

Summary
Most cancer deaths are due to tumor metastasis rather than the primary tumor. Metastasis is a highly complex and dynamic process that requires orchestration of signaling between the tumor, its local environment, distant tissue sites, and immune system. Animal models of cancer metastasis provide the necessary systemic environment but lack control over factors that regulate cancer progression and often do not recapitulate the properties of human cancers. Bioengineered “organs-on-a-chip” that incorporate the primary tumor, metastatic tissue targets, and microfluidic perfusion are now emerging as quantitative human models of tumor metastasis. The ability of these systems to model tumor metastasis in individualized, patient-specific settings makes them uniquely suitable for studies of cancer biology and developmental testing of new treatments. In this review, we focus on human multi-organ platforms that incorporate circulating and tissue-resident immune cells in studies of tumor metastasis.
Subject areas: components of the immune system, bioengineering, cancer, biochemical assay
Graphical abstract
Components of the immune system; bioengineering; cancer; biochemical assay
Tumor metastasis
Despite enormous advances in our understanding of tumor progression, cancer incidence, mortality, and cost of treatment continue to increase (Bray et al., 2018). Recent breakthroughs in cancer modeling and therapy have been primarily directed at early intervention and eradication of the primary tumor, which has limited success in the presence of metastatic disease. Although tumor metastasis accounts for 66.7% of the mortality related to solid tumors (Dillekas et al., 2019), few drugs are designed to specifically target metastatic spreading. This unmet need is due, at least in part, to the lack of predictive human models that recapitulate physiological metastasis and can be used for mechanistic studies of metastatic progression and evaluation of drug candidates that can prevent or treat metastasis in patients.
Metastasis develops as cancer cells are leaving the primary cancer site to find a more suitable niche in the body. This process involves cascades of multidirectional signaling between tumor cells, stromal cells, and immune cells within the primary and secondary tumor sites. Tumor cells co-opt the immune system, enabling escape from tumoricidal immune responses, and alter the primary tumor environment (TME) to promote vascularization (Kitamura et al., 2015). Collectively, these changes facilitate tumor cell invasion through the stroma and migration to nearby vessels, the first step of the metastatic cascade (Figure 1A). Tumor cells then intravasate, via blood or lymphatic vessels, into the circulation, and survive as they traffic to microvessels at distant, metastatic sites that have been primed for colonization. Arrested tumor cells subsequently extravasate into the tissue and proliferate to form a secondary tumor. Two theories have been proposed to explain organ-specific homing and selection of metastatic sites: (1) Paget's “seed and soil” theory, first described in 1889, which posits that circulating cancer cells preferentially “seed” distal organs where the microenvironment (“soil”) favors their survival (Langley and Fidler, 2011); and (2) Ewing's “flow and filter” theory, described in 1929, which posits that circulating tumor cells are mechanically driven to metastatic sites by blood flow patterns that are governed by capillary bed sizes and adhesion. Recent work has demonstrated contributions of both mechanisms to the establishment of metastatic sites (Font-Clos et al., 2020).
Figure 1.

Overview of tumor metastasis
(A) The progression from primary to secondary tumor sites involves (1) remodeling of the primary tumor microenvironment to support (2) intravasation of circulating cancer cells into vascular circulation. Steps (1) and (2) work in conjunction with step (3) inter-organ communication between the primary tumor, bone marrow, and immune organs, to prime the pre-metastatic niche in distant target tissues. The pre-metastatic niche facilitates (4) extravasation of cancer cells from circulation into a tissue bed, and (5) targeted cell engraftment in the secondary metastatic site.
(B) Bioengineered systems can be leveraged to deconstruct each of the steps in metastatic progression and improve our understanding of the dynamic interplay between the primary tumor, targeted tissue sites, and immune cells and organs.
Created with BioRender.com.
Characterizing metastatic progression remains a challenge, both in clinical settings and in the existing tumor models. The most common approaches to investigating tumor progression involve animal models, in vitro culture of cancer cells, and patient-derived organoids and organ-on-a-chip models, with each system having limitations. Animal models offer superior complexity and systemic interactions, but do not necessarily recapitulate human physiology and only rarely develop clinically seen skeletal metastasis (De La Rochere et al., 2018). Compared with tumor cells in patients, cancer cell lines show significant mutational and transcriptional drifts, abnormal ploidy, and loss of heterogeneity when expanded in vitro (Ben-David et al., 2019). Cell lines also lack the pathophysiological milieu and growth conditions of the original tumor, including the three-dimensional (3D) environment with cancer-specific extracellular matrix (ECM), stromal cells, vascular and immune cells, and the associated molecular and physical signals. Over the last decade, bioengineering is increasingly successful in providing physiologically relevant models of cancer growth. However, some of the critical aspects of cancer pathophysiology (i.e., paracrine and endocrine interactions) needed to model the full complexity of cancer progression are still missing in most models.
Although our understanding of tumor metastasis is constantly evolving, we have yet to recapitulate the interactions underlying the systemic effects of tumors on the human body. Despite the growing appreciation for immune cells as key orchestrators of metastasis, only a few models capture the contributions of these cells—a fundamental limitation that must be addressed to develop experimental models of metastasis mimetic of human disease. Bioengineered models provide unique opportunities to decouple the contributions of microenvironmental and systemic components in a controlled, physiologically relevant manner, and thereby advance precision medicine approaches to cancer treatment (Figure 1B). Engineering a predictive body-on-a-chip model of metastasis will depend on the inclusion of (1) primary tissues that are targeted by metastasis, (2) factors regulating tumor cell progression through the metastatic cascade, and (3) immune organs and cells.
We discuss here the recent advances in modeling primary tumors, inclusion of immune cells into these models, and the multi-organ-on-a-chip (OOC) systems designed to recapitulate the progression and organ specificity of tumor metastasis. We focus on areas of cancer metastasis that are critical to mechanistically understand therapeutic interventions and are difficult to experimentally model. Specifically, we highlight how in vitro bioengineered multi-tissue models can be leveraged to recapitulate human immune-oncology models of metastasis for predictive disease modeling and development of treatment regimens.
Tumor microenvironment
Understanding the tumor microenvironment, the tumor-induced changes in the immune landscape, and the events leading to pre-metastatic niche formation is critical for developing effective immunotherapies that can improve patient outcomes. There is growing evidence that, similar to injury, tumor formation systemically alters the body, even before metastasis occurs (Allen et al., 2020). Currently, it is poorly understood how the systemic perturbations, which appear distinct from those occurring at the primary tumor site (Cacho-Diaz et al., 2020; Klemm et al., 2020), contribute to tumor progression. Here, we present a brief overview of the changes occurring locally within the primary tumor environment and peripherally at pre-metastatic niches. Comprehensive reviews on these topics have been provided by others (Gupta and Massague, 2006; Hoye and Erler, 2016; Peinado et al., 2017).
Primary tumor microenvironment
Tumors are heterogeneous tissues containing proliferating cancer cells, stromal cells, vasculature, ECM, and tumor-secreted factors. Unlike healthy cells, cancer cells acquire sustained and unlimited proliferative capacity, while evading negative regulators of cell growth and programmed cell death (Hanahan and Weinberg, 2011). Hyperproliferation of cancer cells causes hypoxia, cellular reprogramming, and activation of a pathological angiogenic cascade that renders the tumor with unstable, leaky vasculature. Cancer cells not only alter the local ECM, leading to increased interstitial pressure and tissue stiffness, but also co-opt the immune system and the surrounding stromal cells that further mediate tumor metastasis. The initiation and progression of tumorigenesis are dependent on tumor-promoting signals that result from a loss of microenvironmental regulation at the primary tumor site. Therefore, the primary TME has been a focus of investigation for anti-cancer therapies.
Pre-metastatic niche
Primary tumors actively contribute to metastasis by secreting soluble factors and extracellular vesicles (EVs) that enter the circulation and traffic to distant tissue sites to form a microenvironment conducive for tumor cell engraftment and secondary tumor growth. These future sites of metastasis are referred to as pre-metastatic niches (Figure 1A) and are known to play critical roles in enabling extravasation and colonization of circulating tumor cells (Bissell and Hines, 2011). For instance, stromal expression of the gene encoding periostin, a component of the ECM, is required for colonization of some metastatic cells (Malanchi et al., 2011). Pre-metastatic niches not only provide physical support enabling the tumor cells to anchor and populate the tissue but also activate pro-survival signaling that prevents tumor cell death (Celia-Terrassa and Kang, 2018). Additionally, the pre-metastatic niches maintain tumor cell stemness and protection against immune surveillance, further enhancing tumor cell survival. Vascular sprouting and ECM remodeling within the pre-metastatic niche facilitate tumor cell proliferation and metastatic lesion outgrowth (Celia-Terrassa and Kang, 2018).
A seminal study by Kaplan et al. (2005) demonstrated that tumors secrete high levels of vascular endothelial growth factor (VEGF) and placental growth factor, which subsequently mobilize VEGFR1-expressing hematopoietic progenitor cells (HPCs) in the bone marrow to initiate pre-metastatic niche formation before tumor cell arrival. These VEGFR1-expressing HPCs also express VLA-4, a fibronectin receptor integrin, which enables adhesion of the bone marrow-derived cells to fibronectin-rich pre-metastatic niches—a result of proliferation and activation of fibroblast-like stromal cells in response to the primary tumor. Binding to fibronectin has been shown to enhance expression of matrix metalloproteinases (MMPs), including MMP9, which degrades basement membranes, releasing ECM-bound factors like VEGF that drive changes in the local environment. For example, increased availability of VEGF can trigger the angiogenic switch that drives endothelial cells from a quiescent state to a metastasis-promoting activated state (Bergers et al., 2000).
In addition to soluble factors, tumor-derived EVs are implicated in generating pre-metastatic niches, in part due to the abundance of integrins on their surfaces (Hamidi and Ivaska, 2018). Upon entry into the distant tissue sites, EVs co-localize with ECM-bound tissue-resident cells to remodel the ECM. Tumor-derived EVs can also reprogram bone marrow-derived cells via transfer of tumor-promoting genes that increase their metastatic behavior (Peinado et al., 2012). Recently, EVs released by non-tumor cells, such as recruited bone marrow-derived cells, have been shown to deliver cargo that regulates pre-metastatic niches (Hsu et al., 2020). Distinct, tissue-specific integrin expression profiles have been observed on EVs, suggesting a link between integrin content and organ-specific metastasis (Hoshino et al., 2015). Consequently, EVs can serve both as predictive markers of metastatic sites and as therapeutic targets.
EVs have also been implicated in changes in vasculature. For instance, micro-RNAs in tumor-secreted EVs induce down-regulation of tight junctions expressed by endothelial cells, leading to increased vascular permeability in distant organs and, consequently, metastatic progression (Zhou et al., 2014). Beyond modulating the vascular barrier, EVs also promote vascular sprouting to promote pre-metastatic niche formation (Grange et al., 2011). However, endothelial remodeling is not limited to EVs. Tumor cells have also been shown to express genes that alter vascular functions to promote tumor cell intravasation, circulation, and extravasation (Gupta et al., 2007). These metastatic gene signatures may work in concert with circulating flow forces, which promote adhesion of circulating tumor cells to the endothelium, endothelial remodeling, and preferential metastasis to regions with lower perfusion (Follain et al., 2018). Recent work (Ghouse et al., 2020) further suggests that angiogenic changes can contribute to premetastatic niche formation, via recruited immune cells, before the arrival of tumor cells.
Hypoxia at the primary tumor site is also an established driver of pre-metastatic niche formation. Building upon the findings that bone marrow-derived cells establish pre-metastatic niches, work by Erler et al. demonstrated that lysyl oxidase (LOX), an enzyme secreted by hypoxic tumor cells, is required for recruitment of bone marrow-derived cells to pre-metastatic niches in an MMP-dependent manner (Erler et al., 2009). LOX cross-links collagen IV in basement membrane, leading to enhanced MMP2 activity, recruitment, and invasion of bone marrow-derived and CD11b+ cells. This activity is regulated by hypoxia-inducible factors (HIFs) (Wong et al., 2011), including HIF-1α, which is thought to promote aggressive tumor cell phenotypes associated with metastasis (Yamamoto et al., 2008). As hypoxia increases the formation of metastases, it also directly influences the metastatic sites, with bone marrow being both one of the most hypoxic environments and a major target of metastasis (Hiraga, 2018).
Immune contributions to tumor metastasis
The tissue-resident and bone marrow-derived cells are key contributors to tumor progression, both locally at the primary tumor site and systemically at pre-metastatic and metastatic sites. Of note, the tumor-promoting or tumor-suppressive roles of these cells are also context-dependent, making it both more important and more difficult to develop suitable in vitro models of the early pre-metastatic niches that may exist in patients before diagnosis. To illustrate the complexity of immune contributions to tumor metastasis, we will highlight some representative recent studies. Several excellent reviews on the roles of immune cells in cancer progression have been recently published (Garner and de Visser, 2020; Hinshaw and Shevde, 2019; Kitamura et al., 2015; Müller et al., 2020; Shelton et al., 2020; Thorsson et al., 2018).
Historically, focus on the tumor-immune microenvironment has been restricted to the primary tumor site and is only recently extending to pre-metastatic niches. A recent report by Allen et al. demonstrates that tumors systemically alter and reshape the immune landscape, possibly due to the increased levels of circulating inflammatory factors interleukin-1α (IL-1α) and granulocyte colony-stimulating factor (G-CSF). Strikingly, tumor resection restored the systemic immune landscape, demonstrating the plasticity of the immune system and revealing an important role of the primary tumor in driving these changes (Allen et al., 2020). This study complements earlier work that showed the potential of G-CSF and macrophage colony-stimulating factor (M-CSF) in directing the lineage commitment of HPCs in the bone marrow (Rieger et al., 2009), where hematopoiesis gives rise to all blood cells (Figure 2). Indeed, tumor-derived G-CSF and other inflammatory factors skewed the circulating HPCs toward the myeloid lineage (Wu et al., 2014), leading to accumulation of myeloid-derived suppressor cells that promote tumor progression, including monocytes, macrophages, and neutrophils (Casbon et al., 2015). Many other tumor-derived factors have been implicated in altering bone marrow-derived cells. For instance, tumor-derived IL-1β induces IL-17 production by γδ T cells, facilitating G-CSF-dependent neutrophil expansion and subsequent suppression of anti-tumoral CD8+ T cells (Coffelt et al., 2015). Likewise, tumor-derived CCL2 cells recruit monocytes that differentiate into metastasis-associated phenotypes and suppress CD8+ T cells (Qian et al., 2011). In addition to tumor-derived signals, activated CD4+ T cells produce tumor necrosis factor-alpha, promoting differentiation and accumulation of myeloid-derived suppressor cells (Al Sayed et al., 2019). Regulatory T cells, responsible for maintaining homeostasis, have also been implicated as drivers of metastasis (Tan et al., 2011).
Figure 2.

Bone marrow-derived blood and immune cells
All mature blood and immune cells originate from the hematopoietic stem cell (HSC) in the bone marrow and differentiate into myeloid and lymphoid progenitors, further maturing in the bone marrow or lymphoid organs (thymus, lymph node, spleen). The traditional in vivo lifetime of each of these cell types ranges from hours to years. For example, neutrophils only remain in circulation for a few hours. We list these timelines and required exogenous factors for each cell type, as well as critical feeder ligand expression (such as Notch delta-like ligand 4 in T lymphocytes). Created with BioRender.com.
Collectively, these reports provide compelling evidence that tumors induce systemic immune cell reprogramming, even before their arrival at the pre-metastatic niche, to promote tumor progression and metastasis. Importantly, Felix Hoyer et al. recently demonstrated systemic, tissue-specific immune cell activation in response to local injury (Hoyer et al., 2019), reinforcing the longstanding notion that tumors resemble wounds that fail to heal (Hua and Bergers, 2019). Nevertheless, the systemic immune contributions to tumor growth are complex, dynamic, and remain incompletely understood. Given the inherent patient-to-patient variability of immune responses, coupled with the patient-specific tumor heterogeneity, there is a growing interest in patient-specific approaches to study immune contributions to the progression and suppression of tumor metastasis. Such bioengineered models of cancer would allow assessing the systems-level effects of tumors and enable identifying checkpoints and master regulators of cancer progression in individualized settings.
Engineered models of cancer
For decades, cancer research has relied on the use of monolayer cultures of cancer cells for studies of cancer biology and drug screening, followed by the use of genetically engineered mice or patient-derived xenotransplants (PdX) for better understanding of the systemic aspects of the disease (Figure 3). The development of engineered cancer models has been motivated by the need for re-creating in vitro many aspects of the disease that are otherwise missed in these established in vitro and in vivo models (Figure 3). While some of the oldest 3D tumor models date back to more than 30 years, there have been major advances in introducing bioengineering methodologies into the field of cancer, toward developing increasingly accurate, useful, practical, and complex models for studying metastasis (Sutherland et al., 1986).
Figure 3.

Model systems for studying primary or metastatic cancers
Traditional models to study primary tumors and resected secondary metastatic cells include 2D cell monolayers and animal models, where cells are either implanted in vivo into an immunodeficient mouse or a transgenic mouse line is created with a known mutational defect. Bioengineered models include 3D multi-cellular tissue-engineered models, patient-derived organoids, organ-on-a-chip microfluidic devices, and recently, multi-organ-on-a-chip devices. We show here the overview of each approach and its advantages and limitations in studying cancer. Created with BioRender.com.
Generally speaking, four approaches are currently utilized to model human cancer: (1) cancer cell culture in 3D scaffolds, (2) patient-derived organoids, (3) single organ-on-a-chip systems, and most recently, (4) multi-organ-on-a-chip systems. Among these approaches, the growth of cancer cells as self-assembled spheroids within hydrogels and scaffolds is most commonly used (Sutherland et al., 1986). However, given that metastasis is a dynamic process that involves multiple tissues and relies on the systemic circulation for its spread, microfluidic single- and multi-tissue OOC platforms have been proved to be most useful for studying metastatic progression.
Cancer cell culture in 3D scaffolds
Arguably the most widely published approach using cancer cell lines has been to grow them in natural and synthetic scaffolds providing a regulatory 3D setting for tumor cells. Complex techniques have since been developed that enable the generation of scaffolds with distinct compositions, structures, and mechanical properties. However, static culture of cancer cells in a 3D microenvironment is inherently limited in its ability to model the multiple stages of metastasis. Several recent reviews on engineering the 3D microenvironment have been provided by others (Jiang et al., 2021; Micek et al., 2020).
Patient-derived organoids
Although practical for laboratory research, cancer cell lines fail to recapitulate the heterogeneity of the primary cancer cell populations. The development of patient-derived tumor organoids representing a wide variety of primary cancer types has overcome this limitation. Critically, these tumor organoids have been shown to preserve inter-patient genetic diversity, both for the primary and metastatic tumor sites (Sachs et al., 2018; Vlachogiannis et al., 2018; Weeber et al., 2015). They have also been proved to be successful at modeling previously difficult to study cancer types, such as pancreatic and prostate cancers (Boj et al., 2015; Gao et al., 2014). CRISPR-Cas9-mediated genetic engineering of healthy epithelial tissue organoids has allowed for the investigation of driver oncogenes and tumor suppressors in tumorigenesis (Matano et al., 2015). Perhaps of most relevance to metastasis and improving the poor clinical outcomes associated with it, the development of patient-derived organoids has the potential for the personalization of cancer treatment. For this reason, there has been wide interest in the use of tumor organoid models for drug sensitivity testing (Driehuis et al., 2020). Unfortunately, most advances in the organoid field were with carcinomas (epithelial cell-derived cancers), given their intrinsic ability to self-assemble into micro-tissues in culture. This concept of using patient-derived materials has begun to be extended to modeling other cancer types. One such case involved multiple myeloma, where bone marrow aspirates were taken from patients and reconstituted in co-cultures of patient cancer cells and stromal cells within patient bone marrow ECM hydrogels (de la Puente et al., 2015).
Single organ-on-a-chip models
Static cultures of cancer cells, whether in scaffolds or as organoids, prevent the incorporation of biophysical stimuli that cancer cells experience in the body. The microfluidic-based OOC systems incorporate fluid flow, to recapitulate metastatic cell circulation and activate the associated biomechanical regulation pathways. For example, the physiologically relevant fluid flow in a model of the human bone perivascular niche allowed for metastatic breast cancer colonization, with recapitulation of shear stress-induced increases in metastatic resistance of breast cancer cells (Marturano-Kruik et al., 2018).
The use of microfluidic OOCs has also enabled investigations into the interactions of cancer cells with the endothelium, which are known to mediate tumor survival and metastatic progression. Perfusion allows re-creating in vitro stable and functional vascular networks that are capable of supporting micro-tumors (Sobrino et al., 2016). Microfluidic models have additionally empowered the ex vivo studies of clinically relevant intravasation and metastasis. Remarkably realistic capillary networks can be formed to study metastatic cell intravasation with high precision, as single cells have been visualized crossing engineered endothelial barriers (Ehsan et al., 2014; Zervantonakis et al., 2012). The downstream processes involved in extravasation have also been modeled using microfluidic OOCs for metastatic breast cancer cell circulation and extravasation into bone, a common secondary tumor site (Jeon et al., 2015). Recent advances in these approaches have even allowed for the evaluation of metastatic propensity of patient-derived cancer cells, as well as for the prediction of treatment efficacies of drugs against tumors formed at metastatic sites (Wang et al., 2020; Yankaskas et al., 2019). Finally, OOCs have been proved to be valuable for drug screening through their ability to re-create biological mass transport of drugs experienced by tumors adjacent to capillaries, and thereby allow studies of the regulatory roles of microvascular endothelial cells (Haase et al., 2020; Shirure et al., 2018).
Multi-organ-on-a-chip systems
As the capabilities of microfluidic OOCs and the confidence in predictability of these systems have increased, so has their complexity, with several groups creating interconnected multi-tissue platforms. The incorporation of several tissue systems allows for studies of systemic effects, such as organ-specific homing and metastasis (Aleman and Skardal, 2019; Skardal et al., 2016). Other advances have been motivated by the need to improve preclinical evaluation of anticancer drugs, which are notorious for the low predictability of both drug toxicity and efficacy. The expectation is that engineered malignant and healthy tissues can better recapitulate human physiology and lead to more accurate preclinical evaluation of candidate therapeutics. Using multi-tissue systems allows physiologically relevant ways of drug testing and measurements of both “target” effects (on tumor tissue) and “off-target” toxicity (on healthy tissues) (Chramiec et al., 2020; McAleer et al., 2019). This approach has shown promise in identifying clinically relevant resistances to experimental small molecule treatments (Chramiec et al., 2020) and has been used to demonstrate metabolism of such drugs, and the subsequent cardiotoxic effects of the metabolites within an in vitro context (McAleer et al., 2019).
Engineered models of cancer with immune cells
Despite the abundance of engineered cancer models described in the literature, the number of reports incorporating immune components pales in comparison. Models that integrate immune cells are generally aimed at either predictive drug studies for personalized immunotherapy screening or at recapitulation of a single stage of the metastatic cascade (reviewed recently by Shelton et al., 2020). This is often achieved using endogenous immune cells contained within resected patient tumors or biopsies, which are then co-cultured with tumor cells in the presence or absence of stroma. Building upon the existing models of cancer, most approaches involve spheroids, organoids, microfluidic culture, and OOC systems, implemented individually or in combination (Table 1).
Table 1.
Engineered models of cancer incorporating immune components
| Model | Description | Purpose | Cancer type | Non-immune cells | Immune cells | Major findings | Advantages | Limitations | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Organotypic culture | Patient- or murine-derived organotypic tumor spheroids in collagen hydrogels |
|
Diverse primary solid tumors | Patient-derived tumor and stromal cells | Endogenous immune cells in excised tumor | Demonstrated using RNA sequencing and CIBERSORT an increase of CD8+ T cells and M0 macrophages in response to αPD-1 + αCTLA-4 | Retains complexity of autologous tumor-infiltrating immune cells | Analysis restricted to immune cells in excised TME; immune cell populations inferred from bulk sequencing | (Aref et al., 2018) |
| Organotypic culture of tumor cells, fibroblasts, and macrophages on collagen gels |
|
Human squamous cell carcinoma | A-5 RT3 cell line; primary human dermal fibroblasts | Human monocytes isolated from blood | Cultures induce M2-like activation after 3 weeks; IL-4 stimulation increased MMP-2 and -9 production, causing reduced thickness of dermal equivalents | Macrophages can be sourced from patient or activated to M1 or M2 before seeding | Exogenous stimuli may alter all cells | (Linde et al., 2012) | |
| Patient-derived organoid | Epithelial tumor organoid co-culture with PBMCs |
|
Human non-small cell lung cancer; colorectal cancer | Patient-derived, surgically resected tumors | Human lymphocytes isolated from blood | Tumor organoids induced autologous CD8+ T cell reactivity, causing tumor cell apoptosis | Autologous circulating T cells are easily sourced; small patient samples needed | Generation of tumor-reactive T cells variable across patients; lack of stroma | (Dijkstra et al., 2018) |
| Air-liquid interface PDOs of primary tumor epithelia and native embedded immune cells in collagen matrix |
|
Diverse primary and metastatic tumors | Patient-derived, surgically resected tumor and stromal cells | Endogenous immune cells in excised tumor | CD3+, CD8+, CD4+ T cells, B cells, NK cells, NKT cells, and macrophages integrated in PDOs, which recapitulated TCR repertoire of tumor biopsy and response to anti-PD-1/anti-PD-L1 immune checkpoint blockade | Streamlined tandem gene expression and immune profiling of single cells | Populations of immune and fibroblast stroma progressively decline over time | (Neal et al., 2018) | |
| Combination tumor-lymph node patient-derived in HA/collagen-based hydrogels |
|
Human Melanoma | Patient-derived, surgically resected heterogeneous tumors | Endogenous immune cells in excised tumor and lymph node; PBMCs | Lymph node immune cells required for checkpoint inhibitor efficacy; clinical responses to inhibitors recapitulated; potential to generate patient-specific adaptive immunity | Rapidly recapitulates patient-specific tumor-immune interactions | Heterogeneous, uncharacterized immune populations tested | (Votanopoulos et al., 2019) | |
| Microfluidic culture | Multiplexed micro-fluidic perfusion system with patient-derived tumor fragments and circulating autologous lymphocytes |
|
Human non-small cell lung cancer | Patient-derived, surgically resected heterogeneous tumors | TILs | TILs infiltrate tumor fragment in response to anti-PD-1 treatment, leading to increased tumor cell death over time | Scalable for high-throughput analysis of diverse patient tumor fragments | Omits effects of inter-fragment cross talk | (Moore et al., 2018) |
| 3D microfluidic model of tumor- DC interactions in collagen |
|
Human colorectal cancer | SW620 cell line | Human dendritic cells isolated from blood | Interferon alpha-stimulated DCs preferentially migrate via CXCR4/CCL12 toward and phagocytose drug-treated cancer cells | Device design mimics spatiotemporal biochemical-driven DC-tumor cell interactions | Proof-of-concept using cell line | (Parlato et al., 2017) | |
| Micro-fluidic co-culture of murine spleen cells and murine melanoma cells |
|
Murine melanoma | B16.F10 metastatic melanoma cells | Splenocytes derived from IRF-8KO or WT mice | Splenocytes lacking IRF-8 displayed uncorrelated random walks not directed toward melanoma cells; WT splenocytes move in a highly coordinated motion toward melanoma cells | Model can be easily applied to study specific immune cell-cancer cell interactions | Lack of biomimetic matrix/tissue components; murine-based | (Agliari et al., 2014) | |
| 3D vascularized microfluidic model of human monocyte migration and tumor cell extravasation |
|
Human melanoma and breast cancer | HUVECs; normal human lung fibroblasts; MDA-MB-231 and MDA-MB-435 cell lines | Human monocytes isolated from blood | Demonstrated extravasation of inflammatory (CCR2+), but not patrolling (CCR2-) monocytes; co-perfusion of monocytes with MDA-MB-231 reduced cancer cell extravasation | Models physiologically relevant human micro-vessels | Absence of flow influences cell arrest on and extravasation across endothelium | (Boussommier-Calleja et al., 2019) | |
| Tissue slice culture on-chip | Dual-tissue slice microfluidic chip with continuous recirculating flow |
|
Murine Breast Cancer | Murine-derived 4T1 tumors, TDLNs & contralateral NDLNs | Endogenous immune cells in excised lymph nodes | Demonstrated dual-slice protein communication and T cell immunosuppression in TDLN and NDLN | Preserves complex microenvironment and enables controlled biomimetic flow | Model limited to paracrine tumor-lymph node; murine-based | (Shim et al., 2019) |
PBMC, peripheral blood mononuclear cells; PDO, patient-derived organoid; PD-1, programmed cell death protein 1; PD-L1, programmed cell death-ligand 1; TIL, tumor-derived infiltrating lymphocytes; DC, dendritic cell, WT, wild-type; HUVEC, human umbilical vein endothelial cells; TDLN, tumor draining lymph nodes; NDLN, non-draining lymph nodes.
For example, several studies have demonstrated the utility of patient-derived tumors that retain autologous immune cells for screening immune checkpoint inhibitors. In one approach, patient-derived tumors were dissociated to generate 40- to 100-μm spheroids, which were subsequently suspended in a collagen hydrogel for ex vivo culture in a passive microfluidic device (Aref et al., 2018; Jenkins et al., 2018). Immune checkpoint blockade via αPD-1 and αCTLA-4 induced immune-mediated tumor cell death and altered the immune cell population landscape, inferred using RNA sequencing and CIBERSORT (Aref et al., 2018). In a study designed to model immune checkpoint blockade, patient-derived tumors were minced and resuspended in a collagen gel layered on top of a pre-solidified collagen gel in a transwell insert, to generate an air-liquid organoid culture (Neal et al., 2018). This approach preserved tumor architecture and stroma, as well as diverse immune cells, although these outcomes declined over time.
In addition to static cultures, microfluidic perfusion systems have been used to model cross talk between patient-derived tumor fragments and circulating autologous tumor-infiltrating lymphocytes that were isolated from the tumor (Moore et al., 2018). In response to αPD-1 treatment, circulating lymphocytes infiltrated the tumor fragments, enhancing tumor cell death. Importantly, although these models recapitulate features of the local tumor-immune microenvironment, the contributions of peripheral immune cells were not captured, despite their critical roles in metastasis. These include circulating cells and cells residing in the hematopoietic bone marrow niche and secondary lymphoid organs, where T cell priming occurs.
To model aspects of tumor-lymph node interactions, Shim et al. developed a dual-compartment microfluidic device with continuous recirculating flow to enable cross talk between ex vivo tissue slices of a murine-derived tumor and a tumor-draining or non-draining lymph node. The use of tissue slices preserved the complex architecture and microenvironment of the excised tissue and captured the contributions of immune cells residing in both the lymph node and TME. Notably, tumor-lymph node communication induced T cell immunosuppression, which was also observed in vivo (Shim et al., 2019), suggesting an important paracrine role for tumor-lymphoid communication. Additional studies are needed to confirm translation of these findings to the human system and to capture cell migration between the tumor and the draining lymph node.
As mentioned earlier (Figure 1A), one of the steps in the metastatic cascade is tumor cell extravasation, during which circulating tumor cells migrate through the vascular endothelium to enter a distant tissue site. To better understand the role of immune cells in this process, Boussommier-Calleja et al. developed a microfluidic model with a perfusable vascular network within fibrin gel, which enabled inflammatory (CCR2-expressing) monocyte trans-endothelial migration from the intravascular space into the surrounding matrix. The authors demonstrated reduction in tumor cell extravasation when tumor cells were perfused together with monocytes, providing evidence for immune cell-mediated extravasation (Boussommier-Calleja et al., 2019).
It is worth noting that although the OOC systems can help elucidate interactions between the tumor and the immune system, they offer an incomplete view of tumor progression and overlook the tumor-induced changes at distant tissue sites that occur before metastasis is diagnosed. To observe such system-wide changes, a new generation of multi-organ models is required. A complete model would (1) include tissues that are targeted as secondary tumor sites, such as the bone, brain, liver, and lung, and the vasculature; (2) enable tumor cell progression through the metastatic cascade, and (3) incorporate physiologically relevant immune components. Considering that metastasis requires extended culture on the order of weeks to months, maintaining the presence of short-lived immune cells (Figure 2) presents a challenge. Leveraging engineered bone marrow niches and current multi-organ OOC models would enable the controlled study of tumor-driven changes in hematopoiesis, recruitment of bone marrow-derived cells to pre-metastatic niches, and long-term modeling of tumor metastasis.
A framework for developing metastasis-on-a-chip systems
The current emphasis in cancer treatment is toward removal of the primary tumor, which fails to consider the systemic inter-organ changes, immune landscape, and circulating tumor cells that drive the subtle progression to metastatic disease. To develop approaches aimed at treating metastasis, we must develop the appropriate tools for modeling this complex, systemic disease that are both accessible and physiologically relevant. By combining previous advances in tumor organoids and organ-on-a-chip engineering, we can design “Metastasis-on-a-Chip” (METoC) systems to fuel the development and translation of successful therapeutics.
Envisioned conceptual design for METoC systems
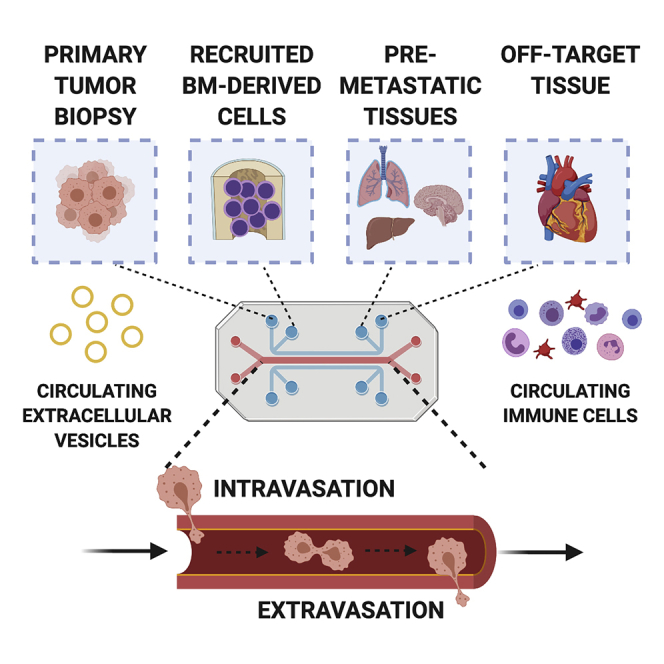
In studying metastasis in vitro, single-tissue perfusion systems mimic only some aspects of intravasation or extravasation, often limited to the primary tumor site and the vascular entry site. In contrast, multi-tissue OOC systems, composed of multiple engineered organ systems connected by vascular perfusion to recapitulate a body-on-a-chip, enable the study of multiple facets of metastatic progression and drug development in vitro using human-equivalent model systems (Figure 4) (Low et al., 2020; Ronaldson-Bouchard and Vunjak-Novakovic, 2018; Sharma et al., 2020). Although the idea of a body- or animal-on-a-chip was first conceptualized in the late 1990s (Aaron et al., 2001), we are only beginning to realize their application in cancer research. Multiple human organs connected in vitro, with a primary tumor and/or circulating tumor cells, provide the basis of the envisioned METoC system—an entirely human environment for studies of pre-metastatic niche conditioning, immune cell cross talk, and metastatic site colonization over time.
Figure 4.

Emerging technologies for metastasis-on-a-chip platforms
Recent advances in bioengineering technologies help in engineering patient-specific organotypic models that are able to recapitulate some aspects of organ-level function in vitro, as well as derivation and maintenance of primary tumor tissue (e.g., organoids). In translational applications, the convergence of bioengineering, immunology, systems biology, and drug development is key to utilizing organs-on-a-chip in both understanding the mechanisms of disease and personalized medicine. Created with BioRender.com.
Human
Human and animal models differ significantly in their immune populations, cytokines, and antibodies expressed in both cancer and immune cell populations. Efforts to “humanize” animals have led to the development of breakthrough treatments (Chambers et al., 2001; Schreiber et al., 2011), whereas the majority of therapeutics developed in mice fails to translate into clinical success (Mak et al., 2014). This paradigm has led to a median of $648 million cost of cancer drug development (Prasad and Mailankody, 2017), which is reasonable compared with other therapeutic areas; however, the resulting clinical trial success rate of only 3.4% is the lowest of all treatment areas (Wong et al., 2019). Recent advances in deriving patient-specific induced pluripotent stem cell (iPSC) and organoids provides human cell sources for both healthy and cancerous cells, enabling a fully isogenic METoC. The use of human cells is critical when developing tools that can be used in parallel with existing animal models to provide complimentary insights.
Systemic
Tumor metastasis is systemic in nature. Consequently, the METoC models should be systemic and include both the primary tumor and the metastatic sites. Additionally, the inclusion of non-metastatic organs, such as the heart, can serve as a negative control to ensure that cancer cell extravasation is driven by factors other than microfluidic flow and also enable modeling of off-target drug effects. In particular, these models need to include the components of immune system, which consists of immune cell organs (bone marrow, thymus, spleen) and the lymphatic circuit (lymph nodes, lymphatic vasculature). Engineering functional immune organs and the differentiation of immune cell types is still in its infancy (Chou et al., 2020; Mengus et al., 2018; Moura Rosa et al., 2016; Sasserath et al., 2020), and we expect this area to be a major focus in the development of physiological METoCs. Although this work is not trivial, collaborative efforts across many fields will enable the realization of these goals toward achieving a true functional METoC model, from which all subsequent goals will be enabled.
Device designs
The development of translational METoC models will rely on their ease of use with minimal training and their ability to provide high-content data. The incorporation of on-line readouts enables spatiotemporal tracking of biological events without disturbing the system, reduces variability, and allows conduction of longitudinal and dynamic (signal-response) studies. Examples include the fluorescent or bioluminescent labeling of both immune and cancer cells and the subsequent tracking of their locations within the system. Monitoring when the immune and cancer cells cross the endothelial barrier and enter/exit the tissue space is necessary for tracking metastatic progression. These same tools can be used to track tumor cell size and immune cell infiltration, both as metastasis progresses and in response to drug treatment. Similarly, supernatant sampling over time enables analysis of secreted factors and biomarkers that can be directly compared with the clinical data and help benchmark and validate these platforms. Design considerations should also include the realistic tumor and tissue environments. In particular, designs that enable hypoxic gradient control and inclusion of advanced biomaterials (i.e., native biomaterials, stiffening biomaterials) will increase the biological fidelity of the model. Of note, increased physiological relevance of the METoC usually increases the device complexity. The trade-off between complexity and biological fidelity can be reassessed according to the specific research question, providing the user with an engineering toolbox to add biological complexity as needed.
METoC models for preclinical screening
Bioengineered multi-organ models can be used to directly determine drug safety, on- and off-target toxicity, and potential immunogenicity. Systemic human models of cancer metastasis (METoC) can greatly accelerate and streamline the therapeutic developments by identifying immune modulating drugs and vaccines that harness the tumoricidal capabilities of the immune response. Recent drug advances are shifting from small molecules or radioactive therapies toward biologically active agents that work in conjunction with the body to achieve therapeutic results. Testing these new therapies requires human models that are systemic and include functional immune components to adequately predict clinical responses. Although PdX models represent the gold standard for patient-specific drug development, the lack of tumor microenvironment, human cell stromal interactions, and human immune cell populations greatly reduces their clinical translation. The practical utility of these models is also an issue, with a range of 2%–40% of injected cells actually taking hold in the host tissue (engrafting). Drugs developed using PdX models fail in Phase II/III clinical trials, the costliest phase, more than 50% of the time. Overall, METoC systems include both on-target organs for evaluating the drug efficacy and off-target organs that may be susceptible to drug toxicity. Similarly, the inclusion of multiple connected tissues facilitates proper pharmacokinetic/pharmacodynamics (PK/PD) modeling, with engineered liver tissues proving effective at metabolizing drugs (Low et al., 2020). Modeling the complex immune profile of the primary tumor and metastatic site will enable insights into how the immune landscape changes to prevent or support metastasis and inform the development of targeted, patient-specific therapies.
METoC models for mechanistic research
Bioengineered models can be leveraged within the METoC to directly control various parameters toward elucidating their distinct mechanistic contributions to disease progression. As new bioinformatic technologies have already demonstrated the power of targeting the molecular mechanisms of cancer progression and reoccurrence, tools like single-cell RNA and DNA sequencing can be used in tandem with patient-specific bioengineered human models to study metastasis and immune cell invasion in a clinically relevant and unbiased context (Figure 4). These tools have already enabled the generation of a human map of organ-specific metastasis (Jin et al., 2020). Likewise, Oliver et al. (2019) recently applied live cell imaging modalities coupled with artificial intelligence to a blood-brain barrier-on-a-chip model for predicting metastatic potential of tumor cells. Ultimately, the marriage of bioengineering with cancer and systems biology can help identify key mechanisms behind metastatic progression and the specific role of each cell type at play and can lead to new therapeutic targets in an entirely human, systemic equivalent to animal models.
Patient-specific METoC models for advancing precision medicine
As the development of human models of metastasis increases our mechanistic standing of the underlying biology, these models can be further leveraged to provide patient-specific models for precision medicine. These tools will enable clinicians to insert patient cells into bioengineered models capable of recapitulating the patient’s specific cancer for drug screening. This approach allows clinicians to identify the therapy for which the patient will have the highest likelihood of success and use this knowledge to personalize each patient’s treatment. The use of patient-specific cells enables development of disease models where animal models fail to recapitulate the disease, thereby enabling mechanistic studies to enhance drug target identification and optimization.
iPSC technologies enable patient-specific tissue engineering, toward “cancer patient-on-a-chip” models. For these models, incorporation of hematopoiesis still remains a challenge (Chou et al., 2020; Chramiec and Vunjak-Novakovic, 2019; Tavakol et al., 2020). As differentiation of many iPSC-derived immune cell types is under development, the inclusion of these cells into an isogenic, human system will further advance studies of metastatic progression in vitro (Figure 4). Patient-specific METoC models can be developed to initially predict which therapeutics will be the most efficacious, and then to subsequently screen for downstream metastatic potential. This new generation of METoCs would help recapitulate patient-specific organ tropism and test the potential of therapeutics to treat or prevent metastasis in these organs. These technologies may also be used more broadly to perform “clinical trials-on-a-chip” and inform the design of subsequent clinical testing by determining the patient populations that will most benefit from the drug and those that may be at risk. This approach can be similarly used to identify which drug combinations are most efficacious, including the specific timing and dosages that maximize therapeutic effect—in both population-specific and individualized manners.
Conclusions
We are only beginning to appreciate the critical role of the systemic immune response in metastatic progression. To further our understanding, bioengineered systems can be leveraged to identify key features of tumor metastasis that are unable to be decoupled in vivo. Integrating the primary tumor, secondary target sites, and immune organs and cells together on a perfusable platform to model metastasis-on-a-chip will have important implications for patient-specific treatment, in terms of efficacy and toxicity, as well as identification of novel drug targets. As researchers learn more about the advantages and limitations of engineered human models, we encourage the continued collaboration between biologists, engineers, and clinicians to solve cancer metastasis.
Acknowledgments
The authors gratefully acknowledge the funding support of their research by the NIH (EB025765, EB027062 and CA249799), NSF (NSF16478), NSF Graduate Research Fellowship (DGE1644869), and NYSTEM (C32606GG).
Author contributions
Conceptualization, P.L.G. and G.V.-N.; Writing – Original Draft, P.L.G., D.N.T., A.C., and K.R.-B.; Writing – Reviewing and Editing, P.L.G., D.N.T., A.C., K.R.-B., and G.V.-N.
Declaration of interests
The authors declare no competing interests.
References
- Aaron S., Gregory T.B., Michael L.S. Animal on a chip: a microscale cell culture analog device for evaluating toxicological and pharmacological profiles. In: Mastrangelo C.H., Becker H., editors. Microfluidics and BioMEMS, Proc. SPIE. Vol. 4560. SPIE; 2001. pp. 98–101. [Google Scholar]
- Agliari E., Biselli E., De Ninno A., Schiavoni G., Gabriele L., Gerardino A., Mattei F., Barra A., Businaro L. Cancer-driven dynamics of immune cells in a microfluidic environment. Sci. Rep. 2014;4:6639. doi: 10.1038/srep06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Sayed M.F., Amrein M.A., Buhrer E.D., Huguenin A.L., Radpour R., Riether C., Ochsenbein A.F. T-cell-Secreted TNFalpha induces emergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res. 2019;79:346–359. doi: 10.1158/0008-5472.CAN-17-3026. [DOI] [PubMed] [Google Scholar]
- Aleman J., Skardal A. A multi-site metastasis-on-a-chip microphysiological system for assessing metastatic preference of cancer cells. Biotechnol. Bioeng. 2019;116:936–944. doi: 10.1002/bit.26871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen B.M., Hiam K.J., Burnett C.E., Venida A., DeBarge R., Tenvooren I., Marquez D.M., Cho N.W., Carmi Y., Spitzer M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020;26:1125–1134. doi: 10.1038/s41591-020-0892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aref A.R., Campisi M., Ivanova E., Portell A., Larios D., Piel B.P., Mathur N., Zhou C., Coakley R.V., Bartels A. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18:3129–3143. doi: 10.1039/c8lc00322j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U., Beroukhim R., Golub T.R. Genomic evolution of cancer models: perils and opportunities. Nat. Rev. Cancer. 2019;19:97–109. doi: 10.1038/s41568-018-0095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Brekken R., McMahon G., Vu T.H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell M.J., Hines W.C. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj S.F., Hwang C.I., Baker L.A., Chio I.I., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussommier-Calleja A., Atiyas Y., Haase K., Headley M., Lewis C., Kamm R.D. The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model. Biomaterials. 2019;198:180–193. doi: 10.1016/j.biomaterials.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Cacho-Diaz B., Garcia-Botello D.R., Wegman-Ostrosky T., Reyes-Soto G., Ortiz-Sanchez E., Herrera-Montalvo L.A. Tumor microenvironment differences between primary tumor and brain metastases. J. Transl. Med. 2020;18:1. doi: 10.1186/s12967-019-02189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casbon A.J., Reynaud D., Park C., Khuc E., Gan D.D., Schepers K., Passegue E., Werb Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. U S A. 2015;112:E566–E575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celia-Terrassa T., Kang Y. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 2018;20:868–877. doi: 10.1038/s41556-018-0145-9. [DOI] [PubMed] [Google Scholar]
- Chambers C.A., Kuhns M.S., Egen J.G., Allison J.P. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- Chou D.B., Frismantas V., Milton Y., David R., Pop-Damkov P., Ferguson D., MacDonald A., Vargel Bolukbasi O., Joyce C.E., Moreira Teixeira L.S. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 2020;4:394–406. doi: 10.1038/s41551-019-0495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chramiec A., Teles D., Yeager K., Marturano-Kruik A., Pak J., Chen T., Hao L., Wang M., Lock R., Tavakol D.N. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab Chip. 2020;20:4357–4372. doi: 10.1039/d0lc00424c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chramiec A., Vunjak-Novakovic G. Tissue engineered models of healthy and malignant human bone marrow. Adv. Drug Deliv. Rev. 2019;140:78–92. doi: 10.1016/j.addr.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt S.B., Kersten K., Doornebal C.W., Weiden J., Vrijland K., Hau C.S., Verstegen N.J.M., Ciampricotti M., Hawinkels L., Jonkers J. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Puente P., Muz B., Gilson R.C., Azab F., Luderer M., King J., Achilefu S., Vij R., Azab A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials. 2015;73:70–84. doi: 10.1016/j.biomaterials.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Rochere P., Guil-Luna S., Decaudin D., Azar G., Sidhu S.S., Piaggio E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018;39:748–763. doi: 10.1016/j.it.2018.07.001. [DOI] [PubMed] [Google Scholar]
- Dijkstra K.K., Cattaneo C.M., Weeber F., Chalabi M., van de Haar J., Fanchi L.F., Slagter M., van der Velden D.L., Kaing S., Kelderman S. Generation of tumor-reactive T cells by Co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174:1586–1598 e1512. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillekas H., Rogers M.S., Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8:5574–5576. doi: 10.1002/cam4.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driehuis E., Kretzschmar K., Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020;15:3380–3409. doi: 10.1038/s41596-020-0379-4. [DOI] [PubMed] [Google Scholar]
- Ehsan S.M., Welch-Reardon K.M., Waterman M.L., Hughes C.C., George S.C. A three-dimensional in vitro model of tumor cell intravasation. Integr. Biol. 2014;6:603–610. doi: 10.1039/c3ib40170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler J.T., Bennewith K.L., Cox T.R., Lang G., Bird D., Koong A., Le Q.T., Giaccia A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follain G., Osmani N., Azevedo A.S., Allio G., Mercier L., Karreman M.A., Solecki G., Garcia Leon M.J., Lefebvre O., Fekonja N. Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev. Cell. 2018;45:33–52 e12. doi: 10.1016/j.devcel.2018.02.015. [DOI] [PubMed] [Google Scholar]
- Font-Clos F., Zapperi S., La Porta C.A.M. Blood flow contributions to cancer metastasis. iScience. 2020;23:101073. doi: 10.1016/j.isci.2020.101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D., Vela I., Sboner A., Iaquinta P.J., Karthaus W.R., Gopalan A., Dowling C., Wanjala J.N., Undvall E.A., Arora V.K. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner H., de Visser K.E. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol. 2020;20:483–497. doi: 10.1038/s41577-019-0271-z. [DOI] [PubMed] [Google Scholar]
- Ghouse S.M., Vadrevu S.K., Manne S., Reese B., Patel J., Patel B., Silwal A., Lodhi N., Paterson Y., Srivastava S.K. Therapeutic targeting of vasculature in the premetastatic and metastatic niches reduces lung metastasis. J. Immunol. 2020;204:990–1000. doi: 10.4049/jimmunol.1901208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange C., Tapparo M., Collino F., Vitillo L., Damasco C., Deregibus M.C., Tetta C., Bussolati B., Camussi G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71:5346–5356. doi: 10.1158/0008-5472.CAN-11-0241. [DOI] [PubMed] [Google Scholar]
- Gupta G.P., Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gupta G.P., Nguyen D.X., Chiang A.C., Bos P.D., Kim J.Y., Nadal C., Gomis R.R., Manova-Todorova K., Massague J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- Haase K., Offeddu G.S., Gillrie M.R., Kamm R.D. Endothelial regulation of drug transport in a 3D vascularized tumor model. Adv. Funct. Mater. 2020;30:2002444. doi: 10.1002/adfm.202002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi H., Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer. 2018;18:533–548. doi: 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hinshaw D.C., Shevde L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79:4557–4566. doi: 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga T. Hypoxic microenvironment and metastatic bone disease. Int. J. Mol. Sci. 2018;19:3523. doi: 10.3390/ijms19113523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A., Costa-Silva B., Shen T.L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder S. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoye A.M., Erler J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016;310:C955–C967. doi: 10.1152/ajpcell.00326.2015. [DOI] [PubMed] [Google Scholar]
- Hoyer F.F., Naxerova K., Schloss M.J., Hulsmans M., Nair A.V., Dutta P., Calcagno D.M., Herisson F., Anzai A., Sun Y. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity. 2019;51:899–914.e7. doi: 10.1016/j.immuni.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.L., Huang M.S., Hung J.Y., Chang W.A., Tsai Y.M., Pan Y.C., Lin Y.S., Tsai H.P., Kuo P.L. Bone-marrow-derived cell-released extracellular vesicle miR-92a regulates hepatic pre-metastatic niche in lung cancer. Oncogene. 2020;39:739–753. doi: 10.1038/s41388-019-1024-y. [DOI] [PubMed] [Google Scholar]
- Hua Y., Bergers G. Tumors vs. Chronic wounds: an immune cell's perspective. Front. Immunol. 2019;10:2178. doi: 10.3389/fimmu.2019.02178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins R.W., Aref A.R., Lizotte P.H., Ivanova E., Stinson S., Zhou C.W., Bowden M., Deng J., Liu H., Miao D. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018;8:196–215. doi: 10.1158/2159-8290.CD-17-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J.S., Bersini S., Gilardi M., Dubini G., Charest J.L., Moretti M., Kamm R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. U S A. 2015;112:214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K., Liang L., Lim C.T. Engineering confining microenvironment for studying cancer metastasis. iScience. 2021;24:102098. doi: 10.1016/j.isci.2021.102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X., Demere Z., Nair K., Ali A., Ferraro G.B., Natoli T., Deik A., Petronio L., Tang A.A., Zhu C. A metastasis map of human cancer cell lines. Nature. 2020;588:331–336. doi: 10.1038/s41586-020-2969-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan R.N., Riba R.D., Zacharoulis S., Bramley A.H., Vincent L., Costa C., MacDonald D.D., Jin D.K., Shido K., Kerns S.A. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T., Qian B.Z., Pollard J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F., Maas R.R., Bowman R.L., Kornete M., Soukup K., Nassiri S., Brouland J.P., Iacobuzio-Donahue C.A., Brennan C., Tabar V. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181:1643–1660 e1617. doi: 10.1016/j.cell.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley R.R., Fidler I.J. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde N., Gutschalk C.M., Hoffmann C., Yilmaz D., Mueller M.M. Integrating macrophages into organotypic co-cultures: a 3D in vitro model to study tumor-associated macrophages. PLoS One. 2012;7:e40058. doi: 10.1371/journal.pone.0040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low L.A., Mummery C., Berridge B.R., Austin C.P., Tagle D.A. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 2020 doi: 10.1038/s41573-020-0079-3. [DOI] [PubMed] [Google Scholar]
- Mak I.W., Evaniew N., Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- Malanchi I., Santamaria-Martinez A., Susanto E., Peng H., Lehr H.A., Delaloye J.F., Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- Marturano-Kruik A., Nava M.M., Yeager K., Chramiec A., Hao L., Robinson S., Guo E., Raimondi M.T., Vunjak-Novakovic G. Human bone perivascular niche-on-a-chip for studying metastatic colonization. Proc. Natl. Acad. Sci. U S A. 2018;115:1256–1261. doi: 10.1073/pnas.1714282115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matano M., Date S., Shimokawa M., Takano A., Fujii M., Ohta Y., Watanabe T., Kanai T., Sato T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- McAleer C.W., Long C.J., Elbrecht D., Sasserath T., Bridges L.R., Rumsey J.W., Martin C., Schnepper M., Wang Y., Schuler F. Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics. Sci. Transl. Med. 2019;11:eaav1386. doi: 10.1126/scitranslmed.aav1386. [DOI] [PubMed] [Google Scholar]
- Mengus C., Muraro M.G., Mele V., Amicarella F., Manfredonia C., Foglietta F., Muenst S., Soysal S.D., Iezzi G., Spagnoli G.C. In vitro modeling of tumor–immune system interaction. ACS Biomater. Sci. Eng. 2018;4:314–323. doi: 10.1021/acsbiomaterials.7b00077. [DOI] [PubMed] [Google Scholar]
- Micek H.M., Visetsouk M.R., Masters K.S., Kreeger P.K. Engineering the extracellular matrix to model the evolving tumor microenvironment. iScience. 2020;23:101742. doi: 10.1016/j.isci.2020.101742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore N., Doty D., Zielstorff M., Kariv I., Moy L.Y., Gimbel A., Chevillet J.R., Lowry N., Santos J., Mott V. A multiplexed microfluidic system for evaluation of dynamics of immune-tumor interactions. Lab Chip. 2018;18:1844–1858. doi: 10.1039/c8lc00256h. [DOI] [PubMed] [Google Scholar]
- Moura Rosa P., Gopalakrishnan N., Ibrahim H., Haug M., Halaas O. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip. 2016;16:3728–3740. doi: 10.1039/c6lc00702c. [DOI] [PubMed] [Google Scholar]
- Müller L., Tunger A., Plesca I., Wehner R., Temme A., Westphal D., Meier F., Bachmann M., Schmitz M. Bidirectional crosstalk between cancer stem cells and immune cell subsets. Front. Immunol. 2020;11:140. doi: 10.3389/fimmu.2020.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal J.T., Li X., Zhu J., Giangarra V., Grzeskowiak C.L., Ju J., Liu I.H., Chiou S.H., Salahudeen A.A., Smith A.R. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175:1972–1988 e1916. doi: 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver C.R., Altemus M.A., Westerhof T.M., Cheriyan H., Cheng X., Dziubinski M., Wu Z., Yates J., Morikawa A., Heth J. A platform for artificial intelligence based identification of the extravasation potential of cancer cells into the brain metastatic niche. Lab Chip. 2019;19:1162–1173. doi: 10.1039/c8lc01387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato S., De Ninno A., Molfetta R., Toschi E., Salerno D., Mencattini A., Romagnoli G., Fragale A., Roccazzello L., Buoncervello M. 3D Microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells. Sci. Rep. 2017;7:1093. doi: 10.1038/s41598-017-01013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H., Aleckovic M., Lavotshkin S., Matei I., Costa-Silva B., Moreno-Bueno G., Hergueta-Redondo M., Williams C., Garcia-Santos G., Ghajar C. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H., Zhang H., Matei I.R., Costa-Silva B., Hoshino A., Rodrigues G., Psaila B., Kaplan R.N., Bromberg J.F., Kang Y. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer. 2017;17:302–317. doi: 10.1038/nrc.2017.6. [DOI] [PubMed] [Google Scholar]
- Prasad V., Mailankody S. Research and development spending to bring a single cancer drug to market and revenues after approval. JAMA Intern. Med. 2017;177:1569–1575. doi: 10.1001/jamainternmed.2017.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger M.A., Hoppe P.S., Smejkal B.M., Eitelhuber A.C., Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325:217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- Ronaldson-Bouchard K., Vunjak-Novakovic G. Organs-on-a-Chip: a fast track for engineered human tissues in drug development. Cell Stem Cell. 2018;22:310–324. doi: 10.1016/j.stem.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386 e310. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- Sasserath T., Rumsey J.W., McAleer C.W., Bridges L.R., Long C.J., Elbrecht D., Schuler F., Roth A., Bertinetti-LaPatki C., Shuler M.L. Differential monocyte actuation in a three-organ functional innate immune system-on-a-chip. Adv. Sci. (Weinh) 2020;7:2000323. doi: 10.1002/advs.202000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Sharma A., Sances S., Workman M.J., Svendsen C.N. Multi-lineage human iPSC-derived platforms for disease modeling and drug discovery. Cell Stem Cell. 2020;26:309–329. doi: 10.1016/j.stem.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton S.E., Nguyen H.T., Barbie D.A., Kamm R.D. Engineering approaches for studying immune-tumor cell interactions and immunotherapy. iScience. 2020;24:101985. doi: 10.1016/j.isci.2020.101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S., Belanger M.C., Harris A.R., Munson J.M., Pompano R.R. Two-way communication between ex vivo tissues on a microfluidic chip: application to tumor-lymph node interaction. Lab Chip. 2019;19:1013–1026. doi: 10.1039/c8lc00957k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirure V.S., Bi Y., Curtis M.B., Lezia A., Goedegebuure M.M., Goedegebuure S.P., Aft R., Fields R.C., George S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip. 2018;18:3687–3702. doi: 10.1039/c8lc00596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skardal A., Devarasetty M., Forsythe S., Atala A., Soker S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol. Bioeng. 2016;113:2020–2032. doi: 10.1002/bit.25950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrino A., Phan D.T., Datta R., Wang X., Hachey S.J., Romero-Lopez M., Gratton E., Lee A.P., George S.C., Hughes C.C. 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 2016;6:31589. doi: 10.1038/srep31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland R.M., Sordat B., Bamat J., Gabbert H., Bourrat B., Mueller-Klieser W. Oxygenation and differentiation in multicellular spheroids of human colon carcinoma. Cancer Res. 1986;46:5320–5329. [PubMed] [Google Scholar]
- Tan W., Zhang W., Strasner A., Grivennikov S., Cheng J.Q., Hoffman R.M., Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavakol D.N., Tratwal J., Bonini F., Genta M., Campos V., Burch P., Hoehnel S., Beduer A., Alessandrini M., Naveiras O. Injectable, scalable 3D tissue-engineered model of marrow hematopoiesis. Biomaterials. 2020;232:119665. doi: 10.1016/j.biomaterials.2019.119665. [DOI] [PubMed] [Google Scholar]
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A. The immune landscape of cancer. Immunity. 2018;48:812–830 e814. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernandez-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votanopoulos K.I., Forsythe S., Sivakumar H., Mazzocchi A., Aleman J., Miller L., Levine E., Triozzi P., Skardal A. Model of patient-specific immune-enhanced organoids for immunotherapy screening: feasibility study. Ann. Surg. Oncol. 2019;27:1956–1967. doi: 10.1245/s10434-019-08143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wu D., Wu G., Wu J., Lu S., Lo J., He Y., Zhao C., Zhao X., Zhang H. Metastasis-on-a-chip mimicking the progression of kidney cancer in the liver for predicting treatment efficacy. Theranostics. 2020;10:300–311. doi: 10.7150/thno.38736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber F., van de Wetering M., Hoogstraat M., Dijkstra K.K., Krijgsman O., Kuilman T., Gadellaa-van Hooijdonk C.G., van der Velden D.L., Peeper D.S., Cuppen E.P. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. U S A. 2015;112:13308–13311. doi: 10.1073/pnas.1516689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.C., Gilkes D.M., Zhang H., Chen J., Wei H., Chaturvedi P., Fraley S.I., Wong C.M., Khoo U.S., Ng I.O. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc. Natl. Acad. Sci. U S A. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.H., Siah K.W., Lo A.W. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20:273–286. doi: 10.1093/biostatistics/kxx069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.C., Sun H.W., Chen H.T., Liang J., Yu X.J., Wu C., Wang Z., Zheng L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. U S A. 2014;111:4221–4226. doi: 10.1073/pnas.1320753111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y., Ibusuki M., Okumura Y., Kawasoe T., Kai K., Iyama K., Iwase H. Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res. Treat. 2008;110:465–475. doi: 10.1007/s10549-007-9742-1. [DOI] [PubMed] [Google Scholar]
- Yankaskas C.L., Thompson K.N., Paul C.D., Vitolo M.I., Mistriotis P., Mahendra A., Bajpai V.K., Shea D.J., Manto K.M., Chai A.C. A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat. Biomed. Eng. 2019;3:452–465. doi: 10.1038/s41551-019-0400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervantonakis I.K., Hughes-Alford S.K., Charest J.L., Condeelis J.S., Gertler F.B., Kamm R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U S A. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Fong M.Y., Min Y., Somlo G., Liu L., Palomares M.R., Yu Y., Chow A., O'Connor S.T., Chin A.R. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]