

Abstract
Menin serves both tumor suppressor and promoter roles in a highly tumor-specific manner. In colorectal cancer (CRC) menin is over-expressed and plays a critical role in regulating transcription of SKP2, and combined treatment with a menin inhibitor (MI) and small molecule EGFR inhibitor (EGFRi) leads to synergistic killing of CRC cells. However, the full spectrum of menin function in CRC remains uncertain. Herein, we demonstrate that menin inhibition increases glycolysis in CRC cells. This MI-induced increase in glycolysis occurs in an mTOR-independent manner and enhances the sensitivity of CRC cells to EGFRis. Additionally, we show that EGFRis induce autophagy in CRC cells, which is important for cell survival in the setting of combined treatment with an EGFRi and MI. Inhibition of autophagy with chloroquine further sensitizes CRCs to treatment with the combination of an EGFRi and MI. Together these findings uncover a novel role for menin in CRC as a repressor of glycolysis and demonstrate that MI-induced increases in glycolysis sensitize CRC to EGFRis. Additionally, these findings illustrate the importance of autophagy as a protective mechanism against EGFRis, especially in the presence of menin inhibition. Ultimately this data opens the possibility of using menin-mediated regulation of glycolysis to potentially improve treatment modalities for CRC.
Introduction
The protein menin, coded by the MEN1 gene, serves primarily as a nuclear epigenetic regulator (1). Menin has divergent roles in cancer that occur in a highly tumor-specific manner (2). In neuroendocrine tumors menin serves as a tumor suppressor (1) while in MLL-fusion leukemia (3) and prostate cancer (4) menin acts as a tumor promoter. Recent work also demonstrated that menin is upregulated in colorectal cancer (CRC) compared to non-cancerous colonic epithelium, where it functions as a contextual tumor promoter (5). However, the full spectrum of menin function in CRC remains uncertain.
Colorectal cancer remains one of the most common cancers in both men and women, and is the third leading cause of cancer-related mortality (6). Five-year survival with metastatic CRC remains less than 15%, indicating the need for improved therapies (7). One class of therapeutics that has been studied extensively in CRC is EGFR inhibitors including both EGFR antibodies as well as small molecule EGFR inhibitors (EGFRis). EGFR antibodies have demonstrated efficacy in metastatic CRC with wild type KRAS (8–10), however EGFRis, while having shown promising results in the treatment of lung cancer with activating EGFR mutations (11), have not been found to be clinically effective for the treatment of CRC (12–14). Therefore, finding mechanisms to sensitize CRC to EGFRis may broaden the treatment options available for metastatic CRC patients.
Similar to other small molecule tyrosine kinase inhibitors, EGFRis are known to have multiple “off-target” effects in cells (15–17). While these effects are unintended based on the predicted target of the EGFRi, these targets may nonetheless be important for inhibiting tumorigenesis. We recently demonstrated that in CRC the EGFRi gefitinib activated the endoplasmic reticulum calcium channel inositol trisphosphate receptor 3 (IP3R3) in an EGFR-independent manner (5). Furthermore, we showed that combining an EGFRi with a menin inhibitor (MI) led to synergistic killing of CRC cells (5). This repression was at least partially due to a synergistic reduction in transcription of SKP2, a key component of the SKP1-Cullin1-F-box (SCF) complex, which serves as an important E3 ubiquitin-ligase that is critical for suppression of cancer cell growth (5). These results highlight the potential to target epigenetic pathways to sensitize CRC cells to EGFRis as well as illustrate how the off-target effects of EGFRis can be exploited to better treat CRC.
EGFRis are known to induce macroautophagy, herein referred to as autophagy (18), which is a mechanism whereby cells degrade and recycle their own cellular content (19). Autophagy is important in cancer growth, as it promotes cell survival through recycling of damaged organelles and unfolded proteins (20). However over-activation of autophagy can be detrimental, leading to recycling of critical cellular components, and subsequent autophagy-induced cell death (18). Inactive EGFR can initiate autophagy in a kinase-independent manner through binding to the protein LAPTM48, leading to Beclin-1 release from the protein Rubicon, which serves as a negative regulator of autophagy (21). Conversely, EGFR activation can inhibit autophagy through several mechanisms, including direct binding of Beclin-1 by phosphorylated EGFR, preventing autophagy initiation (22). Phosphorylated EGFR can also activate PI3K/AKT signaling, resulting in activation of mTORC1, which inhibits the ULK1 complex that is necessary for autophagy initiation (18).
mTORC1 is one of the mammalian target of rapamycin (mTOR) protein complexes. Globally, mTOR regulates many critical cellular growth and metabolic processes (23), which has led to mTOR inhibitors being extensively investigated as potential cancer therapies (24). The activity of the mTORC1 complex is specifically regulated by nutrient availability (23). When active in the presence of a nutrient rich environment, mTORC1 promotes cellular metabolism and at the same time appropriately inhibits autophagy (23). While many factors are known to regulate mTORC1 function, a recent study in CD8 T-cells demonstrated the novel finding that menin inhibited mTORC1 activation, resulting in decreased metabolism including reduced glycolysis and glutaminolysis (25). While harnessing metabolic alterations to target tumor-specific metabolic vulnerabilities in cancers have been a hallmark of cancer treatment for decades (26), given the highly tissue specific functions of menin, whether menin affects metabolism in cancer cells is unclear at present.
Currently there is no known connection between autophagy and menin, and additionally the role of menin in regulation of metabolism in CRC remains uncertain. To address these uncertainties, herein we demonstrate that menin inhibition increases glycolysis in CRC cells, and that de-repression of glycolysis by menin inhibitors is a key step to sensitizing CRC cells to EGFRis. Furthermore, we show that EGFRi-induced autophagy in CRC is important for cell survival, with inhibition of autophagy sensitizing CRC cells to combined treatment with an EGFRi and menin inhibitor.
Methods
Reagents
Gefitinib (#G-4408), erlotinib (#E-4997), lapatinib (#L-4804), everolimus (#E-4040), and rapamycin (#R-5000) were obtained from LC Laboratories. MI-2–2 was obtained from Chemzon Scientific. E64d (#IED-4321-v) was obtained from Peptides International. Pepstatin A (#sc-45036) was obtained from Santa Cruz. 3-MA (#M9281) and chloroquine (#C6628) were obtained from Sigma. MI-463 and MI-503 were obtained from Wuxi Pharma. Spautin-1 was obtained as a gift from the lab of Dr. Costas Koumenis (University of Pennsylvania). C225 was obtained as a gift from the lab of Dr. Mark Greene (University of Pennsylvania).
Cell culture
The human colon adenocarcinoma cell lines HT-29, HCT-15, and SW620 were obtained through the Cell Culture Core of the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases at the University of Pennsylvania. Cell line authentication was performed by short tandem repeat (STR) profiling by American Type Culture Collection. 293T cells were purchased from American Type Culture Collection. All cell lines were used within 10–15 passages of their receipt from the above sources, and are regularly tested for Mycoplasma. Unless otherwise specified, all cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 Units/mL penicillin, and 100 μg/mL streptomycin, and were maintained at 37°C in a humidified 5% CO2 atmosphere.
Protein detection by western blotting
HT-29 and SW620 cells were plated in 10 cm plates at a density of 2 × 106 cells/plate, and HCT-15 cells were plated in 10 cm plates at a density of 106 cells/plate. After attaching overnight, the cells were treated as described for the indicated time. Cells were collected and then lysed with SDS buffer containing protease and phosphatase inhibitors. Protein concentrations were determined using a BCA assay kit (Thermo Scientific). Cell lysates were subjected to polyacrylamide gel electrophoresis on Novex gels (Life Technologies), and protein was transferred to PVDF membranes (Life Technologies). Blocking was performed in TBST containing 5% non-fat dry milk or 5% BSA based on the antibody manufacturer’s blocking instructions. The antibody for p62 (#610497) was purchased from BD Biosciences. Antibodies for LC3B (#2775), p-EGFR (#3777), EGFR (#4267), PARP (#9542), ATG7 (#8558), p-mTOR (#5536), and mTOR (#2983) were purchased from Cell Signaling Technology. The antibody for actin (#A5441) was purchased from Millipore-Sigma. The antibody for menin (#A300–105A) was purchased from Bethyl Laboratories. Anti-rabbit and anti-mouse secondary antibodies were purchased from Bio-Rad. The proteins were visualized by detection with Amersham ECL Western blotting detection reagents (GE Healthcare).
Plasmids and transfections
All plasmids were purified utilizing a GenElute HP plasmid midiprep kit (Sigma) after transformation using DH5α cells with ampicillin. pBABE-puro mCherry-EGFP-LC3B (referred to as GFP-LC3B) was received as a gift from Dr. Costas Koumenis. Retroviral packaging plasmids pVSV-G and pCPG were received as a gift from Dr. Warren Pear. To produce retrovirus, 293T cells were transfected with pVSV-G, pCPG, and GFP-LC3B via calcium phosphate transfection utilizing CaCl2 and HBS.
Lentiviral packing plasmids pMD2.G and psPAX2 were purchased from Sigma. LentiCRISPRv2 was provided as a gift from Dr. Anil Rustgi. The sgRNA TGACCTGCACACCGACTCGC, targeting the MEN1 gene, was cloned into the lentiCRISPRv2 vector using the published protocol (27,28), and the plasmid was sequenced to confirm that the correct insert was in place. shRNA plasmids for EGFR, ATG7, and menin were obtained from the University of Pennsylvania Perelman School of Medicine High-Throughput Screening Core and included EGFR shRNA1 (sense sequence CAGCATGTCAAGATCACAGAT), EGFR shRNA2 (sense sequence CCTCCAGAGGATGTTCAATAA), ATG7 shRNA1 (sense sequence CCCAGCTATTGGAACACTGTA), ATG7 shRNA2 (sense sequence GCCTGCTGAGGAGCTCTCTCCAT), menin shRNA1 (sense sequence GTGCAGATGAAGAAGCAGAAA), and menin shRNA2 (sense sequence GAGTTCTTTGAAGTAGCCAAT), all of which were derived from a pLKO.1-puromycin backbone. To produce lentivirus, 293T cells were transfected with pMD2.G, psPAX2, and the plasmid of interest using Fugene 6 (Promega) according to the manufacturer’s instructions.
For all virus production, after collecting and filtering the virus, cells were then transduced in the presence of 4 μg/ml polybrene (hexadimethrine bromide). 24 hours after completion of transduction, cells were then selected with puromycin for 72 hours.
Fluorescence microscopy
HT-29 cells were plated on sterile coverslips in 6-well plates at a density of 2 × 104 cells/well. After treatment for 48 hours, the cells were fixed with 10% formalin and then mounted using ProLong Gold antifade reagent with DAPI (Life Technologies). These slides were then examined on an inverted Olympus IX81 fluorescent microscope at 200X where pictures were obtained.
Cell proliferation assays
For the MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay, HT-29 cells, were seeded in a 96-well plate at a density of 104 cells/well. HCT-15 cells were seeded in 96-well plates at a density of 5 × 103 cells/well. After adhering overnight, the cells were then treated as described for the indicated time. The MTS assay kit (Promega) was utilized to assess cell growth, and was performed according to the manufacturer’s instructions. Absorbance of each well was recorded at 490 nm using an ELISA plate reader, and after subtracting a background reading, these results were normalized to control well readings. Each experiment was performed in triplicate, with mean values ± standard deviation (SD) reported for each treatment group. P values were calculated using an unpaired 2-tailed t-test.
Clonogenicty assays
HT-29 and HCT-15 cells were plated in 6-well plates at a density of 2 × 103 cells/well and then treated with the described compounds, with new media/compound(s) changed every 3 days. After 10 days, cells were fixed with 10% formalin and then stained with 0.05% crystal violet. Each experiment condition was replicated three times, with a representative image shown for each condition.
YSI assay of glucose consumption and lactate production
For assays performed after small molecule treatment, HT-29 or HCT-15 cells were plated at a density of 2 × 106 cells/10 cm plate in triplicate. The following day, the cells were pre-treated with the indicated small molecule(s) for 24 hours in 10 mL of standard DMEM supplemented with 10% FBS and penicillin/streptomycin. Next, the pre-treatment media was removed, and 13 mL of glucose-free DMEM supplemented with 7 mM exogenous glucose, 10% FBS, and penicillin/streptomycin was added in addition to the indicated small molecule(s). For assays performed with shRNA knockdown of menin, HT-29 cells transfected with either vector or a menin shRNA were plated at a density of 2 × 106 cells/10 cm plate in triplicate. The following day, the media was changed to standard DMEM supplemented with 10% FBS and penicillin/streptomycin. After 24 hours, the media was removed, and 13 mL of glucose-free DMEM supplemented with 7 mM exogenous glucose, 10% FBS, and penicillin/streptomycin was added.
For all experiments, 1.3 mL of media was immediately removed, and an additional 1.3mL of media was removed after 8 hours. Removed media aliquots were centrifuged at 1000 rpm for 3 minutes to pellet any debris and then 1 mL of media was transferred to a new eppendorf and stored at −80°C. At the end of the experiment, cells were trypsinized and counted to allow for calculations on a per cell basis. Media samples were analyzed with a YSI 2950 Biochemistry Analyzer (YSI Incorporated Yellow Springs, OH). For determining the concentrations of glucose and lactate, the sample size was 25 μL and the reaction time was 35 seconds. All analyses were performed in triplicate. Prior to sample analyses, linearity of the instrument response was verified with standards from YSI. Glucose consumption was calculated by subtracting the final glucose reading from the initial glucose reading, dividing by the cell number at the completion of the experiment, and normalizing to the value obtained from vehicle or vector treated control cells. Lactate production was calculated by subtracting the initial lactate reading from the final lactate reading, dividing by the total number of cells, and normalizing to the value obtained from vehicle or vector treated control cells. All values are reported as the mean +/− standard deviation resulting from 3 to 6 separate experiments. P values were calculated using an unpaired 2-tailed t-test.
Glycolytic rate assay
Glycolytic rate assays were performed on an Agilent Seahorse Bioscience XFe96 Extracellular Flux Analyzer. Cells were plated in a 96-well cell culture XF microplate 48-hours prior to the assay (2 × 104 HT-29 cells/well, 3 × 104 HCT-15 cells/well). Twenty-four hours after plating, cells were treated with standard DMEM with 10% FBS containing vehicle (DMSO) or MI. Then 24 hours after this media change the cells were switched to Seahorse medium (DMEM without phenol red containing 7 mmol/L glucose and 4 mmol/L glutamine) and placed in a 37°C, CO2-free incubator 1 hour prior to performing the glycolytic rate assay as per the manufacturer’s instructions. Four measurements were taken at baseline, followed by four measurements after injection of Rotenone/Antimycin A, followed by five measurements after injection of 2-deoxy-glucose as per the manufacturer’s instructions. Each measurement point consisted of 3 minutes of mixing followed by 3 minutes of data acquisition. After lysis with RIPA buffer, BCA assay was performed to determine the protein concentration for each well, with data being normalized to protein concentration. P values were calculated using an unpaired 2-tailed t-test.
Results
Menin inhibition increases glycolysis in CRC cells in an mTOR-independent manner
Recent work demonstrated that menin inhibited glycolysis in T cells (25), however whether menin affects glycolysis in cancer, including CRC, remains unknown. Treatment of the CRC cell line, HT-29 cells, with the small molecule menin inhibitor MI-2–2 (29) led to increased glucose consumption (Fig. 1A) and lactate production (Fig. 1B) compared to vehicle treated control cells, consistent with menin inhibition leading to increased glycolysis. Similar effects were also observed with another menin inhibitor MI-463 (Fig. 1C–D) (30). MI-2–2 and MI-463 also increased glucose consumption and lactate production in HCT-15 CRC cells (Supplementary Fig. 1A–B). Similar to small molecule MIs, knockdown of menin (Fig. 1E, Supplementary Fig. 1C) also increased glycolysis with increased glucose consumption (Fig. 1F, Supplementary Fig. 1D) and lactate production (Fig. 1G, Supplementary Fig. 1E). Real-time measurement of the proton efflux rate further confirmed that menin inhibition increased glycolysis in both HT-29 (Fig. 1H) and HCT-15 cells (Supplementary Fig. 1F).
Figure 1: Menin inhibition increases glycolysis in CRC cells.
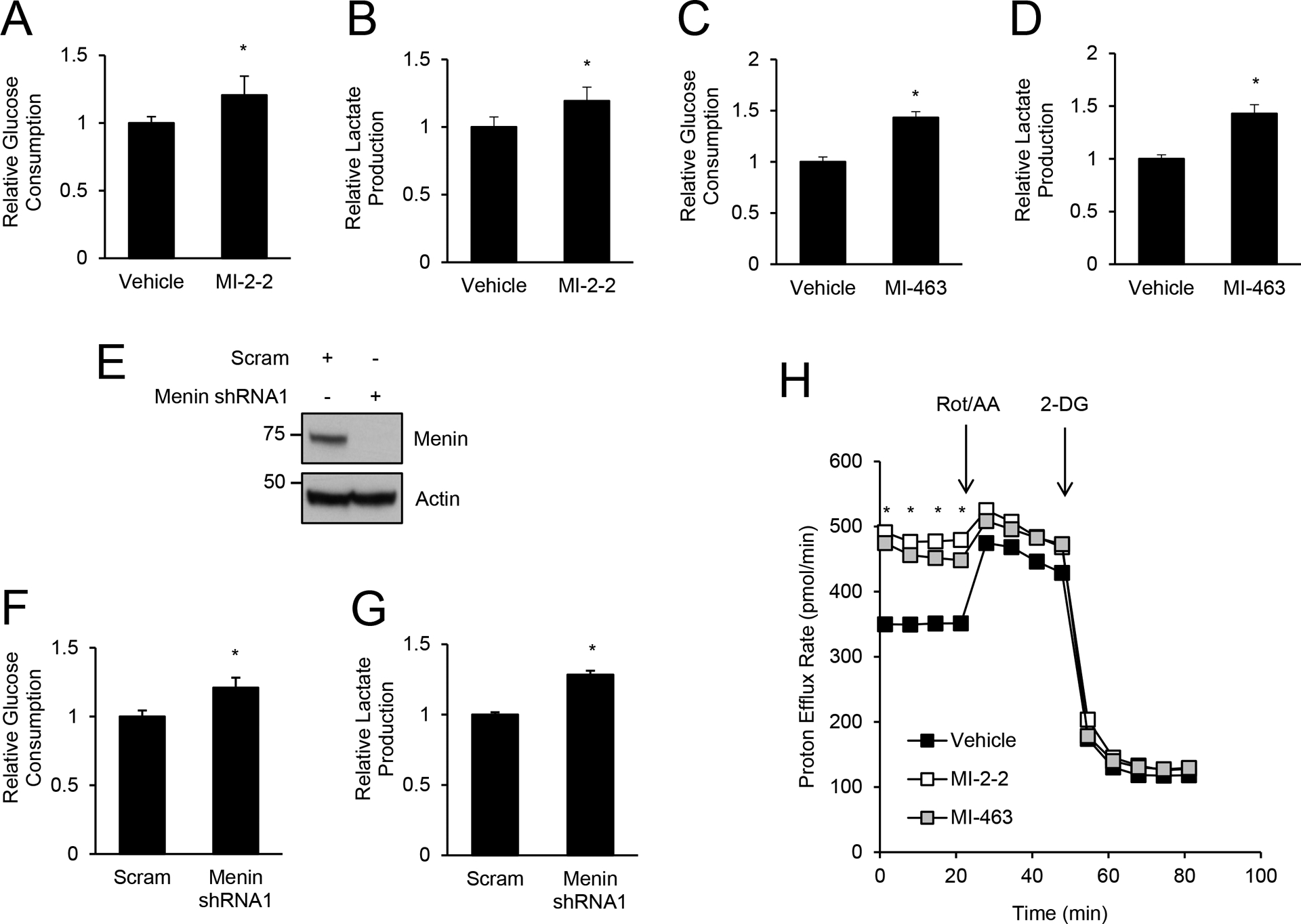
A/B) HT-29 cells were pre-treated with vehicle or 1 μM MI-2–2 for 24 hours, followed by assessment of glucose consumption (A) and lactate production (B) over an 8-hour period. C/D) HT-29 cells were pre-treated with vehicle or 1 μM MI-463 for 24 hours, followed by assessment of glucose consumption (C) and lactate production (D) over an 8-hour period. E/F/G) HT-29 cells transduced with scrambled or menin shRNA1 (E) with assessment of glucose consumption (F) and lactate production (G) over an 8-hour period. H) HT-29 cells were pre-treated with vehicle, 1 μM MI-2–2, or 1 μM MI-463 followed by assessment of the proton efflux rate at baseline and after rotenone/antimycin A (Rot/AA) and 2-deoxyglucose (2-DG). * p < 0.05.
In T cells, menin reduces glycolysis through suppression of mTOR phosphorylation (25). Therefore, we next determined whether menin inhibitors regulated mTOR phosphorylation in CRC cells. Treatment of HT-29 cells with menin inhibitors (Fig. 2A) or menin knockdown (Fig. 2B–C) decreased p-mTOR (Ser2448) levels, which unlike in T cells, shows that menin inhibition does not increase glycolysis through increasing mTOR phosphorylation. To confirm that mTOR is important in glycolysis regulation in CRC cells, HT-29 cells were treated with the mTOR inhibitor everolimus, which effectively reduced mTOR phosphorylation (Fig. 2D) (24). Treatment of CRC cells with everolimus decreased glucose consumption and lactate production consistent with a decrease in glycolysis (Fig. 2E–F), which is not surprising given that mTOR is a critical regulator of glycolysis in cancer cells (31). However, everolimus was unable to decrease menin inhibitor-induced glycolysis to levels achieved with everolimus alone (Fig. 2E–F), indicating that menin inhibition increases glycolysis in CRC cells in an mTOR-independent manner.
Figure 2: Menin inhibitors increase glycolysis independent of mTOR.
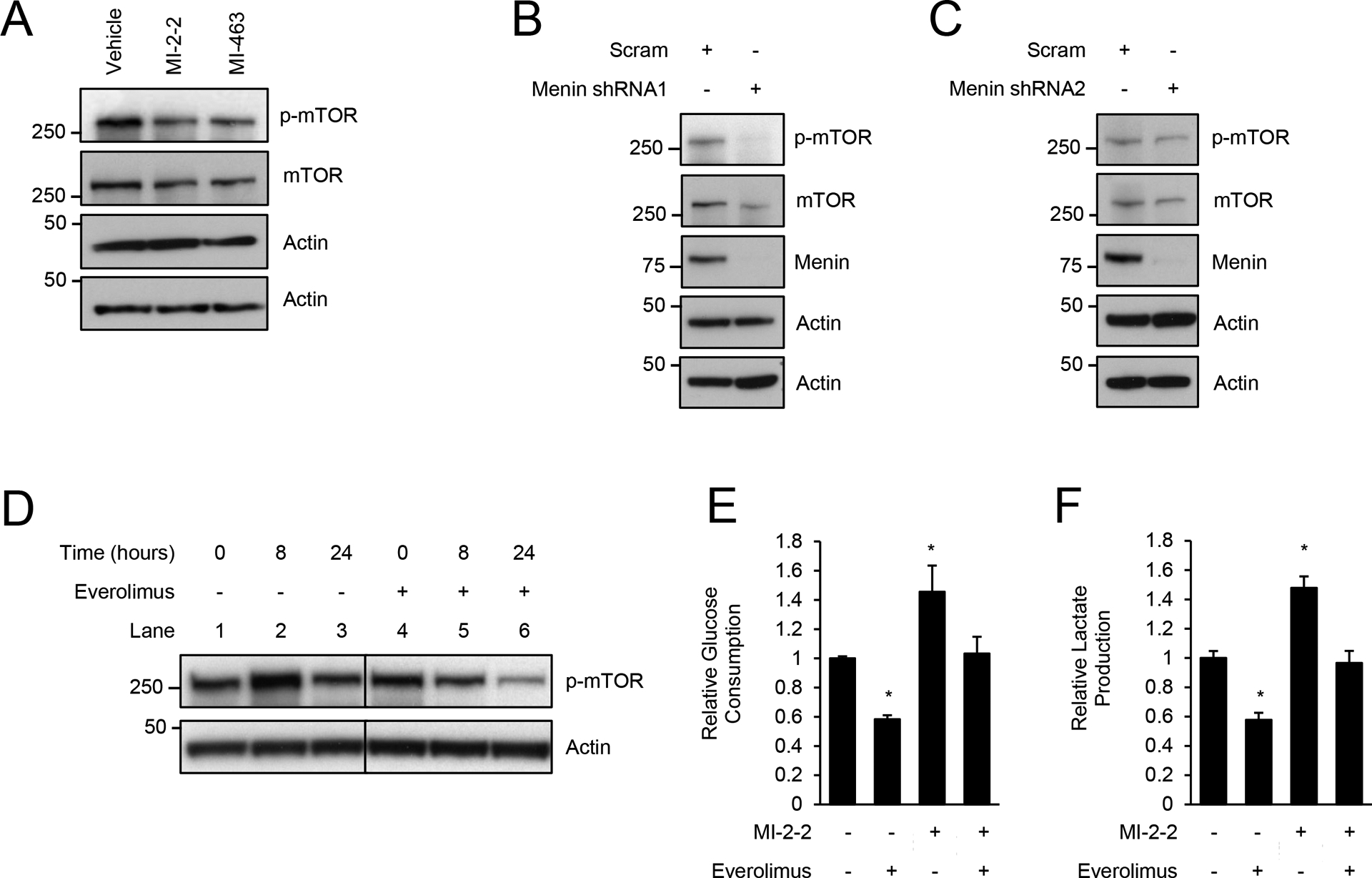
A) Treatment of HT-29 cells for 96 hours, 1 μM MI-2–2, 1 μM MI-463. B/C) HT-29 cells transduced with scrambled, menin shRNA1 (B), or menin shRNA2 (C). D) Treatment of HT-29 cells for various times, 1 μM everolimus. E/F) HT-29 cells were pre-treated with vehicle, 1 μM MI-2–2, and/or 1 μM everolimus for 24 hours, followed by assessment of glucose consumption (E) and lactate production (F) over an 8-hour period. * p < 0.05.
Reducing glycolysis rescues cells from combined treatment with an EGFRi and MI
We previously reported that menin inhibition sensitized CRC to EGFRis, with combined treatment with a MI and EGFRi leading to synergistic cell death (5). To determine if glycolysis may play an important role in this synergy, we first showed that combined treatment with a MI and EGFRi increased glucose consumption and lactate production consistent with increased glycolysis (Fig. 3A–B, Supplementary Fig. 2A–B). Similar to our prior work (5) 10 μM gefitinib was used, and although higher than typical plasma concentrations (32), is comparable to intra-tumor concentrations of gefitinib (33). Similar to the MI-2–2 data, everolimus was unable to decrease glycolysis induced by combined treatment with gefitinib plus MI-2–2 to levels achieved with everolimus alone (Fig. 3A–B, Supplementary Fig. 2A–B). These data suggest that the MI-induced increase in glycolysis may be contributing to the synergistic cell killing observed with combined treatment with an EGFRi and MI.
Figure 3: mTOR inhibition improves survival after combined treatment with EGFR and menin inhibitors.
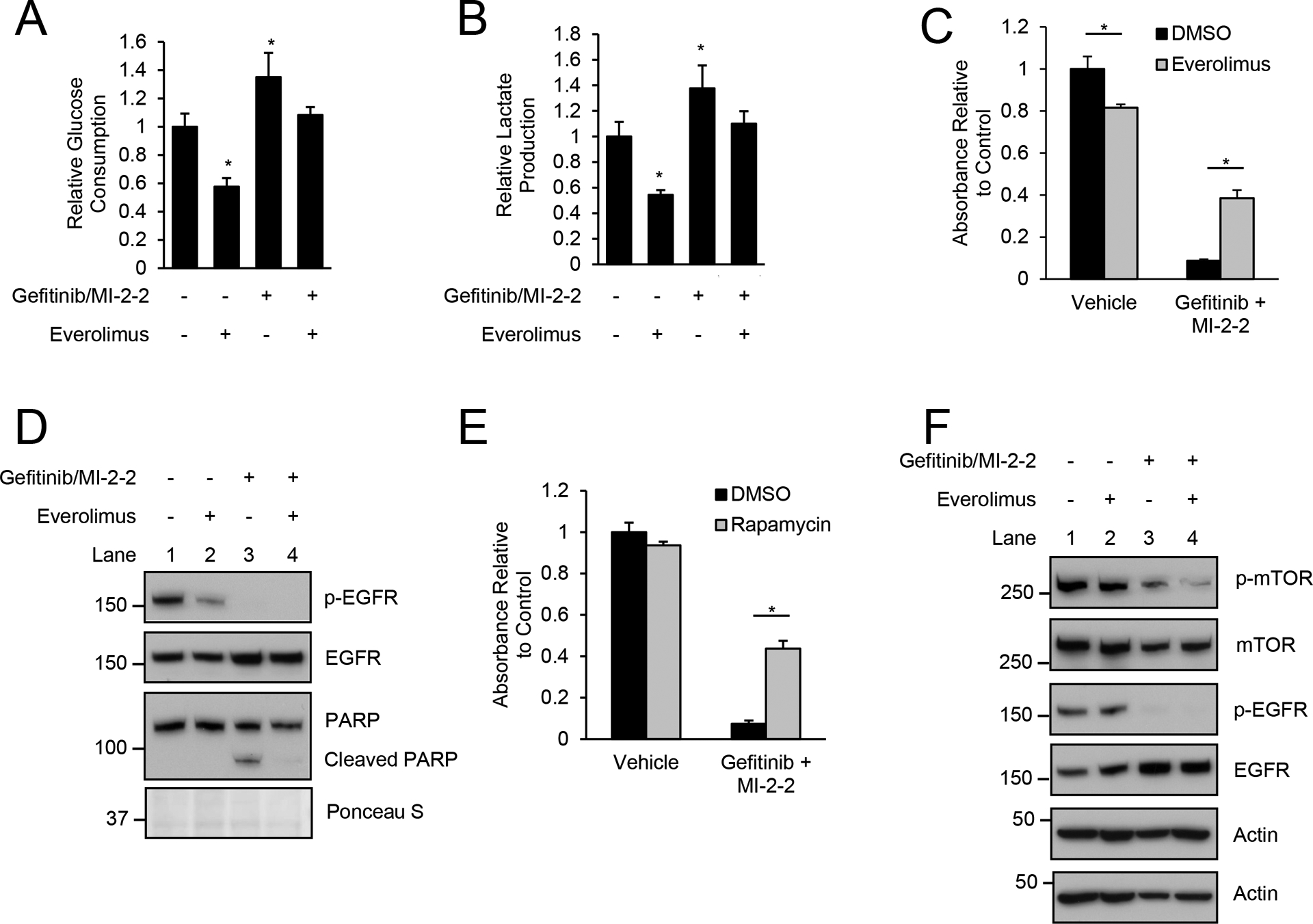
A/B) HT-29 cells were pre-treated with vehicle, 1 μM MI-2–2, and/or 1 μM everolimus for 24 hours, followed by assessment of glucose consumption (A) and lactate production (B) over an 8-hour period after treatment with vehicle or 10 μM gefitinib. C) Treatment of HT-29 cells with cell growth assessed after 96 hours by MTS assay, 10 μM gefitinib, 1 μM MI-2–2, 1 μM everolimus. D) Treatment of HT-29 cells for 48 hours, 10 μM gefitinib, 1 μM MI-2–2, 1 μM everolimus. E) Treatment of HT-29 cells with cell growth assessed after 96 hours by MTS assay, 10 μM gefitinib, 1 μM MI-2–2, 1 μM rapamycin. F) Treatment of HT-29 cells for 48 hours, 10 μM gefitinib, 1 μM MI-2–2, 1 μM everolimus. * p < 0.05.
To determine if overall cellular glycolysis is important in the synergistic killing of CRC by combined EGFRi and MI treatment, we pharmacologically inhibited glycolysis through use of a mTOR inhibitor. Specifically, HT-29 cells were treated with gefitinib and MI-2–2, with or without everolimus, and while gefitinib/MI-2–2 significantly decreased cell growth, addition of everolimus led to partial rescue of cell growth (Fig. 3C) as well as a reduction in apoptosis as assessed by cleaved PARP (Fig. 3D). Similar effects were seen in HCT-15 cells (Supplementary Fig. 2C–D) as well as with the use of another mTOR inhibitor, rapamycin (Fig. 3E). Although gefitinib/MI-2–2 increased glycolysis similar to MI-2–2 alone, this increase in glycolysis was not due to increased mTOR phosphorylation (Fig. 3F, Supplementary Fig. 2E). Addition of everolimus to gefitinib/MI-2–2 decreased mTOR phosphorylation (Fig. 3F, Supplementary Fig. 2E), which coupled with a reduction in glycolysis and improvement in survival, indicates that increased glycolysis may serve as an important mechanism that drives cell killing resulting from combined EGFRi/MI treatment.
Menin inhibition increases EGFRi-induced autophagy
One mechanism that is important for protection of cancer cells under metabolic stress is autophagy (19,20). Given the alterations in glycolysis observed in EGFRi/MI-mediated cell killing, we next investigated the role of autophagy after treatment with EGFRis and MIs. Increases in autophagy can be assessed by monitoring for increases in LC3B-II and reductions in p62 (19). Small molecule EGFR inhibitors, including gefitinib (34) and erlotinib (Supplementary Fig. 3A–B), increase autophagy in a dose dependent manner in CRC cell lines. While the small molecule menin inhibitor MI-2–2 (29) did not induce any autophagy alone, treatment with the combination of the EGFRi gefitinib and MI-2–2 increased autophagy, with increased LC3B-II and decreased p62, in both HT-29 cells (Fig. 4A) and HCT-15 cells (Fig. 4B). Menin inhibition with MI-2–2 increased gefitinib-induced autophagy in a dose dependent manner, with effects noted as low as 10 nM (Fig. 4C). MI-2–2 also increased gefitinib-induced autophagy in a dose dependent manner with increasing gefitinib concentrations (Fig. 4D), and this increase was time dependent, with more autophagy induction after 48 hours compared to after 24 hours of treatment (Fig. 4E).
Figure 4: Menin inhibition increases gefitinib-induced autophagic flux.
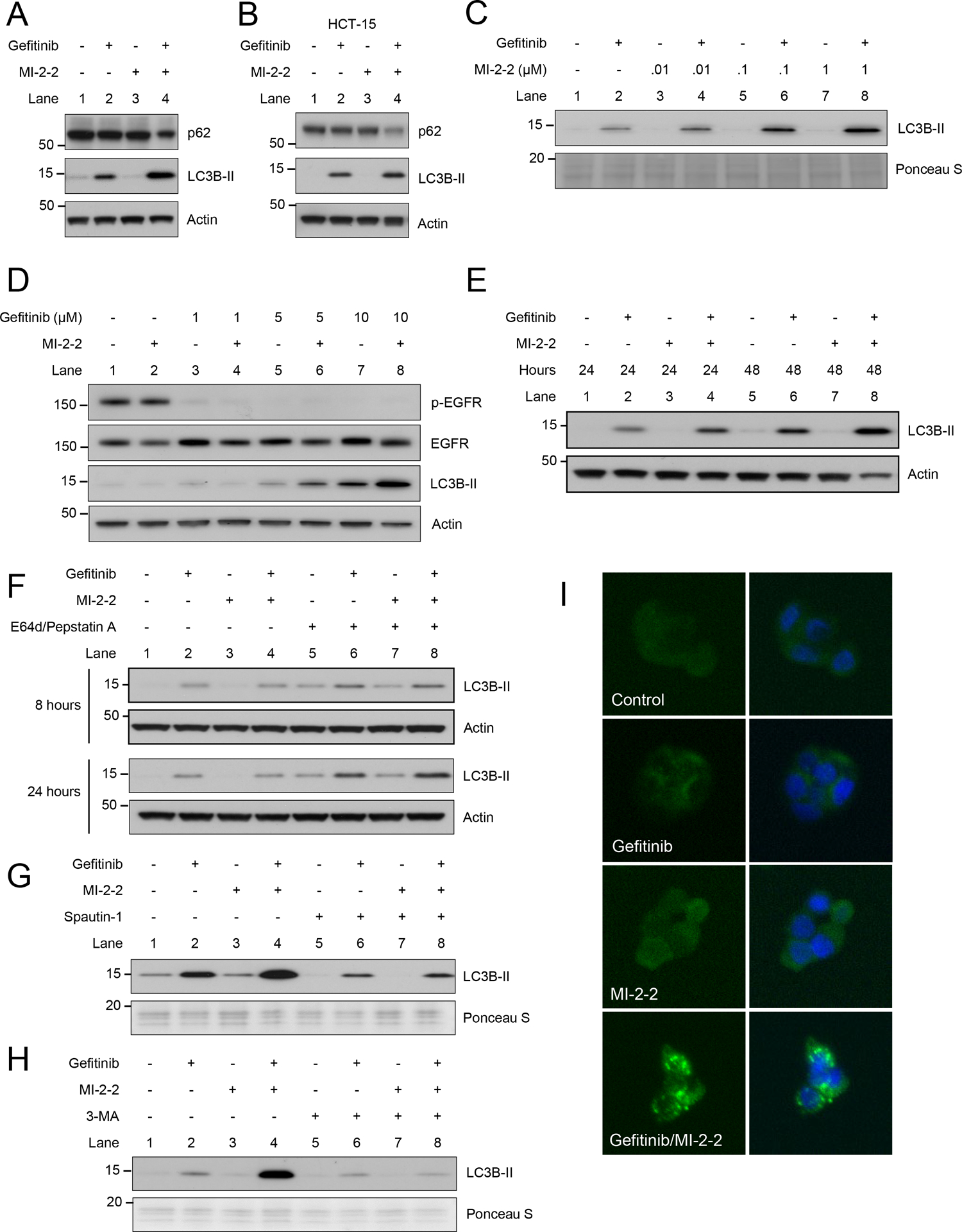
A) HT-29 cells treated for 72 hours, 10 μM gefitinib, 1 μM MI-2–2. B) HCT-15 cells treated for 72 hours, 10 μM gefitinib, 1 μM MI-2–2. C) HT-29 cells treated with varying concentrations of MI-2–2 for 48 hours, 10 μM gefitinib. D) HT-29 cells treated with varying concentrations of gefitinib for 48 hours, 1 μM MI-2–2. E) HT-29 cells treated for either 24 or 48 hours, 10 μM gefitinib, 1 μM MI-2–2. F) HT-29 cells treated for 8 or 24 hours, 10 μg/mL E64d, 10 μg/mL Pepstain A. G) HT-29 cells treated for 72 hours, 10 μM spautin-1. H) HT-29 cells treated for 72 hours, 5mM 3-MA. I) HT-29 cells transfected with GFP-LC3B were treated for 48 hours with images captured at 200X. 10 μM gefitinib, 1 μM MI-2–2.
To determine if menin inhibition with MI-2–2 is increasing autophagic flux, HT-29 cells were treated with the protease inhibitors E64d and pepstatin A (35). Protease inhibitor treatment led to increased LC3B-II accumulation consistent with an increase in autophagy flux (Fig. 4F). To further confirm the increase in autophagy flux, the autophagy inhibitor spautin-1 was used, which inhibits the ubiquitin-specific peptidases USP10 and USP13 leading to decreased Beclin-1 (36). Treatment of HT-29 cells with spautin-1 led to a reduction in LC3B-II levels in gefitinib/MI-2–2 treated cells (Fig. 4G). Next, 3-methyladenine (3-MA), which blocks autophagy flux through inhibition of type III phosphatidylinositol 3-kinases (PI3K) (35), also inhibited the increase in autophagy generated by combined treatment with gefitinib and MI-2–2 (Fig. 4H), further supporting that combined treatment with gefitinib and MI-2–2 increases autophagic flux. HT-29 cells were then transfected with a GFP-tagged LC3B construct, with punctate foci of LC3B noted after the treatment with the combination of gefitinib and MI-2–2, consistent with autophagosome formation in the setting of active autophagy (Fig. 4I).
To confirm that these effects are not specific to gefitinib, other EGFRis were found to have similar results including erlotinib (Fig. 5A) and lapatinib (Supplementary Fig. 4A). Additionally, to confirm that these effects were not specific to MI-2–2, other small molecule MIs were used including MI-463 (Fig. 5B) and MI-503 (Supplementary Fig. 4B) with similar results (30). Removing functional menin using CRISPR/Cas9 led to no increase in autophagy alone, however, treatment of these cells lacking menin expression with gefitinib increased LC3B-II, consistent with the results of MI treatment (Fig. 5C). Taken together, this data demonstrates that EGFRis induce autophagy, and that menin inhibition increases EGFRi-induced autophagy.
Figure 5: Menin inhibition increases EGFRi-induced autophagy in an EGFR-independent manner.
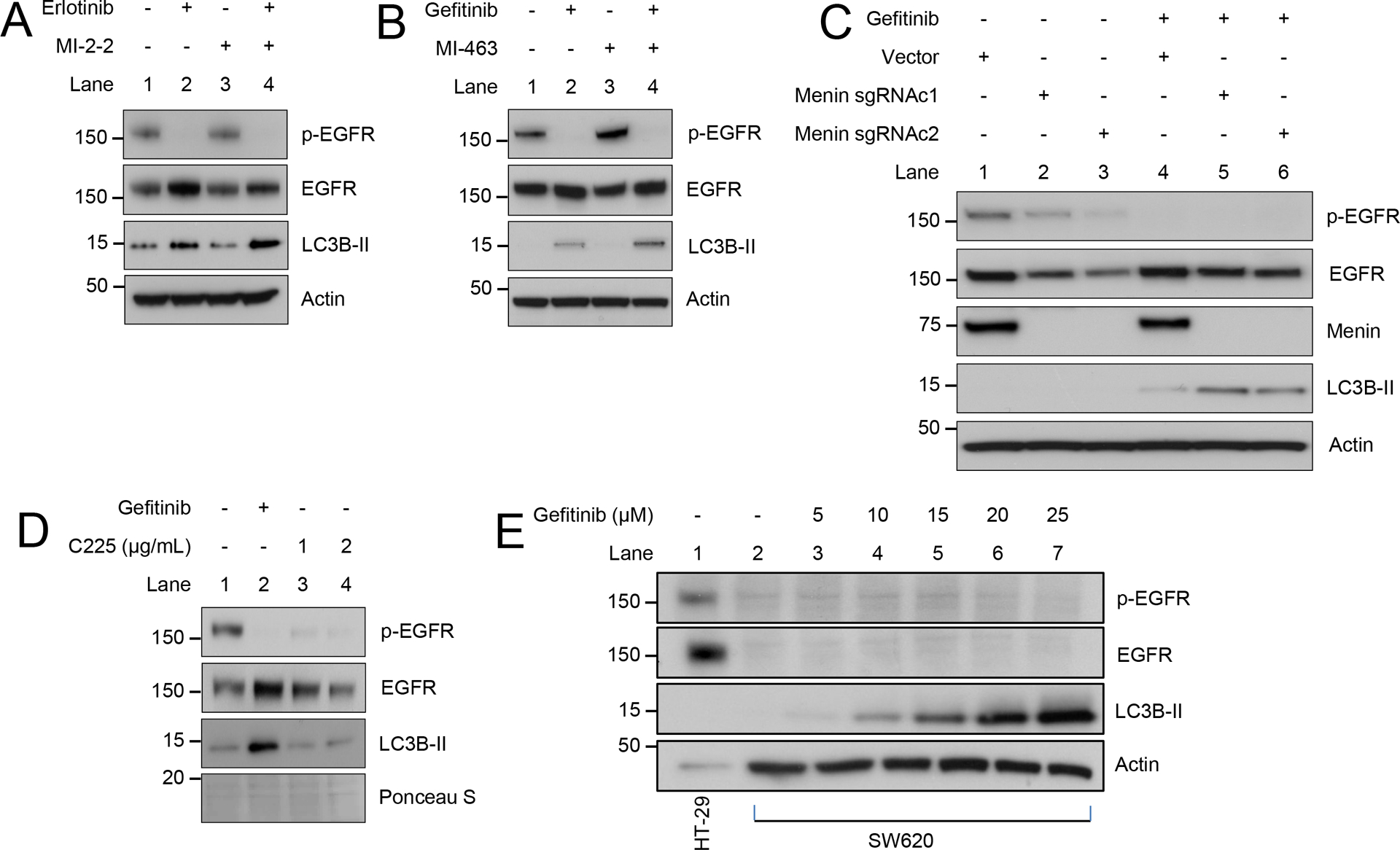
A) HT-29 cells treated for 96 hours, 10 μM erlotinib, 1 μM MI-2–2. B) HT-29 cells treated for 48 hours, 10 μM gefitinib, 1 μM MI-463. C) HT-29 cells transduced with lentiCRISPRv2 vector or menin sgRNA, with clones (c1 and c2) selected lacking menin expression, then treated for 48 hours, 10 μM gefitinib. D) HT-29 cells treated for 24 hours, 10 μM gefitinib. E) SW620 cells treated for 96 hours with varying concentrations of gefitinib, with HT-29 cells as a control for p-EGFR/EGFR expression.
EGFRi-induced autophagy is EGFR-independent
As small molecule EGFRis have EGFR-independent effects (5,15–17), it is important to determine if EGFRi-induced autophagy is in fact EGFR dependent. Unlike gefitinib, the inhibitory EGFR antibody C225 did not increase autophagy in HT-29 cells (Fig. 5D). Furthermore, knockdown of EGFR using two different shRNAs led to a marked reduction in p-EGFR, but no increase in autophagy (Supplementary Fig. 4C). Additionally, EGFR knockdown did not enhance gefitinib-induced autophagy (Supplementary Fig. 4C). To determine if EGFRis induce autophagy in the absence of EGFR, SW620 cells were used which lack expression of EGFR. Treatment of EGFR-null SW620 cells with gefitinib induced autophagy in a dose-dependent manner (Fig. 5E). Together these data support the EGFR-independent ability of EGFRis to induce autophagy in CRC cell lines.
Autophagy inhibition enhances CRC sensitivity to combined EGFRi and MI treatment
As autophagy is often considered a protective mechanism in cancer cells undergoing stress (18,20), we next investigated whether autophagy was important for CRC cell survival after treatment with combined EGFRi and MI. The FDA approved drug chloroquine (CQ) inhibits autophagy flux, potentially through impairment of autophagosome-lysosome fusion (37), and has been tested in numerous recent clinical trials in cancer. CQ alone had no significant toxicity in HT-29 cells (Fig. 6A–B). CQ led to increased LC3B-II accumulation after gefitinib and gefitinib/MI-2–2 treatment consistent with successfully blocking autophagic flux (Fig. 6C). Furthermore, CQ treatment significantly sensitized HT-29 cells to treatment with the combination of gefitinib and MI-2–2, leading to significantly reduced cell growth (Fig. 6A–B) and increased apoptosis (Fig. 6C). Similar results were also observed in HCT-15 cells (Supplementary Fig. 5A–C). Inhibiting autophagy through shRNA knockdown of ATG7 also led to reduced cell growth after combined treatment with gefitinib and MI-2–2 (Fig. 6D and Supplementary Fig. 5D). ATG7 knockdown also led to reduced levels of LC3B-II as well as increased PARP cleavage after treatment with the combination of gefitinib and MI-2–2 (Fig. 6E and Supplementary Fig. 5E), consistent with the results observed with CQ treatment. Finally, long-term treatment of both HT-29 (Supplementary Fig. 5F) and HCT-15 (Fig. 6F) via clonogenicity assays also showed CQ increased sensitivity to combined gefitinib and MI-2–2. Taken together, these results demonstrate that menin inhibition increases EGFRi-induced autophagy, and that this autophagy provides protection for CRC cells, such that autophagy inhibition sensitizes CRC to treatment with the combination of an EGFRi and MI.
Figure 6: Autophagy inhibition sensitizes CRC to combined treatment with gefitinib and MI-2–2.
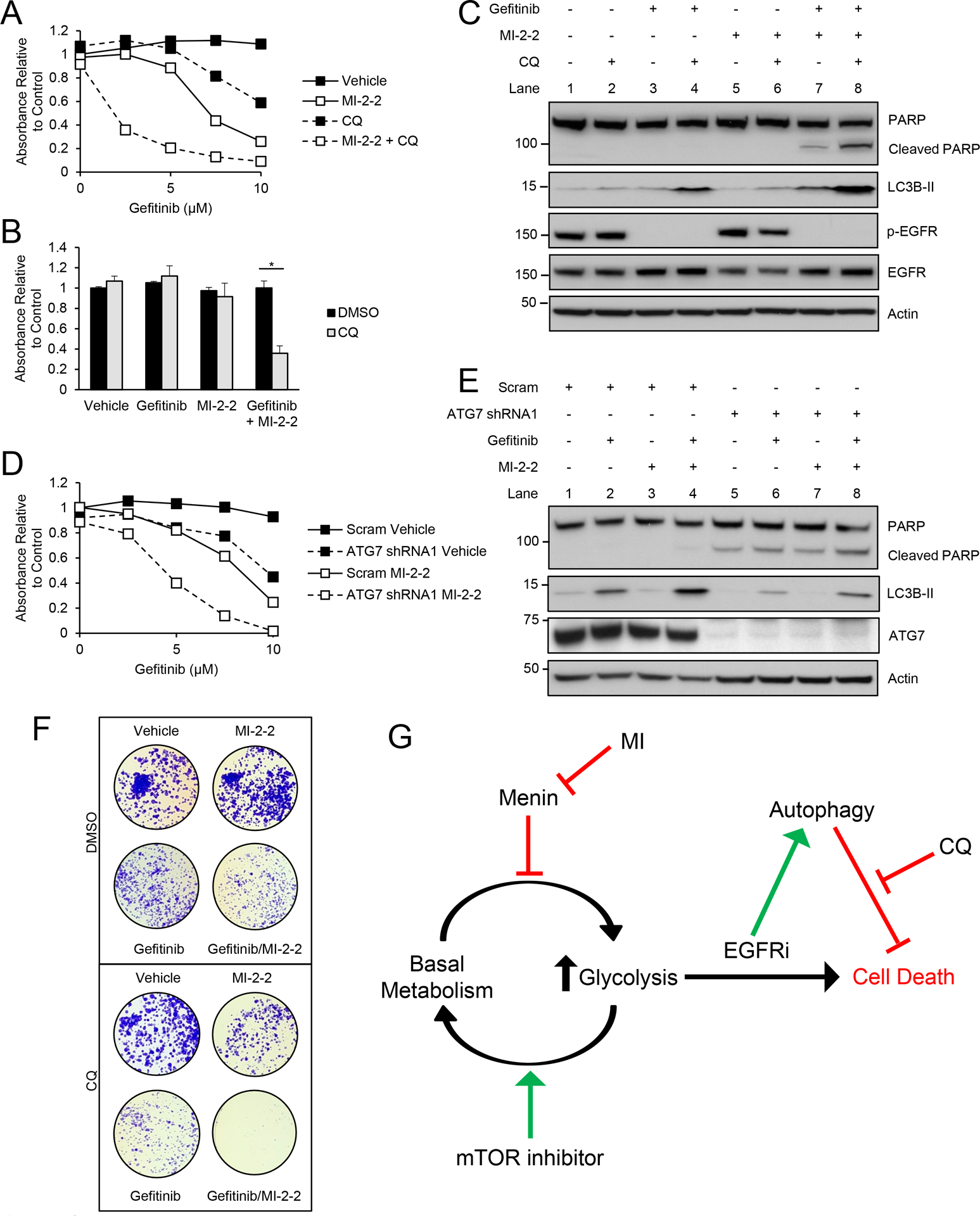
A) Treatment of HT-29 cells with various concentrations of gefitinib, with cell growth assessed after 96 hours by the MTS assay, 1 μM MI-2–2, 1 μM CQ. B) Treatment of HT-29 cells with cell growth assessed after 96 hours by the MTS assay, 2.5 μM gefitinib, 1 μM MI-2–2, 1 μM CQ. C) HT-29 cells treated for 72 hours, 5 μM gefitinib, 1 μM MI-2–2, 1 μM CQ. D) HT-29 cells transduced with scrambled or ATG7 shRNA1, then treated with various concentrations of gefitinib, with cell growth assessed after 96 hours by the MTS assay, 1 μM MI-2–2. E) HT-29 cells transduced with scrambled or ATG7 shRNA1, then treated for 48 hours, 5 μM gefitinib, 1 μM MI-2–2. F) Clonogenicity assay in HCT-15 cells after treatment for 10 days. 2.5 μM gefitinib, 1 μM MI-2–2, 4 μM CQ. G) A representative model of the role of menin, glycolysis, and autophagy in mediating resistance to EGFRis. * p < 0.05
Discussion
In this current work, we expand on the role of menin in CRC, finding that menin regulates glycolysis in CRC, which may open new pathways through which to target CRC. There are several points about this current work that have significance: First, we demonstrate the novel finding that menin suppresses glycolysis in CRC cells through an mTOR-independent mechanism, which is the first report of menin playing a role in CRC metabolism. Second, we show that menin inhibitor-induced increases in glycolysis are important for sensitizing CRC cells to small molecule EGFR inhibitors. Third, we demonstrate that while menin alone does not affect autophagy, menin inhibition increases EGFRi-induced autophagy in CRC. Finally, we show that autophagy is critical for promoting survival in CRC cells after combined treatment with an EGFRi and MI, such that autophagy inhibition sensitizes cells to the combination of an EGFRi and MI. Taken together these findings illustrate that menin acts as a critical regulator of glycolysis in CRC cells, and it is this regulation along with autophagy that serve as important mechanisms for mediating sensitivity to EGFRis (Fig. 6G).
Targeting cancer cell metabolism has long been a key component of cancer therapy. It is well known that even in the presence of oxygen, cancer cells have increased rates of glycolysis, termed aerobic glycolysis or the Warburg effect (31). Although glycolysis produces ATP much less efficiently than oxidative phosphorylation, glycolysis can be advantageous to tumors as it allows for faster ATP production and generates glycolytic intermediates which are important building blocks for rapidly proliferating cells (31). A novel finding in this manuscript is the role of menin in suppressing glycolysis in CRC cells. While a recent report in T cells showed that menin suppressed glycolysis in this cell population (25), there has been no connection between menin and glycolysis in malignant tissues.
Additionally, in T cells it was proposed that menin inhibits glycolysis through inhibition of mTOR phosphorylation, with menin knockout resulting in increased mTOR phosphorylation with a resulting increase in glycolysis (25). In contrast, we show that although menin inhibition increases glycolysis in CRC cells, it did not lead to an increase in mTOR phosphorylation at Ser2448, which is known to activate mTOR and promotes glycolysis (38). This therefore implies that menin is regulating glycolysis downstream of mTOR or through a mTOR-independent pathway. As menin has well-documented tissue-specific roles, as evidenced by its tumor suppressor functions in neuroendocrine tumors (1) compared to its tumor promoter functions in MLL-fusion leukemia (3) and prostate cancer (4), it is not completely surprising that we found menin affects mTOR differently in CRC cells compared to T-cells. However, these results generate numerous interesting questions that will be addressed in the future including investigating the mechanism of menin suppression of glycolysis in CRC. Furthermore, it will also be of great interest to determine how menin regulates glycolysis in other cancers.
Small molecules are a mainstay of cancer therapy, and while they have their predicted - so called “intended” - targets, it is well appreciated that small molecules have many effects through interactions with “unintended” targets as well, frequently deemed as off-target effects. EGFRis are no exception, as these small molecules have been shown to have unintended effects in cancer cells (15–17) including in CRC (5). Although these off-target effects of EGFRi are unintended, they may still be critical for effective tumor suppression (5). Here we demonstrate that EGFRis increase autophagic flux in CRC cells. While EGFR signaling has been shown to regulate autophagy by multiple mechanisms (18,21,22), we demonstrate that EGFRis induce autophagy in an EGFR-independent manner. In fact, EGFR-lacking SW620 cells show a dose-dependent increase in autophagy in response to EGFRi treatment. These findings demonstrate that EGFRi-induced autophagy induction is multi-factorial and further expand the spectrum of the EGFR-independent effects of EGFRis.
Autophagy is critical for maintaining cellular homeostasis, with important roles in both cancerous and non-malignant cells (39). Targeting autophagy for cancer treatment has recently generated substantial interest, with multiple ongoing clinical trials investigating autophagy inhibition as a potential cancer treatment strategy (39). Herein, we show that combined treatment with an EGFRi and MI leads to an increase in autophagy induction, and we further demonstrate that this autophagy is critical for cell survival after combined EGFRi/MI treatment. In fact, inhibition of autophagy with either CQ or ATG7 knockdown significantly enhanced the sensitivity of CRC cells to the combination of an EGFRi and MI, illustrating that autophagy is important for cellular protection against combined treatment with an EGFRi and MI. Additionally, autophagy inhibition was also able to sensitize CRC cells to EGFRis alone, albeit at higher concentrations of the EGFRi. Although the impact of this combination of drugs on CRC in vivo remains to be investigated in future studies, use of FDA-approved drugs such as CQ or hydroxychloroquine (HCQ) to inhibit autophagy has met some success in early phase clinical trials (39). In fact, investigations have begun in CRC; HCQ was combined with the histone deacetylase inhibitor vorinostat in metastatic CRC patients with some initially promising results (40). Our work further supports the continued investigation of targeting autophagy inhibition as a mechanism to enhance the therapeutic efficacy of existing regimens for CRC treatment, as well as new potential treatment regimens on the horizon. Additionally, small molecule menin inhibitors are now heading toward clinical use, with early phase clinical trials using MIs for the treatment of MLL-fusion leukemias being imminent (41), therefore it will only be a matter of time before it is possible to study MIs clinically where they may have potential to combine with other agents to better treat CRC.
In conclusion, these data allow for a model whereby menin represses glycolysis in CRC, with menin inhibition leading to increases in glycolysis. Furthermore, when CRC cells are in a state of higher glycolysis, these cells then become sensitized to the effects of EGFRis (Fig. 6G). Additionally, autophagy serves as an important protective mechanism permitting cell survival in the setting of combined EGFRi and MI treatment. This model is further strengthened by use of mTOR inhibitors, such as everolimus and rapamycin, which decrease CRC glycolysis and subsequently protect CRC cells from combined treatment with an EGFRi and MI. Taken together, this data highlights the complex tumor-specific functions of menin, and highlights how menin repression of glycolysis in CRC may serve to sensitize CRC to the effects of EGFRis and allow for novel future treatment strategies in CRC.
Supplementary Material
Acknowledgements
We would like to thank Dr. Anthony Mancuso at the University of Pennsylvania Laboratory for Quantitative Metabolomics for his assistance with the metabolism experiments. We would like to acknowledge the following support: NIH/NIDDK K08DK106489 (BWK) and R03DK120946 (BWK), NIH/NCI R01NCI563378 (XH), the Institute for Translational Medicine and Therapeutics (ITMAT014001) (XH), and the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases at the University of Pennsylvania (P30DK050306) including its pilot grant funding (BWK) and its core facilities (molecular pathology and imaging, and cell culture).
Footnotes
Conflict of interest: The authors have no conflicts of interest.
References
- 1.Li JWY, Hua X, Reidy-Lagunes D, Untch BR. MENIN loss as a tissue-specific driver of tumorigenesis. Mol Cell Endocrinol 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katona BW, Glynn RA, Hojnacki TA, Hua X. Menin: Expanding and dichotomous roles in cancer. Oncoscience 2019;6:368–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng Z, Ma J, Hua X. Epigenetic regulation by the menin pathway. Endocr Relat Cancer 2017;24:T147–T5928811300 [Google Scholar]
- 4.Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med 2015;21:344–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katona BW, Glynn RA, Paulosky KE, Feng Z, Davis CI, Ma J, et al. Combined Menin and EGFR Inhibitors Synergize to Suppress Colorectal Cancer via EGFR-Independent and Calcium-Mediated Repression of SKP2 Transcription. Cancer Res 2019;79:2195–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.American Cancer Society. Cancer Facts & Figures 2019. Atlanta: American Cancer Society; 2019. [Google Scholar]
- 7.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–93 [DOI] [PubMed] [Google Scholar]
- 8.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408–17 [DOI] [PubMed] [Google Scholar]
- 9.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65 [DOI] [PubMed] [Google Scholar]
- 10.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369:1023–34 [DOI] [PubMed] [Google Scholar]
- 11.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380–8 [DOI] [PubMed] [Google Scholar]
- 12.Mackenzie MJ, Hirte HW, Glenwood G, Jean M, Goel R, Major PP, et al. A phase II trial of ZD1839 (Iressa) 750 mg per day, an oral epidermal growth factor receptor-tyrosine kinase inhibitor, in patients with metastatic colorectal cancer. Invest New Drugs 2005;23:165–70 [DOI] [PubMed] [Google Scholar]
- 13.Townsley CA, Major P, Siu LL, Dancey J, Chen E, Pond GR, et al. Phase II study of erlotinib (OSI-774) in patients with metastatic colorectal cancer. Br J Cancer 2006;94:1136–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kindler HL, Friberg G, Skoog L, Wade-Oliver K, Vokes EE. Phase I/II trial of gefitinib and oxaliplatin in patients with advanced colorectal cancer. Am J Clin Oncol 2005;28:340–4 [DOI] [PubMed] [Google Scholar]
- 15.Verma N, Rai AK, Kaushik V, Brunnert D, Chahar KR, Pandey J, et al. Identification of gefitinib off-targets using a structure-based systems biology approach; their validation with reverse docking and retrospective data mining. Sci Rep 2016;6:33949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, et al. Cellular targets of gefitinib. Cancer Res 2005;65:379–82 [PubMed] [Google Scholar]
- 17.Yamamoto N, Honma M, Suzuki H. Off-target serine/threonine kinase 10 inhibition by erlotinib enhances lymphocytic activity leading to severe skin disorders. Mol Pharmacol 2011;80:466–75 [DOI] [PubMed] [Google Scholar]
- 18.Henson E, Chen Y, Gibson S. EGFR Family Members’ Regulation of Autophagy Is at a Crossroads of Cell Survival and Death in Cancer. Cancers (Basel) 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 2018;19:349–64 [DOI] [PubMed] [Google Scholar]
- 20.White E The role for autophagy in cancer. J Clin Invest 2015;125:42–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015;160:145–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013;154:1269–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017;168:960–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teng QX, Ashar YV, Gupta P, Gadee E, Fan YF, Reznik SE, et al. Revisiting mTOR inhibitors as anticancer agents. Drug Discov Today 2019 [DOI] [PubMed] [Google Scholar]
- 25.Suzuki J, Yamada T, Inoue K, Nabe S, Kuwahara M, Takemori N, et al. The tumor suppressor menin prevents effector CD8 T-cell dysfunction by targeting mTORC1-dependent metabolic activation. Nat Commun 2018;9:3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol 2017;24:1161–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012;120:4461–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015;27:589–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget 2016;7:38908–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol 2002;20:4292–302 [DOI] [PubMed] [Google Scholar]
- 33.McKillop D, Partridge EA, Kemp JV, Spence MP, Kendrew J, Barnett S, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther 2005;4:641–9 [DOI] [PubMed] [Google Scholar]
- 34.Katona BW, Liu Y, Ma A, Jin J, Hua X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther 2014;15:1677–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin 2013;34:625–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011;147:223–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018;14:1435–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014;345:1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer 2018;124:3307–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel S, Hurez V, Nawrocki ST, Goros M, Michalek J, Sarantopoulos J, et al. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 2016;7:59087–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burrows F, Wu T, Kessler L, Li S, Zhang J, Zarrinkar P, et al. A novel small molecule menin-MLL inhibitor for potential treatment of MLL-rearranged leukemias andNPM1/DNMT3A-mutant AML [abstract]. In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2017 Oct 26–30; Philadelphia, PA Mol Cancer Ther 2018;17:Abstract LB–A27 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.