and May, 2013; Seider et al., 2010), as well as establishment of latent infection (Saha et al., 2007). Although the complete set of host factors that regulate intracellular parasitism remain obscure, published reports demonstrate that both “zipper” (receptor-mediated) and “trigger” (membrane ruffle dependent) mechanisms contribute to the internalization of Cn (Guerra et al., 2014). Moreover, interactions between the opsonized or unopsonized pathogen and host cell surface receptors are important to these processes (Shoham et al., 2001; Taborda and Casadevall, 2002). The findings reported here provide a new understanding of phagocytic mechanisms by demonstrating that fungal HA- and host CD44-dependent recruitment of AIC network components to nascent CnCVs play a central role in regulating the internalization of the fungus. Cn cells, opsonized or unopsonized and live or dead, as well as HA-coated beads, efficiently recruited host CD44, components of AIC and its regulatory protein AMPK to forming or formed phagosomes. As such, this report provides the first example of ligand- and receptor-induced recruitment of AIC proteins, including ULK1, FIP200, ATG13, and ATG9, to nascent phagosomes in macrophages.
Host cell galectin 8 is involved in defending against bacterial infection by recruiting the autophagic adaptor NDP52 to damaged Salmonella-containing vacuoles and in activating antibacterial autophagy (Thurston et al., 2012). Different from the defensive autophagy induced by galectin 8, significant recruitment of the danger receptor galectin 8 to forming or formed phagosomes during Cn internalization and/or intracellular replication was not observed, suggesting that a different strategy is employed to recruit elements of the autophagy machinery through interactions between fungal HA and host cell CD44. Besides AMPK and AIC components, the endosomal and lysosomal markers EEA1, M6PR, and Cathepsin D, as well as the ER marker calreticulin, were also recruited to nascent phagosomes (Qin et al., 2011). Interestingly, co-localization of the endocytic marker EEA1 and the AIC component ULK1 was also observed, suggesting that the endosomal and lysosomal pathways, ER-derived membrane and selective autophagy machinery are involved in the internalization of Cn into host cells. How these factors coordinate this process and the involvement of other ubiquitin-binding autophagic adaptors related to the AIC remain to be characterized.
AMPK can be activated by cellular stresses that elevate AMP levels by means of allosteric binding of AMP to sites in the γ subunit AMPK, and by phosphorylation of Thr172 in AMPKα by the tumor suppressor LKB1, CaMKKβ, or the transforming growth factor-β-activated kinase (TAK1) (Hardie et al., 2012; Kola et al., 2006; Zadra et al., 2015). Activation of AMPK can induce autophagy via direct phosphorylation of ULK1 (Egan et al., 2011; Kim et al., 2011; Zhao and Klionsky, 2011). Previous observations showed that HA interactions with CD44 induces Ca2+ elevation in cells (Singleton and Bourguignon, 2002), and that increases in cytoplasmic Ca2+ concentrations induce autophagy through activation of CaMKKβ and AMPK pathways (Feng et al., 2020; Green et al., 2011). Similarly, our findings demonstrate that infection by Cn recruits host cell CD44 to CnCVs, induces intracellular Ca2+-flux in the infected host cells, activates CaMKKβ and AMPK, and recruits AIC components to forming or nascent CnCVs. Cn infection also activates LKB1, and depletion of LKB1 reduced Cn internalization into host cells (Pandey et al., 2017). However, how the interaction of HA and CD44 activates AMPK through the upstream regulatory proteins LKB1 and TAK1 remains to be further characterized.
Our data support a stepwise model in which several sequential molecular events control the internalization of Cn in macrophages. First, interactions between fungal HA and CD44 on the surface of host cells likely stimulates the release of Ca2+ and elevates intercellular Ca2+ levels, which results in the activation of CaMKKβ and AMPK. Second, phosphorylation of ULK1 occurs in an AMPK-dependent fashion. Third, AMPK-dependent activation of ULK1 recruits AIC components, including the ULK1-ATG13-FIP200 complex, ATG9, and LC3, to the site of forming phagocytic cups containing the fungus. Fourth, the coordinated activities of AIC components drive the internalization of the pathogen into host cells. ATG13 and FIP200 facilitate ULK1 to the site of autophagosome formation, kinase activity enhancement, and stability (Ganley et al., 2009). ULK1 kinase functions early in autophagy by regulating the outgrowth of autophagosomal membranes. Direct ULK1 phosphorylation of ATG9 is essential for autophagy and phosphorylated ATG9 is then required for the efficient recruitment of Atg8/LC3 and ATG18 to the sites of autophagosome formation and subsequent expansion of the isolation membrane (Papinski et al., 2014). Besides AIC and LC3, other autophagy proteins, e.g., ATG5, ATG12, may also be recruited to contribute to the elongation of phagosome membranes to envelop Cn in host cells; depletion or conditional-knockout of these proteins results in reduced Cn phagocytosis, intracellular replication, nonlytic exocytosis (Qin et al., 2011) or a less intense pneumonia and later alternative activation (Nicola et al., 2012). Ultimately, AIC component interactions with CnCVs gradually diminish as the pathogen establishes a replicative niche in host cells (Figure for graphical Abstract).
Finally, it is notable that several proteins in AMPKα and AIC regulatory networks are targets of commonly prescribed drugs (e.g., AMPK, a target for metformin) or under development as targets for pharmaceutical intervention (e.g., ULK1) (Egan et al., 2015). Several fungi, including Candida spp. and Histoplasma capsulatum, are capable of intracellular parasitism (Garcia-Rodas et al., 2011; Howard, 1965; Woods, 2003). Therefore, our findings may open up new therapeutic possibilities for preventing cryptococcosis and other infections caused by intracellular pathogens.
Limitations of the study
This study reveals fungal HA and host CD44 interactions activate a Ca2+-CaMKKβ-AMPK-ULK1 signaling pathway that recruits autophagy initiation complex components, including AMPKα, ULK1, ATG13, FIP200 and ATG9, to forming phagosomes to drive fungal internalization. However, our understanding of how CD44 is recruited to phagocytic cups and how it coordinates with other components to promote Cn phagocytosis remains incomplete. It has been noted that the mobility of CD44 is dependent on the state of the actin cytoskeleton and the lipid composition of the plasma membrane (Jong et al., 2008). Our unpublished data imply that Toll-like receptors and mitogen-activated protein (MAP) kinase pathway activity may also be involved in the recruitment of AIC components and/or the induction of autophagy, processes which are mediated by HA-CD44 interactions. Further investigation is needed to address and clarify these remaining questions.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Paul de Figueiredo (pjdefigueiredo@tamu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze datasets or code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We gratefully thank Dr. Arturo Casadevall (Department of Microbiology and Immunology, School of Medicine, Johns Hopkins University) for anti-cryptococcal antibodies, Dr. Jong (University of California, Los Angeles) for Cn strains C177, cps1Δ, and C558, and Steve Fullwood and Kalli Landua (Nikon Instruments) for expert assistance with the microscopy analysis. This work was supported by the Texas A&M Clinical Science Translational Research Institute Pilot Grant CSTR2016-1, the Defense Advanced Research Projects Agency (DARPA) (HR001118A0025-FoF-FP-006), NIH (R21AI139738-01A1, 1 R01AI141607-01A1, 1R21GM132705-01), the National Science Foundation (DBI 1532188, NSF0854684) and the Bill Melinda Gates Foundation to PdF; NIH grant awards NIH 1R01 AI48496-01A1 and NIH 1U54AI057156-0100 to TAF; the National Natural Science Foundation of China (# 81371773) to QMQ. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the funding agencies.
Author contributions
P.D., Q.M.Q., T.A.F. designed the experiments. S.L.D., J.Y., X.F., Q.M.Q, A.P., R.M. D.Z., S.L.B., L.F.d.C. performed experiments. P.D., T.A.F., Q.M.Q., A.R., R.M., R.O.W., K.L.P. provided reagents/analysis tools. P.D., Q.M.Q., S.L.D., X.F., A.P., Y.L., T.A.F. analyzed data. P.D., Q.M.Q. supervised the work and wrote the manuscript. All authors read approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: March 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102192.
Contributor Information
Qing-Ming Qin, Email: qmqin@jlu.edu.cn.
Thomas A. Ficht, Email: tficht@cvm.tamu.edu.
Paul de Figueiredo, Email: pjdefigueiredo@tamu.edu.
Supplemental information
References
- Abada A., Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep. 2014;15:839–852. doi: 10.15252/embr.201439076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case E.D.R., Smith J.A., Ficht T.A., Samuel J.E., de Figueiredo P. Space: a final frontier for vacuolar pathogens. Traffic. 2016;17:461–474. doi: 10.1111/tra.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo P., Dickman M. Plant disease: autophagy under attack. Elife. 2016;5:e14447. doi: 10.7554/eLife.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo P., Ficht T.A., Rice-Ficht A., Rossetti C.A., Adams L.G. Pathogenesis and immunobiology of brucellosis: review of Brucella–Host Interactions. Am. J. Pathol. 2015;185:1505–1517. doi: 10.1016/j.ajpath.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman M., Williams B., Li Y., de Figueiredo P., Wolpert T. Reassessing apoptosis in plants. Nat. Plants. 2017;3:773–779. doi: 10.1038/s41477-017-0020-x. [DOI] [PubMed] [Google Scholar]
- Egan D.F., Chun M.G., Vamos M., Zou H., Rong J., Miller C.J., Lou H.J., Raveendra-Panickar D., Yang C.C., Sheffler D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R.J., Sundaramurthy V., Frickel E.-M. The interplay of host autophagy and eukaryotic pathogens. Front. Cell Dev. Biol. 2018;6:118. doi: 10.3389/fcell.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N., Wang B., Cai P., Zheng W., Zou H., Gu J., Yuan Y., Liu X., Liu Z., Bian J. ZEA-induced autophagy in TM4 cells was mediated by the release of Ca2+ activates CaMKKβ-AMPK signaling pathway in the endoplasmic reticulum. Toxicol. Lett. 2020;323:1–9. doi: 10.1016/j.toxlet.2020.01.010. [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Baehrecke E.H., Ballabio A., Boya P., Bravo-San Pedro J.M., Cecconi F., Choi A.M., Chu C.T., Codogno P., Colombo M.I. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley I.G., Lam D.H., Wang J., Ding X., Chen S., Jiang X. ULK1· ATG13· FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rivera J., Chang Y.C., Kwon-Chung K.J., Casadevall A. Cryptococcus neoformans CAP59 (or Cap59p) is involved in the extracellular trafficking of capsular glucuronoxylomannan. Eukaryot. Cell. 2004;3:385–392. doi: 10.1128/EC.3.2.385-392.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodas R., Gonzalez-Camacho F., Rodriguez-Tudela J.L., Cuenca-Estrella M., Zaragoza O. The interaction between Candida krusei and murine macrophages results in multiple outcomes, including intracellular survival and escape from killing. Infect. Immun. 2011;79:2136–2144. doi: 10.1128/IAI.00044-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodas R., Zaragoza O. Catch me if you can: phagocytosis and killing avoidance by Cryptococcus neoformans. FEMS Immunol. Med. Microbiol. 2012;64:147–161. doi: 10.1111/j.1574-695X.2011.00871.x. [DOI] [PubMed] [Google Scholar]
- Green M.F., Anderson K.A., Means A.R. Characterization of the CaMKKβ–AMPK signaling complex. Cell Signal. 2011;23:2005–2012. doi: 10.1016/j.cellsig.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C.R., Seabra S.H., de Souza W., Rozental S. Cryptococcus neoformans is internalized by receptor-mediated or 'triggered' phagocytosis, dependent on actin recruitment. PLoS One. 2014;9:e89250. doi: 10.1371/journal.pone.0089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S.-i., Natsume T., Takehana K., Yamada N. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard D.H. Intracellular growth of Histoplasma capsulatum. J. Bacteriol. 1965;89:518–523. doi: 10.1128/jb.89.2.518-523.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving A.T., Mimuro H., Kufer T.A., Lo C., Wheeler R., Turner L.J., Thomas B.J., Malosse C., Gantier M.P., Casillas L.N. The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe. 2014;15:623–635. doi: 10.1016/j.chom.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Johnston S.A., May R.C. The human fungal pathogen Cryptococcus neoformans escapes macrophages by a phagosome emptying mechanism that is inhibited by Arp2/3 complex-mediated actin polymerisation. PLoS Pathog. 2010;6:e1001041. doi: 10.1371/journal.ppat.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.A., May R.C. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol. 2013;15:403–411. doi: 10.1111/cmi.12067. [DOI] [PubMed] [Google Scholar]
- Jong A., Wu C.H., Chen H.M., Luo F., Kwon-Chung K.J., Chang Y.C., Lamunyon C.W., Plaas A., Huang S.H. Identification and characterization of CPS1 as a hyaluronic acid synthase contributing to the pathogenesis of Cryptococcus neoformans infection. Eukaryot. Cell. 2007;6:1486–1496. doi: 10.1128/EC.00120-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong A., Wu C.H., Gonzales-Gomez I., Kwon-Chung K.J., Chang Y.C., Tseng H.K., Cho W.L., Huang S.H. Hyaluronic acid receptor CD44 deficiency is associated with decreased Cryptococcus neoformans brain infection. J. Biol. Chem. 2012;287:15298–15306. doi: 10.1074/jbc.M112.353375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong A., Wu C.H., Shackleford G.M., Kwon-Chung K.J., Chang Y.C., Chen H.M., Ouyang Y., Huang S.H. Involvement of human CD44 during Cryptococcus neoformans infection of brain microvascular endothelial cells. Cell Microbiol. 2008;10:1313–1326. doi: 10.1111/j.1462-5822.2008.01128.x. [DOI] [PubMed] [Google Scholar]
- Jung C.H., Jun C.B., Ro S.-H., Kim Y.-M., Otto N.M., Cao J., Kundu M., Kim D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandia R., Dadar M., Munjal A., Dhama K., Karthik K., Tiwari R., Yatoo M., Iqbal H., Singh K.P., Joshi S.K. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8:674. doi: 10.3390/cells8070674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V., Rogov V.V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell. 2019;76:268–285. doi: 10.1016/j.molcel.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Kola B., Boscaro M., Rutter G.A., Grossman A.B., Korbonits M. Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. 2006;17:205–215. doi: 10.1016/j.tem.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Kozubowski L., Heitman J. Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol. Rev. 2012;36:78–94. doi: 10.1111/j.1574-6976.2011.00286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack H.I., Zheng B., Asara J.M., Thomas S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy. 2012;8:1197–1214. doi: 10.4161/auto.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola A.M., Albuquerque P., Martinez L.R., Dal-Rosso R.A., Saylor C., De Jesus M., Nosanchuk J.D., Casadevall A. Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect. Immun. 2012;80:3065–3076. doi: 10.1128/IAI.00358-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara T.R., Alspaugh J.A. The Cryptococcus neoformans capsule: a sword and a shield. Clin. Microbiol. Rev. 2012;25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski M.A., Zhang Y., Huffnagle G.B. Mechanisms of cryptococcal virulence and persistence. Future Microbiol. 2010;5:1269–1288. doi: 10.2217/fmb.10.93. [DOI] [PubMed] [Google Scholar]
- Pandey A., Ding S.L., Qin Q.M., Gupta R., Gomez G., Lin F., Feng X., Fachini da Costa L., Chaki S.P., Katepalli M. Global reprogramming of host kinase signaling in response to fungal infection. Cell Host Microbe. 2017;21:637–649 e636. doi: 10.1016/j.chom.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A., Lin F., Cabello A.L., da Costa L.F., Feng X., Feng H.-Q., Zhang M.-Z., Iwawaki T., Rice-Ficht A., Ficht T.A. Activation of host IRE1α-dependent signaling axis contributes the intracellular parasitism of Brucella melitensis. Front. Cell Infect. Microbiol. 2018;8:103. doi: 10.3389/fcimb.2018.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papinski D., Schuschnig M., Reiter W., Wilhelm L., Barnes C.A., Maiolica A., Hansmann I., Pfaffenwimmer T., Kijanska M., Stoffel I. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell. 2014;53:471–483. doi: 10.1016/j.molcel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q.M., Luo J., Lin X., Pei J., Li L., Ficht T.A., de Figueiredo P. Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog. 2011;7:e1002078. doi: 10.1371/journal.ppat.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D.C., Shpilka T., Elazar Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012;22:R29–R34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Sabiiti W., May R.C. Mechanisms of infection by the human fungal pathogen Cryptococcus neoformans. Future Microbiol. 2012;7:1297–1313. doi: 10.2217/fmb.12.102. [DOI] [PubMed] [Google Scholar]
- Saha D.C., Goldman D.L., Shao X., Casadevall A., Husain S., Limaye A.P., Lyon M., Somani J., Pursell K., Pruett T.L. Serologic evidence for reactivation of cryptococcosis in solid-organ transplant recipients. Clin. Vaccin. Immunol. 2007;14:1550–1554. doi: 10.1128/CVI.00242-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seider K., Heyken A., Luttich A., Miramon P., Hube B. Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr. Opin. Microbiol. 2010;13:392–400. doi: 10.1016/j.mib.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Shoham S., Huang C., Chen J.M., Golenbock D.T., Levitz S.M. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J. Immunol. 2001;166:4620–4626. doi: 10.4049/jimmunol.166.7.4620. [DOI] [PubMed] [Google Scholar]
- Singleton P.A., Bourguignon L.Y. CD44v10 interaction with Rho-kinase (ROK) activates inositol 1, 4, 5-triphosphate (IP3) receptor-mediated Ca2+ signaling during hyaluronan (HA)-induced endothelial cell migration. Cell Motil. Cytoskeleton. 2002;53:293–316. doi: 10.1002/cm.10078. [DOI] [PubMed] [Google Scholar]
- Taborda C.P., Casadevall A. CR3 (CD11b/CD18) and CR4 (CD11c/CD18) are involved in complement-independent antibody-mediated phagocytosis of Cryptococcus neoformans. Immunity. 2002;16:791–802. doi: 10.1016/s1074-7613(02)00328-x. [DOI] [PubMed] [Google Scholar]
- Thurston T.L., Wandel M.P., von Muhlinen N., Foeglein A., Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkentien T., Crum-Cianflone N.F. An update on Cryptococcus among HIV-infected patients. Int. J. STD AIDS. 2010;21:679–684. doi: 10.1258/ijsa.2010.010182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods J.P. Knocking on the right door and making a comfortable home: Histoplasma capsulatum intracellular pathogenesis. Curr. Opin. Microbiol. 2003;6:327–331. doi: 10.1016/s1369-5274(03)00080-8. [DOI] [PubMed] [Google Scholar]
- Yang Y., Kitagaki J., Dai R.M., Tsai Y.C., Lorick K.L., Ludwig R.L., Pierre S.A., Jensen J.P., Davydov I.V., Oberoi P. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- Zadra G., Batista J.L., Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol. Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza O. Basic principles of the virulence of Cryptococcus. Virulence. 2019;10:490–501. doi: 10.1080/21505594.2019.1614383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Klionsky D.J. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 2011;13:119–120. doi: 10.1016/j.cmet.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.