

Abstract
Acromelic dysplasias are a group of rare musculoskeletal disorders that collectively present with short stature, pseudomuscular build, stiff joints, and tight skin. Acromelic dysplasias are caused by mutations in genes (FBN1, ADAMTSL2, ADAMTS10, ADAMTS17, LTBP2, and LTBP3) that encode secreted extracellular matrix proteins, and in SMAD4, an intracellular coregulator of transforming growth factor-β (TGF-β) signaling. The shared musculoskeletal presentations in acromelic dysplasias suggest that these proteins cooperate in a biological pathway, but also fulfill distinct roles in specific tissues that are affectedin individual disorders of the acromelic dysplasia group. In addition, most of the affected proteins directly interact with fibrillin microfibrils in the extracellular matrix and have been linked to the regulation of TGF-β signaling. Together with recently developed knockout mouse models targeting the affected genes, novel insights into molecular mechanisms of how these proteins regulate musculoskeletal development and homeostasis have emerged. Here, we summarize the current knowledge highlighting pathogenic mechanisms of the different disorders that compose acromelic dysplasias and provide an overview of the emerging biological roles of the individual proteins that are compromised. Finally, we develop a conceptual model of how these proteins may interact and form an “acromelic dysplasia complex” on fibrillin microfibrils in connective tissues of the musculoskeletal system.
Keywords: extracellular matrix, fibrillin, ADAMTS, connective tissue, Weill–Marchesani syndrome, geleophysic dysplasia
Introduction
The development and function of most organ systems in the body is dependent on the properties of various types of connective tissues that are composed of extracellular matrix (ECM) interspersed with one or more tissue-specific cell types. The specific composition and organization of connective tissue ECM provides structure and integrity to these organs and defines the dynamic physiological microenvironment that is necessary for cell homeostasis and tissue function.1 Connective tissues also play a major role at biological interfaces, where tissues with very different biomechanical properties intersect, for example, in the connection of skeletal muscle to tendon (myotendinous junction), tendon to bone (enthesis), or in tethering epithelial cell layers to underlying tissues through basement membranes. The ECM of connective tissues is formed by a hierarchical assembly process of its components that is guided by tissue-resident cells, such as fibroblasts, epithelial cells, vascular smooth muscle cells, tenocytes, or skeletal muscle cells. Core components of the ECM comprise different types of fibrillar and nonfibrillar collagens, glycoproteins, proteoglycans, proteases, active and latent growth factors, fibrillin microfibrils, fibronectin, and elastic fibers.1,2 With its diversity and dynamic regulation, the ECM not only provides structural support for connective tissues but also represents a microenvironment that governs the availability of vital biochemical cues to instruct cells to migrate, differentiate, hibernate, undergo programmed cell death, or to secrete bioactive molecules that define tissue function during homeostasis.3 The importance of the different ECM components for connective tissue integrity and organ function is underscored by a large number of genetic and acquired connective tissue disorders, in which individual ECM components are compromised.4,5 Most of these connective tissue disorders result in substantial morbidity and mortality in affected individuals and frequently manifest already during fetal and early postnatal development. Many connective tissue disorders that are caused by mutations in ECM proteins affect the musculoskeletal system, including muscular dystrophies, Ehlers–Danlos syndrome, Marfan syndrome, or acromelic dysplasias.4,6–9 While individual congenital connective tissue disorders are rare, they collectively affect thousands of individuals worldwide.10 Here, we provide an overview of the acromelic dysplasias, a group of syndromic connective tissue disorders that share most of their musculoskeletal phenotypes, which suggests that the affected genes operate in a common pathway in musculoskeletal tissues. Acromelic dysplasias are caused by mutations in genes encoding functionally related ECM proteins or SMAD4.9 Here, we summarize the current knowledge about the proposed molecular pathogenesis of the individual acromelic dysplasias, based on recent reports of novel mouse models for acromelic dysplasias, in vitro data from patient-derived fibroblasts, and biochemical studies. Finally, we will present a model of how the affected proteins may interact in the ECM and explore the possibility of a common pathogenic mechanism for the musculoskeletal presentations in acromelic dysplasias.
The acromelic dysplasia family of rare connective disorders
Acromelic dysplasias (acromelic: affecting the hands or feet) are a group of very rare congenital connective tissue disorders that share characteristic musculoskeletal presentations, that is, short stature, brachydactyly (short digits), joint stiffness, pseudomuscular build, and thick and fibrotic skin (Fig. 1 and Table 1).9,11 The four individual disorders belonging to the acromelic dysplasia group are differentially diagnosed by the specific involvement of additional organ systems, such as the heart or eye, and by the identification of the underlying disease-causing gene mutation, if possible. Although acromelic dysplasias are very rare, deciphering their pathogenic mechanisms will highlight the biological pathways in which the affected genes and their encoded proteins are involved during normal tissue development and homeostasis.
Figure 1.

Signs and symptoms of acromelic dysplasias. Acromelic dysplasias share their musculoskeletal signs and symptoms, but differ in the involvement of other organ systems. All affected genes, with the exception of SMAD4, encode secreted ECM proteins that bind to fibrillin microfibrils and that are implicated in regulating canonical TGF-β and BMP signaling. Several disorders belonging to acromelic dysplasias can be caused by mutations in more than one gene. Panels A–C show a patient diagnosed with GD2 due to an FBN1 mutation at 5 and 15 years of age. Typical features are the “happy face” appearance, short stature, and pseudomuscular build. Panels D and E show a patient with WMS3 due to a mutation in LTBP2. Brachydactyly (D) and upward dislocated lens (E, red arrow) are key features of WMS. Panels A–C are reprinted from Le Goff et al.12 with the permission of Elsevier. Panels D and E are reprinted from Haji-Seyed-Javadi et al.27 with the permission of John Wiley and Sons.
Table 1.
Signs and symptoms in acromelic dysplasias
| Disorder | Affected genes (MIM no.) | Musculoskeletal and skin | Ocular | Cardiovascular and pulmonary | Craniofacial | Cognitive/other |
|---|---|---|---|---|---|---|
| Acromicric dysplasia | FBN1 (#102370) | Short stature Brachydactyly Joint stiffness Thick skin Pseudomuscular build Delayed bone age |
Not reported | Bicuspid aortic valve Atrial septal defect Bronchial and tracheal narrowing |
Round face Defined eyebrows Small mouth Thick lips Prominent philtrum |
No cognitive impairment |
| Geleophysic dysplasia |
FBN1 (#614185) ADAMTSL2 (#231050) LTBP3 (#617809) |
Short stature; brachydactyly Joint stiffness Thick skin Pseudomuscular build Delayed bone age Tip toe walking |
Not reported | Cardiac valve disease Recurrent respiratory infections Bronchial and tracheal narrowing |
“Happy face” Upturned corners of the mouth |
No cognitive impairment Hepatomegaly |
| Weill–Marchesani syndrome |
FBN1 (#608328) ADAMTS10 (#277600) ADAMTS17 (#613195) LTBP2 (#614819) |
Short stature Brachydactyly Joint stiffness Thick skin Pseudomuscular build |
Ectopia lentis Microspherophakia Elevated risk for glaucoma |
Aortic valve stenosis Mitral valve insufficiency Ventricular septal defect |
None reported | Mild cognitive impairment |
| Myhre syndrome | SMAD4 (#139210) | Short stature Brachydactyly Joint stiffness Thick skin Pseudomuscular build |
Not reported | Cardiomyopathy Laryngotracheal abnormalities Progressive, proliferative fibrosis throughout the body |
Hearing impairment Facial dysmorphism |
Significant cognitive impairment |
Note: Not all signs and symptoms are fully penetrant and may vary in severity from individual to individual.
Acromicric dysplasia
Acromicric dysplasia (AD) (acromicric: hypoplasia of the limbs, and often digits, nose, and jaw) is characterized by short stature, short limbs, stiff joints, pseudomuscular build, thick skin, and distinctive facial features, such as a round face, sharply defined eyebrows, and a small mouth accompanied by thick lips.12,13 Other facial features include a prominent philtrum, narrow palpebral fissures, and a bulbous nose. Individuals diagnosed with AD have normal length at birth, but skeletal growth is slower thereafter, ultimately resulting in severe short stature. Most patients with AD do not show any severe cardiac impairments. However, there are some reported cases where individuals have a bicuspid aortic valve or an atrial septal defect.13 Some patients with AD develop respiratory or tracheal complications. On a molecular level, AD can be caused by autosomal dominant mutations in fibrillin-1 (FBN1) or latent transforming growth factor (TGF)-β–binding protein 3 (LTBP3) (Table 2).12,14 All the FBN1 mutations that cause AD are clustered in exons 41 and 42, which encode the TB5 domain of the FBN1 protein (Fig. 2A). TGF-β signaling is elevated in individuals where AD is caused by mutations in FBN1, but not in AD caused by mutations in LTBP3. Individuals with AD have a good prognosis, including a normal life expectancy. There are no current treatments for AD. However, different approaches to alleviate morbidity associated with the musculoskeletal presentations, such as physiotherapy, are offered to individuals with AD. A follow-up study on the orthopedic management of patients with AD showed that surgery for hip dysplasia, limb lengthening, or to treat carpal tunnel syndrome was performed in six out of nine AD patients.15 Two juvenile patients diagnosed with AD were treated with recombinant human growth hormone to promote skeletal growth and to alleviate the severity of short stature.13,16 While one case report showed no effect on final height, the second report showed an accelerated bone growth rate, but final height was not reported.
Table 2.
Mutations that cause acromelic dysplasia
| Gene | Acromelic dysplasia | Total number of mutations | Point mutations | Premature stop codon | Splice site | Deletions | Other |
|---|---|---|---|---|---|---|---|
| FBN1a | All | 30 | 27 | – | – | – | 1b |
| GD | 13 | – | – | – | |||
| AD | 12 | – | – | – | |||
| WMS | 5 | – | – | 2 | |||
| ADAMTSL2 | GD | 29 | 20 | 2 | 2 | 3 | 2c |
| ADAMTS10 | WMS | 12 | 6 | 4 | 2 | – | – |
| ADAMTS17 | WMS | 8 | 2 | 2 | 2 | 1 | 1 |
| LTBP2a | WMS | 1 | 1 | – | – | – | – |
| LTBP3a | AD and GD | 4 | 2 | 1 | 1 | – | – |
| SMAD4a | Myhre syndrome | 4 | 4 | – | – | – | – |
Mutations in these genes can cause other congenital disorders that are not included in this table.
Insertion.
Inversion.
Note: References are included in the text.
Figure 2.

Distribution of mutations in different proteins affected in acromelic dysplasias. Domain organization for FBN1, LTBP2, and LTBP3 (A), ADAMTS10, ADAMTS17, and ADAMTSL2 (B), and SMAD4 (C) are depicted, and the location of individual mutations is indicated with a vertical line. The FBN1 TB5 domain encoded by FBN1 exons 41 and 42 is magnified in panel A, showing individual amino acid residues and the four disulfide bonds. Mutations causing acromelic dysplasias and Marfan syndrome are color coded. The gray box shows amino acid residues where mutations can result in GD and AD, GD and WMS, or even GD, WMS, and Marfan syndrome. (The Marfan syndrome–causing FBN1 mutation is listed in the UMD-FBN1 database as ID 148 and is unpublished.)12,78,112 All SMAD4 mutations associated with Myhre syndrome are gain-of-function mutations and affect two amino acid residues located in the SMAD4 MH2 domain.
Geleophysic dysplasia
Geleophysic dysplasia (GD; geleophysic: happy face) is a rare syndromic disorder, with an estimated prevalence of less than 1 in 1,000,000 individuals and with about 50 cases reported worldwide.17 In addition to the musculoskeletal phenotypes characteristic for acromelic dysplasias, such as brachydactyly, pseudomuscular build, short stature, and delayed radiological development of hands, feet, and other limb bones, distinguishing features of individuals with GD include the “happy face” appearance due to dysmorphic changes in the face, such as upturned corners of the mouth, and tiptoe walking, potentially caused by alterations in the Achilles tendon. Individuals with GD can develop severe respiratory insufficiency and recurrent bronchial infections due to bronchial and tracheal narrowing, as well as progressive cardiac valve disease. As such, GD can be lethal during early childhood due to cardiac and respiratory complications. Similar to AD, GD is typically diagnosed shortly after birth because of reduced postnatal growth and ideally confirmed through genetic testing for pathogenic variants in the three affected genes that are currently known to cause GD, that is, FBN1, ADAMTSL2, and LTBP3. There is no cure for GD, but GD patients may benefit from physiotherapy to manage musculoskeletal morbidity, and they may need cardiac valve replacement or tracheostomy depending on disease progression.18 Treatment of a GD patient with recombinant human growth hormone initiated at 6 years of age showed a moderate improvement in overall height.19 However, two individuals with short stature were diagnosed with GD after they exhibited resistance to growth hormone treatment.20 Future research may resolve if treatment with growth hormone, at least in a subset of individuals with GD, may be a therapeutic strategy to improve final height. GD-causing mutations were identified in three genes encoding ECM proteins, that is, dominant mutations in FBN1 (GD2), all of which are located in exons 41 and 42; dominant mutations in LTBP3 (GD3); or recessive mutations in ADAMTSL2 (GD1).12,14 Interestingly, enhanced TGF-β signaling was only reported in individuals with GD caused by mutations in ADAMTSL2 and FBN1, but not in LTBP3.
Weill–Marchesani syndrome
Weill–Marchesani syndrome (WMS) is characterized by eye abnormalities, such as lens dislocation (ectopia lentis), microspherophakia (smaller, more spherical lens), and a higher risk for cataract formation, as well as musculoskeletal presentations characteristic for acromelic dysplasias.21–23 Other symptoms of WMS include cardiac abnormalities such as aortic valve stenosis, mitral valve insufficiency, and ventricular septal defects. There appears to be a mild level of cognitive impairment in ∼13% of individuals with WMS.24 WMS can be caused by dominant mutations in FBN1 (WMS2), or by recessive mutations in ADAMTS10 (WMS1), ADAMTS17 (WMS4), or LTBP2 (WMS3).21,22,25–27 Patient management for WMS includes ophthalmological procedures to correct vision and replace dislocated lenses, as well as physiotherapy to ameliorate the musculoskeletal symptoms. Overall, the prognosis for patients with WMS is good. However, careful monitoring of the cardiac conditions, especially heart valve function, is required. WMS4, caused by mutations in ADAMTS17, was previously referred to as WMS-like syndrome because of the noticeable lack of joint stif fness and cardiac abnormalities when compared with classical WMS.21,28 However, due to the small overall number of individuals with WMS, it is currently unclear if WMS4 indeed represents a distinct disorder or if WMS4 represents the milder end of the WMS spectrum.
Myhre syndrome
Myhre syndrome was characterized as an acromelic dysplasia due to the presentation with short stature, brachydactyly, stiff joints, muscle hypertrophy, and tight skin.29 However, individuals with Myhre syndrome show involvement of many additional organ systems, including significant cognitive and behavioral impairment, hearing loss, facial dysmorphism, arthropathy, cardiopathy, laryngotracheal abnormalities, and a predisposition to develop fibrosis in many tissues throughout the body.30 The fibrotic response can be elicited by injury or surgical procedures or can be idiopathic. Formation of scar tissue as a consequence of the fibrotic response can compromise the function of vital organs, such as the heart. Myhre syndrome is very rare, with only ∼100 cases documented in the literature. Myhre syndrome is caused by mutations in SMAD4, which is involved in the transduction of canonical TGF-β and bone morphogenetic protein (BMP) signals from the receptor to the nucleus.31,32 Currently, there are no treatments for Myhre syndrome. Patients are symptomatically treated and monitored for the onset and progression of fibrosis to limit organ damage.
Mouse models for acromelic dysplasias
Mouse models for most of the acromelic dysplasias were generated by targeting the individual genes that are mutated in the respective human disorders or by introducing disease-specific human mutations at homologous positions of the mouse gene (Table 3). The analyses of the musculoskeletal phenotypes and the phenotypes in other tissues provide insights into the pathogenic mechanisms that may drive the development of signs and symptoms in acromelic dysplasias in humans. While most mouse models recapitulate many of the aspects of individual acromelic dysplasias in humans, there are different degrees of severity when comparing mouse models with the human disorders. The mechanistic insights derived from the different mouse models require future studies to fully understand the biological functions of the individual genes and their encoded proteins and to understand how mutations in these genes contribute to the pathogenesis of the individual acromelic dysplasias.
Table 3.
Mouse models for acromelic dysplasias
| Targeted gene | Strain-specific gene targeting strategy | Musculoskeletal phenotypes | Ocular phenotype | Other phenotypes |
|---|---|---|---|---|
| Fbn1 | Global exon 10 deletion with Rosa26-Cre | Shortened long bones | Not reported | Thickened and fibrotic skin |
| Adamts10 |
a: Knockout b: Knockin of human WMS mutation |
a, b: Some embryonic/neonatal lethality and reduced weight gain b: Shortened long bones, altered growth plate, and increased skeletal muscle mass |
a, b: Persistence of FBN2 in the adult ciliary zonule |
a: Stiff skin b: Increased skeletal muscle mass |
| Adamts17 | Global deletion of exon 4 with CAG-Cre | Shortened long bones, shortened hypertrophic zone in growth plate, and brachydactyly | Not reported | Thickened skin |
| Adamtsl2 |
a: Global deletion b: Prx1-Cre conditional deletion (limb) c: Scx-Cre conditional deletion (tendon) d: Col2a-Cre conditional deletion (growth plate) |
a: Perinatal lethality b, c, d: Shorter long bones b, c: Shorter Achilles tendon d: Altered growth plate |
Not reported |
a: Bronchial occlusion, ventricular septal defect, and bone shortening in newborns in one model |
| Ltbp2 | Global exon 1 deletion with Cre-Lox | Not reported | Ectopia lentis and fragmented ciliary zone | Not reported |
| Ltbp3 |
a: Deletion of two exons via homologous recombination b: Knockout in 129SvEv/Sw background |
a: Rounded head, shortened snout, domed skull, shorter long bones, osteoarthritis, and osteosclerosis |
Not reported |
b: Involuted spleen and thymus |
| Smad4 |
a:
Tbx18-conditional deletion (cartilage) b: Pax6-conditional deletion (lens) c: Bglap-conditional deletion (bone) |
a: Chrondrodysplasia, limb shortening c: Reduced bone formation and reduced bone mineral density |
b: Microphthalmia and cataract formation |
Not reported |
Fbn1 (WMS2)
Most mouse models targeting Fbn1 have been developed to study pathogenic mechanisms in Marfan syndrome.33 Global or cell type–specific Fbn1 deletion or the knockin of a Marfan syndrome mutation in the homologous position of mouse Fbn1 broadly recapitulates the Marfan syndrome phenotypes, including long bone overgrowth, aortic aneurysm formation and dissection, and alterations in the ciliary zonule.34,35 Recently, a human WMS2–causing mutation was introduced in mouse Fbn1 and caused similar phenotypes as described for human WMS.36 Exon 10–12 of Fbn1 was globally deleted by Rosa26-Cre–mediated recombination, resulting in a WMS-like phenotype. The mice had short stature, with a 6–10% reduction in limb length, which normalized after 5 months of age. In addition, the skin was fibrotic, with increased thickness and excess collagen deposition. The different mouse models targeting Fbn1 clearly emphasize the pleiotropy in the FBN1 gene, as seen in the different human disorders associated with mutations in FBN1.11
Adamtsl2 (GD1)
Global Adamtsl2 deletion by homologous recombination resulted in >90% perinatal lethality due to bronchial occlusion and a ventricular septal defect.37,38 Very few mice survived after several days and they were significantly smaller and had tight skin and stif f limbs that collectively severely restricted movement (unpublished data). In addition, a subset of Adamtsl2 heterozygous mice developed thicker skin with elevated collagen deposition, indicative of a fibrotic response.37 Conditional deletion of Adamtsl2 in the limbs using Prx1-Cre or in the growth plate using Col2a-Cre recapitulated some of the musculoskeletal phenotypes described in individuals with GD, such as limb shortening.38,39 Surprisingly, Scx-Cre–mediated deletion of Adamtsl2 in tendon, the site of the strongest Adamtsl2 expression in the mouse limb, also resulted in shortened limbs, concomitant with a 20–30% reduction in Achilles tendon length.39 Mechanistically, however, different explanations of the short limb phenotypes in the two mouse models were provided. In the Prx1-Cre–mediated deletion of Adamtsl2, normal growth plate histology was observed, but in the Col2a-mediated, chondrocyte-specific Adamtsl2 deletion, growth plate abnormalities and altered TGF-β signaling were noted. Together, both conditional Adamtsl2 deletion strategies targeting the limb or the growth plate showed bone shortening. However, the underlying molecular mechanisms will need to be fully elucidated. In addition to the mouse models of ADAMTSL2 deficiency that mimic GD, there was also a dog model described, where a founder point mutation in Adamtsl2 causes Musladin–Lueke syndrome in beagles.40,41 These dogs suffer from severe joint stiffness and fibrotic skin, but did not show cardiac or pulmonary involvement. The specific ADAMTSL2 founder mutation causing Musladin–Lueke syndrome was also identified in individuals with GD.42 It is evident that different species show different susceptibilities to mutations in ADAMTSL2 gene. Dogs do not show the involvement of the cardiac or pulmonary system, while humans frequently show tracheal narrowing and heart valve alterations. In contrast, deletion of Adamtsl2 in mice is lethal shortly after birth due to bronchial obstruction. The musculoskeletal phenotypes seemed to be common to the two different animal models and human GD.
Adamts10 (WMS1)
Two mouse models have been reported that target Adamts10 with different approaches.43,44 One approach targeted Adamts10 by deleting parts of exon 5 through homologous recombination, resulting in an Adamts10 knockout, whereas the second model was generated by knocking in a WMS1 mutation that introduces a premature termination codon in the homologous position of mouse Adamts10 using CRISPR/Cas9 gene editing. However, the truncated ADAMTS10 peptide comprising the propeptide domain could be detected in mouse embryonic fibroblasts, suggesting that mutated Adamts10 mRNA escaped nonsense-mediated mRNA decay. While the Adamts10 knockout model showed some embryonic or neonatal lethality and developed stiff skin, the characteristic musculoskeletal or cardiac features of WMS1 were not observed.43 By contrast, the knockin mouse model of the WMS1 mutation in Adamts10 showed key features of WMS, such as shortening of the long bones, which was attributed to growth plate abnormalities, and increased skeletal muscle mass, attributed to more but smaller myofibers.44 Mechanistically, mitogen activated protein kinase signaling was altered in the skeletal muscle, and reduced BMP signaling, possibly caused by reduced gene expression of several BMP isoforms, was observed in skin fibroblasts isolated from the Adamts10 knockin mouse model. TGF-β signaling was not changed. Both Adamts10 mouse models showed reduced weight gain and alterations of the ciliary zonule in the eye. The ciliary zonule is formed by cable-like structures based on FBN1 microfibrils that hold the lens in place and mediates accommodation. In both mouse models, FBN2 microfibrils persisted beyond birth, whereas in the respective wild-type strains, only FBN1 microfibrils could be identified.43,44 However, lens dislocation due to a disrupted ciliary zonule, a key feature of WMS, was not observed and the pathological consequences of FBN2 microfibril persistence on ciliary zonule function need to be explored.
Adamts17 (WMS4)
Adamts17 was inactivated by deletion of parts of exon 4 via Cre/Lox-mediated recombination, resulting in a frameshift mutation and subsequent nonsense-mediated mRNA decay.45 Equivalent to human WMS4, these mice developed shorter long bones, brachydactyly, and thickened skin. Similar to the Adamts10 WMS1 knockin model, the shortening of the long bones in ADAMTS17-deficient mice was attributed to alterations in the growth plate. However, ADAMTS17-deficient growth plates showed a shortened hypertrophic zone, while Adamts10 WMS1 knockin growth plates showed an enlarged hypertrophic zone, both resulting in shortening of the long bones. Similar mechanistic alterations in BMP signaling were observed in ADAMTS17-deficient chondrocytes, where canonical BMP signaling was reduced. The skeletal muscle or ocular phenotype, specifically the morphology and molecular composition of the ciliary zonule, was not analyzed in ADAMTS17-deficient mice.
Ltbp2 (WMS3)
Ltbp2 was targeted by deletion of parts of exon 1 using Cre/Lox-mediated recombination.46 The Ltbp2 knockout mice were viable and fertile, which is in contrast to a previously published Ltbp2 knockout strain that showed very early embryonic lethality, and their phenotype was more consistent with the observations in humans with WMS3 caused by mutations in LTBP2.47 Ltbp2 knockout mice developed an ectopia lentis phenotype, which is characteristic for individuals with WMS. The ciliary zonule was fragmented at birth and absent in young adult eyes. However, other phenotypes characteristic for WMS were not described in the Ltbp2 knockout mouse model. It was further demonstrated that LTBP4 can compensate for LTBP2 in many tissues, but since LTBP4 is not expressed in the ciliary zonule, it could not compensate for the absence of LTBP2 in this tissue, resulting in the fragmentation of the ciliary zonule.48
Ltbp3 (AD and GD)
A mouse model of LTBP3 deficiency was created by deleting two exons of Ltbp3 by homologous recombination, causing a reading frame shift. The Ltbp3 knockout displays craniofacial and body malformations reminiscent of aspects of human AD or GD phenotypes.49–51 The offspring were born in a proper Mendelian ratio, indicating no developmental or perinatal lethality due to the absence of LTBP3. Phenotypes in these mice included features, such as a rounded head, shortened snout, domed skull, and curvature of thoracic/cervical vertebrae. These craniofacial malformations were attributed to the lack of a cartilaginous growth plate, which was observed at 3 weeks of age. One week after birth, growth retardation was frequently observed, which ultimately resulted in 10–25% shorter bones. Older mice showed articular cartilage degeneration similar to what is observed in osteoarthritis, resulting in the loss of articular cartilage and the presence of a fibrotic and ossified articular surface by 9 months of age.51 LTBP3-deficient mice also had an involuted thymus and spleen and an impairment in the septation of the alveoli, resulting in lung emphysema.52,53
Smad4 (Myhre syndrome)
Most mouse models targeting Smad4 were developed in the context of studying tumorigenesis because SMAD4 is a known tumor suppressor.54 Since the systemic knockout of Smad4 is lethal during early embryonic development, many of these mouse models focus on phenotypes of Smad4 ablation in specific tissues or cell types using the Cre/Lox system.55–57 For example, Smad4 depletion in chondrocytes using either Tbx18-Cre or Col2a-Cre resulted in chondrodysplasia and subsequent shortening of the limbs.58–60 Multiple defects in the growth plate were observed, including an imbalance in ECM synthesis and degradation, altered chondrocyte polarity, and altered chondrocyte hypertrophy. Postnatal disruption of Smad4 in osteoblasts using Bglap-Cre–mediated recombination affected bone formation, and the mice had a reduced bone mass and bone mineral density.61 In the eye, Smad4 inactivation in the lens using Pax6-Cre resulted in microphthalmia as well as cataract formation.62 The Smad4 ablation strategies described here inactivate SMAD4 and are informative in revealing SMAD4 gene function and its role in mediating TGF-β and BMP signaling. However, SMAD4 mutations causing Myhre syndrome are activating gain-of-function mutations presumably resulting in excessive activation of TGF-β and/or BMP signaling. So far, no knockin of Myhre syndrome–causing mutations in mice have been described.
Structure and function of proteins involved in acromelic dysplasias
With the exception of SMAD4, mutations that cause acromelic dysplasias were identified in genes encoding a group of secreted ECM proteins that are dependent on FBN1 microfibrils for deposition and function in the ECM.11,63 SMAD4 is a signal transducer for canonical TGF-β and BMP signaling. Both signaling pathways appear to be dysregulated in patient-derived fibroblasts and mouse models for some of the acromelic dysplasias, such as the Adamts10 and Adamts17 knockouts and the cartilage-specific deletion ofAdamtsl2.12,38,44,45,64 In the following section, we describe in more detail the structure and function of the proteins that are encoded by the genes mutated in acromelic dysplasias.
Fibrillin-1 (FBN1)
The fibrillin-1 gene (FBN1) encodes a 350-kDa glycoprotein that is composed of 47 epidermal growth factor–like (EGF) domains (43 of which bind calcium), seven TGF-β–binding protein-like/8-cysteine domains (TB), two hybrid domains that share features of both EGF and TB domains, and a proline-rich domain (Fig. 2A).65 Two other fibrillin isotypes, FBN2 and FBN3, are present in the human genome, while FBN3 is absent in rodents.66 Upon secretion, FBN1 assembles into microfibrils, the functional units that are deposited in the ECMs of various connective tissues, either as individual microfibrils or as microfibril bundles. The function and potentially the ultrastructure of microfibrils varies based on the tissue type.67 For example, in the wall of blood vessels, the skin, and the lungs, microfibrils provide a scaffold for the deposition of tropoelastin, which is required for the formation and homeostasis of elastic fibers.68 In the ciliary zonule, microfibrils work independently of elastic fibers to position the lens and mediate accommodation.69,70 Microfibrils can also perform regulatory functions by interacting with cells via integrins or by binding to LTBPs or BMPs to regulate TGF-β and BMP signaling, respectively. Most of the >1800 autosomal dominant mutations in FBN1 cause Marfan syndrome, a connective tissue disorder that affects the musculoskeletal, cardiovascular, and ocular system, resulting in tall stature, disproportionately long limbs and digits, joint laxity, muscle hypoplasia, aortic dilation, and lens dislocation.33,71 Marfan syndrome–causing mutations span the entire length of FBN1, with no established genotype–phenotype correlation and inter and intrafamilial variability in the severity of the Marfan syndrome phenotype.72–74 Recently, a FBN1 missense variant was identified in Peruvians that is located in the neonatal Marfan syndrome region of FBN1 (exon 31, encoding calcium-binding EGF domain #17) that correlated with reduced height and further illustrates the complexity of correlating genotypes with phenotypes in FBN1-related traits.75,76 One of the most definitive genotype–phenotype correlations can be found in the rare FBN1 mutations causing the acromelic dysplasias AD and GD. These mutations are typically located in the fifth TB domain (TB5) of FBN112 (Fig. 2A). Cultured AD and GD patient–derived fibroblasts showed decreased amounts of microfibrils and severe disorganization of the microfibril network. The SMAD2/3 phosphorylation level in these cells was elevated and a 10-fold higher amount of total TGF-β was measured in the conditioned media obtained from these cells compared with the matched controls.12 Collectively, these data indicate that elevated canonical TGF-β signaling may be the underlying pathogenic mechanism for AD and GD. Given that abnormal microfibril deposition and increased TGF-β signaling were also observed in Marfan syndrome, it is difficult to reconcile how the clinically opposite phenotypes of Marfan syndrome and AD or GD can both arise through elevated TGF-β signaling.33,34 One of the FBN1 mutations that causes WMS was identified as an in-frame deletion of 24 nucleotides in exon 41 and also affected the TB5 domain.22 However, a second WMS-causing FBN1 mutation was identified outside that region as an in-frame deletion of exons 9–11, which encode the first 8-cysteine domain, the proline-rich domain, and the fourth EGF-like domain of FBN1.36 Canonical TGF-β signaling was not altered in fibroblasts isolated from these individuals with WMS and no changes in the amount of active or latent TGF-β were detected in the cell culture supernatant. In addition, skin extracts obtained from the corresponding WMS knockin mice showed no alterations in TGF-β signaling or marker gene expression, despite clear evidence for skin fibrosis.36 While the location of the second WMS mutation is outside of the TB5 domain, where the other acromelic dysplasia mutations were identified, both affected domains may come into close proximity when considering the proposed three-dimensional structure of fibrillin microfibrils, and thus may form a functional unit.36,77 The currently reported cases of AD and GD are caused by missense mutations in the TB5 domain of FBN1 and change structurally important residues (Fig. 2A). Among the 22 mutations identified, 10 are specific to GD, 9 are specific to AD, and 2 are found in both GD and AD patients.17 The fact that several mutations can cause GD and AD would suggest the involvement of modifier genes, or that AD and GD are the same disease with different phenotypic expression, or the possibility that some mutations do not cause a loss of function but a pathogenic gain-of-function, depending on the mutated amino acid residue. It is noteworthy that mutations in this domain can also cause classical Marfan syndrome and it is completely unclear how that can be explained, especially when mutations are affecting adjacent amino acid residues. So far, one point mutation has been identified in Marfan syndrome (UMD-ID 148), WMS, and GD.12,78 When FBN1 mutations causing WMS, AD, or GD were introduced into recombinant FBN1 peptides, secretion was not affected.79 However, mutant FBN1 peptides showed decreased binding to heparan sulfate.79 Heparan sulfate interactions mediate cell surface binding of ECM proteins and heparan sulfate–FBN1 interactions are important for fibrillin assembly.80 Thus, acromelic dysplasia mutations in the TB5 domain of FBN1 could result in impaired cell surface anchorage of FBN1 microfibrils and result in reduced microfibrils in the ECM, as observed in patient-derived fibroblasts.12,80
ADAMTS10 and ADAMTS17
ADAMTS10 and ADAMTS17, which are mutated in WMS2 and WMS4, respectively, belong to the 19 member–strong ADAMTS family of proteases. ADAMTS proteases are involved in a wide variety of developmental and homeostatic processes.81 ADAMTS10 and ADAMTS17, which share the same domain organization but have distinct biochemical properties, may work as true proteases or modulate FBN1 microfibril formation independent of their respective proteolytic activities.82 ADAMTS10 and ADAMTS17 are composed of a protease domain, which includes a propeptide domain, a catalytic domain, and a disintegrin-like domain, and an ancillary domain, which includes a thrombospondin-type 1 repeat (TSR), a cysteinerich region, a spacer domain, four additional TSRs, and a PLAC domain.83 Most members of the ADAMTS family are secreted as zymogens that are then activated at the cell surface by furin cleavage of the propeptide domain. ADAMTS10, however, is resistant to furin processing and is present in the ECM largely in its unprocessed form.84 ADAMTS10 binds to FBN1 at the N- and C-terminus and colocalizes with FBN1 microfibrils in the skin and the ciliary zonule.85 In its unprocessed form, ADAMTS10 cleaves FBN1 inefficiently.85 However, upon restoration of furin processing, ADAMTS10 cleaves FBN1 more efficiently and also cleaves FBN2.43 Exogenous addition of ADAMTS10 to cultured fetal bovine nuchal ligament cells enhances the assembly of FBN1 microfibrils.85 As such, ADAMTS10 may have a function in promoting microfibril assembly.85 ADAMTS10 mutations causing WMS2 span the entire length of the gene, although most localize to the signal peptide and the catalytic domain (Fig. 2B). Molecular studies analyzing two missense mutations in the signal peptide of ADAMTS10 showed significantly reduced secretion of the mutant protein compared with the wild-type protein.25 Two mutations resulted in aberrant subcellular distribution of ADAMTS10, with reduced levels in the endoplasmic reticulum and increased levels in the cytoplasm.86,87 A substitution at the –1 position of the signal peptide cleavage site caused a loss of secretion despite normal processing of the propeptide.25 Interestingly, the analysis of the same mutation in C-terminally truncated ADAMTS10 showed normal secretion. This suggests that proper processing and folding of the C-terminal ancillary domain is dependent on upstream domains and is critical for the secretion of ADAMTS10 into the ECM. Analyses of tissue from WMS2 patients showed that the mutated ADAMTS10 transcript is expressed in skin fibroblasts and chondrocytes, which may explain the features of short stature and brachydactyly.26 ADAMTS10 is expressed in the fetal and adult heart, which may explain the cardiac abnormalities in WMS patients. Electron micrographs of skin fibroblasts from a WMS patient showed abnormal extrafibrillar space and abnormally enlarged microfilament bundles inside of the cell, indicating a perturbed connection between the ECM and the cytoskeleton.26 WMS caused by mutations in either ADAMTS10 or FBN1 are clinically indistinguishable, suggesting that the two proteins function in the same biological pathway. Cultured dermal fibroblasts from WMS patients with ADAMTS10 mutations showed reduced FBN1 microfibril deposition compared with cells from unaffected individuals.25 Given this information, WMS2 caused by ADAMTS10 mutations is likely a consequence of disturbed FBN1 microfibril formation in ADAMTS10-expressing tissues.
In contrast with ADAMTS10, ADAMTS17 is furin-processed at the cell surface and undergoes extensive autocatalysis.88 In mice, Adamts17 is expressed in the eye, lungs, bones, tendons, skin, intervertebral disk, and blood vessels, and this pattern coincides with Fbn1 expression. ADAMTS17 colocalized with FBN1 microfibrils in cell culture, but it did not bind to or cleave recombinant FBN1 peptides. So far, no ADAMTS17 substrates other than itself are known. WMS4-causing mutations in ADAMTS17 span the length of the gene and are not localized to any specific domain. Nonsense, splice-site, and deletion mutations are predicted to produce nonfunctional truncated protein products (Table 2). The two ADAMTS17 missense mutations resulted in loss of secretion and retention of ADAMTS17 inside the cell.87,89 Patient-derived fibroblasts showed decreased ECM deposition of FBN1 microfibrils, fibronectin, and collagen type I due to a proposed coretention in the secretory pathway.89 This would suggest that ADAMTS17 has a critical role in ECM formation or as a chaperone aiding the secretion of several ECM molecules. Why do ADAMTS17 mutations not result in joint stiffness and cardiac valve anomalies compared with mutations in FBN1 or ADAMTS10? One possible explanation is that ADAMTS10 could compensate for ADAMTS17 in joints and the heart, but not in other tissues. Curiously, AD and GD share overlapping features with WMS but do not show ocular features, such as ciliary zonule defects. This indicates that although the relevant proteins all contribute to general musculoskeletal and skin homeostasis, they also play additional tissue-specific roles.
ADAMTS-like 2 (ADAMTSL2)
ADAMTSL2 encodes a 105 kDa secreted glycoprotein that contains a TSR repeat, a spacer domain, an N-glycan–rich domain, six additional TSRs, and a PLAC domain. As such, the domain organization of ADAMTSL2 resembles the domain organization of the ancillary domain of ADAMTS proteases, but the seven ADAMTS-like proteins all lack a catalytic domain and thus do not work as proteases in the ECM.90 In situ hybridization of human fetal tissue shows ADAMTSL2 expression in the heart, skin, lungs, tracheal wall, skeletal muscle, and the vasculature.64 Recombinant ADAMTSL2 binds to LTBP1 and FBN1 but its function in the ECM is not well understood.36,37,64 So far, 29 ADAMTSL2 mutations in 43 families affected with GD have been identified. They include 18 missense mutations, 4 nonsense mutations, 2 deletions, and 5 splice-site mutations that together span the entire length of the gene (Fig. 2B and Table 2).91 Cell culture studies have shown that secretion of recombinant mutant ADAMTSL2 is impaired, with no differences in the intracellular levels of the proteins. This suggests that mutations in ADAMTSL2 lead to misfolding of the protein that prevents its secretion into the ECM.64 One potential mechanism by which mutations in ADAMTSL2 cause GD relates to the involvement of ADAMTSL2 in TGF-β signaling.64 Conditioned medium of fibroblasts obtained from GD patients showed a 10-fold increase in total TGF-β compared with matched controls. In addition, cell lysates of GD fibroblasts showed a five-fold higher amount of phosphorylated SMAD2/3 compared with controls, indicative of elevated TGF-β signaling.64 Because ADAMTSL2 binds FBN1 and LTBP1, it may act as a bridge that stabilizes the binding of LTBP1 to FBN1 microfibrils and thus modulates the latency of TGF-β. GD could then arise as a consequence of the perturbation of the FBN1–ADAMTSL2–LTBP1 complex in affected tissues. However, GD can also be caused by mutations in LTBP3, and TGF-β signaling was not altered in fibroblasts from these individuals.14 GD patients with mutations in either ADAMTSL2 or FBN1 share the main features characteristic of GD, including short stature, brachydactyly, laryngeal stenosis, and cardiac valve abnormalities. However, only in GD patients with mutations in ADAMTSL2 were facial dysmorphisms, such as a thin upper lip, a long f lat philtrum, and narrow palpebral fissures, and tiptoe walking consistently observed.42 The reasons for the heterogeneity in these phenotypes have yet to be identified.
LTBP2 and LTBP3
LTBP2 and LTBP3 represent two of the four members of the LTBP protein family, which is evolutionarily related to the fibrillins, both sharing the unique TB domain and a partly similar domain organization (Fig. 2A).92 LTBP1, LTBP3, and LTBP4 can bind latent TGF-β and keep it in an inactive form that can be stored in the ECM. However, LTBP2 does not contain the critical latent TGF-β–binding motif in the third TB domain and therefore has functions other than TGF-β growth factor deposition in the ECM.93 LTBP1 requires fibronectin for ECM deposition and LTBP2, LTBP3, and LTBP4 require FBN1 for deposition in the ECM and as such are functionally linked to FBN1 microfibrils.94,95
LTBP2 encodes a 200 kDa secreted protein, which differs from LTBP3 in the addition of one noncalcium-binding and one calcium-binding EGF-like repeat and the presence of an integrin-binding RGD motif.96 LTBP2 is highly expressed in the trabecular meshwork of the eye and in elastin-rich tissues, such as the large blood vessels and the lungs, but in vitro LTBP2 appears to be a negative regulator of elastogenesis.97 LTBP2 does not bind to latent TGF-β, but it can bind to FGF2 in vitro and inhibit its activity.98 As such, LTBP2 may play a role in wound healing or in fibrotic responses by modulating FGF2 signaling. LTBP2 specifically binds the N-terminus of FBN1 and shows strong colocalization with FBN1 microfibrils, where it is thought to play a role in stabilizing microfibril structure and facilitating the assembly of larger microfibril bundles.46 Most autosomal recessive mutations in LTBP2 cause genetic eye diseases other than WMS, including primary open-angle glaucoma and ectopia lentis.99 Primary open-angle glaucoma manifests within the first year of life and is characterized by high intraocular pressure due to perturbed aqueous humor outflow from the anterior segment of the eye.99 Ectopia lentis describes the dislocation of the crystalline lens of the eye, which can occur as an isolated condition or as part of a syndrome, such as Marfan syndrome or WMS. Since LTBP2-deficient mice develop lens dislocation due to disrupted ciliary zonule formation, it is possible that microfibril formation in the ciliary zonule critically depends on LTBP2, but other matrix proteins, specifically LTBP4, may compensate for the absence of LTBP2 in other tissues.46 The WMS-causing point mutation was identified in exon 24 ofLTBP2, corresponding to the FBN1-binding domain.27 Skin tissue from patients with this mutation showed clumped and fragmented elastic fibers, less collagen fibers, and a more fragmented FBN1 microfibril network compared with control samples. Interestingly, the same LTBP2 mutation that manifested as WMS in one sibling in the affected family manifested as WMS-like syndrome in three other siblings.27 Furthermore, the WMS-like syndrome patients did not exhibit any ocular features, whereas WMS4 caused by ADAMTS17 mutations shows eye defects but lacks joint stiffness and brachydactyly.27 Regardless of the phenotype, LTBP2 mutations are anticipated to disrupt the stability of the fibrillin microfibril network.46 The position of specific mutations in LTBP2 or the presence of modifier genes could be some of the reasons for differences in presentation among individuals with LTBP2 mutations. Analysis of four C-terminal mutations in LTBP2 that retain the FBN1-binding domain showed loss of secretion owing to misfolding of the protein and three of the four mutant proteins also lost the ability to bind FBN1 even though the individual mutations occurred downstream of the FBN1-binding domain.46
LTBP3 encodes the smallest member of the LTBP family. In contrast with LTBP2, LTBP3 can bind all three TGF-β isoforms and in fact requires binding to latent TGF-β for its secretion.100 LTBP3 is highly expressed in the heart, lungs, and bones. LTBP3 requires FBN1 microfibrils for ECM deposition.94 Although previous studies have shown that there is no direct binding of the C-terminal region of LTBP3 to the N-terminal region of FBN1, this does not eliminate the possibility that other domains of LTBP3 or FBN1 bind to each other. It is also possible that LTBP3 binds to microfibrils in the ECM through an adaptor protein.94 Mutations in LTBP3 can cause dental anomalies and short stature (DASS), AD, and GD depending on the type of mutation and the specific location of the mutation in LTBP3. Autosomal recessive mutations cause DASS, a connective tissue disorder characterized by short stature, brachyolmia, and hypoplastic amelogenesis imperfecta, with some patients also exhibiting tooth agenesis or oligodontia.101 Autosomal dominant nonsense and splice-site mutations cause GD, and autosomal dominant missense mutations in the EGF-like domain cause AD.102 The mechanism by which mutations in LTBP3 cause disease is likely related to its association with FBN1 microfibrils. Fibroblasts from AD patients harboring an LTBP3 mutation elaborate a diminished and disorganized microfibrillar network, similar to fibroblasts from AD and GD patients with FBN1 or ADAMTSL2 mutations.12,14,64 Surprisingly, and unlike AD and GD caused by FBN1 or ADAMTSL2 mutations, there was no increase in TGF-β signaling in fibroblasts from individuals with AD due to mutations in LTBP3. Another point of distinction is that GD caused by mutations in LTBP3 showed severe lung anomalies but did not show additional cardiac valve stenosis as it is the cases for GD caused by ADAMTSL2 and FBN1 mutations.14 These similarities and differences indicate overlapping yet distinct roles of LTBP3, ADAMTSL2, and FBN1 in ECM organization and regulation of TGF-β signaling.
SMAD4
SMAD4 encodes a 60 kDa protein that consists of a Mad homology (MH) 1 domain, a DNA-binding domain, a linker region, and an MH2 domain responsible for activation of gene transcription. SMAD4 is a key component of the canonical TGF-β and BMP signaling pathway.91 Once TGF-β or BMP bind to their respective receptors, phosphorylated (p)SMAD2/3 or pSMAD1/5/8, respectively, require dimerization with SMAD4 to translocate from the cytoplasm into the nucleus and to activate or inhibit the expression of TGF-β and BMP target genes.31 These target genes regulate many processes, such as cell proliferation, cell fate determination, cell differentiation, immune regulation, and ECM function cause juvenile polyposis syndrome, characterized by aberrant polyps in the gastrointestinal tract and elevated colorectal cancer risk.31 Mutations in the MH2 domain of SMAD4 lead to increased protein levels and cause Myhre syndrome (Fig. 2C).31 Almost all patients with Myhre syndrome have a dominant missense mutation of the Ile500 amino acid residue mutated into Val, Thr, or Met, with a few patients having a missense mutation of Arg496 to Cys. Most cases occur sporadically, with no occurrence of the disease in other family members, suggesting that the mutations are derived de novo.30,104 Cultured dermal fibroblasts from patients with Myhre syndrome show increased levels of SMAD4 compared with matched control cells. Because the mutated residues are located close to the ubiquitination site of SMAD4, it is possible that the mutated SMAD4 is more stable than the wild-type protein, leading to accumulation of mutant SMAD4. Myhre syndrome fibroblasts show an eight-fold increase in pSMAD2/3 levels and an 11-fold increase in pSMAD1/5/8 levels compared with control cells.31 Evidence for increased mRNA levels of downstream TGF-β target genes suggests that the stabilizing SMAD4 mutations can indeed translate into elevated TGF-β and BMP signaling.31 Transmission electron microscopy showed ultrastructural abnormalities in the ECM of skin tissue from patients with Myhre syndrome. Elastic fibers were fragmented, with aberrant globular elastin deposits, and collagen fibers were more densely packed.30 In addition, Myhre syndrome fibroblasts showed decreased FBN1 microfibril deposition, which is counterintuitive, since elevated TGF-β signaling typically augments ECM gene synthesis and ECM deposition. When Myhre syndrome fibroblasts were treated with losartan, an angiotensin II type 1 receptor antagonist that can disrupt TGF-β signaling, the length, density, and branching of the FBN1 microfibril network were all improved.104 Whether such an improvement can be replicated in appropriate mouse models for Myhre syndrome or in affected individuals has to be investigated.
Conclusions and outlook
Acromelic dysplasias are a fascinating group of rare musculoskeletal disorders in many respects. Studying their underlying molecular pathogenic mechanisms will answer important questions about the biological functions of the proteins that are mutated in acromelic dysplasias and will contribute to the understanding of the fundamental role of the ECM in regulating development and homeostasis of bone, muscle, and skin. Since acromelic dysplasias share their musculoskeletal phenotypes, FBN1, ADAMTS10, ADAMTS17, ADAMTSL2, LTBP2, LTBP3, and SMAD4 are predicted to participate in a common biological pathway in musculoskeletal development and homeostasis (Fig. 3). Because dominant mutations in FBN1 can cause most of the acromelic dysplasias, FBN1 microfibrils very likely play a central role in that pathway. Accordingly, the disease-specific impairment of organs and tissues outside of the musculoskeletal system could then be explained by the tissue-specific functions of ADAMTS10, ADAMTS17, ADAMTSL2, LTBP2, LTBP3, or SMAD4, or combinations thereof. For example, cooperation of ADAMTS10, ADAMTS17, and LTBP2 with FBN1 in the formation and homeostasis of the ciliary zonule could explain why individuals with WMS develop ectopia lentis as a characteristic sign.46,82,85,88 Myhre syndrome, on the other hand, is caused by mutations in SMAD4, a central component of the TGF-β and BMP signaling pathway and, therefore, shows the broadest spectrum of phenotypes.30 TGF-β and BMP signaling is downstream of other ECM genes mutated in acromelic dysplasia, which have been shown or are predicted to be involved to some degree in regulating extracellular TGF-β and BMP bioavailability.105–107
Figure 3.
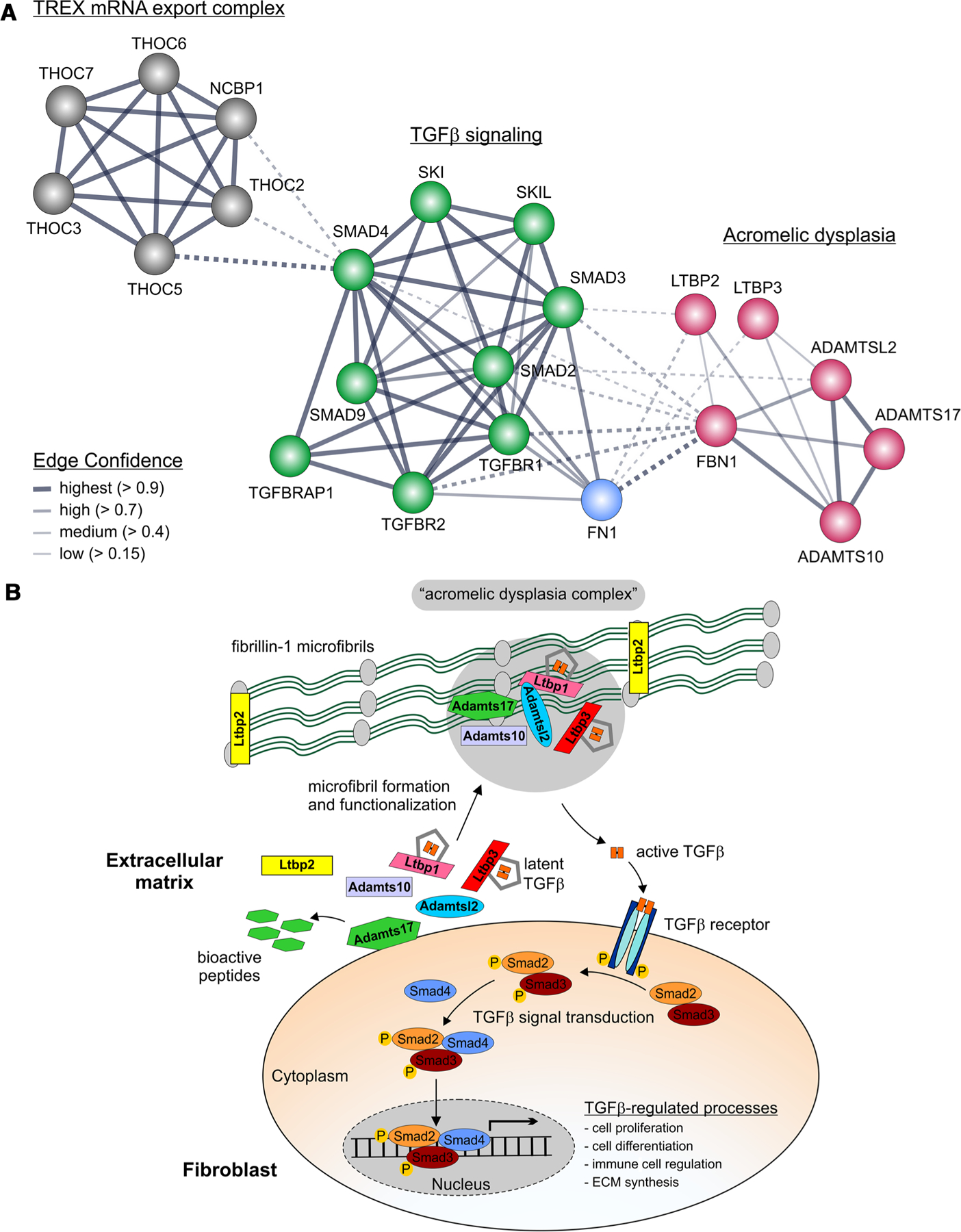
Suggested network of proteins involved in acromelic dysplasias. (A) Gene network surrounding the genes affected in acromelic dysplasias. The genes encoding ECM proteins cluster together and are linked to the TGF-β signaling cluster, which is further connected to a nuclear mRNA export cluster through SMAD4. The edge confidence is indicated by the line thickness. Dashed lines represent connections between clusters. The network was generated using the String database with subsequent Markov clustering.113 (B) The proposed “acromelic dysplasia complex.” Some of the protein–protein interactions are hypothetical, while other interactions were shown experimentally. The common musculoskeletal signs and symptoms in acromelic dysplasias suggest that such a complex could form in these tissues but may lack some of its components, such as ADAMTSL2 in other tisues, for example, the ciliary zonule, which is not affected in GD. ADAMTS10, ADAMTS17, and ADAMTSL2 may cluster at the same region on FBN1 microfibrils, which would explain why mutations in FBN1 can cause AD, GD, and WMS.36 LTBP1 binds to ADAMTSL2 and FBN1 microfibrils. However, no mutations in LTBP1 have been described that cause acromelic dysplasias. We speculate that perhaps an interaction between ADAMTSL2 and LTBP3 is physiologically relevant, based on human genetics and the fact that the LTBP1 domains that bind to ADAMTSL2 show an identical organization in LTBP3.
Many connective tissue disorders, including acromelic dysplasias, can exist on a spectrum of phenotypic severity. However, direct genotype–phenotype correlations for individual acromelic dysplasia mutations are almost impossible, primarily due to the small number of affected individuals that are reported in the literature for each individual acromelic dysplasia. However, in WMS caused by mutations in ADAMTS10 and FBN1, cardiac and joint involvement are more frequent when compared with WMS4 caused by mutations in ADAMTS17.21 In the most extreme case, mutations in the same exons of FBN1 can cause AD, GD, or WMS, as well as Marfan syndrome, a tall stature disorder, which shows the opposite musculoskeletal features of acromelic dysplasia, that is, long bone overgrowth, muscle hypoplasia, and joint laxity.12,78 So far, it is unclear how this can be explained mechanistically, since mutations in adjacent amino acid residues in the FBN1 TB5 domain can result in the distinct and “opposite” disorders of Marfan syndrome and AD or GD (Fig. 2A).12,33 Part of the answer to this question may be the involvement of specific modifier genes, differential expression of the nonmutated versus the wild-type FBN1 allele, or, somewhat less likely, mutation-specific structural and functional alterations in the FBN1 TB5 domain.73,108–110 To our knowledge, only one amino acid residue was mutated in Marfan syndrome and one of the acromelic dysplasias. However, the mutation causing Marfan syndrome is listed in the UMD-FBN1 mutation database, but is so far unpublished.
An unresolved question in understanding the pathogenic mechanisms of acromelic dysplasias revolves around the involvement of alterations in TGF-β or BMP signaling: Is dysregulation of TGF-β or BMP signaling a direct pathogenic driver of disease progression in acromelic dysplasias or are these alterations a consequence of an indiscriminate, default attempt at repairing aberrant ECM by tissue-resident cells?103 TGF-β signaling is elevated in Marfan syndrome (tall stature syndrome) and AD (short stature syndrome) because of FBN1 or ADAMTSL2 mutations, making it somewhat unlikely that elevated canonical TGF-β signaling would drive both bone elongation and bone shortening.8,12,111 In addition, in AD and GD caused by LTBP3 mutations, TGF-β signaling was not elevated.14 Furthermore, two mouse models for GD, one where Adamtsl2 was deleted globally and the other in which Adamtsl2 was deleted in cartilage, showed alterations in TGF-β signaling.37,38 However, treatment with TGF-β–neutralizing antibody could not rescue the severe lung phenotype in the global Adamtsl2 knockout, and TGF-β inhibition to rescue the short limb phenotype in ADAMTSL2-deficient cartilage was not reported. The Adamts17 knockout mouse model, on the other hand, showed alterations in canonical BMP signaling in chondrocytes, but rescue of the phenotype using BMP inhibitors was not attempted.45 Despite the observed alterations in TGF-β and BMP signaling in AD, GD, and WMS, a direct involvement of TGF-β or BMP signaling as the primary pathogenic driver in acromelic dysplasias needs to be established using a more rigorous experimental approach, for example, by performing rescue experiments with inhibitors of TGF-β or BMP signaling in the existing mouse models.
From the human genetic data, it is abundantly clear that FBN1 microfibrils appear to play a central role in the pathogenesis of acromelic dysplasias by acting as an ECM scaffold and by controlling extracellular TGF-β and BMP bioavailability.63 One possibility is that FBN1 microfibrils serve as an ECM hub that brings ADAMTS and LTBPs in close proximity to form a supramolecular “acromelic dysplasia complex” (Fig. 3B).36 The following observations support the existence of such a complex. FBN1 deposition in patient-derived fibroblasts from individuals with AD, GD, and WMS was disrupted and elevated TGF-β signaling was observed.12,89 It was further demonstrated that ADAMTS10, ADAMTS17, and ADAMTSL2 colocalize with FBN1 microfibrils in cell culture and/or in vivo.37,64,85,88 ADAMTSL2 can directly bind to LTBP1, a major carrier of latent TGF-β.37, 64 In addition, LTBP2 binds to FBN1 and LTBP3 requires FBN1 microfibrils for deposition in the ECM.14,46,94 Collectively, these data indicate that all these proteins could be forming a complex on FBN1 microfibrils in the ECM. However, only very few examples have been described where complex formation of more than two of these proteins has been tested. Most notably, it was shown that ADAMTSL2, ADAMTSL3, and ADAMTS10 can bind to a similar region on FBN1 microfibrils, implicating a scaffolding function for FBN1 microfibrils for two of the four proteins altered in WMS.36 It remains to be established if all of the acromelic dysplasia ECM proteins can be localized in one supramolecular complex in musculoskeletal tissue or if a linear pathway involving these proteins or smaller sub-complexes exist. Interestingly, fibroblasts derived from patients with Myhre syndrome due to SMAD4 mutations also showed altered ECM deposition in cell culture.104 We propose a network where the acromelic dysplasia protein complex in the ECM and tissue-resident cells are in constant communication through TGF-β and BMP signaling to relay information and regulate responses to microenvironmental changes, which may involve positive or negative feedback to modulate ECM formation.
In summary, by identifying the molecular protein networks in the ECM and by deciphering how mutations in FBN1 and the other proteins compromised in acromelic dysplasias contribute to the pathogenesis, we may ultimately understand why these disorders share their musculoskeletal phenotypes but also have distinct phenotypes. It could be that all of the proteins compromised in acromelic dysplasia work together and are required in musculoskeletal tissues, but only a subset of these proteins is required for the formation and homeostasis of the additionally affected tissues in the respective individual acromelic dysplasias. It is also quite possible that we have not yet discovered all the genes involved in each of these individual disorders, which may lead to a more advanced and nuanced clinical and molecular understanding of acromelic dysplasias in the future.
Acknowledgments
Research in the Hubmacher laboratory is supported by the NIH/NIAMS (R01AR070748), the Ines Mandl Research Foundation, and the Leni & Peter W. May Department for Orthopedics at the Icahn School of Medicine at Mount Sinai.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Halper J & Kjaer M 2014. Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv. Exp. Med. Biol 802: 31–47. [DOI] [PubMed] [Google Scholar]
- 2.Taye N, Karoulias SZ & Hubmacher D 2020. The “other” 15–40%: the role of non-collagenous extracellular matrix proteins and minor collagens in tendon. J. Orthop. Res 38: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez F & Rifkin DB 2009. Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr. Opin. Cell Biol 21: 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iozzo RV & Gubbiotti MA 2018. Extracellular matrix: the driving force of mammalian diseases. Matrix Biol 71–72: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanakker O, Callewaert B, Malfait F, et al. 2015. The genetics of soft connective tissue disorders. Annu. Rev. Genomics Hum. Genet 16: 229–255. [DOI] [PubMed] [Google Scholar]
- 6.Hubmacher D & Apte SS 2013. The biology of the extracellular matrix: novel insights. Curr. Opin. Rheumatol 25: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callewaert B, Malfait F, Loeys B, et al. 2008. Ehlers–Danlos syndromes and Marfan syndrome. Best Pract. Res. Clin. Rheumatol 22: 165–189. [DOI] [PubMed] [Google Scholar]
- 8.Ramirez F & Dietz HC 2007. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr. Opin. Genet. Dev 17: 252–258. [DOI] [PubMed] [Google Scholar]
- 9.Le Goff C & Cormier-Daire V 2009. Genetic and molecular aspects of acromelic dysplasia. Pediatr. Endocrinol. Rev 6: 418–423. [PubMed] [Google Scholar]
- 10.Klimova B, Storek M, Valis M, et al. 2017. Global view on rare diseases: a mini review. Curr. Med. Chem 24: 3153–3158. [DOI] [PubMed] [Google Scholar]
- 11.Sakai LY & Keene DR 2019. Fibrillin protein pleiotropy: acromelic dysplasias. Matrix Biol 80: 6–13. [DOI] [PubMed] [Google Scholar]
- 12.Le Goff C, Mahaut C, Wang LW, et al. 2011. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet 89: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faivre L, Le Merrer M, Baumann C, et al. 2001. Acromicric dysplasia: long term outcome and evidence of autosomal dominant inheritance. J. Med. Genet 38: 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McInerney-Leo AM, Le Goff C, Leo PJ, et al. 2016. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J. Med. Genet 53: 457–464. [DOI] [PubMed] [Google Scholar]
- 15.Klein C, Le Goff C, Topouchian V, et al. 2014. Orthopedics management of acromicric dysplasia: follow up of nine patients. Am. J. Med. Genet. PartA 164A: 331–337. [DOI] [PubMed] [Google Scholar]
- 16.Jin HS, Song HY, Cho SY, et al. 2017. Acromicric dysplasia caused by a novel heterozygous mutation of FBN1 and effects of growth hormone treatment. Ann. Lab. Med 37: 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng SW, Luk HM, Chu YWY, et al. 2018. A report of three families with FBN1-related acromelic dysplasias and review of literature for genotype–phenotype correlation in geleophysic dysplasia. Eur. J. Med. Genet 61: 219–224. [DOI] [PubMed] [Google Scholar]
- 18.Marzin P & Cormier-Daire V 1993. Geleophysic dysplasia. In GeneReviews((R)) Adam MP, Ardinger HH & Pagon RA, Eds. Seattle, WA: University of Washington. [PubMed] [Google Scholar]
- 19.Li D, Dong H, Zheng H, et al. 2017. A Chinese boy with geleophysic dysplasia caused by compound heterozygous mutations in ADAMTSL2. Eur. J. Med. Genet 60: 685–689. [DOI] [PubMed] [Google Scholar]
- 20.Globa E, Zelinska N & Dauber A 2018. The clinical cases of geleophysic dysplasia: one gene, different phenotypes. Case Rep. Endocrinol 2018: 8212417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales J, Al-Sharif L, Khalil DS, et al. 2009. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet 85: 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faivre L, Gorlin RJ, Wirtz MK, et al. 2003. In frame fibrillin-1 gene deletion in autosomal dominant Weill–Marchesani syndrome. J. Med. Genet 40: 34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evereklioglu C, Hepsen IF & Er H 1999. Weill–Marchesani syndrome in three generations. Eye (Lond.) 13(Pt 6): 773–777. [DOI] [PubMed] [Google Scholar]
- 24.Faivre L, Dollfus H, Lyonnet S, et al. 2003. Clinical homogeneity and genetic heterogeneity in Weill–Marchesani syndrome. Am. J. Med. Genet. Part A 123A: 204–207. [DOI] [PubMed] [Google Scholar]
- 25.Kutz WE, Wang LW, Dagoneau N, et al. 2008. Functional analysis of an ADAMTS10 signal peptide mutation in Weill–Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum. Mutat 29: 1425–1434. [DOI] [PubMed] [Google Scholar]
- 26.Dagoneau N, Benoist-Lasselin C, Huber C, et al. 2004. ADAMTS10 mutations in autosomal recessive Weill–Marchesani syndrome. Am. J. Hum. Genet 75: 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, et al. 2012. LTBP2 mutations cause Weill–Marchesani and Weill–Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum. Mutat 33: 1182–1187. [DOI] [PubMed] [Google Scholar]
- 28.Shah MH, Bhat V, Shetty JS, et al. 2014. Whole exome sequencing identifies a novel splice-site mutation in ADAMTS17 in an Indian family with Weill–Marchesani syndrome. Mol. Vis 20: 790–796. [PMC free article] [PubMed] [Google Scholar]
- 29.Myhre SA, Ruvalcaba RH & Graham CB 1981. A new growth deficiency syndrome. Clin. Genet 20: 1–5. [DOI] [PubMed] [Google Scholar]
- 30.Meerschaut I, Beyens A, Steyaert W, et al. 2019. Myhre syndrome: a first familial recurrence and broadening of the phenotypic spectrum. Am. J. Med. Genet. PartA 179: 2494–2499. [DOI] [PubMed] [Google Scholar]
- 31.Le Goff C, Mahaut C, Abhyankar A, et al. 2011. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat. Genet 44: 85–88. [DOI] [PubMed] [Google Scholar]
- 32.Caputo V, Cianetti L, Niceta M, et al. 2012. A restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre syndrome. Am. J. Hum. Genet 90: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramirez F, Caescu C, Wondimu E, et al. 2018. Marfan syndrome; a connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biol 71–72: 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook JR, Clayton NP, Carta L, et al. 2015. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol 35: 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones W, Rodriguez J & Bassnett S 2019. Targeted deletion of fibrillin-1 in the mouse eye results in ectopia lentis and other ocular phenotypes associated with Marfan syndrome. Dis. Model. Mech 12: dmm037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sengle G, Tsutsui K, Keene DR, et al. 2012. Microenvironmental regulation by fibrillin-1. PLoS Genet 8: e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubmacher D, Wang LW, Mecham RP, et al. 2015. Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia–a novel mouse model providing insights into geleophysic dysplasia. Dis. Model. Mech 8: 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delhon L, Mahaut C, Goudin N, et al. 2019. Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J 33: 2707–2718. [DOI] [PubMed] [Google Scholar]
- 39.Hubmacher D, Taye N, Balic Z, et al. 2019. Limb- and tendon-specific Adamtsl2 deletion identifies a role for ADAMTSL2 in tendon growth in a mouse model for geleophysic dysplasia. Matrix Biol 82: 38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bader HL, Ruhe AL, Wang LW, et al. 2010. An ADAMTSL2 founder mutation causes Musladin–Lueke syndrome, a heritable disorder of beagle dogs, featuring stiff skin and joint contractures. PLoS One 5: e12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Musladin JM, Musladin AM & Lueke A 1998. The New Beagle: A Dog for All Seasons Macmillian General. [Google Scholar]
- 42.Allali S, Le Goff C, Pressac-Diebold I, et al. 2011. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J. Med. Genet 48: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang LW, Kutz WE, Mead TJ, et al. 2019. Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage. Matrix Biol 77: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mularczyk EJ, Singh M, Godwin ARF, et al. 2018. ADAMTS10-mediated tissue disruption in Weill–Marchesani syndrome. Hum. Mol. Genet 27: 3675–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oichi T, Taniguchi Y, Soma K, et al. 2019. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell. Mol. Life Sci 10.1007/s00018-019-03188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue T, Ohbayashi T, Fujikawa Y, et al. 2014. Latent TGF-β binding protein-2 is essential for the development of ciliary zonule microfibrils. Hum. Mol. Genet 23: 5672–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shipley JM, Mecham RP, Maus E, et al. 2000. Developmental expression of latent transforming growth factor beta binding protein 2 and its requirement early in mouse development. Mol. Cell. Biol 20: 4879–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujikawa Y, Yoshida H, Inoue T, et al. 2017. Latent TGF-β binding protein 2 and 4 have essential overlapping functions in microfibril development. Sci. Rep 7: 43714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dabovic B, Levasseur R, Zambuto L, et al. 2005. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone 37: 25–31. [DOI] [PubMed] [Google Scholar]
- 50.Dabovic B, Chen Y, Colarossi C, et al. 2002. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J. Endocrinol 175: 129–141. [DOI] [PubMed] [Google Scholar]
- 51.Dabovic B, Chen Y, Colarossi C, et al. 2002. Bone abnormalities in latent TGF-β binding protein (Ltbp)-3-null mice indicate a role for Ltbp-3 in modulating TGF-β bioavailability. J. Cell Biol 156: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colarossi C, Chen Y, Obata H, et al. 2005. Lung alveolar septation defects in Ltbp-3-null mice. Am. J. Pathol 167: 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, Dabovic B, Colarossi C, et al. 2003. Growth retardation as well as spleen and thymus involution in latent TGF-beta binding protein (Ltbp)-3 null mice. J. Cell. Physiol 196: 319–325. [DOI] [PubMed] [Google Scholar]
- 54.Yang G & Yang X 2010. Smad4-mediated TGF-beta signaling in tumorigenesis. Int. J. Biol. Sci 6: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X, Li C, Xu X, et al. 1998. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA 95: 3667–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sirard C, de la Pompa JL, Elia A, et al. 1998. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 12: 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takaku K, Miyoshi H, Matsunaga A, et al. 1999. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer Res 59: 6113–6117. [PubMed] [Google Scholar]
- 58.Yan J, Li J, Hu J, et al. 2018. Smad4 deficiency impairs chondrocyte hypertrophy via the Runx2 transcription factor in mouse skeletal development. J. Biol. Chem 293: 9162–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitaker AT, Berthet E, Cantu A, et al. 2017. Smad4 regulates growth plate matrix production and chondrocyte polarity. Biol. Open 6: 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Tan X, Li W, et al. 2005. Smad4 is required for the normal organization of the cartilage growth plate. Dev. Biol 284: 311–322. [DOI] [PubMed] [Google Scholar]
- 61.Tan X, Weng T, Zhang J, et al. 2007. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci 120: 2162–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Kawai K, Khashabi S, et al. 2010. Inactivation of Smad4 leads to impaired ocular development and cataract formation. Biochem. Biophys. Res. Commun 400: 476–482. [DOI] [PubMed] [Google Scholar]
- 63.Hubmacher D & Apte SS 2015. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol 47: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le Goff C, Morice-Picard F, Dagoneau N, et al. 2008. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet 40: 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomson J, Singh M, Eckersley A, et al. 2019. Fibrillin microfibrils and elastic fibre proteins: functional interactions and extracellular regulation of growth factors. Semin. Cell Dev. Biol 89: 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corson GM, Charbonneau NL, Keene DR, et al. 2004. Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics 83: 461–472. [DOI] [PubMed] [Google Scholar]
- 67.Eckersley A, Mellody KT, Pilkington S, et al. 2018. Structural and compositional diversity of fibrillin microfibrils in human tissues. J. Biol. Chem 293: 5117–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Godwin ARF, Singh M, Lockhart-Cairns MP, et al. 2019. The role of fibrillin and microfibril binding proteins in elastin and elastic fibre assembly. Matrix Biol 84: 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Y, Tu Y, De Maria A, et al. 2013. Development, composition, and structural arrangements of the ciliary zonule of the mouse. Invest. Ophthalmol. Vis. Sci 54: 2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Maria A, Wilmarth PA, David LL, et al. 2017. Proteomic analysis of the bovine and human ciliary zonule. Invest. Ophthalmol. Vis. Sci 58: 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinson PN, Arteaga-Solis E, Baldock C, et al. 2006. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet 43: 769–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakai LY, Keene DR, Renard M, et al. 2016. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene 591: 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franken R, Teixido-Tura G, Brion M, et al. 2017. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart 103: 1795–1799. [DOI] [PubMed] [Google Scholar]
- 74.Maeda J, Kosaki K, Shiono J, et al. 2016. Variable severity of cardiovascular phenotypes in patients with an early-onset form of Marfan syndrome harboring FBN1 mutations in exons 24–32. Heart Vessels 31: 1717–1723. [DOI] [PubMed] [Google Scholar]
- 75.Faivre L, Collod-Beroud G, Callewaert B, et al. 2009. Clinical and mutation-type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. Eur. J. Hum. Genet 17: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asgari S, Luo Y, Akbari A, et al. 2020. A positively selected FBN1 missense variant reduces height in Peruvian individuals. Nature 582: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuo CL, Isogai Z, Keene DR, et al. 2007. Effects of fibrillin-1 degradation on microfibril ultrastructure. J. Biol. Chem 282: 4007–4020. [DOI] [PubMed] [Google Scholar]
- 78.Cecchi A, Ogawa N, Martinez HR, et al. 2013. Missense mutations in FBN1 exons 41 and 42 cause Weill–Marchesani syndrome with thoracic aortic disease and Marfan syndrome. Am. J. Med. Genet. PartA 161A: 2305–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cain SA, McGovern A, Baldwin AK, et al. 2012. Fibrillin-1 mutations causing Weill–Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS One 7: e48634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tiedemann K, Batge B, Muller PK, et al. 2001. Interactions of fibrillin-1 with heparin/heparan sulfate, implications for microfibrillar assembly. J. Biol. Chem 276: 36035–36042. [DOI] [PubMed] [Google Scholar]
- 81.Mead TJ & Apte SS 2018. ADAMTS proteins in human disorders. Matrix Biol 71–72: 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karoulias SZ, Taye N, Stanley S, et al. 2020. The ADAMTS/fibrillin connection: insights into the biological functions of ADAMTS10 and ADAMTS17 and their respective sister proteases. Biomolecules 10. 10.3390/biom10040596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Somerville RP, Jungers KA & Apte SS 2004. Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation. J. Biol. Chem 279: 51208–51217. [DOI] [PubMed] [Google Scholar]
- 84.Hubmacher D & Apte SS 2011. Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function. Cell. Mol. Life Sci 68: 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kutz WE, Wang LW, Bader HL, et al. 2011. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J. Biol. Chem 286: 17156–17167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Steinkellner H, Etzler J, Gogoll L, et al. 2015. Identification and molecular characterisation of a homozygous missense mutation in the ADAMTS10 gene in a patient with Weill–Marchesani syndrome. Eur. J. Hum. Genet 23: 1186–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Evans DR, Green JS, Fahiminiya S, et al. 2020. A novel pathogenic missense ADAMTS17 variant that impairs secretion causes Weill–Marchesani syndrome with variably dysmorphic hand features. Sci. Rep 10: 10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hubmacher D, Schneider M, Berardinelli SJ, et al. 2017. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci. Rep 7: 41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karoulias SZ, Beyens A, Balic Z, et al. 2020. A novel ADAMTS17 variant that causes Weill–Marchesani syndrome4 alters fibrillin-1 and collagen typeI deposition in the extracellular matrix. Matrix Biol 88: 1–18. [DOI] [PubMed] [Google Scholar]
- 90.Apte SS 2009. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J. Biol. Chem 284: 31493–31497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Piccolo P, Sabatino V, Mithbaokar P, et al. 2019. Geleophysic dysplasia: novel missense variants and insights into ADAMTSL2 intracellular trafficking. Mol. Genet. Metab. Rep 21: 100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Robertson IB, Horiguchi M, Zilberberg L, et al. 2015. Latent TGF-β-binding proteins. Matrix Biol 47: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saharinen J, Taipale J & Keski-Oja J 1996. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J 15: 245–253. [PMC free article] [PubMed] [Google Scholar]
- 94.Zilberberg L, Todorovic V, Dabovic B, et al. 2012. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J. Cell. Physiol 227: 3828–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vehvilainen P, Hyytiainen M & Keski-Oja J 2009. Matrix association of latent TGF-beta binding protein-2 (LTBP-2) is dependent on fibrillin-1. J. Cell. Physiol 221: 586–593. [DOI] [PubMed] [Google Scholar]
- 96.Rifkin DB 2005. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J. Biol. Chem 280: 7409–7412. [DOI] [PubMed] [Google Scholar]
- 97.Sideek MA, Menz C, Parsi MK, et al. 2014. LTBP-2 competes with tropoelastin for binding to fibulin-5 and heparin, and is a negative modulator of elastinogenesis. Matrix Biol 34: 114–123. [DOI] [PubMed] [Google Scholar]
- 98.Menz C, Parsi MK, Adams JR, et al. 2015. LTBP-2 has a single high-affinity binding site for FGF-2 and blocks FGF2-induced cell proliferation. PLoS One 10: e0135577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rauf B, Irum B, Khan SY, et al. 2020. Novel mutations in LTBP2 identified in familial cases of primary congenital glaucoma. Mol. Vis 26: 14–25. [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Y, Dabovic B, Annes JP, et al. 2002. Latent TGF-beta binding protein-3 (LTBP-3) requires binding to TGF-beta for secretion. FEBS Lett 517: 277–280. [DOI] [PubMed] [Google Scholar]
- 101.Dugan SL, Temme RT, Olson RA, et al. 2015. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. Part A 167: 1396–1399. [DOI] [PubMed] [Google Scholar]
- 102.Intarak N, Theerapanon T, Thaweesapphithak S, et al. 2019. Genotype–phenotype correlation and expansion of orodental anomalies in LTBP3-related disorders. Mol. Genet. Genomics 294: 773–787. [DOI] [PubMed] [Google Scholar]
- 103.Horiguchi M, Ota M & Rifkin DB 2012. Matrix control of transforming growth factor-β function. J. Biochem 152: 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Piccolo P, Mithbaokar P, Sabatino V, et al. 2014. SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. Eur. J. Hum. Genet 22: 988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ramirez F & Sakai LY 2010. Biogenesis and function of fibrillin assemblies. Cell Tissue Res 339: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wohl AP, Troilo H, Collins RF, et al. 2016. Extracellular regulation of bone morphogenetic protein activity by the microfibril component fibrillin-1. J. Biol. Chem 291: 12732–12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neptune ER, Frischmeyer PA, Arking DE, et al. 2003. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet 33: 407–411. [DOI] [PubMed] [Google Scholar]
- 108.Hutchinson S, Furger A, Halliday D, et al. 2003. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum. Mol. Genet 12: 2269–2276. [DOI] [PubMed] [Google Scholar]
- 109.Aubart M, Gross MS, Hanna N, et al. 2015. The clinical presentation of Marfan syndrome is modulated by expression of wild-type FBN1 allele. Hum. Mol. Genet 24: 2764–2770. [DOI] [PubMed] [Google Scholar]
- 110.Aubart M, Gazal S, Arnaud P, et al. 2018. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet 26: 1759–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Habashi JP, Judge DP, Holm TM, et al. 2006. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312: 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Newell K, Smith W, Ghoshhajra B, et al. 2017. Cervical artery dissection expands the cardiovascular phenotype in FBN1-related Weill–Marchesani syndrome. Am. J. Med. Genet. Part A 173: 2551–2556. [DOI] [PubMed] [Google Scholar]
- 113.Szklarczyk D, Gable AL, Lyon D, et al. 2019. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47: D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]