

Abstract
A new end-on low-spin ferric heme peroxide, [(PIm)FeIII─(O22−)]− (PIm-P), and subsequently formed hydroperoxide species, [(PIm)FeIII─(OOH)] (PIm-HP) are generated utilizing the iron-porphyrinate PIm with its tethered axial base imidazolyl group. Measured thermodynamic parameters, the ferric heme superoxide [(PIm)FeIII─(O2•−)] (PIm-S) reduction potential (E°’) and the PIm-HP pKa value, lead to the finding of the OO─H bond dissociation free energy (BDFE) of PIm-HP as 69.5 kcal/mol, using a thermodynamic square scheme and Bordwell relationship. The results are validated by the observed oxidizing ability of PIm-S via hydrogen atom transfer (HAT) compared to that of the F8 superoxide complex, [(F8)FeIII─(O2•−)] (S) (F8 = tetrakis(2,6-difluorophenyl)porphyrinate, without an internally appended axial base imidazolyl), as determined from reactivity comparison of superoxide complexes PIm-S and S with the hydroxylamine (O─H) substrates TEMPO-H and ABNO-H.
Thermodynamic comparisons for O2-derived iron-porphyrinate interrelated ferric heme superoxide, peroxide and hydroperoxide analogs in the presence and absence of an appended imidazolyl axial base are presented.
Keywords: end-on peroxide, bond dissociation free energy, BDFE, hydrogen atom transfer, square scheme, thermodynamic parameters
Graphical Abstract
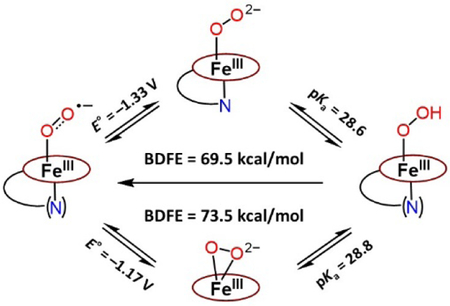
Introduction
Heme-containing enzymes show diverse biological functions such as substrate monooxygenation, oxygen reduction in the respiratory chain, nitric oxide synthesis, oxygen storage and delivery, electron transfer, and H2O2 activation and dismutation.[1] Heme enzyme active sites contain a proximal ligand; cytochrome P450s (CYP450s) as well as NO synthase and chloroperoxidase utilize a cysteinate residue, whereas other peroxidases include a histidyl proximal ligand and catalases possess a tyrosinate ligand. The identity and nature of the axial ligand contributes to or defines spectroscopic properties, coordination, iron reduction potentials and thus the reactivity. These proximal ligand donors contribute the so-called “push effect”, due to their electron donation.[1] In particular, the “push effect” of the anionic thiolate ligand in CYP450s leads to a much more negative FeIII/II reduction potential[2] and helps effect the heterolytic O─O bond cleavage of a hydroperoxide species, forming Compound I (Cmpd I, (P•+)FeIV=O) as the reactive species which hydroxylates substrates.[1]
CYP450 monooxygenases transfer one atom of O2 to a substrate, giving hydroxylated, epoxidized, or sulfoxidized products.[1] The reaction cycle involves formation of heme FeIII─superoxide (the initial FeII/O2 adduct), with subsequent transformation to FeIII─peroxide and FeIII─hydroperoxide intermediates prior to O─O cleavage to give Cmpd I.[1,4]
For these three initial FeIII CYP450 intermediates, radiolytic cryoreduction/spectroscopic studies and theoretical calculations lead to the conclusion that all have an end-on binding geometry, with FeIII-coordination only to the O2-derived proximal oxygen atom.[5] Synthetic bioinorganic groups have been interested in the characterization of model compounds for the superoxide, peroxide, or hydroperoxide intermediates, as relevant to O2 or H2O2 activating heme enzymes. There are many known ferric heme superoxide[6] and hydroperoxide synthetic compounds.[3,6g,6i,6j,7] Valentine and co-workers[8] originally synthesized and characterized a series of ferric heme peroxides possessing a side-on bound O22− ligand (η2, with both peroxide O-atoms equivalently bound to FeIII). To date, there has been only one report of an end-on (η1) bound low-spin peroxide synthetic complex, [(P)FeIII─(η1:O22−)]− (P possesses an internal imidazolyl axial ligand), that by Naruta and co-workers.[6g] Recently, we[3,6d] reported on an end-on ferric heme superoxide, a corresponding hydroperoxide species, and the related ferric heme peroxide, where the O2-derived O22− ligand is side-on bound (Figure 1, upper right); the chemistry was carried out in tetrahydrofuran (THF), which serves as an axial base ligand. In that work,[3] thermodynamic parameters, the [(F8)FeIII─(O2•−)] (S) reduction potential and [(F8)FeIII─(OOH)] (HP) pKa, led to the determination of the ferric heme hydroperoxide BDFE to be 73.5 kcal/mol.
Figure 1.

Stepwise generation of superoxide, peroxide, and hydroperoxide heme analogues and relevant thermodynamic square scheme (E° as determined vs Fc+/0 and pKa) and OO─H BDFE of ferric heme hydroperoxide. ArF = 2,6-difluorophenyl group. Upper right: the previously studied ferric heme peroxide [(F8)FeIII─(η2:O22−)]−.[3]
Herein, the newly generated end-on low-spin peroxide, [(PIm)FeIII─(O22−)]− (PIm-P), and its subsequent conversion to hydroperoxide species, [(PIm)FeIII─(OOH)] (PIm-HP) are described. The PIm porphyrinate possesses an appended axial base strongly donating imidazolyl group (Figure 1), in partial analogy to the strongly donating cysteinate base in CYP450s; FeIII is bound to only one oxygen atom of the peroxide moiety. Also, a reduction potential for the [(PIm)FeIII─(O2•−)] (PIm-S)/ [(PIm)FeIII─(O22−)]− (PIm-P) couple and a pKa for the [(PIm)FeIII─(OOH)] (PIm-HP) acid-base pair are established here. These thermodynamic parameters and use of the Bordwell relationship square scheme (Figure 1) allow the determination of OO─H bond dissociation free energy (BDFE) for PIm-HP. We compare and contrast our thermodynamic findings and HAT reactivity with substrates to those obtained with a previously studied system employing the F8 porphyrinate (Figure 1).
Results and Discussion
The ferric heme superoxide [(PIm)FeIII─(O2•−)] (PIm-S) generated by bubbling O2 to a solution of [(PIm)FeII] (Figure 1), was previously characterized (νO─O, 1180 (Δ18O2, −56) cm−1; νFe-O, 575 (Δ18O2, −23) cm−1).[6h,9] Addition of cobaltocene (CoCp2) to PIm-S at −80 ℃ in THF results in the formation of the ferric heme peroxide complex [(PIm)FeIII─(O22−)]− (PIm-P); UV-vis spectral shifts occurring are 423 to 424 nm (Soret) and 532 to 535, 567 nm (Figure 2A). These product electronic absorption spectra are very similar to those of Naruta’s end-on low-spin ferric heme peroxide, as mentioned the first of its kind.[6g] The UV-vis features of PIm-P and Naruta’s analog exhibit higher energy Soret λmax values than the side-on bound ferric heme peroxides (also lacking a trans axial base ligand), as in [(F8)FeIII─(η2:O22−)]− (Figure 1, upper right).[3,6d,6f,8] The low-spin end-on formulation is confirmed by the EPR spectra which display signals at g = 2.25, 2.14, and 1.95 (Figure 2A), matching Naruta’s analog,[6g] as well as for those end-on ferric peroxide obtained by cryoreduction methods in heme enzymes.[5] Such EPR signatures also are distinct from side-on heme peroxide intermediates which are intermediate spin species, g = 4.2 (S = 3/2).[3,6d,6f,8]
Figure 2.

(A) Electronic absorption spectra of [(PIm)FeIII─(O2•−)] (PIm-S) (red) to [(PIm)FeIII─(O22−)]− (PIm-P) (blue) at −80 ℃ in THF. Inset: Frozen THF solution EPR (10 K) spectrum of PIm-P. The small feature (g = 1.99) corresponds to an excess of cobaltocene.[6d] (B) Resonance Raman spectra of [(PIm)FeIII─(O22−)]− (PIm-P) in frozen THF obtained at 77 K with 413 nm excitation. The 16O2─18O2 difference spectrum is shown in blue.
Further, [(PIm)FeIII─(O22−)]− (PIm-P) was characterized using rR spectroscopy. Two isotopic sensitive bands were observed, at 810 (Δ18O2 = −34) cm−1 and 578 (Δ18O2 = −26) cm−1 corresponding to the νO─O and νFe─O stretches, respectively (Figure 2B). This latter stretching frequency value matches the expected value (ΔFe-O,calc (16O2/18O2) = −26 cm−1) using the harmonic oscillator approximation. The magnitude of the isotope shift for νO─O is smaller than the calculated value (ΔO-O,calc (16O2/18O2) = −46 cm−1); however, the same trend is found for Naruta’s complex [(P)FeIII─(η1:O22−)]− (νO─O, 808 (Δ18O2, −37) cm−1; νFe-O, 585 (Δ18O2, −25) cm−1).[6g] It is notable that νFe─O in [(PIm)FeIII─(η1:O22−)]− (PIm-P) is much larger than that found for the side-on peroxide species which has no internally appended axial base imidazolyl, [(F8)FeIII─(η2:O22−)]− (Figure 1, upper right; νO─O, 806 (Δ18O2, −46) cm−1; νFe-O, 466 (Δ18O2, −19) cm−1).[3] The increased Fe─O stretching frequency is likely a result of both the monodentate coordination and the σ-symmetry O2 donation into the fully unoccupied low-spin Fe dz2 orbital which is stronger than the π-symmetry donation into the half-occupied Fe dxz/dyz orbital of the side-on peroxide complex. These effects would strengthen the Fe─O bond of PIm-P, and favor the end-on binding mode, giving a low-spin six-coordinate species [(PIm)FeIII─(η1:O22−)]− (PIm-P).
When excess triflic acid (HOTf) is added to [(PIm)FeIII─(O22−)]− (PIm-P), Fe─O bond cleavage occurs, releasing H2O2 (89 % yield; horseradish peroxidase assay, see Figure S1), corroborating the complex’s peroxide formulation. PIm-P is singly protonated by adding 1 equiv of 2,6-lutidinium triflate [(LutH+)](OTf) at −80 ℃ in THF, giving [(PIm)FeIII─(OOH)] (PIm-HP) with absorptions at 423 and 533 nm (Figure 3A). EPR spectra of PIm-HP confirm an end-on low-spin ferric heme hydroperoxide structure (g = 2.25, 2.14, and 1.95) as shown in Figure 3B. These values correspond closely to those already known for synthetically derived end-on low-spin hydroperoxide complexes[3,6f,6g,6i,6j,7a,7b] as well as for ferric heme hydroperoxides generated within hemoglobin and myoglobin.[10a,b] A high yield of H2O2 (90.1 %) is also obtained when PIm-HP is acidified using HOTf (see Figure S1). Further, we performed rR spectroscopy; while only noisy spectra could be obtained (Figure S2), νO─O (808 (Δ18O2, −48) cm−1) and νFe─O (579 (Δ18O2, −36) cm−1) resonances could be well discerned. These parameters, for PIm-HP, are quite similar to those for the other synthetic or protein low-spin ferric heme hydroperoxide complexes (vide supra).[3,6g,7c,10]
Figure 3.

(A) UV–vis spectra illustrating the conversion of [(PIm)FeIII─(O22−)]− (PIm-P) to [(PIm)FeIII─(OOH)] (PIm-HP) by addition of [(LutH+)](OTf) at −80 ℃ in THF. (B) Low-spin EPR spectrum (10 K) of PIm-HP. Note that there is g = 1.99 signal from excess of cobaltocene used for generation of the peroxide complex. An extra signal at g > 2.00 (compared to the EPR spectrum shown in Figure 2) is attributed to an unknown impurity.
We find [(PIm)FeIII─(O2•−)] (PIm-S) and [(PIm)FeIII─(O22−)]− (PIm-P) are interconvertible (Figure 1) using redox reagents. Addition of a ferrocenium reagent (Fc+; E1/2 = 0 V vs Fc+/0) to a solution of PIm-P leads to the reappearance of the optical bands (423 and 532 nm) associated with PIm-S (Figure S3). This result is supported by the diamagnetic behaviour (i.e., EPR silent; 10 K, THF) of the resulting PIm-S solution. The further addition of CoCp2 leads to re-reduction giving the peroxide species PIm-P (Figure S4).
Interestingly, titration of [(PIm)FeIII─(O2•−)] (PIm-S) with aliquots of CoCp2 (E1/2 = −1.33 V vs Fc+/0 in THF)[11] at −80 ℃ in THF gives equilibrium mixtures; UV-vis monitoring (at either 423 or 567 nm) (Figure 4A, Table S1, Figure S5), allowed determination of Keq for each aliquot added. From these results and utilizing the Nernst equation, the reduction potential for the couple, [(PIm)FeIII─(O2•−)] (PIm-S)/[(PIm)FeIII─(O22−)]− (PIm-P) is calculated to be −1.33 V ± 0.01 V vs Fc+/0. This value is more negative by ~160 mV than that reported for [(F8)FeIII─(O2•−)] (−1.17 V vs Fc+/0) (Figure S6) which has no internally appended axial base in the porphyrinate complex.[3] Consistent with this finding is the observation that in order to fully reduce [(F8)FeIII─(O2•−)] to its peroxide analogue, one equiv CoCp2 was used, while multiple equiv of CoCp2 are required to fully reduce the presently described superoxide complex PIm-S. These findings suggest a “push effect” due to the axial imidazolyl ligand which is covalently appended to the heme of PIm iron-porphyrinate complexes.
Figure 4.

UV-vis spectroscopic monitoring of the incremental addition of (A) CoCp2 to a solution of [(PIm)FeIII─(O2•−)] (PIm-S) (red). Inset: monitoring of the absorbance at 567 nm (blue). (B) EtP2(dma) addition to a solution of [(PIm)FeIII─(OOH)] (PIm-HP) (pink). Inset: monitoring of the absorbance at 567 nm (blue).
Dey and co-workers[2] reported the reduction potentials of iron-porphyrinate complexes (FeIII/II) which have thiolate and imidazolyl linkers bound to the iron center of porphyrinate complexes. A thiolate ligated porphyrin complex shows 500 mV more negative FeIII/II reduction potential compared to an imidazolyl ligated porphyrinate analogue; the thiolate is a much better donor than imidazolyl. Never-the-less, in our case the greater charge donation by an axial imidazolyl ligand in PIm reduces the reduction potential of [(PIm)FeIII─(O2•−)] (PIm-S) in comparison with that of [(F8)FeIII─(O2•−)].
To complete the thermodynamic square scheme analysis (Figure 1),[12] the pKa value of ferric heme hydroperoxide [(PIm)FeIII─(OOH)] (PIm-HP) was evaluated at −80 ℃ in THF. Full deprotonation of PIm-HP was accomplished by spectral titration using the derivatized phosphazene base EtP2(dma) (see Table S2 for the chemical structure; conjugate base pKa = 28.1; THF solvent, RT).[13] The concentrations of each species (PIm-P, PIm-HP, EtP2(dma), and EtP2(dma)H+) were evaluated by examination of the observed absorbance at either 423 or 567 nm in the UV-vis spectra (Figure 4B, Table S2, Figure S7). Such titration experiments lead to the establishment of Keq constants for the protonation of PIm-P to give PIm-HP, allowing the calculation of the pKa of the ferric heme hydroperoxide to be 28.6 ± 0.5 (−80 °C, THF). The reversibility of acid-base reaction was further demonstrated by adding [(LutH+)](OTf) to the solution generated in this titration, which gives back PIm-HP, (Figure S8).
This allows the completion of the thermodynamic square scheme for electron and proton transfer reactions (Figure 1) using the presently determined thermodynamic parameters, E°’ (−1.33 V vs. Fc+/0, vide supra) and pKa (28.6, vide supra), and determination of the BDFE of the OO─H in the ferric heme hydroperoxide complex [(PIm)FeIII─(OOH)] (PIm-HP). According to eq. 1 (Bordwell relationship, CG stands for the H+/H• standard reduction potential in a particular solvent),[12] where CG = 61 kcal/mol in THF, the PIm-HP BDFE is calculated to be 69.5 kcal/mol (eq. 2).
| (1) |
| (2) |
Our own previous report is the only other experimental evaluation of the thermodyanmics and use of the square shcme as applied to ferric heme superoxide, peroxide (Figure 1, upper right) and hydroperoxide relatives, i.e., for the F8 heme containing superoxide, (side-on) peroxide and hydroperoxide, [(F8)FeIII─(OOH]] BDFEOO─H = 73.5 kcal/mol.[3] Our OO─H BDFE value presented here with PIm is shown to be ~ 4 kcal/mol less, and the difference resides primarily in the variation in the superoxide/peroxide redox couple (Figure 5).
Figure 5.

Comparison of PIm and F8 iron-porphyrinate complex for thermodynamic parameters and the geometry of peroxide species.
The pKa values for [(F8)FeIII─(OOH)] vs [(PIm)FeIII─(OOH)] (PIm-HP) are essentially identical,[14] but the E°’ reduction potential is measurably more negative for the (PIm-S) / (PIm-P) couple (Figure 5). This makes chemical sense as the PIm system possesses the strong imidazolyl axial base ligand donor, that not present in the F8 heme system. Also, the dissimilarity of BDFE values ([(PIm)FeIII─(OOH)] BDFEOO─H = 69.5 kcal/mol and [(F8)FeIII─(OOH)] BDFEOO─H = 73.5 kcal/mol) is supported by their hydrogen atom transfer (HAT) reactivities toward H atom donors (vide infra). As we noted previously[3] our BDFE results are also roughly in accord with computationally derived ferric heme hydroperoxides (those modeling proteins), where a proximal ligand is either an imidazolyl or ─SH group, BDEs (64–66 kcal/mol).[15]
HAT reactivity (shown in the diagonal of Figure 1) studies were performed with [(PIm)FeIII─(O2•−)] (PIm-S) toward external substrates to support our experimentally determined BDFE value. Addition of an excess of substrates such as 9,10-dihydroanthracene (76 kcal/mol in DMSO)[12] and xanthene (72.2 kcal/mol in THF)[13] to the solutions of PIm-S at −80 ℃ in THF led to no reaction. These observations are consistent with our BDFE determination of 69.5 kcal/mol for [(PIm)FeIII─(OOH)] (PIm-HP). In contrast, TEMPO-H (66.5 (THF), 72.6 (aq. buffer), and 69.6 (benzene) kcal/mol)[13,16] does react with complex PIm-S. The final spectra (UV-vis and EPR; Figure 6A, Figure S9 and S10) reveal a high yield of TEMPO•, while PIm-HP seems to only form in small amounts (~20% yield), probably due to side-reactions. The results support the conclusion that an initial H-atom abstraction from TEMPO-H to give PIm-HP occurred. Based on these reactivity studies, the BDFE of PIm-HP can be bracketed to be, 66.5 < BDFEOO─H < 72 kcal/mol, which is consistent with our experimentally determined BDFE value, 69.5 kcal/mol.
Figure 6.

(A) Hydrogen atom transfer (HAT) reaction by [(PIm)FeIII─(O2•−)] (PIm-S) from TEMPO-H and ABNO-H at −80 ℃ in THF. (B) HAT from [(F8)FeIII─(O2•−)] (S) toward ABNO-H at −80 ℃ in THF.
As we previously showed that [(F8)FeIII─(O2•−)] (S) reacts with TEMPO-H to cleanly give the TEMPO• and corresponding hydroperoxide complex [(F8)FeIII─(OOH)] (HP), we now see that S and [(PIm)FeIII─(O2•−)] (PIm-S) reactions toward TEMPO-H are comparable. Thus, we sought to find a different substrate that might differentiate between the oxidizing capabilities of the two superoxide complexes, which is what we have found here based on our thermodynamic parameter analysis. The superoxide complex S should be a somewhat stronger oxidant for HAT chemistry as based on our finding of the ~ 4 kcal/mol difference in HP versus PIm-HP complexes’ BDFEs (vide supra).
Thus, we turned to the use of the hydroxylamine substrate ABNO-H (9-azabicyclo[3.3.1]nonane-N-hydroxide, BDFE = 76.0 (aq. buffer) and 76.2 (benzene) kcal/mol)[16] whose BDFE value is in the range 3.4-to-6.6 kcal/mol higher than that of TEMPO-H. As summarized in Figure 6, there was no reaction between [(PIm)FeIII─(O2•−)] (PIm-S) and ABNO-H (Figure 6A and Figure S11), while spectroscopic monitoring (UV-vis and EPR) of the corresonding [(F8)FeIII─(O2•−)] (S) plus ABNO-H reaction (Figure 6B and Figure S12) revealed both the generation of the organic radical product (ABNO•) along with the formation of the low-spin end-on hydroperoxide species, HP (See Figure S12 for further details). The HAT reaction of ABNO-H with PIm-S is thermodynamically uphill. In contrast, the oxidizing ability of S via HAT from ABNO-H is favored. ABNO-H is a borderline substrate which enables us to discern the clear difference in the reactivities between PIm-S and S; the results are fully consistent with our findings of heme-hydroperoxide OO─H BDFE’s via thermodynamic analyses, here and previously.[3]
Conclusion
In conclusion, using the PIm iron-porphyrinate which includes an imidazolyl moiety appended to the periphery of a fluorinated tetraphenylporphyrin, we here generated a new (only the second one known) end-on low-spin ferric heme peroxide [(PIm)FeIII─(O22−)]− (PIm-P) and also a hydroperoxide [(PIm)FeIII─(OOH)] (PIm-HP) species which are relevant to the catalytic cycle of CYP450s and many other heme enzymes which activate O2 or H2O2. The end-on peroxide binding mode with the PIm heme contrasts with that of the F8 heme (without the appended axial imidazolyl ligand) which forms a side-on peroxide species. The reversibility of the [(PIm)FeIII─(O2•−)] (PIm-S)/ [(PIm)FeIII─(O22−)]− (PIm-P) redox process and the [(PIm)FeIII─(O22−)]− (PIm-P)/ [(PIm)FeIII─(OOH)] (PIm-HP) acid-base equilibrium allowed for the calculation of reduction potential and pKa, respectively. With the measured thermodynamic parameters, the ferric heme OO─H BDFE was determined to be 69.5 kcal/mol, contrasting with the value of 73.5 kcal/mol found for [(F8)FeIII─(OOH)] (HP).
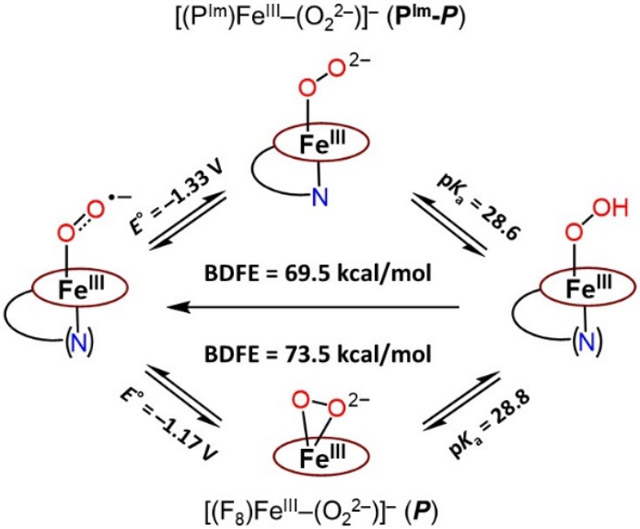
This difference is a result of the existence of the appended axial imidazolyl ligand in the PIm system. Its “push effect” appears to contribute significantly to the lower reduction potential found for (PIm-S)/(PIm-P) compared to that measured for the (S)/(P) couple with F8-heme. Confirmation of our thermodynamically derived parameters comes from comparison of the [(PIm)FeIII─(O2•−)] (PIm-S) versus [(F8)FeIII─(O2•−)] (S) reactivity results employing the hydroxylamine substrates TEMPO-H and/or ABNO-H. Future studies will explore further similar thermodynamic relationships to determine how ligand effects may control such properties and chemical reactivity.
Supplementary Material
Acknowledgements
This research was supported by the U.S. National Institutes of Health (GM60353 to K.D.K. and GM040392 to E.I.S.).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
Institute and/or researcher Twitter usernames: @KarlinLab
References
- [1].a) Sono M, Roach MP, Coulter ED, Dawson JH, Chem. Rev 1996, 96, 2841–2887; [DOI] [PubMed] [Google Scholar]; b) Denisov IG, Makris TM, Sligar SG, Schlichting I, Chem. Rev 2005, 105, 2253–2277; [DOI] [PubMed] [Google Scholar]; c) Ortiz de Montellano PR, Chem. Rev 2010, 110, 932–948; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Huang X, Groves JT, Chem. Rev 2018, 118, 2491–2553; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Adam SM, Wijeratne GB, Rogler PJ, Diaz DE, Quist DA, Liu JJ, Karlin KD, Chem. Rev 2018, 118, 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Das PK, Chatterjee S, Samanta S, Dey A, Inorg. Chem 2012, 51, 10704–10714. [DOI] [PubMed] [Google Scholar]
- [3].Kim H, Rogler PJ, Sharma SK, Schaefer AW, Solomon EI, Karlin KD, J . Am. Chem. Soc 2020, 142, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dubey KD, Shaik S, Acc. Chem. Res 2019, 52, 389–399. [DOI] [PubMed] [Google Scholar]
- [5].a) Davydov R, Macdonald IDG, Makris TM, Sligar SG, Hoffman BM, J. Am. Chem. Soc 1999, 121, 10654–10655; [Google Scholar]; b) Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM, J. Am. Chem. Soc 2001, 123, 1403–1415; [DOI] [PubMed] [Google Scholar]; c) Davydov R, Satterlee JD, Fujii H, Sauer-Masarwa A, Busch DH, Hoffman BM, J. Am. Chem. Soc 2003, 125, 16340–16346; [DOI] [PubMed] [Google Scholar]; d) Garcia-Serres R, Davydov RM, Matsui T, Ikeda-Saito M, Hoffman BM, Huynh BH, J. Am. Chem. Soc 2007, 129, 1402–1412; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W, Chem. Rev. 2010, 110, 949–1017. [DOI] [PubMed] [Google Scholar]
- [6].a) Collman JP, Gagnet RR, Christopher A, Robinson WT, Proc. Natl. Acad. Sci. U. S. A 1974, 71, 1326–1329; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Momenteau M, Reed CA, Chem. Rev 1994, 94, 659–698; [Google Scholar]; c) Collman JP, Sunderland CJ, Berg KE, Vance MA, Solomon EI, J. Am. Chem. Soc 2003, 125, 6648–6649; [DOI] [PubMed] [Google Scholar]; d) Chufan EE, Karlin KD, J. Am. Chem. Soc. 2003, 125, 16160–16161; [DOI] [PubMed] [Google Scholar]; e) Liu J-G, Naruta Y, Tani F, Angew. Chem. Int. Ed 2005, 44, 1836–1840; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 1870–1874; [Google Scholar]; f) Liu J-G, Ohta T, Yamaguchi S, Ogura T, Sakamoto S, Maeda Y, Naruta Y, Angew. Chem. Int. Ed. Engl 2009, 48, 9262–9267; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 9426–9431; [Google Scholar]; g) Liu J-G, Shimizu Y, Ohta T, Naruta Y, J. Am. Chem. Soc 2010, 132, 3672–3673; [DOI] [PubMed] [Google Scholar]; h) Li Y, Sharma SK, Karlin KD, Polyhedron 2013, 58, 190–196; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Nagaraju P, Ohta T, Liu J-G, Ogura T, Naruta Y, Chem. Commun 2016, 52, 7213–7216; [DOI] [PubMed] [Google Scholar]; j) Singha A, Dey A, Chem. Commun 2019, 55, 5591–5594; [DOI] [PubMed] [Google Scholar]; k) Kim H, Sharma SK, Schaefer AW, Solomon EI, Karlin KD, Inorg. Chem 2019, 58, 15423–15432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Tajima K, Oka S, Edo T, Miyake S, Mano H, Mukai K, Sakurai H, Ishizu K, J. Chem. Soc., Chem. Commun, 1995, 1507–1508; [Google Scholar]; b) Oliveira R, Zouari W, Herrero C, Banse F, Schöllhorn B, Fave C, Anxolabéhère-Mallart E, Inorg. Chem 2016, 55, 12204–12210; [DOI] [PubMed] [Google Scholar]; c) Sengupta K, Chatterjee S, Samanta S, Dey A, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 8431–8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) McCandlish E, Miksztal AR, Nappa M, Spreger AQ, Valentine JS, J. Am. Chem. Soc 1980, 102, 4268–4271; [Google Scholar]; b) Selke M, Sisemore MF, Valentine JS, J. Am. Chem. Soc 1996, 118, 2008–2012; [Google Scholar]; c) Wertz DL, Valentine JS, Struct. Bonding (Berlin) 2000, 97, 37–60. [Google Scholar]
- [9].Sharma SK, Schaefer AW, Lim H, Matsumura H, Moënne-Loccoz P, Hedman B, Hodgson KO, Solomon EI, Karlin KD, J. Am. Chem. Soc 2017, 139, 17421–17430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Mak PJ, Denisov IG, Victoria D, Makris TM, Deng T, Sligar SG, Kincaid JR, J. Am. Chem. Soc 2007, 129, 6382–6383; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR, J. Phys. Chem. A 2008, 112, 13172–13179; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM, J. Am. Chem. Soc 1999, 121, 10656–10657; [Google Scholar]; d) Ibrahim M, Denisov IG, Makris TM, Kincaid JR, Sligar SG, J. Am. Chem. Soc 2003, 125, 13714–13718. [DOI] [PubMed] [Google Scholar]
- [11].Yandulov DV, Schrock RR, Inorg. Chem 2005, 44, 1103–1117. [DOI] [PubMed] [Google Scholar]
- [12].Warren JJ, Tronic TA, Mayer JM, Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quist DA, Ehudin MA, Schaefer AW, Schneider GL, Solomon EI, Karlin KD, J. Am. Chem. Soc 2019, 141, 12682–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].We are somewhat surprised by the observation that PIm-HP and HP have very similar pKa values. However, explanations for this seem to not now exist the literature and further detailed experimental and computational investigations are required. To compare hydroperoxide pKa's is not straightforward since the conjugate base peroxide complexes PIm-P and P have different structures, end-on vs. side-on.
- [15].Chung LW, Li X, Hirao H, Morokuma K, J. Am. Chem. Soc 2011, 133, 20076–20079. [DOI] [PubMed] [Google Scholar]
- [16].Gerken JB, Pang YQ, Lauber MB, Stahl SS, J. Org. Chem 2018, 83, 7323–7330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.