

Summary
Phosphatidylethanolamine (PE) made in mitochondria has long been recognized as an important precursor for phosphatidylcholine production that occurs in the endoplasmic reticulum (ER). Recently, the strict mitochondrial localization of the enzyme that makes PE in the mitochondrion, phosphatidylserine decarboxylase 1 (Psd1), was questioned. Since a dual localization of Psd1 to the ER would have far-reaching implications, we initiated our study to independently re-assess the subcellular distribution of Psd1. Our results support the unavoidable conclusion that the vast majority, if not all, of functional Psd1 resides in the mitochondrion. Through our efforts, we discovered that mutant forms of Psd1 that impair a self-processing step needed for it to become functional are dually localized to the ER when expressed in a PE-limiting environment. We conclude that severely impaired cellular PE metabolism provokes an ER-assisted adaptive response that is capable of identifying and resolving nonfunctional mitochondrial precursors.
Subject areas: Molecular Physiology, Cell Biology, Proteomics
Graphical abstract
Highlights
-
•
Functional Psd1, the enzyme that makes PE, is mitochondrial localized
-
•
When cellular PE metabolism is impaired, mutant Psd1 is also targeted to the ER
-
•
Mutant Psd1 targeted to the ER is ubiquitinated and rapidly degraded
-
•
Impaired PE metabolism activates a reversible stress response
Molecular Physiology; Cell Biology; Proteomics
Introduction
Mitochondria are vital cellular components that generate energy, phospholipids, amino acids, reducing equivalents, and other essential metabolites. Nearly 99% of the mitochondrial proteome (∼1,000 proteins in yeast and ∼1,500 in mammals) is encoded in the nucleus, translated in the cytosol, and imported into one of four mitochondrial compartments—the outer membrane (OM), intermembrane space (IMS), inner membrane (IM), or matrix. Mitochondrial biogenesis is mediated by a series of dedicated import machineries in each compartment and is safeguarded by the collaborative efforts of an emerging network of factors including those that operate in other cellular compartments (Boos et al., 2019; Bykov et al., 2020; Hansen et al., 2018; Martensson et al., 2019; Matsumoto et al., 2019; Shpilka and Haynes, 2018; Wang and Chen, 2015; Weidberg and Amon, 2018; Wrobel et al., 2015). A small proportion of the nuclear-encoded mitochondrial proteome mediates and/or facilitates lipid biosynthetic or trafficking steps critical for the proper functioning of this organelle (Acoba et al., 2020; Sam et al., 2019). Mitochondrial lipid biosynthesis is dependent on the acquisition of phospholipid precursors provided by the endoplasmic reticulum (ER)/vacuole that fuel the generation of phosphatidic acid, phosphatidylglycerol, cardiolipin, and phosphatidylethanolamine (PE) (Elbaz-Alon et al., 2014; Honscher et al., 2014; Kawano et al., 2017; Kojima et al., 2016; Lu and Claypool, 2015). Mitochondrial PE is produced by phosphatidylserine decarboxylase-1 (Psd1), an enzyme that is embedded in the IM with its catalytic site in the IMS and which removes the carboxyl group from phosphatidylserine (PS) in either the IM or potentially the OM (Aaltonen et al., 2016; Tamura et al., 2012b). The newly produced PE can then be exported from mitochondria and tri-methylated to phosphatidylcholine (PC) in the ER (Cui et al., 1993; Horvath et al., 2012; Samborski et al., 1990; Tamura et al., 2012b).
The Psd1 precursor contains a bipartite N-terminus that consists of a mitochondrial targeting signal (MTS) followed by a hydrophobic transmembrane domain (TM). Upon engaging the TIM23 translocase in the IM, the TM located carboxy terminal to the MTS stops the translocation process and promotes lateral release of the Psd1 precursor into the IM (Horvath et al., 2012). Following two processing steps performed by matrix resident peptidases, membrane-anchored Psd1 performs a final self-processing event termed autocatalysis (Horvath et al., 2012; Onguka et al., 2015; Tamura et al., 2012b). Autocatalysis is mediated by an evolutionarily conserved catalytic triad that provides a single-use serine protease activity (Choi et al., 2015; Ogunbona et al., 2017; Watanabe et al., 2020). This self-processing event occurs at the conserved Leucine-Glycine-Serine (LGS) motif and results in the formation of two subunits, α and β that remain non-covalently attached, and an N-terminal pyruvoyl prosthetic group on the α subunit that is essential for Psd1 activity (Li and Dowhan, 1988; Recsei and Snell, 1984). Interestingly, overexpression of mitochondrial precursor proteins with N-terminal bipartite signals, including Psd1, evokes a novel stress response called the mitochondrial compromised protein import response or mito-CPR (Weidberg and Amon, 2018).
Yeasts lacking Psd1 have decreased respiratory capacity (Baker et al., 2016; Bottinger et al., 2012; Calzada et al., 2019) similar to that of PISD-deficient mammalian cells (Heden et al., 2019; Tasseva et al., 2013). Deletion of Psd1 also impairs autophagic capacity (Nebauer et al., 2007), decreases cell growth (Birner et al., 2001), reduces membrane fusion (Chan and McQuibban, 2012), and impairs OM protein biogenesis (Becker et al., 2013). In yeast, some of these defects can be completely or partially rescued through alternative PE biosynthetic pathways that reside in the ER, such as lyso-PE supplementation (Riekhof et al., 2007) or PE generation through the Kennedy pathway (Calzada et al., 2019; Riekhof and Voelker, 2006). Still, the limitations of these non-mitochondrial PE pathways to replace mitochondrial Psd1 are highlighted by the fact that murine whole body pisd-/- mice die in utero and that rare pathogenic PISD mutations are associated with mitochondrial disease (Fullerton et al., 2007; Girisha et al., 2019; Peter et al., 2019; Selathurai et al., 2019; Steenbergen et al., 2005; Zhao et al., 2019).
Phosphatidylserine decarboxylases (PSDs) are conserved from bacteria to humans, and type I PSDs have been documented to localize to the mitochondrial IM in Arabidopsis (Rontein et al., 2003), Saccharomyces cerevisiae (Horvath et al., 2012; Tamura et al., 2012b), rats (Dennis and Kennedy, 1972; van Golde et al., 1974; Zborowski et al., 1983), hamster cells (Kuge et al., 1996; Voelker, 1985), and humans (Keckesova et al., 2017). Although there are Psd1 paralogs, classified as type II PSDs, that localize to endosomes in yeast (Psd2) (Gulshan et al., 2010) and to the tonoplast (PSD2) or ER (PSD3) in Arabidopsis (Nerlich et al., 2007), only the mitochondrial type I PSD is present in humans (PISD). The yeast mitochondrial Psd1 protein (Psd1) shares 48% identity with human PISD, while it only shares 24% identity with the non-mitochondrial Psd2 (Altschul et al., 1997, 2005).
Tools to monitor phospholipid trafficking in real time are limited (Wills et al., 2018). As a surrogate, definitive information on the localization of enzymes in a sequential phospholipid metabolic pathway provides information on the trafficking steps that must have occurred. This paradigm has been extensively used to define steps involved in the PS to PE to PC pathway (Achleitner et al., 1995; Daum et al., 1986; Hovius et al., 1992; Kojima et al., 2016; Lahiri et al., 2014; Tamura et al., 2012a; Vance, 1991; Voelker, 1985). Further, it has served as the foundational premise for work leading to the molecular identification of the first mitochondria-ER contact site structure, ER and Mitochondria Encounter Structures (ERMES) (Kornmann et al., 2009; Nguyen et al., 2012), and was exploited to confirm the functional importance of contact sites between mitochondria and vacuoles (Elbaz-Alon et al., 2014; Honscher et al., 2014).
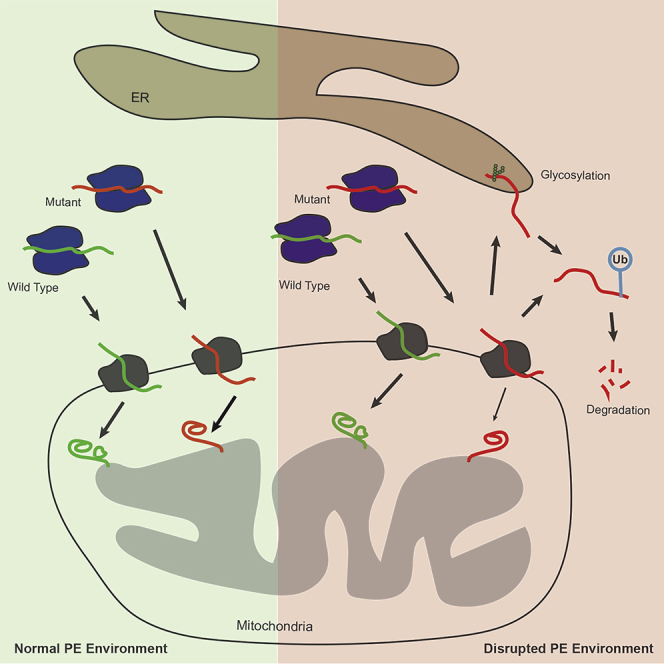
Recently, the foundation of this paradigm has been called into question. It was reported that a functionally significant fraction of Psd1 is N-glycosylated and thus targeted to the endomembrane system (Friedman et al., 2018). Moreover, the relative abundance of glycosylated Psd1 was reported to be sensitive to metabolic growth conditions, being most abundant when yeast cells are grown on fermentable carbon sources, like dextrose. Combined with studies that employed Psd1 chimeras targeted to either the ER or IM, the metabolically sensitive distribution of Psd1 was taken as evidence that Psd1 produces two distinct subcellular pools of PE that have non-redundant functions (Friedman et al., 2018).
Given how a dual localization of Psd1 would impact the interpretation of numerous studies relying on its strict mitochondrial localization, here, using yeast and mammalian models, we re-examined its subcellular localization and determined that wild-type (WT) Psd1 co-fractionates and co-localizes exclusively with mitochondria. Multiple Psd1 chimeras targeted to the IM via sequence elements provided by other topologically similar IM proteins functionally replace WT Psd1. Interestingly, we confirmed that a significant proportion of an autocatalytic mutant of Psd1, which is enzymatically non-functional, is N-glycosylated and thus ER targeted (Friedman et al., 2018). Additional mutations that impair autocatalysis are also partially directed to the ER. In contrast to the mitochondrial targeted autocatalytic mutant Psd1, the glycosylated mutant Psd1 present in the ER is ubiquitinated and rapidly degraded. The accumulation of glycosylated non-functional Psd1 in the ER only occurs when cellular PE metabolism is significantly disrupted and can be prevented by supplements that stimulate different lipid biosynthetic pathways that reside in the ER and that do so via distinct mechanisms. In addition to re-establishing the strict mitochondrial localization of functional Psd1, our work supports that Psd1 quality assurance is in part monitored during its import and enforced with the assistance of the ER and ubiquitin-proteasome system. We conclude that severely impaired cellular PE metabolism provokes an adaptive response that is capable of identifying and resolving nonfunctional mitochondrial precursors.
Results
Functional Psd1 is a mitochondrial resident
Given the recent suggestion that Psd1 has dual residences (Friedman et al., 2018), we decided to thoroughly re-evaluate its subcellular localization using multiple distinct yeast strain backgrounds. First, we compared the specificity of two in-house generated Psd1-specific antibodies raised in two different rabbits (4,077 and 4,078) with similar sensitivities (Figure 1A). Notably, although each antiserum recognized the Psd1 β subunit in mitochondria, they each cross-reacted with several closely migrating background bands that were importantly not the same. Thus, each antiserum specifically recognizes Psd1 β but also reacts with different non-Psd1 proteins.
Figure 1.

Endogenous and overexpressed Psd1 is localized to mitochondria
(A) Psd1 detection range was determined for antisera raised in two rabbits 4,077 (green) and 4,078 (blue) using a standard curve of recombinant Psd1. Bleeds number 4 and 5 were analyzed, respectively. The amount of Psd1 detected in WT mitochondria was calculated (n = 3).
(B) Cell extracts derived from pairs of WT and psd1Δ yeast of the indicated backgrounds grown at 30°C in synthetic complete dextrose (SCD) were treated with EndoH as listed and analyzed by immunoblot using the designated anti-Psd1 antisera; Aac2 served as loading control. psd2Δpsd1Δ yeast stably transformed with WT (::WT) or ER-targeted Psd1 (::ER-Psd1) acted as overexpression and glycosylation controls, respectively. Due to its high relative expression, different exposures of ::ER-Psd1 are shown versus all other samples (indicated by frames) which were resolved on the same gel (n = 3).
(C) Following growth in SCD medium to late log phase, fractions were collected from the indicated yeast strains by differential gravity centrifugation. Equal protein amounts from each collected fraction were resolved by SDS-PAGE and immunoblotted for Psd1 and mitochondrial (Qcr6), ER (Sec62), and cytosolic (Hsp70) markers. SM, starting material, P13, pellet of 13,000xg; P21, pellet of 21,000xg; P40, pellet of 40,000xg; and S40, supernatant of 40,000xg (n = 3).
(D) Mitochondria (P13) and ER (P40) fractions were mock or EndoH treated prior to immunoblot analysis (n = 3).
(E) Tandem mass tag (TMT) comparison of ER (P40) proteomes from psd2Δpsd1Δ (n = 3 preps) and psd2Δpsd1Δ::WT yeast (n = 4 preps).
(F) Representative images of HEK293 cells overexpressing FLAG-tagged WT PISD obtained via immunofluorescence to visualize PISD (anti-FLAG; green), mitochondria (anti-TOMM20; red), and ER (anti-calnexin; gray). Bottom panels are a magnification of the white-boxed areas shown in the upper panel. Intensity profile for PISD (green), mitochondria (red), and ER (gray) along the pixels indicated by a solid white line. Scale bars, 5 μm.
Next, we tested the N-glycosylation status of endogenous Psd1 using endoglycosidase H (EndoH) in cell extracts derived from four different yeast strain backgrounds—GA74-1A, BY-4741, D273-10B, and W303—grown in synthetic complete dextrose (SCD) medium (Figure 1B). Neither the same antiserum (4,077) as used by (Friedman et al., 2018) nor our second polyclonal Psd1 antibody (4,078) detected an EndoH-sensitive form of Psd1 in any of the tested WT backgrounds. As a glycosylation control, we analyzed a psd2Δpsd1Δ strain expressing an ER-targeted Psd1 chimera (designated ::ER-Psd1) which as expected (Onguka et al., 2015) was glycosidase sensitive. Notably, a glycosylated form of Psd1 was also not detected in psd2Δpsd1Δ yeast expressing WT Psd1 (noted as ::WT) at higher than endogenous levels or in WT and psd1Δ strains employed in the study by Friedman et al (Figure S1 (Friedman et al., 2018)).
If Psd1 is glycosylated, it should accumulate in membrane fractions that are lighter than mitochondria. However, endogenous Psd1 in GA74-1A yeast grown in rich dextrose media (yeast extract peptone dextrose (YPD)) did not co-fractionate with the microsomal fraction (P40) following differential centrifugation, in contrast to ER-Psd1 (Figure 1C). Consistently, we did not detect an EndoH-sensitive form of WT Psd1 in either the P13 (mitochondria and ER) or P40 fractions (Figure 1D). In contrast, ER-Psd1, which co-fractionated with ER-associated mitochondria (P13) and microsome-enriched membranes (P40), was sensitive to the glycosidase. Further, a tandem mass tag (TMT) proteomics comparison of P40 fractions from ::WT and psd2Δpsd1Δ failed to detect Psd1-derived tryptic peptides in the microsome fractions from the yeast overexpressing WT Psd1 (Figure 1E). Our failure to detect Psd1, glycosylated or otherwise, in microsomes is consistent with the lack of significant Psd1 activity in this compartment when the non-mitochondrial Psd2 is missing (Onguka et al., 2015; Trotter and Voelker, 1995).
Finally, to extend our analysis to human cells, the localization of human PISD was determined by super-resolution microscopy in HEK293 cells over-expressing PISD harboring a C-terminal FLAG tag (Figure 1F). The anti-FLAG signal for PISD (green) overlapped with the mitochondrial marker, TOMM20 (red), but not with the ER marker, calnexin (Figure 1F). Immunofluorescence signal intensity plots of super-resolution images quantitatively showed that WT human PISD sharply overlapped with a mitochondrial marker. Collectively, our results support the unavoidable conclusion that the vast majority, if not all, functional Psd1 resides in the mitochondrion.
Psd1 targeted to, and embedded in, the IM via non-Psd1 signals are functional
Friedman et al. used a chimeric strategy in which Psd1 was either directed to the ER or the IM, the latter by virtue of replacing the MTS and TM of Psd1 with the equivalent information from Mic60, and concluded that Psd1 targeted to either the mitochondria or the ER produces two distinct subcellular pools of PE that have non-redundant functions (Friedman et al., 2018). Specifically, when chimeric Psd1 is forced to exclusively localize to the IM, it is fully capable of supporting respiratory growth that requires mitochondrial energy production; however, it is unable to promote robust growth on dextrose that does not depend on energy from oxidative phosphorylation (Friedman et al., 2018). Given the significance of the reported growth phenotype with respect to Psd1 function and the underlying lipid trafficking steps needed to support its activity, we decided to adopt and expand upon this chimeric strategy. To this end, we designed a series of constructs, each containing a C-terminal 3XFLAG tag to monitor autocatalysis, in which the N-terminal MTS and TM domains of Psd1 were replaced by equivalent components from other inner membrane proteins with the same topology (Tim50, Yme1, Tim54, Mic60, and Ccp1) (Figure 2A). Each chimera, similar to previously characterized OM-Psd1 (Calzada et al., 2019) and ER-Psd1 (Onguka et al., 2015), produced Psd1 β subunits that predictably varied in molecular weight and α subunits that were the same size, indicating that they each retained self-processing activity (Figure 2B). Consistent with their autocatalytic status, each chimera rescued the psd2Δpsd1Δ ethanolamine auxotrophy (Figure 2C) and generated mitochondrial PE levels that were indistinguishable from ::WT, with only one exception (Figures 2D and 2E). The elevated mitochondrial PE in ::54-Psd1 mitochondria is highly reminiscent of what we previously observed when Psd1 is re-directed to the OM (OM-Psd1 (Calzada et al., 2019). As such, we determined the subcellular and submitochondrial localization of each chimera in comparison to WT Psd1. The β and α subunits of each chimera co-fractionated with the mitochondrial inner membrane marker, Qcr6, and not the endosomal marker, Sec62 (Figure S2A). Using a well-established protease protection assay in isolated mitochondria, we determined that like WT Psd1, each chimera, with the possible exceptions of 54-Psd1 and Ccp1-Psd1, was protected from protease in intact mitochondria (Mito), similar to IM (Tim54) and matrix (Abf2) proteins, and unlike proteins in the OM (Tom70) (Figure S2B). Of note, a modest fraction of the 54-Psd1 chimera was degraded in intact mitochondria treated with protease, which could indicate that a proportion of this chimera is stuck in the OM and thus explain its elevated mitochondrial PE levels (Figure 2E).
Figure 2.

Re-localized Psd1 constructs are stable and functional
(A) Schematic of chimeric constructs. MTS (mitochondrial targeting signal) and TM (transmembrane domain) of residues are indicated. Psd subunits β, α, and LGS motif are shown. All constructs have a 3XFLAG tag at the C-terminus.
(B) The indicated strains were pre-cultured at 30°C in YPD, and after isolation of whole-cell extracts, the α and β subunits of Psd1 were analyzed by immunoblotting. Pic1 served as a loading control (n = 3).
(C) The indicated strains pre-cultured at 30°C in YPD were spotted onto SCD with (+) or without (−) 2 mM ethanolamine and incubated at 30°C for 3 days (n = 3).
(D) Mitochondrial phospholipids from the indicated strains were labeled overnight in rich lactate medium spiked with 32Pi and separated by TLC. The migration of phosphatidylcholine (PC), phosphatidylinositol (PI), PS, phosphatidylglycerol (PG), PE, phosphatidic acid (PA), and CL is indicated (n = 6).
(E) The relative abundance of PE was determined for each strain (mean ± SEM for n = 6). Statistical differences (2 symbols p ≤ 0.01; 3 symbols p ≤ 0.001 compared to ::WT (∗) or psd2Δpsd1Δ (#)) were calculated by unpaired Student's t-test (for both ::Yme1 comparisons) or Mann-Whitney rank-sum test (all the rest).
(F) Serial dilutions of the indicated strains were spotted onto SCD with or without 2 mM ethanolamine, synthetic complete lactate (SC Lactate), or synthetic complete ethanol-glycerol (SCEG) plates and incubated at 30°C or 37°C for the indicated duration (n = 3).
Finally, we determined the growth phenotypes of psd2Δpsd1Δ yeast expressing the assorted chimeras in comparison to WT Psd1, OM-Psd1, and ER-Psd1 on synthetic media containing dextrose with or without ethanolamine, lactate, or ethanol glycerol (Figure 2F). Each chimera, including Mic60-Psd1, grew similar to each other and to WT Psd1 in SCD with or without ethanolamine at 30°, but some chimeras (50-Psd1, Yme1-Psd1, 60-Psd1, and OM-Psd1) displayed a slight temperature-sensitive phenotype at 37°C (Figure 2F). With the sole exception of 54-Psd1, each chimera rescued the severe respiratory growth phenotype of psd2Δpsd1Δ yeast similar to WT Psd1. In contrast, ::54-Psd1 growth was delayed compared to WT Psd1 (and the other chimeras) in respiratory conditions (synthetic complete lactate and synthetic complete ethanol-glycerol) (Figure 2F). This growth phenotype correlates with the previous observation that strains with elevated mitochondrial PE levels (such as OM-Psd1 and ER-Psd1) have diminished respiratory capacity (Calzada et al., 2019). Our combined results indicate that Psd1 targeted to and embedded in the inner membrane via non-Psd1 elements functions for the most part the same as WT Psd1. Based on the absence of any overt phenotype, we conclude that Psd1 contained within mitochondria is capable of supporting all activities, mitochondrial and non-mitochondrial, that depend on the PE that it produces.
Glycosylation of mutant Psd1 requires severe cellular membrane dyshomeostasis
Psd1 autocatalysis is necessary for PE synthesis, and amino acids required for this process have been functionally characterized (Choi et al., 2015; Ogunbona et al., 2017) including residues at the site of cleavage (LGS motif) and catalytic residues in its vicinity (Watanabe et al., 2020). With the goal of determining whether or not the putative glycosylated Psd1 and assorted IM- and ER-directed chimeras were capable of autocatalysis, non-functional, S463A mutant forms of Psd1, unable to perform autocatalysis, were used as immunoblotting controls and yielded Psd1 products that indeed migrated slower than their fully processed counterparts (Friedman et al., 2018). Notably, the relative amount of the glycosylated non-functional Psd1-S463A was significantly higher than that of the WT protein.
Therefore, to confirm and potentially extend this previous observation, we analyzed the glycosylation status of Psd1 mutants for each amino acid of the catalytic triad—S463A, H345A, and D120A. Consistent with (Friedman et al., 2018), we observed both EndoH-resistant (Mito β-α; blue arrow) and EndoH-sensitive (ER β-α; purple brackets) unprocessed forms for all three catalytic triad mutants (Figure 3A). These higher molecular weight protein bands were not detected in ::WT where only mature Psd1 β (Mito β; pink arrow) and the released Psd1 α were detected.
Figure 3.

Glycosylation of endogenous and overexpressed mutant, nonfunctional Psd1 requires severe disruption of cellular PE metabolism
(A) Cell extracts from the indicated strains grown at 30°C in YPD were treated with EndoH as listed and analyzed by immunoblot using the designated anti-Psd1 antisera; Aac2 served as loading control. ER β-α, glycosylated mutant Psd1; Mito β-α, mutant Psd1 not glycosylated (n = 3).
(B) TMT comparison of ER (P40) proteomes from psd2Δpsd1Δ::LGS (n = 3 preps) and psd2Δpsd1Δ::WT yeast (n = 4 preps). LGS; autocatalytic mutant Psd1.
(C) Cell extracts from the indicated strains grown at 30°C in YPD were immunoblotted for the Psd1 β subunit; Kgd1, Aac2, and Qcr6 served as loading controls. LGS was knocked into endogenous PSD1 of the listed strains via Hi-CRISPR (n = 3).
(D) Serial dilutions of same strains as in (C) were spotted onto SCD with or without 2 mM ethanolamine (+Eth) and incubated at 30°C for 3 days (n = 3).
(E) EndoH sensitivity assay of cell extracts from indicated strains grown in YPD at 30°C. Immunoblots were probed for Psd1 β (4,077.4) and Psd1 α (FLAG); Pic1 served as loading control (n = 3).
(F) PSD1 mRNA level in the indicated strains determined by two-step reverse transcription-quantitative PCR of PSD1 and normalized to ACT1 (means ± SEM, n = 4). Statistical differences (1 symbol, p ≤ 0.05; 2 symbols p ≤ 0.01; 3 symbols p ≤ 0.001 compared WT (PSD2 +, PSD1 +) were calculated with unpaired Student's t-test.
(G) Mitochondrial phospholipids from the indicated strains were labeled overnight in YPD medium spiked with 14C-Acetate and separated by TLC (n = 6).
(H) Cell extracts derived from the indicated strains grown at 30°C in YPD were analyzed by immunoblot for Psd1 β (4,077.4), Psd1 α (FLAG), Cho1 (PS synthase), and Kar2 (ER chaperone); Aac2 acted as loading control (n = 5).
(I) The relative abundance of ER β-α and Mito β-α in LGS mutant Psd1 expressing yeast of the indicated genotype was determined (means ± SEM for n = 5). Statistical differences (2 symbols p ≤ 0.01; 4 symbols p ≤ 0.0001) were determined by one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test.
Next, we performed a TMT proteomics comparison of the microsome-enriched P40 fractions from ::WT versus those purified from psd2Δpsd1Δ::Psd1-LGS/AAA (noted as ::LGS and Psd1-LGSFlag as it also has C-terminal 3XFLAG). Psd1, which was not elevated in P40 fractions from ::WT versus those isolated from Psd-lacking yeast (Figure 1E), was detected at significantly increased levels in Psd1-LGSFlag (::LGS) versus ::WT microsome-enriched fractions (Figure 3B). Consistently, cell fractionation analysis of Psd1-LGSFlag showed ER β-α in all fractions except the cytosol/S40 (Figure S3A). Notably, the unprocessed Psd1 Mito β-α band from Psd1-LGSFlag had a similar fractionation profile to Mito β from ::WT with each being highly enriched in the Qcr6-containing mitochondrial P13 fraction. As only two additional mitochondrial proteins (Mpm1 and Tom5) were significantly elevated in the Psd1-LGSFlag microsomes (Figure 3B), we conclude that the accumulation of Psd1-LGSFlag in the ER is specific to this nonfunctional form of Psd1.
Since overexpression of WT Psd1 induces the mito-CPR import stress response (Weidberg and Amon, 2018), we considered the possibility that the accumulation of glycosylated autocatalytic mutants of Psd1 was a by-product of their overexpression. Therefore, we used homology-integrated (HI) clustered regularly interspaced short palindromic repeats (CRISPR) to engineer two strains with the LGS/AAA mutation inserted into the PSD1 gene (designated as Psd1-LGS as they lack appended 3XFLAG tag). This mutation was edited in strains with and without PSD2. Consistent with psd2Δpsd1Δ strains transformed with autocatalytic Psd1 mutants (Figure 3A), two unprocessed forms of Psd1-LGS were detected in the absence of PSD2, Mito β-α and ER β-α, the latter of which was EndoH sensitive (Figures 3C and 3E). Surprisingly, when PSD2 was present, only one unprocessed form of Psd1-LGS was detected, Mito β-α, which was not sensitive to glycosidase (Figures 3C and 3E). Importantly, in the absence of PSD2, Psd1-LGS failed to support growth without ethanolamine supplementation (Figure 3D). We noticed that the steady-state level of Psd1-LGS was increased in the absence versus the presence of PSD2 (Figure 3C). Since PSD1 mRNA levels were similar between these two strains (Figure 3F), the basis for this difference is post-transcriptional. As expected (Calzada et al., 2019), mitochondrial PE was reduced or essentially absent when Psd1-LGS was expressed in the presence or absence of PSD2, respectively (Figures 3G and S3D).
Our failure to detect the ER β-α form of Psd1-LGS in the context of a functional Psd2 suggested that its accumulation may require severe perturbation of cellular PE metabolism. Consistent with this possibility, when Psd1-LGSFlag was overexpressed in an otherwise WT yeast strain (PSD1+, PSD2+), only one unprocessed form of Psd1-LGSFlag was detected which co-fractionated with the Qcr6- and endogenous Psd1-containing mitochondrial P13 fraction (Figure S3A). Interestingly, when expressed in WT yeast, Psd1-LGSFlag was resistant to protease in intact mitochondria and only became accessible upon osmotic disruption of the OM or addition of detergent (Figure S3B). In contrast, when expressed in psd2Δpsd1Δ yeast, a noticeable amount of Psd1-LGSFlag was resistant to protease upon OM rupture (Figure S3B) and which was not related to any changes in its aggregation status (Figure S3C).
To directly test if the accumulation of autocatalytic mutant ER β-α depends on a severely compromised phospholipid metabolism, we performed an epistasis analysis in which Psd1-LGSFlag was integrated into strains lacking Psd1, Psd2, or Cho1, singly or in combination (Figure 3H). CHO1 encodes the ER-resident PS synthase (Vance, 1990) that provides the substrate, PS, that is, decarboxylated to PE by Psd1 and Psd2. Of consideration is that the cho1Δpsd2Δpsd1Δ::LGS strain was generated through Hi-CRISPR-based deletion of CHO1 in the psd2Δpsd1Δ::LGS parental strain; as such the abundance of Psd1-LGSFlag forms in these strains is directly comparable. As observed for LGS knock-in strains (Figure 3C), glycosylated ER β-α was not detected in the absence of Psd1 (psd1Δ) or Psd2 (psd2Δ) alone but was detected in their combined absence (Figures 3H and 3I). Further, glycosylated ER β-α accumulated in the absence of Cho1, and consistent with its role upstream of Psd1 and Psd2, the amount of ER β-α was not further increased upon its deletion in psd2Δpsd1Δ yeast (Figures 3H and 3I). These results indicate that the accumulation of the glycosylated, non-functional Psd1 precursor in the ER requires severe disturbance in cellular PE metabolism.
Additional autocatalytic Psd1 mutants are also glycosylated
We screened a series of 33 alanine mutants of evolutionarily conserved residues by fluorescence immunoblotting with the goal of identifying additional mutations that either yield glycosylated forms of Psd1, regardless of their impact on autocatalysis, or that impair autocatalysis but are not partially glycosylated (Figures 4A and 4B). In this assay, Psd1 constructs that have successfully performed autocatalysis form mature Psd1 β (red) and Psd1 α (green) subunits that migrate separately, as for WT Psd1. In contrast, any mutation that impairs or ablates autocatalysis results in unprocessed forms of Psd1 that are co-detected by the Psd1 β and Psd1 α antisera (yellow/orange), as observed for autocatalytic null Psd1-LGSFlag. For Psd1 mutants in which autocatalysis was not impeded, only a single Psd1 β band was detected that was not recognized by the Psd1 α antibody. Thus, no mutants that performed autocatalysis were glycosylated. We identified several additional mutations (R196A, R385A, 402-VGAT/AAAA-405, and 407-VGSI/AAAA-411) beyond the autocatalytic triad (D210, H345, and S463) that disrupted autocatalysis, each of which generated both unprocessed forms of Psd1, Mito β-α, and ER β-α (Figures 2A and 2B). Intriguingly, three mutations (R189A, K230A, and H346A) that only partially impaired autocatalysis and rescued the psd2Δpsd1Δ ethanolamine auxotrophy (Figures 4C and 4D) generated all three forms of Psd1, Mito β (pink arrow), Mito β-α (blue arrow), and ER β-α (purple arrow) (Figures 4A and 4B). It is worth mentioning that the co-detected (yellow) Psd1 bands migrating below Mito β are proteolytic fragments derived from unprocessed Mito β-α that are generated by mitochondrial proteases and accumulate when autocatalysis is impaired (Ogunbona et al., 2017). From this screen, we conclude that in the context of impaired cellular PE metabolism, any mutation that perturbs autocatalysis results in the accumulation of glycosylated ER β-α.
Figure 4.

Mutations that indirectly impair autocatalysis are also glycosylated
(A and B) Cell extracts derived from the indicated strains grown at 30°C in YPD were analyzed by fluorescent immunoblot for Psd1 β (red) and Psd1 α (green); Pic1 acted as loading control (n = 3).
(C and D) The same strains in (A and B) were pre-cultured at 30°C in YPD, spotted onto SCD with (+) or without (−) 2 mM ethanolamine and incubated at 30°C for 3 days (n = 3).
(E) Homology model structure of Psd1 based on the E. coli PSD structure (PDB code: 6L06)—view from the side of membrane. The α subunit and β subunit are colored in red and cyan, respectively. The right panel is a magnified view around S463 (pyruvoyl group at N-terminus of α subunit). Residues whose mutation to alanine impairs autocatalysis are indicated and colored in magenta. Autocatalytic triad residues are underlined and shown in stick form.
(F) The distances were measured between the carbonyl carbon of the pyruvoyl group (S463) and the alpha carbon of the residues whose mutation to alanine impairs autocatalysis.
To gain further insight, we built a homology model of yeast (Saccharomyces cerevisiae) Psd1 based on the Escherichia coli crystal structure (PDB code: 6L06) (Watanabe et al., 2020) using SWISS-MODEL (Figure 4C). Mutations that were not compatible with autocatalysis cluster in the immediate vicinity of S463 (cleavage occurs between G462 and S463). Similar to D210 (10.7 Å) and H345 (7.8 Å), H346 (10.1 Å), R385 (10.3 Å), VGAT (5.5–9.0 Å), and VGSI (6.5–9.9Å) are in close proximity to S463, the residue onto which the pyruvoyl prosthetic group is attached post-autocatalysis (Figure 4D). On the other hand, R189, R196, and K230 are greater than or equal to 15 Å away from S463 (Figure 4D). As R189 and R196 are close to the catalytic triad residues H345 and D210, respectively (Figure 4C), we speculate that their mutation to alanine may secondarily impact the positioning of these important residues that form the base and acid of the triad. K230, whose mutation also incompletely impairs autocatalysis (Figure 4A), is relatively distant from the catalytic site and thus may contribute to structural stabilization of an autocatalytic-competent form of the Psd1 precursor.
Supplements of phospholipid synthesizing pathways decrease glycosylated mutant Psd1
Next, we asked if supplements that feed PE-biosynthetic pathways outside of the mitochondrion could prevent the accumulation of the glycosylated, non-functional mutant Psd1 protein in the ER. Indeed, supplementation of YPD with either ethanolamine, which promotes PE production via the ER-resident Kennedy pathway, or lyso-PE (LPE), which is converted to PE by Ale1, significantly diminished the relative amount of the glycosylated precursor protein (ER β-α; Figures 5A and 5B). LPE supplementation, which was shown to fully rescue mitochondrial defects when Psd1 is absent (Riekhof et al., 2007), decreased the detection of both Mito β-α and ER β-α precursor bands to a stronger degree than ethanolamine supplementation, which incompletely rescues the mitochondrial defects when Psd1 is missing (Calzada et al., 2019).
Figure 5.

Supplements for distinct ER-resident phospholipid biosynthetic pathways decrease accumulation of glycosylated nonfunctional Psd1
(A) Pre-cultures of the indicated strains were inoculated in YPD medium supplemented with 1% (v/v) Tergitol (−), 0.5mM lyso-phosphatidylethanolamine in 1% (v/v) Tergitol (L), or 2mM ethanolamine (E). Following overnight growth at 30°C, cell extracts were harvested and immunoblotted for Psd1 β, Psd1 α, Cho1, and Kar2; Aac2 acted as loading control (n = 5).
(B) The relative abundance of LGS mutant Psd1 ER β-α and Mito β-α was determined from yeast analyzed in (A) (mean ± SEM, n = 5).
(C) Pre-cultures of the indicated strains were inoculated in YPD medium alone or supplemented with 2mM ethanolamine (E) or 2mM choline (C). Following overnight growth at 30°C, cell extracts were harvested and immunoblotted as listed.
(D) The relative abundance of LGS mutant Psd1 ER β-α and Mito β-α was determined from yeast analyzed in (C) (mean ± SEM, n = 5). For (B) and (D), statistical differences (3 symbols p ≤ 0.001; 4 symbols p ≤ 0.0001) compared to psd2Δpsd1Δ were calculated by one-way ANOVA with Dunnett's multiple comparison test.
(E) Cellular phospholipids from the indicated strains grown in YPD alone or supplemented with choline, ethanolamine, or lyso-phosphatidylethanolamine were labeled overnight with 14C-Acetate and separated by TLC (n = 6).
(F) Cellular PE abundance was determined for each strain in each growth condition (mean ± SEM for n = 6). Statistical differences (1 symbol p ≤ 0.05; 3 symbols p ≤ 0.001) relative to growth in YPD alone were determined by one-way ANOVA with Holm-Sidak pairwise comparison (for :WT samples) or one-way ANOVA by ranks (:LGS samples).
(G) Steady-state abundance of Kar2 and Aac2 in indicated strains grown in absence or presence of lyso-PE or ethanolamine relative to psd2Δpsd1Δ:WT yeast grown in YPD alone (mean ± SEM for n = 4).
(H) Steady-state abundance of Kar2 and Aac2 in indicated strains grown in absence or presence of ethanolamine or choline relative to psd2Δpsd1Δ::WT yeast grown in YPD alone (mean ± SEM for n = 5). For (G) and (H), statistical differences (1 symbol p ≤ 0.05; 2 symbols p ≤ 0.01; 3 symbols p ≤ 0.001; 4 symbols p ≤ 0.0001) compared to WT in YPD alone (∗) or YPD alone for a given strain (#) were calculated with unpaired Student's t-test.
To determine if the ability of lyso-PE and ethanolamine to decrease the relative amount of ER β-α was specific to ER-resident PE biosynthetic pathways, we used choline, which promotes PC biosynthesis in the ER (Walkey et al., 1998). Surprisingly, the addition of choline also decreased the proportion of nonfunctional ER β-α (Figures 5C and 5D). Lipid analyses confirmed that cellular PE levels were significantly increased when cultures were supplemented with LPE and ethanolamine but not choline (Figures 5E and 5F). Kar2, a marker of ER stress (Hsu et al., 2012; Lajoie et al., 2012), was increased in psd2Δpsd1Δ, as seen previously (Calzada et al., 2019), and Psd1-LGSFlag (::LGS) relative to ::WT (Figures 5G and 5H). Interestingly, the addition of LPE or ethanolamine, but not choline, to YPD medium drastically reduced steady-state Kar2 levels in both psd2Δpsd1Δ and Psd1-LGSFlag yeast. In combination, these results indicate that supplements that feed distinct ER-resident lipid biosynthetic pathways reduce the accumulation of glycosylated non-functional Psd1-LGSFlag by a mechanism(s) that is not strictly related to their capacity to promote PE production.
Glycosylated Psd1 mutant is short lived and ubiquitinated
We reasoned that the ability of the ER lipid-boosting supplements to reduce the steady state amount of ER β-α could reflect their ability to decrease its generation or alternatively increase its removal. As such, we performed cycloheximide-based in vivo degradation assays to determine the relative stabilities of both unprocessed forms of Psd1-LGSFlag in comparison to the separated α and β subunits of WT Psd1 and whether or not addition of choline, ethanolamine, or LPE exerted any affect (Figures 6A, S4A, and S4B). In the absence or presence of choline, the WT Psd1 α and β subunits were stable for the 2-hr incubation following inhibition of cytosolic translation with cycloheximide (Figures 6A and 6B). In contrast, the steady state amounts of ER β-α and Mito β-α from Psd1-LGSFlag each decreased significantly over time, and strikingly, ER β-α decreased much faster than Mito β-α (Figures 6A and 6C). Moreover, the relative instability of Psd1-LGSFlag was not impacted by choline, ethanolamine, or LPE supplementation (Figures 6C and S4B). Given that none of these supplements significantly altered PSD1-LGS transcript levels (Figure S5A), this suggests that they decrease ER β-α abundance by a post-transcriptional mechanism that does not reflect their ability to stimulate ER β-α degradation.
Figure 6.

Glycosylated non-functional mutant Psd1 is short lived and ubiquitinated
(A) In vivo degradation assay. Cell extracts from designated strains were isolated at the indicated times following growth in YPD containing cycloheximide (CHX) only or CHX and choline. Samples were resolved by SDS-PAGE and immunoblotted as indicated (n = 5). ∗, nonspecific bands.
(B) The percentages of WT α and β subunits remaining at each time point were quantified (mean ± SEM for n = 5).
(C) The percentages of nonfunctional glycosylated ER β-α and mitochondrial β-α remaining at each time point were quantified (mean ± SEM for n = 5). Statistical differences (1 symbol p ≤ 0.05; 2 symbols p ≤ 0.01; 3 symbols p ≤ 0.001; 4 symbols p ≤ 0.0001) between ER β-α and Mito β-α were determined at each time point by unpaired student t-test.
(D) An overnight YPD culture of the psd2Δpsd1Δpdr5Δ:LGS strain was resuspended in SCD with 2mM choline and further spiked with vehicle (DMSO) or the proteosomal inhibitor MG132. After a 1hr incubation at 30°C, CHX was added and cell extracts harvested following growth at 30°C for the indicated times. Samples were resolved by SDS-PAGE and immunoblotted as indicated (n = 5).
(E) The percentages of nonfunctional glycosylated ER β-α and mitochondrial β-α remaining at each time point were quantified (mean ± SEM for n = 5). Statistical differences (1 symbol p ≤ 0.05) between vehicle and MG132 treated samples for ER β-α and Mito β-α were determined at each time point by unpaired Student's t-test.
(F) In vivo ubiquitination assay. Crude lysate was prepared from the indicated strains and lysates were ultracentrifuged into soluble, cytosolic (cyt.) and membrane (mem.) fractions, the latter of which was solubilized with digitonin. FLAG-tagged WT and mutant Psd1 were immunoprecipitated from the soluble and digitonin-extracted membrane fractions and recovered Mito β, Mito α, ER β-α, and Mito β-α detected by immunoblot using 4,077.4 antisera; ubiquitin antibody detected ubiquitination (n = 3).
(G) In vivo re-translocation assay. Ubiquitin removal with Usp2Core, quenched with SUME, immunoprecipitated with anti-FLAG resin, and immunoblotted for Psd1 β and ubiquitin. SM, starting material before anti-FLAG immunoprecipitation (n = 3).
Given its atypical localization and comparatively short half-life, we postulated that the cytosolic proteasomal system may be responsible for the rapid degradation of ER β-α. As such, we cultured Psd1-LGSFlag (::LGS) cells lacking the multi-drug exporter Pdr5 (psd2Δpsd1Δpdr5Δ::LGS) in SCD medium supplemented with 2 mM choline (included to support growth of psd2Δpsd1Δ which are auxotrophs for ethanolamine or choline (Atkinson et al., 1980; Birner et al., 2001; Burgermeister et al., 2004)) in the absence (dimethyl sulfoxide (DMSO)) or presence of the proteasomal inhibitor MG132. While the effect was modest, MG132 slightly stabilized both ER β-α and Mito β-α following proteasomal inhibition for 2 hr (Figures 6D and 6E). As expected, ubiquitinated proteins accumulated when Psd1-LGSFlag yeast were treated with MG132 but not DMSO (Figure 6D). The fact that Mito β-α was stabilized by MG132 to a similar extent as ER β-α suggests a possible role for the cytosolic proteasomal system in the turnover of both unprocessed forms of Psd1-LGSFlag. The transcript levels for numerous plasma membrane ATP binding cassette transporters were significantly increased in Psd1-LGSFlag versus ::WT (Figure S5B). As such, we speculate that the relatively weak ability of MG132 to stabilize Psd1-LGSFlag could reflect the increased expression of multi-drug pumps in this yeast strain. Thus, to definitively implicate the proteasome in Psd1-LGSFlag degradation, genetic tools and approaches are needed that circumvent the caveats associated with using small molecules in yeast studies.
Proteasomal degradation of a subset of substrates requires removal from their organellar homes—in a process known as retro-translocation—for degradation by the cytosolic proteasome (Neal et al., 2018). As such, we examined the ubiquitination status of Psd1-LGSFlag compared to WT Psd1 and analyzed whether membrane-residing ubiquitinated Psd1-LGSFlag retro-translocated to the cytosol for proteasomal degradation (Figure 6F). Cell extracts were ultracentrifuged to separate the soluble cytosol from membrane-containing organelles, the latter of which was then solubilized with digitonin, a mild detergent that preserves the non-covalent association of Psd1 α and β subunits (Ogunbona et al., 2017). Next, Psd1 was immunoprecipitated from each fraction using anti-FLAG conjugated beads and bound material analyzed by immunoblot. While both subunits of WT Psd1 and both unprocessed forms (ER β-α and Mito β-α) of Psd1-LGSFlag were immunoprecipitated from digitonin-extracted membrane pellets, only the anti-FLAG bound material from Psd1-LGSFlag was robustly ubiquitinated (Figure 6F). The associated ubiquitin signal was even stronger following FLAG immunoprecipitation of Psd1-LGSFlag from the cytosolic supernatant fractions, even though neither ER β-α or Mito β-α was detected by the Psd1 antibody (Figure 6F). To confirm that the ubiquitin signal resulted from Psd1-LGSFlag, the supernatant fraction was incubated with human recombinant Usp2Core, a broadly active ubiquitin protease, prior to FLAG immunoprecipitation (Figure 6G). Following Ups2 pretreatment, the ubiquitin signal was decreased and a single unprocessed form of Psd1-LGSFlag observed. These data indicate that, under standard growth conditions, there is a measurable amount of retro-translocated non-functional mutant Psd1-LGSFlag in the cytosol that has been deglycosylated and that is likely en route to the proteasome for degradation.
Supplements that diminish glycosylated mutant Psd1 act by distinct mechanisms
When grown in dextrose, the expression of many mitochondrial genes is repressed, including those involved in oxidative phosphorylation, and the vast majority of cellular energy is generated by glycolysis. One consequence of a less active electron transport chain is a proportionally weaker membrane potential across the IM which serves as a critical driving force for import and assembly of mitochondrial precursors destined for the IM or matrix. Therefore, we asked whether the relative proportion of the Mito β-α and ER β-α unprocessed forms of Psd1-LGSFlag is influenced by metabolic growth condition (Figure S5C). Indeed, the relative abundance of ER β-α was significantly decreased, and Mito β-α proportionately increased, following growth in YPEG, which requires mitochondrial function (Figures S5C–S5E). However, in rich lactate, which contains a small amount of dextrose, ER β-α and Mito β-α were only moderately altered.
Yeasts lacking Psd1 have a respiratory growth defect that is at least in part attributed to a significant reduction in respiratory complex IV activity and a modest decline in complex III function (Calzada et al., 2019). In the combined absence of Psd1 and Psd2, there is an additive effect such that the psd1Δ respiratory growth phenotype is exacerbated and the activities of respiratory complexes III and IV are almost completely ablated (Calzada et al., 2019). Therefore, we reasoned that the inability to detect ER β-α when Psd2 is expressed (Figure 3H) could reflect an increased IM membrane potential secondary to this strain's higher respiratory capacity. To test this idea, we utilized the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a pharmacological uncoupler that collapses the proton gradient across the IM which in turn blocks protein import into and across this membrane. In the presence of Psd2 but absence of CCCP, only one unprocessed form of Psd1-LGSFlag, Mito β-α, was detected (Figure 7A). However, in presence of Psd2 and CCCP, a small amount of ER β-α accumulated (Figure 7A), indicating that severe import stress can stimulate the accumulation of glycosylated Psd1-LGSFlag. Notably, only a single, fully processed Mito β was detected for WT Psd1 in either the absence or presence of IM uncoupling.
Figure 7.

Supplements that diminish amount of glycosylated mutant Psd1 act by distinct mechanisms
(A) The indicated strains were cultured overnight at 30°C in YPD without or with 10 μM CCCP. Cell extracts were harvested and immunoblotted as designated. OE indicates overexposed blot in which the Mito β-α signals are saturated. (n = 3).
(B) CIS1 mRNA levels in the indicated strains grown in YPD at 30°C were determined by two-step reverse transcription-quantitative PCR and normalized to ACT1 (mean ± SEM, n = 5). Statistical differences (2 symbols p ≤ 0.01; 4 symbols p ≤ 0.0001) versus WT were calculated with unpaired Student's t-test.
(C) Cell extracts from the indicated strains, untransformed or transformed with the autocatalytic LGS Psd1 mutant, grown overnight in YPD were immunoblotted for Psd1 β (4,077.4), Psd1 α (FLAG), Cox2 (encoded by mtDNA), and Tam41; Kgd1 acted as loading control (n = 3). ∗, nonspecific bands.
(D) TMT comparison of ER (P40) proteomes from psd2Δpsd1Δ::LGS and psd2Δpsd1Δ yeast (n = 3 preps each). LGS; autocatalytic mutant Psd1.
(E and F) Heatmaps of gene expression analysis from RNAseq with the indicated strains grown in YPD medium alone or YPD supplemented with lyso-PE (L), ethanolamine (E), or choline (C). padj values that were significantly different from YPD medium alone (p ≤ 0.05) are designated (∗). (E) The top 20 upregulated genes in ::LGS vs ::WT and how each supplement does or does not affect their expression relative to ::LGS grown in YPD alone. (F) The top 20 downregulated genes in ::LGS vs ::WT and how each supplement does or does not affect their expression relative to ::LGS grown in YPD alone.
It was previously demonstrated that mitochondrial import stress activates the mito-CPR pathway whose hallmark feature is an increase in CIS1 mRNA levels (Weidberg and Amon, 2018). Thus, we asked whether the glycosylated Psd1-LGSFlag mutant induces CIS1 transcription. As previously reported (Weidberg and Amon, 2018), CIS1 transcript levels, which are normally low, were elevated in rho- and tam41Δ yeast (Figure 7B), strains with mitochondrial import defects stemming from the loss of mitochondrial DNA or defective cardiolipin biosynthesis, respectively. Compared to WT yeast, CIS1 mRNA levels were also significantly increased in psd2Δpsd1Δ and Psd1-LGSFlag strains but not in the psd2Δpsd1Δ strain rescued with WT Psd1 (::WT). These results indicate that mito-CPR is activated, likely due to impaired mitochondrial protein import, when cellular PE metabolism is severely compromised regardless of whether or not a nonfunctional form of Psd1 is expressed. Surprisingly, CIS1 transcript levels were not reduced when Psd1-LGSFlag yeast were supplemented with ethanolamine (Figure 7B, pink bar). This implies that ethanolamine reduces the relative abundance of ER β-α (Figure 5A) in a mito-CPR independent manner. Consistent with this interpretation, when Psd1-LGSFlag was integrated into rho- and tam41Δ yeast, only a single unprocessed form, Mito β-α, was detected (Figure 7C). From these results, we conclude that mito-CPR is not itself sufficient to drive the accumulation of glycosylated nonfunctional Psd1 mutants.
As mito-CPR was similarly activated in the psd2Δpsd1Δ and Psd1-LGSFlag (::LGS) strains and unaffected by ethanolamine, we next asked whether the expression of the Psd1-LGSFlag mutant imposes any overt changes beyond that typically observed in the already severely PE-deficient psd2Δpsd1Δ strain. To this end, we compared the microsome-enriched P40 proteomes of Psd1-LGSFlag (::LGS) versus psd2Δpsd1Δ (Figure 7D). This comparison showed that numerous mitochondrial proteins in addition to Psd1 were significantly increased in the Psd1-LGSFlag (::LGS) versus psd2Δpsd1Δ P40 fractions, indicating that additional mitochondrial proteins are mislocalized. Given that non-Psd1 mitochondrial proteins were not significantly enriched in microsomes from either psd2Δpsd1Δ (Figure 1E) or Psd1-LGSFlag (Figure 3B) relative to ::WT, these results raise the possibility that the combination of compromised cellular PE metabolism and expression of Psd1-LGSFlag, which is both difficult to import and non-functional, impinge upon mitochondrial biogenesis in distinct but partially overlapping manners.
To gain additional insight into the molecular underpinnings for how the ER lipid-boosting supplements reduce the steady state amount of ER β-α, we determined their impact on the transcriptomes of ::WT and Psd1-LGSFlag (::LGS) yeast (Figures 7E and 7F). When cultured in dextrose alone, PDR5, which encodes the major drug pump, and CIS1, the mito-CPR marker (Weidberg and Amon, 2018), were the two most upregulated genes in Psd1-LGSFlag versus ::WT (Figure 7E). RPN4 and PDR3, genes in the recently defined mitoprotein-induced stress response that function upstream of CIS1 (Boos et al., 2019, 2020), were also significantly upregulated in Psd1-LGSFlag (Figure S5F). Many of the other top upregulated genes in dextrose-grown Psd1-LGSFlag yeast have roles related to cytosolic translation (e.g. RSA4, TOD6, TRM11, RRN11, NOG1, NOG2, IMT1, RCL1, and REI1). In contrast, many of the top downregulated genes in dextrose-grown Psd1-LGSFlag yeast have diverse functions in metabolism such as amino acid biosynthesis (STR3, HIS4, ARG1, ARG4, ARG5,6, and MET6), other biosynthetic processes (ADH5 and GLG1), vacuolar amino acid transport (RTC2 and DIP5), reactive oxygen detoxification (TSA2 and CTT1), and mitochondrial fuel utilization (MPC3) and efficiency (COX26) (Figure 7F). Many, including CIS1, but not all, of these changes were corrected when Psd1-LGSFlag yeast were grown in dextrose spiked with LPE (Figures 7E and 7F). In contrast, ethanolamine, which like LPE increased cellular PE levels (Figure 5F), albeit to a lesser extent, and reduced the amount of Kar2 (Figures 5G and 5H), was comparatively ineffective at normalizing the altered transcripts in Psd1-LGSFlag. With one exception (ALD4), choline supplementation did not impact the altered transcriptional profile of Psd1-LGSFlag yeast grown in dextrose. Overall, our combined results provide strong evidence supporting the conclusion that LPE, ethanolamine, and choline reduce the relative abundance of ER β-α by distinct mechanisms.
Discussion
Our data indicate that the vast majority, if not all, of functional Psd1 localizes to the mitochondrion. This conclusion is based on results from multiple yeast strain backgrounds, overexpression studies in yeast and mammalian cells, and a powerful combination of chimeric constructs and biochemical and immunofluorescence approaches. Further, it is consistent with the failure to detect significant Psd activity in microsomes when the non-mitochondrial Psd2 is missing (Onguka et al., 2015; Trotter and Voelker, 1995) and a recent high-confidence proteomics study, which failed to annotate Psd1 as being localized to organelles other than mitochondria (Morgenstern et al., 2017). Based on this overwhelming confluence, we conclude that a functionally relevant population of Psd1 does not localize to the ER, as recently suggested (Friedman et al., 2018). As such, the myriad of studies in which the strict mitochondrial localization of Psd1 served as a foundational premise (Baker et al., 2016; Birner et al., 2001; Bykov et al., 2020; Cui et al., 1993; Daum et al., 1986; Heden et al., 2019; Kornmann et al., 2009; Lahiri et al., 2014; Li and Dowhan, 1988; Steenbergen et al., 2005; Tamura et al., 2012a; Tasseva et al., 2013) do not need to be re-evaluated in consideration of a dual localization for this mitochondrial enzyme, as contended (Friedman et al., 2018).
While the final mature and functional enzyme localizes to mitochondria, there is clearly an interesting and perhaps unexpected relationship between Psd1 and the ER. First, proximity-specific ribosome profiling indicates that Psd1 translation occurs in the vicinity of both mitochondria and the ER, similar to Osm1, a protein that is dually localized to these two compartments (Williams et al., 2014). In fact, this observation motivated the study by (Friedman et al., 2018). Second, when expressed in yeast with severely compromised cellular PE metabolism, autocatalytic Psd1 mutants do engage the ER where they are glycosylated and quickly degraded. Together, these two observations suggest that some fraction of Psd1 biogenesis normally occurs in the context of the ER and when problems with its ability to be properly imported and fully matured arise, it can again be directed to the ER which now exerts a quality control functionality. Classically, mitochondrial precursors are viewed as being translated in the cytosol and, with the help of cytosolic chaperones, directly targeted to the mitochondrial translocase of the OM (TOM), the common entry gate for most nuclear-encoded mitochondrial proteins (Hansen and Herrmann, 2019). Recently, an ER-assisted pathway for a subset of mitochondrial proteins termed ER-SURF (ER-surface-mediated targeting) was discovered in yeast that functions in parallel to the classic direct import pathway (Hansen et al., 2018). Whether or not Psd1 is an ER-SURF substrate and if so, whether or not the components of this pathway, most notably Djp1, can discriminate between functional and non-functional Psd1 precursors are interesting questions for future studies. Further, it must be acknowledged that given this intimate relationship with the ER, there is the opportunity that there may be biological contexts and situations in which it is harnessed such that functional Psd1 is targeted to this organelle.
Although the effect was modest, proteasomal inhibition stabilized both unprocessed forms of Psd1-LGSFlag to the same extent (Figure 6E). When considered in the context of the striking ubiquitination of Psd1-LGSFlag in both membrane-associated and soluble cytosolic fractions, this implicates the ubiquitin proteasome system in mediating the removal of both ER β-α and Mito β-α, although based on the relative kinetics of their degradation (Figure 6C), ER β-α is the easier substrate to resolve. The ubiquitin-proteasome system has a documented role in policing mitochondrial precursor biogenesis and ensuring that non-functional and/or mis-localized proteins are degraded. Recently, the mitochondrial protein translocation-associated degradation (mito-TAD) pathway was identified that monitors the routine import of precursors through TOM (Martensson et al., 2019). When precursor translocation is stalled, Ubx2, which also participates in the ER-associated degradation (ERAD) of misfolded proteins (Neuber et al., 2005), recruits Cdc48 together with its cofactors Npl4 and Ufd1 to deliver unwanted proteins to the proteasome for degradation (Martensson et al., 2019). When mitochondrial biogenesis is challenged, which according to our data occurs when cellular PE metabolism is knee capped (Figure 7B), mito-CPR is activated to enhance the normal maintenance provided by mito-TAD (Weidberg and Amon, 2018). Mito-CPR induces the expression of Cis1 through Pdr3 to recruit the highly conserved OM-anchored and cytosol-facing, AAA-ATPase Msp1 which functions as an extractase that removes clogged protein precursors from the TOM complex and targets them for proteasomal degradation directly or after first sending them to the ER (Matsumoto et al., 2019). It is tempting to speculate that when expressed in a PE-limited setting, Psd1-LGS stimulates Msp1 activity, which then contributes to the increased abundance of multiple mitochondrial proteins in microsomes from Psd1-LGSFlag (::LGS) versus psd2Δpsd1Δ (Figure 7D). In this context, the fact that only one form of unprocessed Psd1-LGSFlag, Mito β-α, was readily detected in the cardiolipin-deficient tam41Δ is particularly interesting (Figure 7C). While cardiolipin and cellular PE deficiency each causes import stress that activates mito-CPR, there are at least nuanced differences in how they are registered by the cell and in the responses that they generate. Ongoing experiments are being performed to dissect the contribution of these assorted quality control pathways and factors.
The ability of LPE and ethanolamine to diminish the accumulation of ER β-α in psd2Δpsd1Δ was initially not surprising as each compound feeds a different ER-resident PE biosynthetic pathway. Based on the literature and our prior work, the fact that LPE provided a bigger reduction in ER β-α than ethanolamine was taken to reflect its capacity to completely (Riekhof et al., 2007), instead of only partially as in the case of ethanolamine (Calzada et al., 2019), rescue the mitochondrial defects that occur in the absence of Psd1. The initial surprise was that choline, which we used as a negative control to demonstrate the specificity of the rescue to PE production, actually worked better in this regard than ethanolamine (Figure 5D). This was the first indication that the stress response activated by Psd1-LGSFlag as expressed in a PE-deficient setting involved processes beyond PE and/or that can be reversed/overcome by PE-independent means. The sum of our results using these three supplements provides strong evidence that each reduces the amount of ER β-α through distinct mechanisms. The most likely mode of action for LPE is through its ability to significantly increase PE levels. If correct, then many of the transcriptional changes that occur in Psd1-LGSFlag (::LGS), especially those that are reversed by LPE, are the direct consequence of PE-depleted membranes. Ethanolamine, which rescued PE to ∼50% of LPE levels, did decrease Kar2-associated ER stress to a similar degree as LPE. Thus, it is possible that the ability of ethanolamine and LPE to decrease ER β-α reflects their impact on ER stress. According to this possibility, their different capacities to revert the transcriptional changes in Psd1-LGSFlag could indicate that a threshold level of PE was not achieved by ethanolamine but was with LPE. Recently, it was demonstrated that ethanolamine rescues mitochondrial respiration of cardiolipin mutant yeast by a PE-independent mechanism (Basu Ball et al., 2018). Thus, it is certainly possible that the underlying mechanism for the reduction provided by ethanolamine is only partially due to its ability to augment PE. The mechanism by which choline decreases ER β-α is the most mysterious and perhaps most interesting. Choline does not decrease Kar2-associated ER stress and in fact may be expected to increase it (Thibault et al., 2012). Moreover, choline does not increase the rate of ER β-α disappearance (Figure 6C) nor reduce the elevated CIS1 levels in Psd1-LGSFlag. Moving forward, it will be exciting to mechanistically distinguish how these supplements reduce ER β-α accumulation in the context of cellular PE deficiency and perhaps more broadly in additional models of mitochondrial import stress.
Limitations of the study
The failure to detect a glycosylated form of WT Psd1 does not formally demonstrate that it does not exist either normally or in specific situations. While this is a noted limitation of this study, our results do forcefully demonstrate that a significant population of functional Psd1 is not likely to reside in the ER under the conditions employed here. The effect of MG132 on Psd1-LGSFlag stability was modest. As such, to definitively implicate the proteasome in Psd1-LGS degradation, genetic tools and approaches are needed that circumvent the caveats associated with using small molecules in yeast studies. Also, our work with autocatalytic Psd1 mutants was limited to yeast models. It will be important to ascertain whether some of the principles established in yeast are conserved in other eukaryotic systems. Finally, the ability of the ER-assisted adaptive response that is activated by dysfunctional cellular PE metabolism to detect other mitochondrial precursors was not formally tested beyond the proteomics comparison of Psd1-LGSFlag (::LGS) and psd2Δpsd1Δ microsomal fractions (Figure 7D).
Resource availability
Lead contact
Requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Steven Claypool (sclaypo1@jhmi.edu).
Material availability
All unique reagents and materials generated in this study are available from the lead contact without restriction, except for the possible completion of a Materials Transfer Agreement.
Data and code availability
The RNASeq data sets are available at the Gene Expression Omnibus under accession number GSE162987.
The TMT proteomics data set is available at the MassIVE repository massive.ucsd.edu and can be accessed using the link (ftp://massive.ucsd.edu/MSV000086558/).
Original/source data for all figures presented in the paper are available from the lead contact upon request.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We would like to thank Drs. Jonathan Friedman (UT Southwestern, USA) and Jodi Nunnari (UC Davis, USA) for sharing W303-JF WT and Δpsd1 strains, Drs. Susan Michaelis (JHMI, USA), George Carman (Rutgers University, USA), and Carla Koehler (UCLA, USA) for antibodies, Dr. Hilla Weidberg (University of British Columbia, Canada) for sharing her protocol for detecting CIS1 transcript levels, and Linhao Ruan and Drs. Selvaraju Kandasamy, Oluwaseun B. Ogunbona, Eric Spear, and Samuel Jayakanthan for technical assistance. This work was supported by the National Institutes of Health (Grant R01GM111548 to S.M.C. and R01GM111548-03S1 to P.N.S.), the National Science Foundation Graduate Research Fellowship Program (DGE1746891 to P.N.S.), a Biochemistry, Cellular, and Molecular Biology Program training grant (T32GM007445 to E.C.), a predoctoral fellowship from the American Heart Association (16PRE31140006 to M.G.A.), the Indiana University Precision Health Grand Challenge Initiative to J.C.T., NIH grant (1R35GM133565 to S.E.N) and Pew Biomedical Award (to S.E.N.), the Japan Society for the Promotion of Science (JSPS) KAKENHI (20K15734 to Y.W.), and the Canadian Institutes of Health Research (T.E.S.).
Author contributions
S.M.C conceived the project; P.N.S., E.C., T.Z., A.N., T.S., S.E.N., and S.M.C. designed research, performed experiments, and made strains; P.N.S., M.G.A., and S.M.C. analyzed data; J.C.T performed the mass spectrometry-based proteomics; Y.W. developed and analyzed the homology models of Psd1; and P.N.S., E.C., and S.M.C. wrote the paper with the feedback and approval from all authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identify as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: March 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102196.
Supplemental information
References
- Aaltonen M.J., Friedman J.R., Osman C., Salin B., di Rago J.P., Nunnari J., Langer T., Tatsuta T. MICOS and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J. Cell Biol. 2016;213:525–534. doi: 10.1083/jcb.201602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achleitner G., Zweytick D., Trotter P.J., Voelker D.R., Daum G. Synthesis and intracellular transport of aminoglycerophospholipids in permeabilized cells of the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 1995;270:29836–29842. doi: 10.1074/jbc.270.50.29836. [DOI] [PubMed] [Google Scholar]
- Acoba M.G., Senoo N., Claypool S.M. Phospholipid ebb and flow makes mitochondria go. J. Cell Biol. 2020;219:e202003131. doi: 10.1083/jcb.202003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Wootton J.C., Gertz E.M., Agarwala R., Morgulis A., Schaffer A.A., Yu Y.K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson K.D., Jensen B., Kolat A.I., Storm E.M., Henry S.A., Fogel S. Yeast mutants auxotrophic for choline or ethanolamine. J. Bacteriol. 1980;141:558–564. doi: 10.1128/jb.141.2.558-564.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C.D., Basu Ball W., Pryce E.N., Gohil V.M. Specific requirements of nonbilayer phospholipids in mitochondrial respiratory chain function and formation. Mol. Biol. Cell. 2016;27:2161–2171. doi: 10.1091/mbc.E15-12-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu Ball W., Baker C.D., Neff J.K., Apfel G.L., Lagerborg K.A., Zun G., Petrovic U., Jain M., Gohil V.M. Ethanolamine ameliorates mitochondrial dysfunction in cardiolipin-deficient yeast cells. J. Biol. Chem. 2018;293:10870–10883. doi: 10.1074/jbc.RA118.004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T., Horvath S.E., Bottinger L., Gebert N., Daum G., Pfanner N. Role of phosphatidylethanolamine in the biogenesis of mitochondrial outer membrane proteins. J. Biol. Chem. 2013;288:16451–16459. doi: 10.1074/jbc.M112.442392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birner R., Burgermeister M., Schneiter R., Daum G. Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol. Biol. Cell. 2001;12:997–1007. doi: 10.1091/mbc.12.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos F., Kramer L., Groh C., Jung F., Haberkant P., Stein F., Wollweber F., Gackstatter A., Zoller E., van der Laan M. Publisher Correction: mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat. Cell Biol. 2019;21:793–794. doi: 10.1038/s41556-019-0326-1. [DOI] [PubMed] [Google Scholar]
- Boos F., Labbadia J., Herrmann J.M. How the mitoprotein-induced stress response safeguards the cytosol: a unified view. Trends Cell Biol. 2020;30:241–254. doi: 10.1016/j.tcb.2019.12.003. [DOI] [PubMed] [Google Scholar]
- Bottinger L., Horvath S.E., Kleinschroth T., Hunte C., Daum G., Pfanner N., Becker T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 2012;423:677–686. doi: 10.1016/j.jmb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgermeister M., Birner-Grunberger R., Nebauer R., Daum G. Contribution of different pathways to the supply of phosphatidylethanolamine and phosphatidylcholine to mitochondrial membranes of the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2004;1686:161–168. doi: 10.1016/j.bbalip.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Bykov Y.S., Rapaport D., Herrmann J.M., Schuldiner M. Cytosolic events in the biogenesis of mitochondrial proteins. Trends Biochem. Sci. 2020;45:650–667. doi: 10.1016/j.tibs.2020.04.001. [DOI] [PubMed] [Google Scholar]
- Calzada E., Avery E., Sam P.N., Modak A., Wang C., McCaffery J.M., Han X., Alder N.N., Claypool S.M. Phosphatidylethanolamine made in the inner mitochondrial membrane is essential for yeast cytochrome bc1 complex function. Nat. Commun. 2019;10:1432. doi: 10.1038/s41467-019-09425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.Y., McQuibban G.A. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1) J. Biol. Chem. 2012;287:40131–40139. doi: 10.1074/jbc.M112.399428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.Y., Duraisingh M.T., Marti M., Ben Mamoun C., Voelker D.R. From protease to decarboxylase: the molecular metamorphosis of phosphatidylserine decarboxylase. J. Biol. Chem. 2015;290:10972–10980. doi: 10.1074/jbc.M115.642413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z., Vance J.E., Chen M.H., Voelker D.R., Vance D.E. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 1993;268:16655–16663. [PubMed] [Google Scholar]
- Daum G., Heidorn E., Paltauf F. Intracellular transfer of phospholipids in the yeast, Saccharomyces cerevisiae. Biochim. Biophys. Acta. 1986;878:93–101. doi: 10.1016/0005-2760(86)90347-4. [DOI] [PubMed] [Google Scholar]
- Dennis E.A., Kennedy E.P. Intracellular sites of lipid synthesis and the biogenesis of mitochondria. J. Lipid Res. 1972;13:263–267. [PubMed] [Google Scholar]
- Elbaz-Alon Y., Rosenfeld-Gur E., Shinder V., Futerman A.H., Geiger T., Schuldiner M. A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell. 2014;30:95–102. doi: 10.1016/j.devcel.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Friedman J.R., Kannan M., Toulmay A., Jan C.H., Weissman J.S., Prinz W.A., Nunnari J. Lipid homeostasis is maintained by dual targeting of the mitochondrial PE biosynthesis enzyme to the ER. Dev. Cell. 2018;44:261–270 e266. doi: 10.1016/j.devcel.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton M.D., Hakimuddin F., Bakovic M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2) Mol. Cell Biol. 2007;27:3327–3336. doi: 10.1128/MCB.01527-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girisha K.M., von Elsner L., Neethukrishna K., Muranjan M., Shukla A., Bhavani G.S., Nishimura G., Kutsche K., Mortier G. The homozygous variant c.797G>A/p.(Cys26C6Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum. Mutat. 2019;40:299–309. doi: 10.1002/humu.23693. [DOI] [PubMed] [Google Scholar]
- Gulshan K., Shahi P., Moye-Rowley W.S. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol. Biol. Cell. 2010;21:443–455. doi: 10.1091/mbc.E09-06-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.G., Aviram N., Laborenz J., Bibi C., Meyer M., Spang A., Schuldiner M., Herrmann J.M. An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science. 2018;361:1118–1122. doi: 10.1126/science.aar8174. [DOI] [PubMed] [Google Scholar]
- Hansen K.G., Herrmann J.M. Transport of proteins into mitochondria. Protein J. 2019;38:330–342. doi: 10.1007/s10930-019-09819-6. [DOI] [PubMed] [Google Scholar]
- Heden T.D., Johnson J.M., Ferrara P.J., Eshima H., Verkerke A.R.P., Wentzler E.J., Siripoksup P., Narowski T.M., Coleman C.B., Lin C.T. Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Sci. Adv. 2019;5:eaax8352. doi: 10.1126/sciadv.aax8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honscher C., Mari M., Auffarth K., Bohnert M., Griffith J., Geerts W., van der Laan M., Cabrera M., Reggiori F., Ungermann C. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev. Cell. 2014;30:86–94. doi: 10.1016/j.devcel.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Horvath S.E., Bottinger L., Vogtle F.N., Wiedemann N., Meisinger C., Becker T., Daum G. Processing and topology of the yeast mitochondrial phosphatidylserine decarboxylase 1. J. Biol. Chem. 2012;287:36744–36755. doi: 10.1074/jbc.M112.398107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovius R., Faber B., Brigot B., Nicolay K., de Kruijff B. On the mechanism of the mitochondrial decarboxylation of phosphatidylserine. J. Biol. Chem. 1992;267:16790–16795. [PubMed] [Google Scholar]
- Hsu C.L., Prasad R., Blackman C., Ng D.T. Endoplasmic reticulum stress regulation of the Kar2p/BiP chaperone alleviates proteotoxicity via dual degradation pathways. Mol. Biol. Cell. 2012;23:630–641. doi: 10.1091/mbc.E11-04-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano S., Tamura Y., Kojima R., Bala S., Asai E., Michel A.H., Kornmann B., Riezman I., Riezman H., Sakae Y. Structure-function insights into direct lipid transfer between membranes by Mmm1-Mdm12 of ERMES. J. Cell Biol. 2017;217:959–974. doi: 10.1083/jcb.201704119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keckesova Z., Donaher J.L., De Cock J., Freinkman E., Lingrell S., Bachovchin D.A., Bierie B., Tischler V., Noske A., Okondo M.C. LACTB is a tumour suppressor that modulates lipid metabolism and cell state. Nature. 2017;543:681–686. doi: 10.1038/nature21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima R., Endo T., Tamura Y. A phospholipid transfer function of ER-mitochondria encounter structure revealed in vitro. Sci. Rep. 2016;6:30777. doi: 10.1038/srep30777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Currie E., Collins S.R., Schuldiner M., Nunnari J., Weissman J.S., Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge O., Saito K., Kojima M., Akamatsu Y., Nishijima M. Post-translational processing of the phosphatidylserine decarboxylase gene product in Chinese hamster ovary cells. Biochem. J. 1996;319(Pt 1):33–38. doi: 10.1042/bj3190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S., Chao J.T., Tavassoli S., Wong A.K., Choudhary V., Young B.P., Loewen C.J., Prinz W.A. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014;12:e1001969. doi: 10.1371/journal.pbio.1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P., Moir R.D., Willis I.M., Snapp E.L. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol. Biol. Cell. 2012;23:955–964. doi: 10.1091/mbc.E11-12-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.X., Dowhan W. Structural characterization of Escherichia coli phosphatidylserine decarboxylase. J. Biol. Chem. 1988;263:11516–11522. [PubMed] [Google Scholar]
- Lu Y.W., Claypool S.M. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson C.U., Priesnitz C., Song J., Ellenrieder L., Doan K.N., Boos F., Floerchinger A., Zufall N., Oeljeklaus S., Warscheid B. Mitochondrial protein translocation-associated degradation. Nature. 2019;569:679–683. doi: 10.1038/s41586-019-1227-y. [DOI] [PubMed] [Google Scholar]
- Matsumoto S., Nakatsukasa K., Kakuta C., Tamura Y., Esaki M., Endo T. Msp1 clears mistargeted proteins by facilitating their transfer from mitochondria to the ER. Mol. Cell. 2019;76:191–205 e110. doi: 10.1016/j.molcel.2019.07.006. [DOI] [PubMed] [Google Scholar]
- Morgenstern M., Stiller S.B., Lubbert P., Peikert C.D., Dannenmaier S., Drepper F., Weill U., Hoss P., Feuerstein R., Gebert M. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 2017;19:2836–2852. doi: 10.1016/j.celrep.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S., Jaeger P.A., Duttke S.H., Benner C., C K.G., Ideker T., Hampton R.Y. The Dfm1 derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol. Cell. 2018;69:306–320.e4. doi: 10.1016/j.molcel.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebauer R., Rosenberger S., Daum G. Phosphatidylethanolamine, a limiting factor of autophagy in yeast strains bearing a defect in the carboxypeptidase Y pathway of vacuolar targeting. J. Biol. Chem. 2007;282:16736–16743. doi: 10.1074/jbc.M611345200. [DOI] [PubMed] [Google Scholar]
- Nerlich A., von Orlow M., Rontein D., Hanson A.D., Dormann P. Deficiency in phosphatidylserine decarboxylase activity in the psd1 psd2 psd3 triple mutant of Arabidopsis affects phosphatidylethanolamine accumulation in mitochondria. Plant Physiol. 2007;144:904–914. doi: 10.1104/pp.107.095414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O., Jarosch E., Volkwein C., Walter J., Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005;7:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- Nguyen T.T., Lewandowska A., Choi J.Y., Markgraf D.F., Junker M., Bilgin M., Ejsing C.S., Voelker D.R., Rapoport T.A., Shaw J.M. Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic. 2012;13:880–890. doi: 10.1111/j.1600-0854.2012.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunbona O.B., Onguka O., Calzada E., Claypool S.M. Multitiered and cooperative surveillance of mitochondrial phosphatidylserine decarboxylase 1. Mol. Cell Biol. 2017;37:e00049-17. doi: 10.1128/MCB.00049-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onguka O., Calzada E., Ogunbona O.B., Claypool S.M. Phosphatidylserine decarboxylase 1 autocatalysis and function does not require a mitochondrial-specific factor. J. Biol. Chem. 2015;290:12744–12752. doi: 10.1074/jbc.M115.641118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter V.G., Quinodoz M., Pinto-Basto J., Sousa S.B., Di Gioia S.A., Soares G., Ferraz Leal G., Silva E.D., Pescini Gobert R., Miyake N. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene. Genet. Med. 2019;21:2734–2743. doi: 10.1038/s41436-019-0595-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recsei P.A., Snell E.E. Pyruvoyl enzymes. Annu. Rev. Biochem. 1984;53:357–387. doi: 10.1146/annurev.bi.53.070184.002041. [DOI] [PubMed] [Google Scholar]
- Riekhof W.R., Voelker D.R. Uptake and utilization of lyso-phosphatidylethanolamine by Saccharomyces cerevisiae. J. Biol. Chem. 2006;281:36588–36596. doi: 10.1074/jbc.M608851200. [DOI] [PubMed] [Google Scholar]
- Riekhof W.R., Wu J., Jones J.L., Voelker D.R. Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 2007;282:28344–28352. doi: 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- Rontein D., Wu W.I., Voelker D.R., Hanson A.D. Mitochondrial phosphatidylserine decarboxylase from higher plants. Functional complementation in yeast, localization in plants, and overexpression in Arabidopsis. Plant Physiol. 2003;132:1678–1687. doi: 10.1104/pp.103.023242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam P.N., Avery E., Claypool S.M. Proteolytic control of lipid metabolism. ACS Chem. Biol. 2019;14:2406–2423. doi: 10.1021/acschembio.9b00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samborski R.W., Ridgway N.D., Vance D.E. Evidence that only newly made phosphatidylethanolamine is methylated to phosphatidylcholine and that phosphatidylethanolamine is not significantly deacylated-reacylated in rat hepatocytes. J. Biol. Chem. 1990;265:18322–18329. [PubMed] [Google Scholar]
- Selathurai A., Kowalski G.M., Mason S.A., Callahan D.L., Foletta V.C., Della Gatta P.A., Lindsay A., Hamley S., Kaur G., Curtis A.R. Phosphatidylserine decarboxylase is critical for the maintenance of skeletal muscle mitochondrial integrity and muscle mass. Mol. Metab. 2019;27:33–46. doi: 10.1016/j.molmet.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T., Haynes C.M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- Steenbergen R., Nanowski T.S., Beigneux A., Kulinski A., Young S.G., Vance J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y., Onguka O., Hobbs A.E., Jensen R.E., Iijima M., Claypool S.M., Sesaki H. Role for two conserved intermembrane space proteins, Ups1p and Ups2p, [corrected] in intra-mitochondrial phospholipid trafficking. J. Biol. Chem. 2012;287:15205–15218. doi: 10.1074/jbc.M111.338665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y., Onguka O., Itoh K., Endo T., Iijima M., Claypool S.M., Sesaki H. Phosphatidylethanolamine biosynthesis in mitochondria: phosphatidylserine (PS) trafficking is independent of a PS decarboxylase and intermembrane space proteins UPS1P and UPS2P. J. Biol. Chem. 2012;287:43961–43971. doi: 10.1074/jbc.M112.390997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasseva G., Bai H.D., Davidescu M., Haromy A., Michelakis E., Vance J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013;288:4158–4173. doi: 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault G., Shui G., Kim W., McAlister G.C., Ismail N., Gygi S.P., Wenk M.R., Ng D.T. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell. 2012;48:16–27. doi: 10.1016/j.molcel.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter P.J., Voelker D.R. Identification of a non-mitochondrial phosphatidylserine decarboxylase activity (PSD2) in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1995;270:6062–6070. doi: 10.1074/jbc.270.11.6062. [DOI] [PubMed] [Google Scholar]
- van Golde L.M., Raben J., Batenburg J.J., Fleischer B., Zambrano F., Fleischer S. Biosynthesis of lipids in Golgi complex and other subcellular fractions from rat liver. Biochim. Biophys. Acta. 1974;360:179–192. doi: 10.1016/0005-2760(74)90168-4. [DOI] [PubMed] [Google Scholar]
- Vance J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Vance J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991;266:89–97. [PubMed] [Google Scholar]
- Voelker D.R. Disruption of phosphatidylserine translocation to the mitochondria in baby hamster kidney cells. J. Biol. Chem. 1985;260:14671–14676. [PubMed] [Google Scholar]
- Walkey C.J., Yu L., Agellon L.B., Vance D.E. Biochemical and evolutionary significance of phospholipid methylation. J. Biol. Chem. 1998;273:27043–27046. doi: 10.1074/jbc.273.42.27043. [DOI] [PubMed] [Google Scholar]
- Wang X., Chen X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature. 2015;524:481–484. doi: 10.1038/nature14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Watanabe Y., Watanabe S. Structural basis for phosphatidylethanolamine biosynthesis by bacterial phosphatidylserine decarboxylase. Structure. 2020;28:799–809.e5. doi: 10.1016/j.str.2020.04.006. [DOI] [PubMed] [Google Scholar]
- Weidberg H., Amon A. MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science. 2018;360:eaan4146. doi: 10.1126/science.aan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.C., Jan C.H., Weissman J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 2014;346:748–751. doi: 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills R.C., Goulden B.D., Hammond G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell. 2018;29:1526–1532. doi: 10.1091/mbc.E17-12-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L., Topf U., Bragoszewski P., Wiese S., Sztolsztener M.E., Oeljeklaus S., Varabyova A., Lirski M., Chroscicki P., Mroczek S. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015;524:485–488. doi: 10.1038/nature14951. [DOI] [PubMed] [Google Scholar]
- Zborowski J., Dygas A., Wojtczak L. Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett. 1983;157:179–182. doi: 10.1016/0014-5793(83)81141-7. [DOI] [PubMed] [Google Scholar]
- Zhao T., Goedhart C.M., Sam P.N., Sabouny R., Lingrell S., Cornish A.J., Lamont R.E., Bernier F.P., Sinasac D., Parboosingh J.S. PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes. Life Sci. Alliance. 2019;2:e201900353. doi: 10.26508/lsa.201900353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNASeq data sets are available at the Gene Expression Omnibus under accession number GSE162987.
The TMT proteomics data set is available at the MassIVE repository massive.ucsd.edu and can be accessed using the link (ftp://massive.ucsd.edu/MSV000086558/).
Original/source data for all figures presented in the paper are available from the lead contact upon request.