

Abstract
Multilocus variable-number tandem repeat analysis (MLVA) is widely used for genotyping of Bordetella pertussis, the causative bacteria for pertussis. However, MLVA genotyping is losing its discriminate power because prevalence of the epidemic MT27 strain (MLVA-27) is increasing worldwide. To address this, we developed a single nucleotide polymorphism (SNP) genotyping method for MT27 based on multiplexed single-base extension (SBE) assay. A total of 237 MT27 isolates collected in Japan during 1999–2018 were genotyped and classified into ten SNP genotypes (SG1 to SG10) with a Simpson’s diversity index (DI) of 0.79 (95% CI 0.76–0.82). Temporal trends showed a marked increase in the genotypic diversity in the 2010s: Simpson’s DI was zero in 1999–2004, 0.16 in 2005–2009, 0.83 in 2010–2014, and 0.76 in 2015–2018. This indicates that the SNP genotyping is applicable to the recently circulating MT27 strain. Additionally, almost all outbreak-associated MT27 isolates were classified into the same SNP genotypes for each outbreak. Multiplexed SBE assay allows for rapid and simple genotyping, indicating that the SNP genotyping can potentially be a useful tool for subtyping the B. pertussis MT27 strain in routine surveillance and outbreak investigations.
Subject terms: Clinical microbiology, Bacterial techniques and applications, Infectious-disease epidemiology
Introduction
Bordetella pertussis, a highly transmissible gram-negative coccobacillus, is the etiologic agent of pertussis (whooping cough), a severe acute respiratory illness among children and a persistence cough among adolescents and adults. Vaccination is the most effective method for preventing and controlling pertussis; however, pertussis vaccines are unable to provide lifelong immunity1–3. The waning of vaccine-acquired immunity is thus a major contributor to pertussis resurgence. In most industrialized countries, the incidence of pertussis has increased despite high vaccination coverage, and the number of pertussis epidemics have also occurred4–8. In Japan, the number of reported pertussis cases of adolescents and adults significantly increased in the 2000s, and a nationwide epidemic of pertussis occurred during 2008–2010. Molecular strain typing revealed that certain strains of B. pertussis (genotype, MT27d) were associated with the pertussis epidemic4.
Molecular strain typing of infectious disease agents has become an important tool for their epidemiological surveillance and outbreak investigations. Strain typing methods currently used for B. pertussis are pulsed-field gel electrophoresis (PFGE)9,10, multilocus sequence typing (MLST)11,12, multilocus variable-number tandem repeat analysis (MLVA)4,13,14, and single nucleotide polymorphism (SNP) typing15–20. While PFGE has greater discriminatory power, it has the disadvantage in that the inter-laboratory comparison of PFGE patterns is difficult. MLST targets virulence-associated allelic genes of B. pertussis (commonly, ptxP, ptxA1, prn, and fim3 alleles), whereas MLVA targets six variable number tandem repeat loci of B. pertussis. The MLST and MLVA methods allow for direct comparisons of B. pertussis isolates in different laboratories, with the MLVA method being the most widely used to study molecular epidemiology of this organism. However, MLST and MLVA are losing their discriminate power, since the MLVA type 27 (MT27) strain carrying the allele profile ptxP3/ptxA1/prn2/fim3A has become the predominant strain in B. pertussis population worldwide4,10,21–24. Currently, SNP genotyping based on whole-genome sequencing (WGS) is the most accurate method, and there are several reports of B. pertussis populations analyzed using whole-genome SNP analysis15–18,21,25,26. Recently, a novel WGS-based genotyping, called core genome MLST, was developed for global comparisons of B. pertussis isolates27. However, WGS has the disadvantage in that it is laborious, costly, and time-consuming for use in routine surveillance, especially in outbreak investigations.
Previously, a simple SNP genotyping method with 38 SNP targets was developed for B. pertussis using a single-base extension (SBE) assay28. The SBE assay is based on the incorporation of fluorescently labeled dideoxynucleotides (ddNTPs) into the 3′ end of allele-specific extension primers with a distinct length, and subsequent analysis with a capillary DNA sequencer. This assay allows for rapid and high-throughput genotyping. However, the SBE-based SNP genotyping had low discriminatory power for B. pertussis epidemic strain MT27, since the 38 SNP targets were selected based on the WGS data of various B. pertussis strains28. This limitation identifies the need for selecting polymorphic SNP markers among MT27 isolates in order to improve the discriminatory power for the MT27 strain.
In the present study, in order to easily subtype B. pertussis epidemic MT27 strain, we screened for polymorphic SNPs among MT27 isolates using their genome data and developed a novel SNP genotyping with 20 SNP targets based on multiplexed SBE assay. This genotyping system was evaluated with 237 B. pertussis MT27 isolates, and its applicability to outbreak investigations was assessed with outbreak-associated MT27 isolates.
Methods
Isolates and DNA preparation
We studied 237 B. pertussis MT27 isolates collected in Japan during 1999–2018. They were all of the MT27 isolates stored in the National Institute of Infectious Diseases (NIID) strain collection, which included epidemiologically related outbreak-associated isolates (Supplementary Table S1). The isolates were cultured on cyclodextrin solid medium (CSM) agar29 and incubated at 36 °C for 2–3 days. DNAs were extracted from the isolates by boiling and stored at − 20 °C. For whole-genome sequencing (WGS), DNAs were purified using the NucleoSpin Tissue kit (Macherey–Nagel, Germany). Novogene (Beijing, China) performed the whole-genome sequencing experiment.
WGS
Twenty MT27 isolates collected during 2011–2018 were sequenced on the Illumina HiSeq X Ten platform with 150-bp paired-end reads performed by Novogene Co. (Supplementary Table S1). The average coverage depth of the sequencing was more than 200 × for each isolate. The sequence data were submitted to the DDBJ Sequence Read Archive (DRA) (accession no. DRA007914). All the isolates were epidemiologically unrelated cases of pertussis.
Identification and selection of SNPs
WGS reads were mapped to the reference genome sequence of B. pertussis Tohama I (accession no. NC_002929.2) by the Burrows-Wheeler Aligner (BWA) software, and SNPs were detected by using the SAMtools software. A total of 269 SNPs were identified in coding sequences (including pseudogenes) between 20 sequenced isolates and the reference strain Tohama. Among the 269 SNPs, we selected a set of 20 informative SNPs (representing SNP frequencies of 7.8–49.0%) for single-base extension (SBE) assay based on 51 genome sequences of MT27 isolates: 20 sequences from this study and 31 from a previous study17 (Supplementary Table S2). General information on the 20 SNPs is given in Table 1.
Table 1.
SNP markers selected for SNP genotyping using single-base extension assay.
| SNP name | Positiona | Locus ID | Gene | SNP variation | Amino acid change | Gene description | Reference |
|---|---|---|---|---|---|---|---|
| SNP2 | 62,982 | BP0063 | C/T | A100T | Hypothetical protein | 17 | |
| SNP8 | 870,614 | BP0841 | nuoA | G/T | V15L | NADH-quinone oxidoreductase subunit A | 17 |
| SNP11 | 980,942 | BP0944 | gyrA | A/G | D87G | DNA gyrase subunit A | 17,30 |
| SNP12 | 1,182,054 | BP1122 | A/G | E47G | AsnC family transcription regulator | 17 | |
| SNP14 | 1,459,272 | BP1383 | flgL | A/G | D237G | Flagellar hook-associated protein FlgL | 17 |
| SNP15 | 1,470,281 | BP1394 | fliM | C/T | Silent | Flagellar motor switch protein FliM | 17,18,28 |
| SNP16 | 1,692,984 | BP1610 | C/T | Pseudo | 17,25 | ||
| SNP17 | 1,806,441 | BP1722 | G/A | Silent | Hypothetical protein | 17 | |
| SNP18 | 1,824,667 | BP1740 | cphA | C/T | Silent | Cyanophycin synthetase | 17 |
| SNP19 | 1,845,571 | BP1758 | C/T | Silent | Fatty acid desaturase | 17 | |
| SNP20 | 1,890,117 | BP1799 | iscS | C/T | L131F | Cysteine desulfurase | 17,25 |
| SNP22 | 2,308,009 | BP2187 | G/A | Silent | Transferase | 17 | |
| SNP24 | 2,395,350 | BP2274 | G/A | Silent | ABC transporter substrate-binding protein | 17 | |
| SNP25 | 2,436,106 | BP2310 | hemF | C/T | T136I | Coproporphyrinogen III oxidase | 17 |
| SNP26 | 2,657,330 | BP2509 | dapB | A/C | T5P | 4-Hydroxy-tetrahydrodipicolinate reductase | 17 |
| SNP28 | 2,884,401 | BP2718 | T/C | K276R | Biotin synthase | 17 | |
| SNP30 | 3,326,160 | BP3121 | G/A | Silent | GTP-binding protein | 17 | |
| SNP31 | 3,436,938 | BP3224 | C/T | Silent | Cytochrome oxidase | 17,18 | |
| SNP32 | 3,523,618 | BP3303 | C/T | G56R | ADP-dependent (S)-NAD(P)H-hydrate dehydratase | 17,18 | |
| SNP34 | 3,758,827 | BP3547 | G/A | Silent | GntR family transcriptional regulator | 17 |
aPositions according to the plus strand on Bordetella pertussis Tohama genomic sequence: NC_002929.2.
Of the 20 SNP targets, 10 were coding SNPs, 9 were silent SNPs, and the remaining one was a genome SNP located in the pseudogene BP1610 (Table 1). SNP11 located in gyrA was associated with quinolone resistance in B. pertussis30, while SNP16 and SNP26 were previously identified as unique SNPs to epidemic isolates of Australian B. pertussis25. All 20 SNP targets were found in previous studies17,18,25,28.
SNP genotyping with SBE assay
Twenty selected SNPs were divided into two groups and typed in two 10-plex PCR assays termed SNP20A and SNP20B panels. The SNP20A panel targeted SNP2, SNP11, SNP12, SNP14, SNP16, SNP18, SNP19, SNP20, SNP25, and SNP26, whereas the SNP20B targeted SNP8, SNP15, SNP17, SNP22, SNP24, SNP28, SNP30, SNP31, SNP32, and SNP34 (Table 2). Each 10-plex PCR was performed in a 15-μl reaction volume containing 7.5 μl of 2 × PCR buffer for KOD FX, 0.33 U of KOD-FX DNA polymerase (TOYOBO, Co., Ltd., Japan), 3.3 µl of 2 mM dNTPs, 1 µl of DNA sample, and 1.5 µl of primer mixture (each one at 2 µM). The primer sequences for SNP20A and SNP20B panels are shown in Table 2. PCR conditions were as follows: denaturation for 2 min at 94 °C; 35 cycles with denaturation at 98 °C for 10 s, primer annealing for 30 s at 60 °C (SNP20A) or 63 °C (SNP20B), and extension at 68 °C for 30 s; and final extension at 68 °C for 5 min. Following the PCR assay, the PCR products were treated with 5 U of shrimp alkaline phosphatase (New England Biolabs) and 2 U of exonuclease I (New England Biolabs) at 37 °C for 60 min, followed by incubation at 75 °C for 15 min for enzyme inactivation.
Table 2.
PCR primers used for the multiple PCR panels, SNP20A and SNP20B.
| Multiplex PCR panel | SNP name | PCR primer sequence (5′ → 3′) | Amplicon size (bp) | |
|---|---|---|---|---|
| Forward | Reverse | |||
| SNP20A | SNP2 | GTGGAGAGGCGACGCCAAAG | CTGGTGGTAACGCGTGCGAA | 112 |
| SNP11 | TATCGTCGGGGACGTCATCGG | GCCAGGCGGATTTCGGTGTAG | 180 | |
| SNP12 | ATGCCAGCCTCACCAACGTC | AACCGCTCCAGCTCGCTTTC | 202 | |
| SNP14 | GCCGCGATTTCCGTATCGAGTT | CGCCGTTCATGTCGATTACGGAG | 150 | |
| SNP16 | GTTGGGTGCAGGAACTCAAGG | GTACGACGCGTACAGATTGTGG | 143 | |
| SNP18 | CCCAATTGGCGCAATCGCCT | CGCTCCATCGTCGACCACCT | 139 | |
| SNP19 | GTATCGCGAGGAGTCCGCCAA | GTCGATGGCCAGCATGGTGAG | 130 | |
| SNP20 | AGGGCTTTGAAGTGACCTACCT | GGATGACGCCGATCTCGTTG | 132 | |
| SNP25 | ATGCCGAATCCGACGTGTTCTG | CGGGTTTCGTTGCGGTGCTT | 190 | |
| SNP26 | GGACAACCCCACCCTCGACTAC | CGAGGACGGCTTCGATCAGCA | 143 | |
| SNP20B | SNP8 | TGAACCTGCACCCGTATTTCCCC | TCGAACTTCATCCGCGCGTCT | 172 |
| SNP15 | GCAGCACGTTGCGCATGTGG | CGGCGAAAGCGACGAGAAGCAG | 152 | |
| SNP17 | CGAGGTGTCCGACCTGGAAGTG | GGAAACCCTGGTCGTCGCGAAA | 134 | |
| SNP22 | GCCTTTCCGGATCCAGGCTTCA | GAAGGGGCATTCGCAGGCCA | 130 | |
| SNP24 | GGATCGCCAGCACGGCGAAG | GAAGCCGTGTTCCCGTCCGAAG | 160 | |
| SNP28 | CACAGCGCCTGCTCGCTGTC | TGCCCATCAACAACCTGGTGCAA | 172 | |
| SNP30 | CTTGCCTTCGGCGCGCATCT | GCCGGTGTCATCCACACCGACT | 129 | |
| SNP31 | CATCATGCGGCCGCGCTCTT | GCCCCGACGATCCCTCGTTCT | 149 | |
| SNP32 | TCGATGCCCAGGCCCGCTT | AGCACCTGCCCGCCCTGTT | 256 | |
| SNP34 | GAACGTGGGCGACAAATTGCCT | CCGACGTTCTGCACGTCGCT | 192 | |
Each 10-plex SBE assay was carried out in a 10-µl reaction volume containing 2.5 µl of SNaPshot Multiplex Ready Reaction Mix (Applied Biosystems), 2 µl of 5 × sequencing buffer (Applied Biosystems), 0.7 µl of cleaned PCR products, and optimized concentrations of ten SBE primers. The sequences and concentrations of SBE primers for SNP20A and SNP20B panels are listed in Table 3. Each SBE reaction was performed for 25 cycles of denaturation at 94 °C for 10 s, primer annealing at 50 °C for 5 s, and extension at 60 °C for 30 s. Unincorporated fluorescently labeled ddNTPs were removed by addition of 1 U of shrimp alkaline phosphatase and incubation at 37 °C for 60 min, followed by incubation at 75 °C for 15 min to inactivate the enzyme. The SBE products (0.5 µl) were mixed with 9 µl of Hi-Di formamide and 0.5 µl of GeneScan 120 LIZ size standard (Applied Biosystems). After heat denaturation for 5 min at 95 °C and rapid cooling on ice, the SBE products were separated using the Applied Biosystems 3130xl Genetic Analyzer with dye set E5. The data analysis was performed with GeneMapper software (Applied Biosystems), and the resulting data were concatenated to produce a 20-position SNP profile for each isolate tested.
Table 3.
Single-base extension (SBE) primers used for the multiplex PCR panels, SNP20A and SNP20B.
| Multiplex PCR panel | SNP name | SBE primer sequence (5′ → 3′)a | Orientationb | Primer size (nt) | Primer conc (µM) |
|---|---|---|---|---|---|
| SNP20A | SNP2 | ATTTCAGATCTTGTCGCAGCG | R | 21 | 0.05 |
| SNP11 | ctgactACGGCGACCAGTCGGTATACG | F | 27 | 0.05 | |
| SNP12 | actgactgactgactCCCGCGTCAAGGCCCTGG | F | 33 | 0.2 | |
| SNP14 | gactgactgactgactgactACGCGCGATGCGGTGACCG | F | 39 | 0.2 | |
| SNP16 | actgactgactgactgactgactgactCGCGCCCAGGTCGGTACG | R | 45 | 0.6 | |
| SNP18 | gactgactgactgactgactgactgactgactTCTGCACCAGGCGCGCCAC | F | 51 | 0.2 | |
| SNP19 | actgactgactgactgactgactgactgactgactGGCTCGAACGCAACATCTATTC | F | 57 | 0.2 | |
| SNP20 | actgactgactgactgactgactgactgactgactgactgactATGTCCAGGACGATGGTCTG | F | 63 | 0.8 | |
| SNP25 | actgactgactgactgactgactgactgactgactgactgactgactgactGCGGCGGGCTCGACCTCA | F | 69 | 0.2 | |
| SNP26 | ctgactgactgactgactgactgactgactgactgactgactgactgactgactCTCAAGTAAATGACGCAAGCC | F | 75 | 0.3 | |
| SNP20B | SNP8 | ctgactgactgactgactgactgactgactgactgactgactgactgactgactgactgactCCCGTCCTGCTGTTTATCCTG | F | 83 | 0.3 |
| SNP15 | ctgactgactAGCTGAGGTCGTAGGCGCG | F | 29 | 0.5 | |
| SNP17 | ctgactgactgactGTGCAGGGGCAGGAAGTCGTG | R | 35 | 0.4 | |
| SNP22 | actgactgactgactgactGCCATTCGAGGACTACGACAGC | R | 41 | 0.1 | |
| SNP24 | actgactgactgactgactgactgactGTCAAGGGCACCAAGCATCT | R | 47 | 0.1 | |
| SNP28 | ctgactgactgactgactgactgactgactgactATCACCATGCCGCTGGCCA | R | 53 | 0.2 | |
| SNP30 | tgactgactgactgactgactgactgactgactgactCGATGAAGTCTTCGTAGGCGAT | F | 59 | 0.1 | |
| SNP31 | tgactgactgactgactgactgactgactgactgactgactgactAACAGCCCGACGAAAACCAG | F | 65 | 0.2 | |
| SNP32 | tgactgactgactgactgactgactgactgactgactgactgactgactTTGCCGGCCCCCACTTTCAGTC | F | 71 | 0.4 | |
| SNP34 | gactgactgactgactgactgactgactgactgactgactgactgactgactgactCTGCGAGAAATTCGGCGTGTC | F | 77 | 0.6 |
aThe sequences in uppercase and lowercase represent the target specific sequence and neutral sequence, respectively.
bOrientation of primer according to the genomic sequence of Bordetella pertussis Tohama: NC_002929.2. F forward; R reverse.
Analysis of allele sequence for virulence-associated genes
Four virulence-associated allelic genes, ptxP, ptxA, prn, and fim3, were analyzed using PCR-based sequencing, as previously described4. The allele profiles were expressed as ptxP/ptxA/prn/fim3 based on the combination of the allelic genes.
MLVA genotyping
PCR-based MLVA targeting VNTR1, VNTR3a, VNTR3b, VNTR4, VNTR5, and VNTR6 loci was performed as previously described4,31,32. The MT27 strain was defined as carrying eight tandem repeats in VNTR1, seven in VNTR3a, zero in VNTR3b, seven in VNTR4, six in VNTR5 and seven in VNTR6 as described previously14.
Data analysis
Analysis of B. pertussis genome data was performed with CLC Genomics Workbench version 8.5.4 (CLC Bio, Denmark) using the reference genome sequence of B. pertussis Tohama I (accession no. NC_002929.2). CSI Phylogeny 1.4 was used to call SNPs from WGS data and infer phylogeny based on the concatenated alignment of SNPs33. Maximum parsimony tree was constructed using MEGA7 version 7.0.2634. Simpson’s diversity index (DI) was calculated as described by Hunter and Gaston35 using the online tool available at http://www.comparingpartitions.info/.
Results
Characteristics of MT27 isolates
Among the 237 B. pertussis MT27 isolates tested, 208 (88%) carried the allele profile of ptxP3/ptxA1/prn2/fim3A, and were the most predominant subtype. The remaining 29 isolates carried the minor allele profiles, ptxP3/ptxA1/prn2/fim3B (n = 19, 8%), ptxP3/ptxA1/prn9/fim3A (n = 5, 2%), ptxP3/ptxA1/prn9/fim3B (n = 2, 0.8%) ptxP3/ptxA1/prn3/fim3A (n = 1, 0.4%), ptxP3/ptxA1/prn14/fim3A (n = 1, 0.4%), and ptxP1/ptxA1/prn15/fim3A (n = 1, 0.4%). The B. pertussis MT27 isolate carrying ptxP1 allele was identified for the first time in Japan.
Discriminatory power and reliability of the SNP genotyping
A total of 237 MT27 isolates were divided into 10 SNP genotypes (SGs) with a Simpson’s DI of 0.79 (95% CI 0.76–0.82) (Fig. 1). Four SGs, SG1, SG2, SG7, and SG10 were the prevalent genotypes, representing 35%, 11%, 22% and 13% of the isolates, respectively. Other SGs, SG3–SG6, SG8, and SG9 were minor genotypes (representing 0.4–7.2% of the isolates). SG2 had a unique 20-position SNP profile because six SNPs (SNP2, SNP8, SNP11, SNP19, SNP20, and SNP34) were specifically associated with SG2. In contrast, five SGs (SG6–SG10) formed a phylogenetic cluster on the maximum parsimony tree (Fig. 1).
Figure 1.

Maximum parsimony tree of Bordetella pertussis MT27 isolates based on 20-position SNP profile. A total of 237 isolates collected in Japan were analyzed by SNP genotyping and were divided into 10 SNP genotypes (SGs). The phylogenetic tree was constructed using MEGA7. The branch length is proportional to the number of SNP differences. The 20-position SNP profiles are shown on the right.
In reference to the 51 sequenced isolates, all 20-position SNP profiles identified by the SNP genotyping were identical to those obtained from WGS data. The SBE-based SNP genotyping showed high reliability.
Comparison with whole-genome SNP genotyping
Figure 2 shows the phylogenetic tree of the 51 MT27 isolates based on whole-genome SNPs. The tree was constructed based on 254 SNPs that were identified among the genome-sequenced isolates using CSI Phylogeny 1.4 (Supplementary Table S3), and it was compared with SBE-based SNP genotyping with 20 SNP targets. Almost all isolates (49/51) were distinguished by the whole-genome SNP genotyping. Twenty-two SG1 isolates collected mainly in the 2000s were not grouped into a small cluster, but other isolates (SG2, SG5–SG8, and SG10, which were mostly collected in the 2010s) were grouped into each category of the SBE-based SNP genotypes. The SG1 isolates had a mean of 20.3 SNP differences between them (intra-clade SNP distance, 2–36 SNPs), whereas other isolates had small SNP differences (SG2, 8.2 differences; SG5, 7.8; SG6, 1.8; SG7, 5.9; SG8, 5.0; SG10, 3.8). Whole-genome SNP genotyping illustrated that the old SG1 strain had high SNP diversity. Except for SG1 isolates, there was good agreement between the SBE-SNP genotyping and the whole-genome SNP genotyping. The reference strain Tohama I (MLVA type, MT83) belonged to SG1.
Figure 2.

Maximum parsimony tree of 51 Bordetella pertussis MT27 isolates based on whole-genome SNPs. A total of 254 SNPs were identified among 51 genome-sequenced MT27 isolates using CSI Phylogeny 1.4. The radial tree was constructed using MEGA7. SBE-based SNP genotypes are highlighted by different colors. Scale bar indicates SNPs.
Temporal changes in SNP genotypes and genotypic diversity
Figure 3 shows the temporal trend of the frequency of SGs during four time periods. The MT27 isolates from 1999 to 2004 (n = 39) belonged to only SG1, whereas those from 2005 to 2009 (n = 24) belonged to SG1, SG2, and SG7. Further, the isolates from 2010 to 2014 (n = 115) belonged to nine SGs (SG1, SG2, and SG4–SG10), and those from 2015 to 2018 (n = 59) belonged to six SGs (SG1, SG3, SG5, SG7, SG8, and SG10). The frequency of the SG1 strain markedly decreased in the 2010s, while the frequency of the SG7 strain significantly increased. Simpson’s DI for the SGs was zero in 1999–2004, 0.16 in 2005–2009, 0.83 in 2010–2014, and 0.76 in 2015–2018. The genotypic diversity of MT27 isolates increased markedly in the 2010s.
Figure 3.
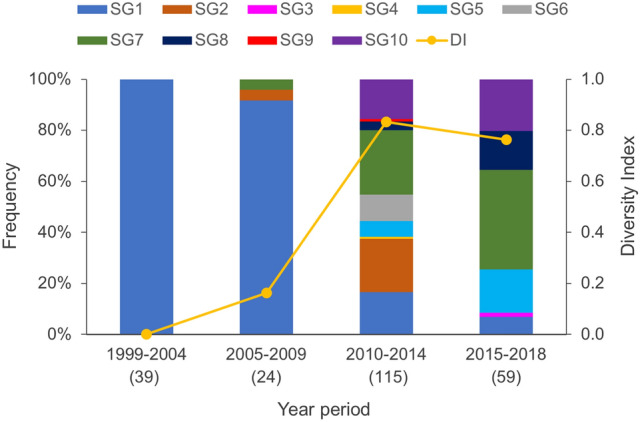
Frequency of SNP genotypes and genotypic diversity of Bordetella pertussis MT27 isolates collected in Japan during 1999–2018. Simpson’s diversity index (DI) was examined with the frequencies of SNP genotypes (SGs) within four time periods. Numbers in parentheses indicate the number of isolates analyzed.
Relationship between SNP genotypes and virulence-associated allelic genes
Seven allele profiles were identified in the MT27 isolates tested. The isolate carrying ptxP3/ptxA1/prn2/fim3A was found to be predominant in all SGs (Table 4). The isolates carrying other allele profiles were in SG1, SG7, and SG10, but the numbers of isolates were much smaller compared with isolates carrying the predominant allele profile. There were no SGs predominantly related to the minor allele profiles; however, all isolates carrying fim3B were in SG1.
Table 4.
Relationship between SNP genotypes and virulence-associated allelic gene profiles in 237 Bordetella pertussis MT27 isolates.
| SNP genotype | No. of isolates | No. of isolates with the allele profile | ||||||
|---|---|---|---|---|---|---|---|---|
| ptxP3/ptxA1/prn2/fim3A | ptxP3/ptxA1/prn2/fim3B | ptxP3/ptxA1/prn9/fim3A | ptxP3/ptxA1/prn9/fim3B | ptxP3/ptxA1/prn3/fim3A | ptxP3/ptxA1/prn14/fim3A | ptxP1/ptxA1/prn15/fim3A | ||
| SG1 | 84 | 61 | 19 | 1 | 2 | 1 | ||
| SG2 | 25 | 25 | ||||||
| SG3 | 1 | 1 | ||||||
| SG4 | 1 | 1 | ||||||
| SG5 | 17 | 17 | ||||||
| SG6 | 12 | 12 | ||||||
| SG7 | 53 | 51 | 1 | 1 | ||||
| SG8 | 13 | 13 | ||||||
| SG9 | 1 | 1 | ||||||
| SG10 | 30 | 26 | 4 | |||||
Application of SNP genotyping to outbreak-associated isolates
MT27 isolates from pertussis outbreaks were genotyped (Table 5). Fifteen outbreak-associated isolates were collected in Miyazaki prefecture36, and all exhibited the same genotype of SG2. Similarly, all isolates from Toyama and Niigata prefectures (n = 4 each) belonged to the same SG10. Of the 12 isolates from Nagano prefecture, 1 and 11 were SG5 and SG7, respectively. The SG5 strain was not closely related to the SG7 strain since the SG5 strain had eight different SNPs in the 20-position SNP profile as compared with the SG7 strain. Together, all except one isolate were classified into the same SNP genotypes for each outbreak.
Table 5.
SNP genotypes of Bordetella pertussis MT27 isolates collected from pertussis outbreaks.
| Location (Prefecture) | Year | Allelic gene profile of isolates | No. of isolates with the SNP genotype | |||
|---|---|---|---|---|---|---|
| SG2 | SG5 | SG7 | SG10 | |||
| Miyazaki | 2010–2011 | ptxP3/ptxA1/prn2/fim3A | 15 | |||
| Toyama | 2015 | ptxP3/ptxA1/prn2/fim3A | 4 | |||
| Nagano | 2016 | ptxP3/ptxA1/prn2/fim3A | 1 | 11 | ||
| Niigata | 2018 | ptxP3/ptxA1/prn9/fim3A | 4 | |||
SNP genotyping for non-MT27 isolates
A total of 104 non-MT27 isolates were classified into 4 SGs (SG1, SG3, SG7, and SG10) with a Simpson’s DI of 0.21 (95% CI 0.11–0.31) (Supplementary Table S4). Ninety-two isolates (89%) belonged to SG1 and the remainder (11%) to SG3, SG7, and SG10. The isolates belonging to SG3, SG7, and SG10 were recent isolates collected in the 2010s. Overall, the genotypic diversity was much lower than that of MT27 isolates.
SNP genotyping for Taiwanese isolates
We genotyped B. pertussis isolates collected in Taiwan during 2010–2019. Of 48 isolates, 30 (62.5%) were MT27 and 18 (37.5%) were non-MT27 (Supplementary Table S5). The MT27 isolates were subdivided into 5 SGs (SG1, SG3, SG5, SG7, and SG11) with a Simpson’s DI of 0.69 (95% CI 0.56–0.82). Fifteen MT27 isolates belonged to SG1 and the remainder to SG3 (n = 4), SG5 (n = 1), SG7 (n = 3), and SG11 (n = 7). Similarly, the non-MT27 isolates were classified into 4 SGs (SG1, SG3, SG7, and SG11) with a Simpson’s DI of 0.73 (95% CI 0.61–0.85). The SG11 was not found in the Japanese isolates tested including non-MT27 isolates, and its 20-position SNP profile was CGAAATTGTCCGGCCTGTTG.
SNP genotypes of US isolates
We determined 20-position SNP profiles of MT27 isolates collected in the US during 2000–2013, based on their complete genome sequences available in the GenBank database16. One hundred twenty-two MT27 isolates were classified into the 6 SGs, i.e., SG1 (n = 89), SG3 (n = 5), SG4 (n = 6), SG7 (n = 20), SG11 (n = 1), and SG12 (n = 1). The minor SG12 was novel and its 20-position SNP profiles was CGAAATCGCCCAGCATGTTG. Simpson’s DI for the SGs was zero in 2000–2004, zero in 2005–2009, and 0.64 in 2010–2013 (Supplementary Fig. S1). The genotypic diversity of the US isolates markedly increased in the 2010s, similar to that of Japanese MT27 isolates.
Discussion
We developed an SBE-based SNP genotyping for the B. pertussis epidemic strain MT27 and evaluated its applicability. The data presented here show that Japanese MT27 isolates were subdivided into ten SNP genotypes and that the genotypic diversity of MT27 isolates markedly increased in the 2010s. Moreover, almost all outbreak-associated MT27 isolates were classified into the same SNP genotypes for each outbreak. The SNP genotyping method allows for subtyping of the recently circulating MT27 strain in routine surveillance and outbreak investigations. This is supported by analyses of Taiwanese and US isolates.
In this study, we demonstrated that the genotypic diversity with 20 SNP targets rapidly increased among Japanese MT27 isolates in the 2010s. One possible cause for the increased diversity is the pertussis epidemic. In Japan, a nationwide epidemic of pertussis occurred between 2008 and 2010, and the frequency of the MT27 strain increased during the epidemic period4. In this study, the SG1-MT27 strain was predominant in 2005–2010, and the genotypic diversity of the MT27 strain increased markedly in the 2010s (Fig. 3). These observations indicate that the SG1–MT27 strain was rapidly replaced with other genotypes (non-SG1) during and after the epidemic. We therefore speculate that B. pertussis has evolved by increasing diversity during pertussis epidemics. A previous study showed that the B. pertussis population (genotype prevalence) increased with changes in vaccine usage (coverage and schedule)17. In Japan, acellular pertussis vaccines (ACVs) were introduced in 1981 instead of whole-cell pertussis vaccines, and there were no changes in the vaccine usage after 1996. Therefore, ACVs were not directly associated with the increased genotypic diversity.
Interestingly, the rapid increase in SNP diversity was also observed among the US MT27 isolates in the early 2010s (Supplementary Fig. S1). In Japan and the US, the frequency of the SG1 strain decreased with time, whereas that of the SG7 strain markedly increased. The Japanese SG7 isolates (n = 53) were collected from 7 out of 8 districts, indicating that the emergence of the SG7 strain occurred nationwide. Similarly, the US SG7 isolates (n = 20) were collected from 13 states16. These observations suggest that the SBE-based SNP genotyping may also apply to recently circulating MT27 strain collected in countries other than Japan. In fact, we confirmed here that the SBE-based SNP genotyping was applicable to recent Taiwanese MT27 isolates. For the Japanese and US SG7 strains, our preliminary analysis with whole-genome SNPs shows that most US SG7 isolates are slightly different from Japanese SG7 isolates (inter-clade SNP distance, 4 SNPs), but one isolate was found to be 100% identical to Japanese SG7 isolates. Further genetic studies are needed to characterize the increasing non-SG1 strains, especially SG7.
The B. pertussis population has significantly changed worldwide in the last 60 years18. Strains carrying the virulence-associated allelic genes ptxP3 and fim3A have emerged and expanded, and those carrying ptxP1 and fim3A had decreased. More recently, ptxP3 strains carrying fim3B (alias fim3-2) have expanded. In this study, most Japanese MT27 isolates carried ptxP3 and fim3A and were classified into 10 SGs based on the 20-position SNP profile (Supplementary Fig. S2). In contrast, all MT27 isolates carrying fim3B (n = 21) were interestingly grouped into only SG1, implying that the fim3B strains are not included in non-SG1 groups. The fim3B strain has increased globally and has the potential to cause recent pertussis epidemics4,23. Our SBE-based SNP genotyping could be helpful to identify the emerging fim3B strain.
Here, whole-genome SNP genotyping showed that non-SG1 isolates clustered into each group of the SBE-based SNP genotypes, but SG1 isolates did not (Fig. 2). Most SG1 isolates were collected in the 2000s, whereas non-SG1 isolates were in the 2010s (Fig. 3). This suggests that the new non-SG1 strains have clonally expanded, but their genetic diversity may further increase with time, similar to the old SG1 strain. Thus, continuous genome surveillance is required for accurate subtyping of B. pertussis MT27 strain, especially for non-SG1 strains.
In the present study, we applied the SBE-based SNP genotyping to outbreak-associated MT27 isolates. Among 12 isolates from the Nagano outbreak, one isolate had a genotype (SG5) different from those of the other 11 isolates (SG7) (Table 5). In the 20-position SNP profile, 8 SNPs were different between the SG5 and SG7 isolates. This indicates that the SG5 isolate was collected from a sporadic case that was not associated with the outbreak. In outbreak investigations, strain typing contributes to understanding bacterial transmission route(s), and rapid typing is important to take countermeasures to prevent further spread of the bacteria. Our SBE-based SNP genotyping is amenable to high-throughput analysis using 96-well plates. Starting from DNA extracts, the SBE-based SNP genotyping was able to analyze 96 isolates within two days, contributing to rapid genotyping, which would be critical in outbreak investigations involving large numbers of isolates.
Culture of B. pertussis has limited sensitivity for previously vaccinated persons, older children, adolescents, and adults37,38. Therefore, molecular strain typing (MLVA and/or MLST) is performed not only on the bacterial isolates, but also on DNA extracts from clinical specimens such as nasopharyngeal swabs22,39–41. Our SBE-based SNP genotyping includes an initial PCR step that amplifies the region around each SNP target. We, therefore, tested the applicability of the SNP genotyping to clinical specimens. Of the 20 clinical specimens that were positive for B. pertussis by a nucleic acid amplification test, 13 (65%) were genotyped by the direct SNP genotyping (Supplementary Table S6). The remaining seven clinical specimens had lower bacterial loads than those of the genotyped specimens (mean Ct values, 23.7 versus 20.4; P < 0.01, Mann Whitney U test), indicating that improvements in the initial PCR conditions (including primer design) are required to increase the analytical power. Although there is room for improvement, SBE-based SNP genotyping has the potential to be directly applicable to clinical specimens.
In this study, we also tested the applicability of the SBE-based SNP genotyping to B. pertussis non-MT27 strains. Our data demonstrated that most Japanese non-MT27 isolates (89%) belonged to SG1, showing low genotypic diversity (Simpson’s DI, 0.21) (Supplementary Table S4). In contrast, high genotypic diversity was seen for Taiwanese non-MT27 isolates (Simpson’s DI, 0.73), although the number of isolates tested was small (Supplementary Table S5). Further analyses are needed for the applicability of the SBE-based SNP genotyping to non-MT27 isolates. A limitation of our SNP genotyping targeting 20 SNPs is that its discriminatory power is lower than that of whole-genome SNP genotyping (targeting over 250 SNPs). Therefore, our simple SNP genotyping cannot provide true genetic relationships among MT27 isolates, especially for SG1 strain of MT27; whole-genome SNP genotyping is required for this purpose.
In conclusion, the present study describes the successful development of a simple and rapid SNP genotyping for the subtyping of B. pertussis MT27 isolates. Since the MT27 strain is the predominant strain in most industrialized countries, our SNP genotyping can serve as a novel alternative to whole-genome SNP genotyping in routine surveillance and outbreak investigations.
Supplementary Information
Acknowledgements
We thank all researchers for providing B. pertussis isolates to the NIID strain collections, especially S. Yoshino (Miyazaki Prefectural Institute for Health and Environment), H. Ohya (Kanagawa Prefectural Institute of Public Health), Y. Arakawa (Nagoya University Graduate School of Medicine), Y. Kimura (Niigata Prefectural Institute of Public Health and Environmental Sciences), and E. Maeda (Nanto Central Hospital). This study was supported by a grant for Research Program on Emerging and Reemerging Infectious Diseases from the Japan Agency for Medical Research and Development, AMED (No. JP18fk0108049).
Author contributions
K.Ka. designed the study and wrote the manuscript. S-M.Y, C-S.C. and N.O. collected B. pertussis samples. K.Ka. and K.K. performed experiments and analyzed data. K.S. discussed the data and supervised the project. All authors reviewed the manuscript.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-84409-0.
References
- 1.McGirr A, Fisman DN. Duration of pertussis immunity after DTaP immunization: A meta-analysis. Pediatrics. 2015;135:331–343. doi: 10.1542/peds.2014-1729. [DOI] [PubMed] [Google Scholar]
- 2.Hallander HO, Gustafsson L, Ljungman M, Storsaeter J. Pertussis antitoxin decay after vaccination with DTPa. Response to a first booster dose 3 1/2–6 1/2 years after the third vaccine dose. Vaccine. 2005;23:5359–5364. doi: 10.1016/j.vaccine.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Wendelboe AM, Van Rie A, Salmaso S, Englund JA. Duration of immunity against pertussis after natural infection or vaccination. Pediatr. Infect. Dis. J. 2005;24:S58–61. doi: 10.1097/01.inf.0000160914.59160.41. [DOI] [PubMed] [Google Scholar]
- 4.Miyaji Y, Otsuka N, Toyoizumi-Ajisaka H, Shibayama K, Kamachi K. Genetic analysis of Bordetella pertussis isolates from the 2008–2010 pertussis epidemic in Japan. PLoS ONE. 2013;8:e77165. doi: 10.1371/journal.pone.0077165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Maas NA, et al. Pertussis in the Netherlands, is the current vaccination strategy sufficient to reduce disease burden in young infants? Vaccine. 2013;31:4541–4547. doi: 10.1016/j.vaccine.2013.07.060. [DOI] [PubMed] [Google Scholar]
- 6.Lam C, et al. Rapid increase in pertactin-deficient Bordetella pertussis isolates Australia. Emerg. Infect. Dis. 2014;20:626–633. doi: 10.3201/eid2004.131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amirthalingam G. Strategies to control pertussis in infants. Arch. Dis. Child. 2013;98:552–555. doi: 10.1136/archdischild-2012-302968. [DOI] [PubMed] [Google Scholar]
- 8.Cherry JD. Epidemic pertussis in 2012–the resurgence of a vaccine-preventable disease. N. Engl. J. Med. 2012;367:785–787. doi: 10.1056/NEJMp1209051. [DOI] [PubMed] [Google Scholar]
- 9.Cassiday PK, Skoff TH, Jawahir S, Tondella ML. Changes in predominance of pulsed-field gel electrophoresis profiles of Bordetella pertussis isolates, United States, 2000–2012. Emerg. Infect. Dis. 2016;22:442–448. doi: 10.3201/eid2203.151136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barkoff AM, et al. Surveillance of circulating Bordetella pertussis Strains in Europe during 1998 to 2015. J. Clin. Microbiol. 2018 doi: 10.1128/JCM.01998-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Loo IH, Heuvelman KJ, King AJ, Mooi FR. Multilocus sequence typing of Bordetella pertussis based on surface protein genes. J. Clin. Microbiol. 2002;40:1994–2001. doi: 10.1128/jcm.40.6.1994-2001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowden KE, et al. Molecular epidemiology of the pertussis epidemic in Washington State in 2012. J. Clin. Microbiol. 2014;52:3549–3557. doi: 10.1128/JCM.01189-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petridou E, et al. Molecular epidemiology of Bordetella pertussis in Greece, 2010–2015. J. Med. Microbiol. 2018;67:400–407. doi: 10.1099/jmm.0.000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schouls LM, van der Heide HG, Vauterin L, Vauterin P, Mooi FR. Multiple-locus variable-number tandem repeat analysis of Dutch Bordetella pertussis strains reveals rapid genetic changes with clonal expansion during the late 1990s. J. Bacteriol. 2004;186:5496–5505. doi: 10.1128/JB.186.16.5496-5505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Z, et al. Genomic epidemiology of erythromycin-resistant Bordetella pertussis in China. Emerg. Microbes Infect. 2019;8:461–470. doi: 10.1080/22221751.2019.1587315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigand MR, et al. Genomic Survey of Bordetella pertussis Diversity, United States, 2000–2013. Emerg. Infect. Dis. 2019;25:780–783. doi: 10.3201/eid2504.180812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zomer A, et al. Bordetella pertussis population dynamics and phylogeny in Japan after adoption of acellular pertussis vaccines. Microb. Genom. 2018 doi: 10.1099/mgen.0.000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bart MJ, et al. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio. 2014;5:e01074. doi: 10.1128/mBio.01074-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Octavia S, et al. Insight into evolution of Bordetella pertussis from comparative genomic analysis: Evidence of vaccine-driven selection. Mol. Biol. Evol. 2011;28:707–715. doi: 10.1093/molbev/msq245. [DOI] [PubMed] [Google Scholar]
- 20.van Gent M, et al. SNP-based typing: A useful tool to study Bordetella pertussis populations. PLoS ONE. 2011;6:e20340. doi: 10.1371/journal.pone.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sealey KL, et al. Genomic analysis of isolates from the United Kingdom 2012 pertussis outbreak reveals that vaccine antigen genes are unusually fast evolving. J. Infect. Dis. 2015;212:294–301. doi: 10.1093/infdis/jiu665. [DOI] [PubMed] [Google Scholar]
- 22.Moriuchi T, et al. Molecular epidemiology of Bordetella pertussis in Cambodia determined by direct genotyping of clinical specimens. Int. J. Infect. Dis. 2017;62:56–58. doi: 10.1016/j.ijid.2017.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Schmidtke AJ, et al. Population diversity among Bordetella pertussis isolates, United States, 1935–2009. Emerg. Infect. Dis. 2012;18:1248–1255. doi: 10.3201/eid1808.120082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Gent M, et al. Analysis of Bordetella pertussis clinical isolates circulating in European countries during the period 1998–2012. Eur. J. Clin. Microbiol. Infect. Dis. 2015;34:821–830. doi: 10.1007/s10096-014-2297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safarchi A, et al. Genomic dissection of Australian Bordetella pertussis isolates from the 2008–2012 epidemic. J. Infect. 2016;72:468–477. doi: 10.1016/j.jinf.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, et al. Whole-genome sequencing reveals the effect of vaccination on the evolution of Bordetella pertussis. Sci. Rep. 2015;5:12888. doi: 10.1038/srep12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchez V, et al. Genomic sequencing of Bordetella pertussis for epidemiology and global surveillance of whooping cough. Emerg. Infect. Dis. 2018;24:988–994. doi: 10.3201/eid2406.171464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeddeman A, et al. Studying Bordetella pertussis populations by use of SNPeX, a simple high-throughput single nucleotide polymorphism typing method. J. Clin. Microbiol. 2015;53:838–846. doi: 10.1128/JCM.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoyama T, et al. Comparison of blood-free medium (cyclodextrin solid medium) with Bordet-Gengou medium for clinical isolation of Bordetella pertussis. J. Clin. Microbiol. 1986;23:1046–1048. doi: 10.1128/JCM.23.6.1046-1048.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohtsuka M, et al. Emergence of quinolone-resistant Bordetella pertussis in Japan. Antimicrob. Agents Chemother. 2009;53:3147–3149. doi: 10.1128/AAC.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurniawan J, et al. Bordetella pertussis clones identified by multilocus variable-number tandem-repeat analysis. Emerg. Infect. Dis. 2010;16:297–300. doi: 10.3201/eid1602.081707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otsuka N, et al. Prevalence and genetic characterization of pertactin-deficient Bordetella pertussis in Japan. PLoS ONE. 2012;7:e31985. doi: 10.1371/journal.pone.0031985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE. 2014;9:e104984. doi: 10.1371/journal.pone.0104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: An application of Simpson's index of diversity. J. Clin. Microbiol. 1988;26:2465–2466. doi: 10.1128/JCM.26.11.2465-2466.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamiya H, et al. Transmission of Bordetella holmesii during pertussis outbreak, Japan. Emerg. Infect. Dis. 2012;18:1166–1169. doi: 10.3201/eid1807.120130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Zee A, Schellekens JF, Mooi FR. Laboratory diagnosis of pertussis. Clin. Microbiol. Rev. 2015;28:1005–1026. doi: 10.1128/CMR.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Konig CH, Halperin S, Riffelmann M, Guiso N. Pertussis of adults and infants. Lancet Infect. Dis. 2002;2:744–750. doi: 10.1016/S1473-3099(02)00452-8. [DOI] [PubMed] [Google Scholar]
- 39.Du Q, et al. Direct molecular typing of Bordetella pertussis from nasopharyngeal specimens in China in 2012–2013. Eur. J. Clin. Microbiol. Infect. Dis. 2016;35:1211–1214. doi: 10.1007/s10096-016-2655-3. [DOI] [PubMed] [Google Scholar]
- 40.Galit SR, et al. Molecular epidemiology of Bordetella pertussis in the Philippines in 2012–2014. Int. J. Infect. Dis. 2015;35:24–26. doi: 10.1016/j.ijid.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Litt DJ, Jauneikaite E, Tchipeva D, Harrison TG, Fry NK. Direct molecular typing of Bordetella pertussis from clinical specimens submitted for diagnostic quantitative (real-time) PCR. J. Med. Microbiol. 2012;61:1662–1668. doi: 10.1099/jmm.0.049585-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its Supplementary Information files.