

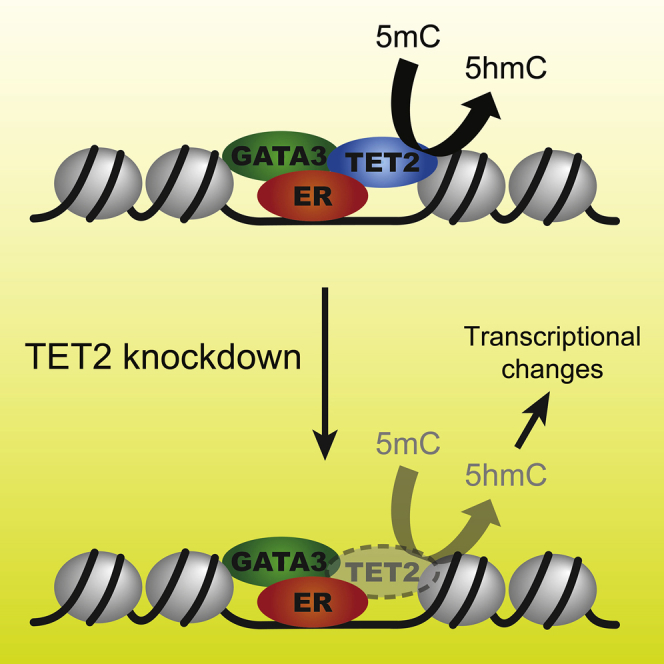
Summary
Estrogen receptor-α (ER) drives tumor development in ER-positive (ER+) breast cancer. The transcription factor GATA3 has been closely linked to ER function, but its precise role in this setting remains unclear. Quantitative proteomics was used to assess changes to the ER complex in response to GATA3 depletion. Unexpectedly, few proteins were lost from the ER complex in the absence of GATA3, with the only major change being depletion of the dioxygenase TET2. TET2 binding constituted a near-total subset of ER binding in multiple breast cancer models, with loss of TET2 associated with reduced activation of proliferative pathways. TET2 knockdown did not appear to change global methylated cytosine (5mC) levels; however, oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) was significantly reduced, and these events occurred at ER enhancers. These findings implicate TET2 in the maintenance of 5hmC at ER sites, providing a potential mechanism for TET2-mediated regulation of ER target genes.
Keywords: TET2, estrogen receptor, GATA3, 5hmC, gene regulation, enhancers
Graphical Abstract
Highlights
-
•
TET2 is identified as a component of the ER complex through depletion of GATA3
-
•
TET2 global chromatin binding tracks that of ER/GATA3 in multiple breast cancer models
-
•
Loss of TET2 is linked to dysregulated expression of ER target genes
-
•
Concurrent loss of 5hmC at ER sites provides insights into TET2’s role in ER activity
Broome et al. use quantitative proteomics and chromatin immunoprecipitation to explore the contribution of TET2 to ER/GATA3 transcriptional activity. TET2 tracks ER chromatin binding in breast cancer models and plays an essential role in the maintenance of 5-hydroxymethylcytosine at ER enhancers, revealing a role for TET2 in ER-driven gene expression.
Introduction
Estrogen receptor-α (ER) drives tumor development in ∼75% of breast cancer cases, and treatments directly targeting ER (e.g., aromatase inhibitors, tamoxifen, and fulvestrant) are currently the standard of care. ER operates as part of a transcriptional complex, coordinating with several other proteins to access chromatin and in turn regulate gene expression and tumor development. GATA3 is a transcription factor that has been closely linked to ER function and is a signature gene of ER-positive (ER+) breast cancer. GATA3 is highly expressed and frequently mutated in ER+ breast cancers (Perou et al., 2000; Sorlie et al., 2003; Kouros-Mehr et al., 2006; Cancer Genome Atlas Network, 2012), and estrogen-induced growth of ER+ breast cancer cells has been shown to be dependent on GATA3 (Eeckhoute et al., 2007; Kong et al., 2011). GATA3 motifs are enriched around ER binding sites (Carroll et al., 2005; Lin et al., 2007), and ER and GATA3 co-localize at a large proportion (∼45%) of ER binding sites in ER+ breast cancer cells (Kong et al., 2011; Theodorou et al., 2013). Therefore, it appears there may be direct functional interplay between these two proteins that contributes to the breast cancer phenotype. However, despite suggestions that GATA3 may modulate enhancer accessibility (Theodorou et al., 2013; Takaku et al., 2018), the precise contribution of GATA3 to ER biology is yet to be fully elucidated.
TET2 is an Fe(II)/α-ketoglutarate-dependent dioxygenase that acts on methylated cytosine (5mC), a reversible epigenetic mark implicated in genome stability and transcriptional control (Bird and Wolffe, 1999; Klose and Bird, 2006; Tahiliani et al., 2009). TET2 oxidizes 5mC in an iterative process that successively produces the DNA modifications 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (Tahiliani et al., 2009; He et al., 2011; Ito et al., 2011). Ongoing research suggests that these 5mC oxidation products may also influence transcriptional outcomes, and these discoveries have further fueled research into TET proteins in both development and cancer. In this study, quantitative multiplexed rapid immunoprecipitation mass spectrometry of endogenous proteins (qPLEX-RIME) revealed TET2 as a key component of the ER signaling complex, with its participation in this complex shown to be regulated by the key ER-associated factor GATA3. ER and TET2 were further shown to occupy shared gene regulatory regions across the genome, with TET2 appearing important for both proper recruitment of ER to chromatin and expression of ER and GATA3 target genes, indicating functional importance of the interaction between these three proteins. Mechanistically, loss of TET2 did not detectably alter global 5mC levels in ER+ breast cancer cells but rather led to a robust and dramatic drop in the levels of 5hmC, including at ER cis-regulatory elements. These data show that TET2 is important for maintenance of this specific mark as part of the ER complex and provide a potential mechanism for TET2-mediated regulation of ER target genes.
Results
In order to identify the contribution of GATA3 to the ER interactome, the ER complex was purified in the presence and absence of GATA3. GATA3 was robustly depleted in MCF7 cells after 48 h of small interfering RNA (siRNA)-mediated knockdown with no significant effect on total ER protein levels, confirmed by both western blot and parallel reaction monitoring (PRM) (Figures S1A–S1C). Subsequently, ER qPLEX-RIME was conducted by comparing the ER interactome under control or GATA3-silenced conditions, with a total of four biological replicates. The most significantly changing ER interactor was GATA3 itself, which was depleted in the GATA3-silenced condition, validating the knockdown approach (Figure 1A). Beyond this finding, a small number of significant changes were observed in response to GATA3 knockdown. The proteins significantly enriched in the ER complex in response to loss of GATA3 included the transcription factors LIM-homeobox 4 (LHX4) and zinc finger and BTB domain containing protein 34 (ZBTB34). Concurrent with this result, the only protein that was significantly depleted in response to GATA3 knockdown (other than GATA3 itself) was TET2. RNA sequencing (RNA-seq) demonstrated that decreased TET2 association with the ER complex in response to GATA3 knockdown is likely due to decreased expression (Figure S1D), and PRM analysis further confirmed that TET2 protein levels are reduced in response to GATA3 knockdown (Figure S1E). It has been suggested that both LHX4 and the BTB/POZ protein family (of which ZBTB34 is a member) may be capable of binding to methylated DNA (Filion et al., 2006; Qi et al., 2006; Yin et al., 2017). The recruitment of LHX4 and ZBTB34 to the ER complex may partly reflect their regulation in response to GATA3 knockdown (also examined at the RNA level in Figure S1D). Nevertheless, because TET2 is known to regulate DNA modifications, it was reasoned that the loss of this enzyme from the ER complex may potentially induce a shift in the normal profile of DNA modifications at ER enhancers, influencing the recruitment of LHX4/ZBTB34 to the complex. Indeed, it has been suggested that TET2 may protect against hypermethylation of enhancers (Rasmussen et al., 2015). These results suggest a potential role for GATA3 in modulating reading and writing of DNA modifications at ER enhancer elements.
Figure 1.

TET2 is recruited to the ER complex by GATA3
(A) ER qPLEX-RIME in MCF7 cells showing changes to the ER complex after GATA3 knockdown (48 h). Four replicates of ER RIME and one pooled immunoglobulin G (IgG) control RIME for each condition were included in the 10plex tandem mass tag (TMT) mass spectrometry (MS) run. Significantly enriched or depleted proteins according to the adjusted p value are highlighted in red (adjusted p value [p adj] ≤ 0.05, after multiple testing correction using Benjamini-Hochberg procedure).
(B) Overlap of lists of Uniprot IDs of specific interactors for ER, GATA3, and TET2. Specific interactors were defined as those occurring in at least two out of three independent replicates. Proteins that appeared in any one of the three IgG control RIME experiments were excluded. ER, GATA3, and TET2 shared a total of 379 common interactors by RIME. Several key ER complex proteins were among them, highlighted in the central portion of the diagram.
See also Figures S1, S2, and S5A–S5C.
Given the capacity of TET2 for regulating DNA modifications, and the recently developing focus on this factor in transcriptional regulation (Wang et al., 2015, 2018; Chen et al., 2018; Rasmussen and Helin, 2016; Rasmussen et al., 2019), the putative genomic link between TET2 and the ER complex was pursued. Few studies have successfully mapped endogenous TET2-chromatin interactions by using chromatin immunoprecipitation (ChIP), and the challenges surrounding the availability of robust ChIP-grade antibodies for this protein have been documented (Wang et al., 2018; Rasmussen et al., 2019). Consequently, the performance of various TET2 antibodies was evaluated using both ChIP and non-quantitative RIME, and the specificity of the chosen candidate (Abcam ab94580) was further validated through confirmation of genome-wide depletion of ChIP sequencing (ChIP-seq) enrichment in response to TET2 silencing (Figures S5A–S5C). Non-quantitative RIME experiments were then performed against ER, GATA3, and TET2 (Figure 1B; Figure S2). All three proteins were reciprocally detected as associating with one another, implying that these factors are reproducibly associated with the ER complex. ER, GATA3, and TET2 were also found to share a large number of common associated proteins, including key ER cofactors such as FOXA1, GREB1, and RARα, as well as the ER co-activators NCOA3 and CARM1 (Anzick et al., 1997; Chen et al., 2000; Deschênes et al., 2007; Ross-Innes et al., 2010; Mohammed et al., 2013), suggesting that TET2 associates with central components of the ER machinery.
ChIP-seq for TET2 was conducted in MCF7 and ZR-75-1 cells, with a total of 4 biological replicates, resulting in 16,884 and 13,423 binding sites, respectively. ER ChIP-seq demonstrated that these TET2 sites constitute a near-total subset of ER binding events (Figure 2A), and these findings were validated in two ER+ patient-derived xenograft (PDX) models, namely, STG195 and AB555 (Figure 2B; Bruna et al., 2016). Genomic annotation of all ChIP-seq datasets showed that, as expected, the distribution of TET2 binding appears to largely mimic that of ER, with the majority of binding sites occupying non-promoter regions (Figure 2E). This result is consistent with previous findings indicating that both ER and TET2 preferentially localize to enhancers (Carroll et al., 2006, Rasmussen et al., 2015, Wang et al., 2018). These endogenous TET2 mapping approaches therefore show that in ER+ breast cancer models, the binding of TET2 tracks that of the driving transcription factor ER. Motif analysis of TET2 binding regions further confirmed the association between TET2 and ER, with ER and FOXA1 motifs being the two most significantly enriched sequences within TET2 peaks (data not shown). In addition to confirming that ER and TET2 co-bind at key ER target genes (representative examples GREB1 and RARA shown in Figures 2C and 2D), inspection of individual ChIP-seq tracks indicated that the TET2 gene itself possesses ER binding sites 20–30 kb upstream of its transcription start site (TSS) in all four ER+ breast cancer models (Figure 2F). This finding suggests that TET2 may be an ER target gene in ER+ breast cancer and is supported by recent studies in MCF7 cells demonstrating that TET2 expression is induced by estrogen (Wang et al., 2018) and robustly repressed by tamoxifen (Papachristou et al., 2018). These results indicate that TET2 is a common target gene of both ER and GATA3 and is a central component of the ER complex on chromatin. Consistent with this finding, higher TET2 expression associates with improved relapse-free survival in ER+ breast cancer (Figure S3), implying that TET2 may help to sustain ER-regulated transcription. To assess whether TET2 was required for ER-mediated gene expression, RNA-seq was performed following TET2 knockdown in asynchronous MCF7 cells, revealing repression of 2,269 genes and activation of 2,144 genes (p ≤ 0.05) (Figure S4A). To compare the gene regulatory program of TET2 with that of ER and GATA3, RNA-seq was also performed after knockdown of ER or GATA3 (Figures S4B and S4C). As expected, TET2 mRNA levels were robustly and significantly repressed by both ER and GATA3 knockdown (to 23% of control levels by ER knockdown, and 43% of control levels by GATA3 knockdown), and 60% of the genes significantly regulated by TET2 knockdown (2,656 out of 4,413 genes; p ≤ 0.05) were also significantly regulated by both GATA3 and ER silencing, suggesting common gene targets. Taking the 500 most induced and 500 most repressed genes in response to TET2 silencing (according to log2 fold change), 60% of these most differentially regulated genes were modulated following ER knockdown, and these changes were in the same direction as observed in TET2-silenced cells (Figure 3A). This included repression of key ER target genes such as PGR, CCND1, XBP1, and CXCL12. In agreement with ER, GATA3, and TET2 regulating a similar set of genes, a clear positive correlation was observed between the most highly regulated genes in response to depletion of each factor (Figure 3B). Importantly, ER/TET2 co-bound sites were seen to be enriched adjacent to TET2 target genes (Figure 3C), further implying genomic cooperation between these two proteins. To assess whether the genes regulated by TET2 show a tendency toward modulation of any particular pathway, Gene Ontology (GO) analysis was performed using DAVID (Database for Annotation, Visualization and Integrated Discovery) (Huang et al., 2007). Genes repressed by TET2 knockdown showed significant enrichment of functional categories linked to cell division and cell cycle processes (Figure 3D, left panel). In contrast, the genes induced by TET2 knockdown demonstrated significant enrichment of only two functional categories, linked to cell communication and signal transduction. When solely examining the TET2-regulated genes that also changed in response to GATA3 and ER knockdown, enrichment of the same functional categories was observed (Figure 3D, right panel). This result demonstrates that the pathways most strongly affected by loss of TET2 in these cells are the same as those regulated by GATA3 and ER. This suggests that TET2 cooperates with ER to promote cell proliferation in ER+ breast cancer cells, consistent with the findings of Wang et al. (2018) that showed TET2 is required for effective estrogen-induced growth of MCF7 cells.
Figure 2.

TET2 binds to ER enhancers in breast cancer cells
(A and B) Venn diagrams indicating positional overlap of ER and TET2 ChIP-seq peaks in ER+ breast cancer cell lines MCF7 and ZR-75-1 (A) and two ER+ PDX models, namely, STG195 and AB555 (B). Heatmaps below each Venn diagram illustrate the ChIP-seq signal intensity for ER and TET2 at ER/TET2 shared sites (top) and “TET2 low” sites where TET2 peaks were not called (bottom). Mouse schematic was created with BioRender.com.
(C and D) UCSC genome browser tracks indicating overlap of TET2 and ER peaks at ER target genes RARA and GREB1 in ER+ breast cancer cell lines MCF7 and ZR-75-1 and ER+ PDX models STG195 and AB555, respectively.
(E) Pie charts show classification of ER and TET2 binding sites according to genomic location for all the models tested. Promoters were defined as regions inclusive of 1 kb downstream and 2 kb upstream of the TSS.
(F) UCSC genome browser tracks demonstrating ER binding sites upstream of the TET2 TSS in two ER+ breast cancer cell lines (MCF7 and ZR-75-1) and two ER+ PDX models (STG195 and AB555). Scale bar indicates 5 kb. Cell line ChIPs were performed in biological quadruplicate.
See also Figures S3 and S5A–S5C.
Figure 3.

TET2, ER, and GATA3 regulate similar genes
(A) Heatmaps depicting the top 500 induced and top 500 repressed TET2-regulated genes according to log2 fold change. Color scale represents the relative expression (Z score) of genes across the two conditions (control and knockdown), calculated separately within each comparison (TET2 knockdown [siTET2] versus non-targeting control siRNA [siNT], siESR1 versus siNT, and siGATA3 versus siNT). Hierarchical clustering of genes in the leftmost (siTET2) heatmap is preserved across all three heatmaps. Columns represent independent biological replicates (n = 6). siRNA treatments were performed for 48 h.
(B) Pairwise correlations of data used for the heatmaps in (A).
(C) Graph showing the cumulative fraction of total ER/TET2 shared binding sites (n = 15,945, MCF7 cells) within up to 100 kb of the TSSs of the following three groups of genes: genes upregulated by siTET2 (n = 2,144, red line), genes downregulated by siTET2 (n = 2,269, blue line) (p ≤ 0.05), and genes unchanging in response to siTET2 (constant genes, gray lines). Constant genes were randomly selected from those with p > 0.5 and mean expression > 1.0. Grey lines indicate analysis based on constant genes: the dotted line indicates analysis matched to the number of downregulated genes, and the solid line indicates analysis matched to number of upregulated genes.
(D) Left: bar plot displaying –log10(false discovery rate [FDR]) for GO analysis of the top 500 induced and top 500 repressed TET2-regulated genes according to log2 fold change. Only categories with FDR ≤ 0.05 (threshold indicated by dotted line) are shown. Right: bar plot displaying –log10(FDR) for GO analysis of the top 500 induced and top 500 repressed TET2-regulated genes according to log2 fold change, sub-selected from genes also significantly (p ≤ 0.05) regulated by both GATA3 and ER silencing. The top 6 enriched categories are shown for repressed genes and the top 2 for induced genes. Dotted line indicates FDR of 0.05. Enriched processes were identified using the biological process category level 3 of the GO hierarchy (GOTERM_BP_3).
See also Figures S4A–S4F.
To further investigate the relationship between TET2 and ER gene expression, the impact of TET2 loss on ER binding to chromatin was assessed, with three replicates of ER ChIP-seq performed in both TET2-depleted and control-treated cells. A drop in overall ER binding was observed in response to TET2 knockdown (Figures 4A–4C), as has previously been shown (Wang et al., 2018), despite no change in total ER protein levels (Figure S4H), implying a role for TET2 in stabilizing ER-chromatin interactions. ER binding appeared to be depleted at a substantial proportion of ER sites, despite TET2 binding occurring at only a subset of these sites (Figure 2). This may be a result of the TET2 ChIP failing to effectively capture the full range of TET2 sites. If this is the case, TET2 is likely to be present at a greater fraction of ER binding sites than detected. Alternatively, this result could suggest an indirect mechanism for TET2 in stabilizing ER-chromatin interactions. Overall, these findings imply that the loss of ER binding as a result of TET2 knockdown contributes to impaired regulation of ER target genes under TET2-depleted conditions.
Figure 4.

ER is required to recruit TET2 to a subset of enhancer elements
(A) MA plot showing log2 fold change in ER binding under control versus siTET2 conditions against the log2 mean intensity of ChIP-seq signal for all ER sites (20,386 peaks).
(B) Normalized tag density of ER ChIP-seq signal under control (siNT) and siTET2 (siTET2) conditions within all ER peaks. ∗∗∗∗p ≤ 0.0001.
(C) Average plot showing normalized signal enrichment of ER ChIP-seq under control (siNT) or siTET2 conditions within all ER peaks. siRNA treatments were performed for 72 h. ChIPs were performed in biological triplicate.
(D) MA plot showing log2 fold change in TET2 binding in response to fulvestrant treatment (100 nM, 3 h) against the log2 mean intensity of TET2 ChIP-seq signal for all TET2 sites (20,599 peaks). “Lost” sites (n = 1,810) and “gained” sites (n = 64) according to DiffBind analysis (p ≤ 0.05) are highlighted in red.
(E) Normalized tag density of ER ChIP-seq signal at unchanging (common) (n = 18,725) and lost (n = 1,810) TET2 sites in response to fulvestrant treatment.
(F) Motif frequency (number of motifs divided by the total number of peaks in each category) of ER, FOXA1, and GATA3 motifs for lost, common, and background sites. Background values were obtained using random open chromatin regions from an MCF7 MNase dataset (EBI Array Express E-MTAB-1958) and are expressed as the average ± SD of two separate background values calculated matched to the number of sites in the lost and common cohorts. Significance against background is indicated; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001.
(G) UCSC genome browser tracks showing TET2 binding in response to treatment with vehicle (ethanol, 3 h) or fulvestrant (100 nM, 3 h) at significantly (p ≤ 0.05) depleted sites according to Diffbind analysis, within 50 kb of the TSS of two key ER target genes (PGR and XBP1). For the gene schematics below each track, lines indicate introns, boxes indicate exons, and arrowheads indicate the direction of transcription. Scale bar indicates 10 kb.
(H) UCSC genome browser tracks showing TET2 binding in response to treatment with vehicle (ethanol, 3 h) or fulvestrant (100 nM, 3 h) at unchanged sites according to Diffbind analysis. ChIPs were performed in biological triplicate. For the gene schematics below each track, lines indicate introns, boxes indicate exons, and arrowheads indicate the direction of transcription. Scale bar indicates 10 kb.
See also Figures S4G–S4I and S5.
Unlike TET family members TET1 and TET3, which possess CXXC domains facilitating their targeting to CG-rich sequences (Xu et al., 2018), TET2 lacks a sequence-targeted DNA binding domain (Ko et al., 2013). It thus remains poorly understood how it is recruited to specific genomic regions. In contrast to TET1 and TET3, which favor promoters (Williams et al., 2011; Jin et al., 2016; Rasmussen et al., 2015), the preference of TET2 for enhancers reinforces the notion that this protein may use context-dependent mechanisms to access its target sites. Several studies have revealed insights into how TET2 might be recruited to chromatin, with Wang et al., (2015) showing that the transcription factor WT1 recruits TET2 to regulate gene expression in the human leukemia cell line HL-60, and with Chen et al. (2018) showing a role for SNIP1 in bridging the interaction between TET2 and the transcription factor c-MYC to aid expression of target genes in U2OS cells. To assess whether the extent of TET2 and ER genomic co-localization implicates a role for ER in targeting TET2 to chromatin in ER+ breast cancer cells, MCF7 cells were treated with the selective estrogen receptor degrader (SERD) fulvestrant for 3 h, with the assumption that promoting active degradation of existing ER protein would rapidly deplete ER-chromatin interactions while minimizing protein-level changes in target genes, including TET2. After 3 h of fulvestrant treatment, PRM analysis indicated a significant drop in ER protein levels (Figure S5D, left panel), and ChIP-qPCR at several key ER regulatory sites confirmed effective depletion of ER-chromatin interactions (Figure S5E). Four out of the five TET2 peptides detected showed no significant reduction with fulvestrant treatment, suggesting that there was not a robust decline in total TET2 protein levels (Figure S5D, right panel). Despite the fact that total TET2 protein levels did not appreciably change when ER was degraded, ER depletion resulted in significantly reduced TET2 binding at a distinct subset of TET2 binding events according to ChIP-seq (1,810 out of 20,599 peaks, representing ∼9% of all binding sites) (Figure 4D; Figure S5F). TET2 binding remained unchanged at the majority (∼90%) of sites, implying that ER recruits TET2 to a small subset of sites. TET2 “lost” sites corresponded to the high-affinity ER and TET2 co-bound regions, which was supported by the observation that these peaks had the highest frequencies of ER motifs and motifs for key ER cofactors FOXA1 and GATA3, relative to the TET2 binding sites that did not change in response to treatment (Figures 4E and 4F). Furthermore, genes in proximity to these lost sites included key ER target genes that are also repressed in response to TET2 knockdown, including PGR and XBP1 (Figure 4G), CCND1, and members of the EGR and E2F protein families. Contrasting examples of TET2 sites unchanged in response to fulvestrant treatment, proximal to the MIPOL1 and RARG genes, are shown in Figure 4H. These results demonstrate that the sites where TET2 is recruited by ER are the highest affinity ER regulatory elements, adjacent to classic ER target genes.
Depletion of GATA3 leads to a loss of TET2 from the ER complex (Figure 1). However, as GATA3 knockdown impairs TET2 protein levels, assessing the contribution of GATA3 to TET2 chromatin recruitment in a physiologically relevant manner presents challenges. To circumvent this complication, a clinically relevant GATA3 mutant cell line was generated for which a single base pair insertion was introduced into GATA3, resulting in a mutant protein with a frameshift at position 409 and an additional 62 amino acids (Figure 5A). As commercially available GATA3 antibodies are unable to distinguish between the wild-type and mutant variants, PRM analysis was used to confirm the successful generation of the GATA3 mutant, with a mutant-specific peptide used to validate the presence of the longer GATA3 C-terminal sequence in the edited cell line (Figure 5B). Importantly, when GATA3 chromatin binding was investigated by ChIP-seq in wild-type MCF7s versus the mutant cell line, mutant-specific GATA3 enrichment was observed at 450 stringently defined binding sites (Figure 5C). As the only difference between these two cell lines is the single nucleotide insertion in GATA3, the gained binding sites are a direct result of mutant GATA3 expression. When ER and TET2 chromatin binding profiles were mapped at these mutant-enriched GATA3 binding sites, a clear enrichment over background was observed for both of these factors in the mutant versus wild-type GATA3 cell line. GATA3, ER, and TET2 ChIP-seq signal enrichment was subsequently visualized at GATA3 binding sites found in both mutant and wild-type cells (“Common GATA3,” Figure 5D, top panel) or at the newly defined GATA3 mutant sites (“Gained GATA3,” Figure 5D, bottom panel). Both ER and TET2 were found to be enriched at the genomic binding sites bound by mutant GATA3. Together with the TET2 dependency on ER as revealed by fulvestrant treatment (Figure 4), these results indicate GATA3 is responsible for recruiting ER and ER is then subsequently required to recruit TET2 to these shared binding sites, thereby providing a clear hierarchy for these transcription factors (GATA3 > ER > TET2).
Figure 5.

ER and TET2 are recruited to GATA3 mutant-specific binding sites
(A) Schematic representation of the human GATA3 transcription factor (Uniprot: P23771), with the two transactivation (TA1 and TA2) and the two zinc finger (ZnF1 and ZnF2) domains illustrated. The insertion resulting in a frameshift (“409fs”) mutation at amino acid 409 (COSMIC genomic mutation ID: COSV60515158) generates the variant GATA3 that is extended by 62 amino acids.
(B) The GATA3 409 frameshift mutant was generated using CRISPR-based gene editing. PRM-based proteomics using a peptide common to both wild-type (WT) and mutant GATA3 (GATA3 WT amino acids 389–399, sequence NSSFNPAALSR) or a peptide specific to the elongated GATA3 mutant (GATA3 mutant amino acids 489–496, sequence IMFATLQR) was used to confirm the presence of the elongated GATA3 mutant. MCF7 wild-type (WT) cells were run in parallel as controls. The endogenous (light) peptide peak (where found) is shown in the top two chromatograms of each sub-panel, while the peak of the spiked-in (heavy) standard peptide is shown in the bottom two chromatograms of each sub-panel.
(C) Heatmaps illustrating the ChIP-seq signal intensity for GATA3, ER, and TET2 in WT and GATA3 mutant (MUT) MCF7 cells, focusing on the sites in which mutant GATA3 is specifically enriched (n = 450 sites). ChIPs were performed in biological triplicate.
(D) Average plots showing normalized signal enrichment of GATA3, ER, or TET2 ChIP-seq at GATA3 sites common between mutant and WT cells (top, n = 34,845 sites) or at the GATA3 sites specifically gained in the mutant cells (bottom, n = 450 from C). Lines illustrate the signal enrichment for the respective factors in GATA3 mutant cells, and dotted lines indicate the enrichment in MCF7 WT cells.
To investigate the extent to which TET2’s capacity for regulating DNA modifications might be relevant to ER biology, total levels of 5mC and the most abundant TET2-catalyzed oxidation product 5hmC were investigated in response to TET2 silencing. Using mass spectrometry, global levels of 5hmC were shown to be decreased by ∼50% in three different ER+ breast cancer cell lines in response to TET2 knockdown for 48 h (Figure 6A; Figure S6). These effects were additionally measured in MCF7 cells at 72 h and 96 h TET2 knockdown, with the decline in 5hmC sustained over this time course. Despite the robust drop in 5hmC in response to TET2 knockdown, no significant change in 5mC was detected in any of the cell lines tested (Figure 6A; Figure S6).
Figure 6.

TET2 is required for maintaining 5hmC globally and at ER enhancer elements
(A) Mass spectrometry was used to assess levels of 5mC or 5hmC in genomic DNA isolated from MCF7 cells treated with either siNT or siTET2 for various durations. Results represent mean ± SD (n ≥ 4). Results are expressed as % of total cytosines.
(B) MA plots showing log2 fold change in 5mC + 5hmC (left, MMS readout) and 5hmC exclusively (right, RRHP readout) under control versus siTET2 conditions. Each datapoint represents an individual 5mC or 5hmC residue. siRNA treatments were performed for 72 h. Results represent biological duplicates.
(C) MMS signal (5mC + 5hmC) and RRHP signal (5hmC) were assessed at ER peak regions (left) or ER/TET2 overlapping peak regions (right) under control (siNT) or siTET2 conditions. The total numbers of sites analyzed within each category are as follows: ER MMS sites = 8,463, ER RRHP sites = 10,104, ER/TET2 MMS sites = 3,762, and ER/TET2 RRHP sites = 4,512.
To further probe TET2-mediated regulation of 5mC/5hmC in a site-specific manner, a form of bisulfite sequencing, Methyl-MiniSeq (MMS), was used to profile total 5mC and 5hmC under control and TET2 knockdown conditions. In addition, 5hmC was uniquely profiled using reduced representation hydroxymethylation profiling (RRHP) (Petterson et al., 2014). Although no global changes were observed in the MMS readout, implying no alteration to overall 5mC levels, a substantial and global drop in 5hmC was observed at the vast majority of sites where this modification was profiled using RRHP (Figure 6B). Figure 6C shows MMS and RRHP readouts under control or TET2 knockdown conditions at all ER sites and at the subset of ER sites co-occupied by TET2. Interestingly, the overall drop in 5hmC as measured by RRHP was the same at both groups of sites; hence, the magnitude of 5hmC loss may not rely on the robust physical binding of TET2 at ER regions. This is somewhat consistent with the findings of Rasmussen et al. (2019), who showed that most differentially methylated regions identified in TET2−/− mESCs did not demonstrate detectable TET2 binding. This result could imply that the TET2 antibodies used for ChIP in both our investigation and the Rasmussen et al. (2019) study may not have fully captured all TET2 sites, or that TET2 may regulate DNA modifications at some sites in the absence of robust and persistent binding. An alternative possibility is that these DNA modifications might change due to events secondary to TET2 depletion, pointing to an indirect role for TET2 in regulating 5mC/5hmC levels. Overall, these findings show that TET2 is required for deposition of 5hmC at ER enhancer elements, where the ER-associated transcription factor GATA3 is essential for effective TET2 expression and chromatin association. TET2 forms part of the ER/GATA3 complex and is an essential component required for effective ER transcriptional activity.
Discussion
A complex relationship exists between TET2 and components of the ER transcriptional machinery in ER+ breast cancer cells. Previous work has shown that TET2 co-binds with ER at enhancer elements and that TET2 is required for efficient ER binding (Wang et al., 2018), such that TET2 is an ER target gene but also plays a functional role as a mediator of ER-chromatin interactions. This work confirms these findings and shows that TET2 is both regulated by the ER/GATA3 pathway and required for optimal binding of ER to chromatin. These findings suggest that TET2 expression is dependent on GATA3, and when GATA3 is depleted from ER+ breast cancer cells, the ER interactome stays largely unaffected, with the exception of a significantly reduced association of TET2 with the ER complex. Because GATA3 is frequently mutated in breast cancer (Usary et al., 2004), TET2 function in these tumors is likely to be altered. This work also shows that TET2 chromatin binding exhibits dependence on ER because short-term ER depletion (when global TET2 levels were not grossly affected) resulted in diminished TET2 binding at several key ER regulatory elements. Previous work has suggested that the Complex Proteins Associated with Set1 (COMPASS) complex protein MLL3 is essential for TET2 expression and chromatin binding (Wang et al., 2018), although other work has suggested that TET2 is responsible for recruitment of the COMPASS complex to chromatin (Deplus et al., 2013). Work from our lab previously showed that MLL3 is recruited to ER enhancers by the pioneer factor FOXA1 (Jozwik et al., 2016), implying a functional connection between the three core ER transcription factor proteins (ER, GATA3, and FOXA1) and the enzymes that regulate both histone modifications and DNA modification events. The previous work showing a role for MLL3 in TET2 function might be a result of decreased TET2 expression in the absence of a functional ER complex (when MLL3 is silenced), akin to what is observed with GATA3 inhibition, where TET2-ER interactions are diminished as a result of decreased TET2 expression in GATA3-silenced cells.
What has become clear from this cell line and PDX ChIP-seq analysis is that in ER+ breast cancer cells, TET2 binding occurs almost exclusively at ER enhancer elements, regardless of the source of the cell line/tissue. Unlike other TET family members, TET1 and TET3, which possess CXXC domains enabling association with CG-rich sequences (Xu et al., 2018), TET2 appears to lack a sequence-targeted DNA binding domain (Ko et al., 2013). In contrast to TET1 and TET3, which tend to bind to promoter proximal regions (Jin et al., 2016; Rasmussen et al., 2015; Rasmussen and Helin, 2016; Williams et al., 2011), the preference of TET2 for enhancers supports the conclusion that TET2 recruitment involves cell-specific transcription factors (Wang et al., 2015; Chen et al., 2018); and in ER+ breast cancer cells, this appears to be mediated by the ER/GATA3 complex. Importantly, a role for TET2 in ER+ breast cancer cell function is evidenced by the global change in gene expression in the absence of TET2 (Wang et al., 2018), where loss of TET2 largely mimics the gene expression changes observed following either depletion of ER or GATA3, suggesting that these three proteins contribute to the same gene expression program.
TET2 is known to convert 5mC to 5hmC (He et al., 2011; Ito et al., 2011; Tahiliani et al., 2009). It was speculated that the recruitment of TET2 to ER enhancers might contribute to altered DNA modification dynamics at these regulatory elements. Initial mass-spectrometry-based global analysis revealed a decline in 5hmC but no appreciable change in 5mC when TET2 was depleted. To further support this result, RRHP analysis was conducted in the presence or absence of TET2; and when we focused specifically on ER enhancer elements throughout the genome, the same findings were recapitulated, with 5mC not detectably altered, but significant declines in 5hmC were observed at these sites.
TET2 mutations have been observed in different cancer types, most notably in hematological malignancies including chronic myelomonocytic leukemia, acute myeloid leukemia, and T cell lymphomas (Kosmider et al., 2009; Patnaik et al., 2016), but mutations in breast cancer are relatively rare (Stephens et al., 2012; Scourzic et al., 2015). However, changes in TET2 expression levels in ER+ breast cancer are associated with distinct clinical outcomes, possibly a result of changes in TET2’s contribution to ER-mediated gene expression. TET2 is required for efficient ER binding (Wang et al., 2018; this paper), but it has been shown in other cell types that TET2 is associated with active enhancers (Hon et al., 2014) and open chromatin regions, and that altered TET2 function is linked with changes in gene expression. Given the observations that conversion of 5mC to higher oxidation levels (including 5hmC and 5fC) are associated with the recruitment of distinct transcriptional regulators (Iurlaro et al., 2013, 2016) and altered transcriptional activity (Wu et al., 2011; Raiber et al., 2012; Lin et al., 2017), changes in the ratio of 5hmC to 5mC at ER enhancers likely alter gene expression potential. This could influence the expression of adjacent coding genes or expression of localized enhancer RNAs (eRNAs) that are abundantly produced in ER+ breast cancer models (Hah et al., 2011). These findings implicate TET2 as a GATA3-dependent factor required for effective ER transcriptional activity, which is associated with a TET2-dependent accumulation of 5hmC at ER enhancer elements that control cell cycle progression.
STAR★methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-β-actin (13E5) (used for Western Blot) | Cell Signaling Technology | Cat# 4970; RRID:AB_2223172 |
| Mouse monoclonal anti-ERα (6F11) (used for Western Blot) | Leica | Cat# NCL-L-ER-6F11; RRID:AB_563706 |
| Rabbit polyclonal anti-ERα (used for ChIP) | Abcam | Cat# ab3575; RRID:AB_303921 |
| Rabbit polyclonal anti-ERα (used for ChIP) | Millipore | Cat# 06-935; RRID:AB_310305 |
| Mouse monoclonal anti-GATA3 (HG3-31) (used for ChIP, Western Blot) | Santa Cruz Biotechnology | Cat# sc268; RRID:AB_2108591 |
| Rabbit polyclonal anti-GATA3 (used for ChIP, RIME) | Abcam | Cat# ab106625; RRID:AB_10887935 |
| Rabbit polyclonal anti-IgG isotype control (used for RIME) | Abcam | Cat# ab171870; RRID:AB_2687657 |
| Rabbit polyclonal anti-TET2 (used for ChIP, RIME) | Abcam | Cat# ab94580; RRID:AB_10887588 |
| IRDye 800 CW Goat anti-Mouse IgG | Li-Cor | Cat# 925-32210; RRID:AB_2687825 |
| IRDye 680LT Goat anti-Rabbit | Li-Cor | Cat# 926-68021; RRID:AB_10706309 |
| Bacterial and virus strains | ||
| One Shot TOP10 chemically competent E. coli | Thermo Fisher (Invitrogen) | Cat #C404010 |
| Biological samples | ||
| Patient-derived breast cancer xenograft models AB555 and STG195 | Caldas Lab, University of Cambridge (Bruna et al., 2016) | https://caldaslab.cruk.cam.ac.uk/bcape/ |
| Chemicals, peptides, and recombinant proteins | ||
| Fulvestrant (ICI-182780, ZD 9238) | Selleckchem | Cat# S1191 |
| Disuccinimidyl glutarate (DSG) | Santa Cruz Biotechnology | Cat# CAS79642-50-5 |
| SpikeTides peptides for targeted proteomics (Parallel Reaction Monitoring), see Method details for sequences | Custom-designed, synthesized by JPT Peptide Technologies | N/A |
| Critical commercial assays | ||
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# RS-122-2101 |
| Methyl Midi-seq (MMS) | Zymo Research (outsourced) | N/A |
| Reduced Representation Hydroxymethylation Profiling (RRHP) | Zymo Research (outsourced) | N/A |
| Panomics Nuclear Extraction Kit for Use with Transcription Factor Assays | Panomics | Cat# 13938, AY2002 |
| Ultra-Micro C18 Spin Columns | Harvard Apparatus | Cat# 74-7226 |
| iST 96x Sample Preparation Kit | Preomics | Cat# P.O.00027 |
| ThruPlex DNA-seq kit | Rubicon Genomics | Cat# R400407 |
| DNA HT Dual Index Kit - 96N Set A | Takara | Cat# R400660 |
| Deposited data | ||
| ChIP-seq, RNA-seq, MMS and RRHP datasets | This paper | GEO: GSE153255 |
| RIME, qPLEX-RIME and whole proteome datasets | This paper | ProteomeXchange Consortium via PRIDE (Perez-Riverol et al., 2019) partner repository: PXD019438 |
| PRM datasets | This paper | Panorama Public database: PXD019726 (also available via https://panoramaweb.org/TET2_Project.url) |
| Experimental models: cell lines | ||
| Human: MCF7 | ATCC | HTB-22 |
| Human: T-47D | ATCC | HTB-133 |
| Human: ZR-75-1 | ATCC | CRL-1500 |
| Oligonucleotides | ||
| TET2 forward primer for qRT-PCR 5′- ATTCTCGATTGTC TTCTCTAGTGAG-3′ |
This paper | N/A |
| TET2 reverse primer for qRT-PCR 5′- CATGTTTGGACTT CTGTGCTC-3′ |
This paper | N/A |
| UBC forward primer for qRT-PCR 5′- ATTTGGGTCGCG GTTCTTG-3′ |
This paper | N/A |
| UBC reverse primer for qRT-PCR 5′- TGCCTTGACATTC TCGATGGT-3′ |
This paper | N/A |
| RARα forward primer for ChIP-qPCR 5′- GCTGGGTCCT CTGGCTGTTC-3′ |
This paper | N/A |
| RARα reverse primer for ChIP-qPCR 5′- CCGGGATAAAGCCACTCCAA-3′ | This paper | N/A |
| GREB1 forward primer for ChIP-qPCR 5′- GAAGGGCAGAGCTGATAACG-3′ | This paper | N/A |
| GREB1 reverse primer for ChIP-qPCR 5′- GACCCAGTTGCCACACTTTT-3′ | This paper | N/A |
| MYC forward primer for ChIP-qPCR 5′- GCTCTGGGCACACACATTGG-3′ | This paper | N/A |
| MYC reverse primer for ChIP-qPCR 5′- GGCTCACCCTTGCTGATGCT-3′ | This paper | N/A |
| Negative control region forward primer for ChIP-qPCR 5′- GCCACCAGCCTGCTTTCTGT-3′ | This paper | N/A |
| Negative control region reverse primer for ChIP-qPCR 5′- CGTGGATGGGTCCGAGAAAC-3′ | This paper | N/A |
| ON-TARGETplus SMARTpool siRNAs against ER | Dharmacon (Horizon Discovery) | Cat# L-003401-00 |
| ON-TARGETplus SMARTpool siRNAs against GATA3 | Dharmacon (Horizon Discovery) | Cat# L-003781-00 |
| ON-TARGETplus SMARTpool siRNAs against TET2 | Dharmacon (Horizon Discovery) | Cat# L-013776-03 |
| ON-TARGETplus SMARTpool non-targeting control siRNAs | Dharmacon (Horizon Discovery) | Cat# D-001810-10 |
| Guide RNA (target sequence) targeting wild-type GATA3, including 5 base-pair 3′ overhang facilitating ligation into the GeneArt CRISPR Nuclease Vector: 5′- AGTGGCTGAAGGGCGAGATGGTTTT-3′ (PAM = TGG) | This paper | N/A |
| Guide RNA (reverse complement) targeting wild-type GATA3, including 5 base-pair 3′ overhang facilitating ligation into the GeneArt CRISPR Nuclease Vector: 5′- CATCTCGCCCTTCAGCCACTCGGTG-3′ (PAM = TGG) | This paper | N/A |
| U6 Forward Primer for Sanger sequencing 5′- GGACTA TCATATGCTTACCG −3′ |
Standard sequence | N/A |
| Custom primer for sequencing CRISPR/Cas9-edited clones: sense 5′-GCATCCAGACCAGAAACCGA-3′ | This paper | N/A |
| Custom primer for sequencing CRISPR/Cas9-edited clones: antisense 5′-TGAAACCCTCAACGGCAACT-3′ | This paper | N/A |
| Recombinant DNA | ||
| GeneArt CRISPR Nuclease Vector | GeneArt | Cat# A21174 |
| Software and algorithms | ||
| STAR v. 2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al., 2014 | https://github.com/mikelove/DESeq2 |
| Skyline-daily software v.19.0.9.190 | MacCoss Lab, University of Washington | https://skyline.ms/project/home/software/Skyline/begin.view |
| Proteome Discoverer v. 1.4 or 2.1 | Thermo Scientific | Cat# OPTON-30945 |
| qPLEXanalyzer | Papachristou et al., 2018 | http://www.bioconductor.org/packages/release/bioc/html/qPLEXanalyzer.html |
| Image Studio v. 4.0 software | Li-Cor | N/A |
| BioRad CFX Maestro software v. 1.1 | BioRad | N/A |
| bowtie2 v. 2.2.6 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 v. 2.0.10.20131216 | Zhang et al., 2008 | N/A |
| DiffBind | Stark and Brown, 2011 | http://www.bioconductor.org/packages/release/bioc/html/DiffBind.html |
| R v. 3.5.1 or later | R Project | https://www.r-project.org/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| MEME Suite (v. 4.9.1): FIMO | Grant et al., 2011 | https://meme-suite.org/ |
| MEME Suite (v. 4.9.1): MEME | Bailey and Elkan, 1994 | https://meme-suite.org/ |
| MEME Suite (v. 4.9.1): DREME | Bailey, 2011 | https://meme-suite.org/ |
| MEME Suite (v. 4.9.1): TOMTOM | Gupta et al., 2007 | https://meme-suite.org/ |
| Prism v. 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jason Carroll (jason.carroll@cruk.cam.ac.uk).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
hIP-seq, RNA-seq, Methyl Midi-Seq and Reduced Representation Hydroxymethylation Profiling datasets have been deposited to Gene Expression Omnibus (GEO) and are available under the accession number GSE153255. RIME, qPLEX-RIME and whole proteome data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019438. PRM datasets have been deposited to the Panorama Public database and can be accessed at http://panoramaweb.org/Panorama%20Public/2020/CRUK%20Proteomics%20Core%20-%20TET2_Project/project-begin.view? or using the dataset identifier PXD019726.
Experimental model and subject details
ER+ luminal breast cancer cell lines MCF7, T-47D and ZR-75-1 were obtained from ATCC. MCF7 cells were grown in DMEM (GIBCO), T-47D cells and ZR-75-1 cells in in RPMI 1640 (GIBCO). All media was supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin (GIBCO) and 2 mM L-glutamine (GIBCO). Cells were genotyped by short-tandem repeat (STR) profiling using the PowerPlex 16HS Cell Line panel and analyzed using Applied Biosystems Gene Mapper ID v 3.2.1 software by the external provider Genetica DNA Laboratories (LabCorp Specialty Testing Group) at least every six months and around every major experiment. Cells were routinely mycoplasma tested using MycoProbe Mycoplasma detection kit (R&D).
PDX material was kindly provided by Carlos Caldas and colleagues (Bruna et al., 2016). Frozen PDX tissue was propagated in immune-compromised mice. Briefly, tumor pieces (1 mm3) were implanted into the mammary pad of NOD scid gamma (NSG) mice. All mice were supplemented with estrogen, using silastic E2 pellets (made in-house) inserted into the dorsal scruff. Tumors were measured twice weekly. Once tumors reached ∼1000 mm3, mice were sacrificed by cervical dislocation under deep, isoflurane-induced anesthesia. Tumors were resected and either snap frozen in liquid nitrogen, fixed in 10% neutral buffered formalin solution for subsequent paraffin embedding, embedded in Optimal Cutting Temperature compound (OCT), or viably frozen in fetal calf serum (FCS) supplemented with 5% dimethyl sulphoxide (DMSO). STG195 possesses a Y537S mutation in the ESR1 gene; AB555 is ER wild-type.
Method details
Transfections and drug treatments
Control small interfering RNAs (siRNAs) (D-001810-10), and those used to knock down GATA3 (L-003781-00), ER (L-003401-00), and TET2 (L-013776-03) were obtained from Dharmacon (Horizon Discovery). For knockdowns, cells were transfected with siRNA using Lipofectamine RNAiMax Transfection Reagent (Invitrogen) according to manufacturer’s instructions. All siRNAs were used at a final concentration of 10 nM. For cell treatments, fulvestrant (Selleckchem) was used at a final concentration of 100 nM. For the purposes of obtaining protein, RNA or DNA samples, cells were washed twice in cold PBS and harvested in PBS containing protease inhibitors (Roche). For growth assays, cells were left in transfection medium for the duration of the assay.
Guide RNA design
Guide RNAs were designed with the GeneArt CRISPR gRNA Design Tool. Initial guide sequences were selected based on the Homo sapiens GATA binding protein 3 (GATA3) gene (NCBI Reference Sequence: NM_001002295.1) and ranked according to the number of potential off-target sites to select designs that minimized the possibility of off-target cleavage. Three pairs of single-stranded oligonucleotides (Sigma) were ordered based on these 19–20 base pair target sequences adjacent to an NGG proto-spacer adjacent motif (PAM) sequence on the 3′ end, with compatible 3′ overhangs added to complement the linearized GeneArt CRISPR Nuclease Vector (Cat# A21174). The gRNA sequence resulting in successful generation of the desired GATA3 mutation is shown in the Key Resources table.
Generation of CRISPR nuclease constructs
Equal amounts of each single-stranded oligonucleotide were annealed to generate a double-stranded (ds) oligonucleotide and cloned into the GeneArt CRISPR Nuclease Vector using T4 DNA Ligase according to manufacturer’s instructions. In parallel, the supplied ds control oligonucleotide was used as a positive ligation control. A separate ligation reaction, omitting the ds oligonucleotide, was performed as a negative control. One Shot TOP10 chemically competent E. coli (Invitrogen) were transformed with the resulting CRISPR nuclease constructs. 50 μL from each transformation reaction was spread on a pre-warmed LB agar plate containing 100 μg/ml ampicillin and incubated overnight at 37°C. Ten colonies from each ligation were selected for further analysis and cultured overnight in LB medium containing 100 μg/mL ampicillin at 37°C. To confirm the identity and correct orientation of the ds oligonucleotide insert, a QIAprep Spin Miniprep Kit (QIAGEN) was used to isolate plasmid DNA, and Sanger sequencing verification was performed using the U6 Forward Primer (5′- GGACTATCATATGCTTACCG −3′).
Transfection and isolation of positive cells
MCF7 cells were maintained in culture medium as described above. 24 hours prior to transfection, cells were plated in growth medium without antibiotics, such that they were 60%–65% confluent on the day of transfection. Cells were transfected with the CRISPR/Cas9 expression vector using Lipofectamine 2000 Transfection Reagent (Invitrogen) according to manufacturer’s instructions. As a parallel control, cells were transfected with the CRISPR/Cas9 ‘empty’ expression vector. 48 hours post-transfection, cells were harvested, resuspended in fluorescence activated cell sorting (FACS) buffer and subjected to high-throughput FACS using the orange fluorescent protein (OFP) reporter to enrich for the Cas9-expressing live cell population. Live OFP-positive cells were plated by limiting dilution in full media and monitored daily for single cell clonal isolation. Multiple colonies were harvested via disruption with a pipette tip, and sequentially transferred to increasingly larger culture vessels until sufficient material was collected for detection of the engineering event in endogenous GATA3.
Screening for CRISPR/Cas9 engineering event by Sanger sequencing
Genomic DNA was extracted using a QIAamp DNA Mini Kit (QIAGEN). Custom-designed primers (sense 5′-GCATCCAGACCAGAAACCGA-3′, antisense 5′-TGAAACCCTCAACGGCAACT-3′) (Sigma) were used to amplify the gRNA-targeted region through polymerase chain reaction (PCR) using Platinum Taq DNA Polymerase (Invitrogen). An amplicon of the predicted size was gel-excised and purified using a QIAquick Gel Extraction Kit (QIAGEN). For each clone, forward and reverse sequences were determined by Sanger sequencing using the same primers as for the PCR.
Analysis of cell growth
Cell growth after siRNA transfection was assessed using the IncuCyte ZOOM Live Cell Analysis System (Essen BioScience). Cells were seeded in 96-well plates and transfected in at least quadruplicate, upon which plates were immediately placed in the IncuCyte ZOOM Live Cell Analysis System (37°C with 5% CO2) and growth was monitored for at least 120 hours via phase-contrast images taken every 3 hours. Confluence was assessed using default settings of the IncuCyte ZOOM software.
RNA isolation and quantification
Total RNA was extracted using an RNeasy kit (QIAGEN) according to manufacturer’s instructions, and quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific).
Quantitative real-time PCR
Total RNA (1 μg) was used for cDNA conversion and qRT-PCR analysis. cDNA was synthesized using the SuperScript III Reverse Transcriptase kit (Invitrogen) according to manufacturer’s instructions. For subsequent qPCR analysis, cDNA was diluted 1:10. Reactions were performed in triplicate using 1X Power SYBR Green PCR Master Mix (Applied Biosystems) and run on the BioRad CFX Connect RealTime System and analyzed using BioRad CFX Maestro software v. 1.1.
RNA sequencing (RNA-seq)
RNA libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina) and samples were sequenced on a HiSeq 4000 to approximately 30 million reads per sample. 50 bp single-end reads were aligned to the Human Reference Genome (assembly hg38) using STAR v. 2.5.2b (Dobin et al., 2013). Read counts were normalized and tested for differential gene expression using DESeq2 (Love et al., 2014).
Mass spectrometry measurements of 5mC and 5hmC
Cells were harvested and DNA extracted using a DNeasy Blood and Tissue Kit (QIAGEN) according to manufacturer’s instructions. DNA was RNase (Sigma) treated on-column (20 μl of 20 mg/ml) for 15 minutes at room temperature. Purified DNA was quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific). Subsequently, 5mC and 5hmC were measured according to Bachman et al. (2014). Nucleotides were quantified using a Thermo Q-Exactive mass spectrometer. 5mC levels are expressed as a percentage of total cytosines.
Methyl Midi-seq (MMS) sample preparation and sequencing
Library preparation, sequencing and initial data processing for genomic analysis of 5mC was undertaken using the Methyl Midi-seq (MMS) service provided externally by Zymo Research (Irvine, California). Cells were harvested and DNA extracted using a DNeasy Blood and Tissue Kit (QIAGEN) according to manufacturer’s instructions. DNA was RNase (Sigma) treated on-column (20 μl of 20 mg/mL) for 15 minutes at room temperature. Purified DNA was quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific). Subsequently, libraries were prepared from 500 ng of genomic DNA sequentially digested with 60 U of TaqαI and 30 U of MspI (NEB) and then extracted with a DNA Clean & Concentrator kit (Zymo Research). Fragments were ligated to pre-annealed adapters containing 5′-methylcytosine instead of cytosine according to Illumina’s specified guidelines (https://www.illumina.com). Adaptor-ligated fragments of 150–250 bp and 250–350 bp were recovered from a 2.5% NuSieve 1:1 agarose gel using a Zymoclean Gel DNA Recovery Kit (Zymo Research). The fragments were then bisulfite-treated using the EZ DNA Methylation-Lightning Kit (Zymo Research). Preparative-scale PCR was performed and the resulting products were purified using a DNA Clean & Concentrator kit (Zymo Research) for sequencing on an Illumina HiSeq.
MMS analysis
Sequence reads from bisulfite-treated EpiQuest libraries were identified using standard Illumina basecalling software and then analyzed using a Zymo Research proprietary analysis pipeline, which is written in Python and uses Bismark to perform the alignment (Krueger and Andrews, 2011). Index files were constructed using the bismark_genome_preparation command and the entire reference genome. The –non_directional parameter was applied while running Bismark. All other parameters were set to default. Filled-in nucleotides were trimmed off when doing methylation calling. The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. The genome was partitioned into non-overlapping tiles of length 1 kb and 5mC was profiled within these tiles. Regions with low read coverage (less than 4 reads in any sample) were discarded from the analysis. The differential 5mC analysis was carried out by methylKit using Fisher Exact test (Akalin et al., 2012). For site-specific 5mC/5hmC analysis, shared ER/TET2 regions were defined using the intersect function in R to generate completely overlapping regions.
Reduced Representation Hydroxymethylation Profiling (RRHP) sample preparation and sequencing
Library preparation, sequencing and initial data processing for genomic analysis of 5hmC was undertaken using the Reduced Representation Hydroxymethylation Profiling (RRHP) service provided externally by Zymo Research (Irvine, California), as described in Petterson et al. (2014). Cells were harvested and DNA extracted using a DNeasy Blood and Tissue Kit (QIAGEN) according to manufacturer’s instructions. DNA was RNase (Sigma) treated on-column (20 μl of 20 mg/ml) for 15 minutes at room temperature. Purified DNA was quantified using a Qubit 3.0 Fluorometer (Thermo Fisher Scientific). Subsequently, genomic DNA was fragmented overnight at 37°C with a hydroxymethyl-insensitive enzyme, MspI, and purified using the DNA Clean & Concentrator kit (Zymo Research). Modified Illumina TruSeq P5 and P7 adapters containing 5′-CG overhangs were ligated onto the digested DNA using T4 DNA ligase (2 hours at 16°C). Libraries were then strand extended at 72°C with Taq DNA Polymerase. The adapters were designed to regenerate the 5′-CCGG site at the P5 junction while the P7 adaptor generates a 5′-TCGG junction, making it insensitive to MspI digestion. Adapterised libraries were treated with β-glucosyltransferase to label 5hmC modifications and purified using a DNA Clean & Concentrator kit (Zymo Research). The glucosylated libraries were then subjected to an overnight MspI digestion at 37°C, cutting any fragments not containing a glucosyl-5hmC site at the P5 CCGG junction. After incubation, the libraries were size-selected from 100 bp to 500 bp and purified using a Zymoclean Gel DNA Recovery Kit (Zymo Research). The fragments were amplified using OneTaq 2X Master Mix (NEB), with PCR conditions including an initial denaturation of 94°C for 30 s followed by 12 cycles of 94°C for 30 s, 58°C for 30 s, and 68°C for 1 minute. Fragments containing 5hmC were positively selected during PCR amplification with adaptor-specific indexing primers whereas fragments lacking glucosylated-5hmC at the P5 junction were cleaved and, therefore, not amplified by PCR. Amplified libraries were purified using the DNA Clean and Concentrator kit, and multiplexed using equal volumes of the libraries. All adapters and primers used were synthesized by Integrated DNA Technologies.
RRHP analysis
Sequence reads from RRHP libraries were first processed to trim off the low quality bases and the P7CG adaptor at the 3′ end of the reads. Reads were then aligned to the reference genome using bowtie (Langmead and Salzberg, 2012) default parameters and the ‘–best’ setting. Aligned reads with the MspI tag (CCGG) were counted. The correlation analysis between different RRHP libraries was performed by comparing the presence of the tagged reads at each profiled MspI site, and Pearson’s coefficient was calculated accordingly. The genome was partitioned into non-overlapping tiles of length 1 kb and 5hmC was profiled within these tiles. Regions with low read coverage (those with less than 10 reads in any sample) were discarded from the analysis. The differential 5hmC analysis was carried out using DESeq2 (Love et al., 2014). For site-specific 5mC/5hmC analysis, shared ER/TET2 regions were defined using the intersect function in R to generate completely overlapping regions.
Western blot
Cells were harvested in RIPA buffer (Pierce) and the lysate sonicated using a Bioruptor Plus (Diagenode) for 2 minutes (30 s on/30 s off) to degrade the DNA. Protein was then quantified using a Direct Detect Spectrometer (Millipore). Samples were denatured and run on NuPAGE 4%–12% bis-Tris gels (Invitrogen). Proteins were transferred to nitrocellulose membranes using the iBlot 2 Dry Transfer System (Invitrogen), and membranes were blocked using Odyssey Blocking Buffer (Li-Cor) for 1 hour at room temperature then incubated with primary antibody overnight at 4°C. After washing, membranes were incubated with fluorescent secondary antibodies (IRDye 800 CW Goat anti-Mouse IgG 1:5000 or IRDye 680LT Goat anti-Rabbit 1:20000, both Li-Cor) before imaging using the Odyssey CLx Imaging System (Li-Cor). Images were taken with the automated capture option of the Image Studio v. 4.0 software.
Parallel Reaction Monitoring (PRM) sample preparation and sequencing
Surrogate peptides unique to the target proteins of interest (ER, TET2, GATA3, mutant GATA3 and Actin) were chosen and stable-isotope-labeled versions (SIS) of these peptides were synthesized as SpikeTides peptides by JPT Peptide Technologies, GmbH (Berlin, Germany). Peptide sequences are shown in the table below. PRM assays were first characterized by performing a response curve assay to identify the lower limit of quantification (LLOQ), limit of detection (LOD) and linear range for each surrogate peptide. Briefly, a background matrix consisting on a MCF7 cell lysate was digested with trypsin. Reverse curves were prepared in triplicate by varying SIS peptide concentration over nine concentration points (1000, 200, 66.6, 22.2, 7.4, 2.46, 0.82, 0.27, 0.09, fmol/μg). Light peptide was added at a constant concentration of 100 fmol/μg. Blanks contained no SIS peptide. Peak areas of all transitions of a particular peptide were first summed and then peak area ratios heavy/light were calculated and averaged for the three replicates of the curve. The limit of detection (LOD) was obtained by using the average of the three blank measurements plus three times the standard deviation of the blank signal. The lower limit of quantification (LLOQ) was reported as the lowest point in the response curve measured with CV ≤ 20%. Liner regression was used to fit the data and the coefficient of determination (R-squared) was calculated; data was considered linear if R-squared was > 0.95. Peptides that did not follow a linear behavior were not considered for further experiments. For the quantitative analysis of proteins of interest, cells were washed twice in cold PBS and harvested in PBS containing protease inhibitors (Roche). Cells were lysed and peptides were digested with trypsin, and a mix of stable isotope-labeled peptide standards was added to the mixture at a concentration dependent on their LLOQ. Mixtures were desalted using either Ultra-Micro C18 Spin Columns (Harvard Apparatus) or cartridges from an iST Sample Preparation Kit (Preomics) and reconstituted in either 3% acetonitrile/0.1% formic acid or the iST Sample Preparation Kit load buffer (Preomics). A Pierce Peptide Retention Time Calibration Mixture containing 15 synthetic heavy peptides mixed at an equimolar ratio (Thermo Scientific) was added to each sample at a final concentration of 20 fmol of peptides per 2 μg of total protein to assess chromatography performance and optimize scheduled MS acquisition windows. Diluted peptide mixtures were analyzed by liquid chromatography mass spectrometry (LC-MS) on a Dionex Ultimate 3000 UHPLC system coupled to a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific). Scheduled PRM transitions used a retention time window of 120 s. All samples were analyzed in triplicate in the mass spectrometer.
PRM analysis
All raw files were processed using Skyline-daily software v.19.0.9.190 (MacCoss Lab, University of Washington) for the generation of extracted-ion chromatograms and peak integration. Peak integrations were reviewed manually and transitions from analyte peptides were confirmed by the same retention times of the endogenous peptides and heavy stable isotope-labeled peptides time in a pre-selected retention time window. At least three transition ion peak areas were integrated and summed for each peptide (heavy and endogenous). The ratio of endogenous/heavy peak areas was calculated and the average of three independent injections of every sample was calculated to obtain a final quantification value for each peptide. Data were exported from Skyline for analysis and plotting using an in-house R script to calculate fold changes and p values between different experimental conditions. Quantitative values obtained for actin peptides were used to normalize the data between different conditions.
| Protein (UniProt ID) | Peptide sequence | Peptide sequence (modified) | Isotope | Mass [m/z] |
|---|---|---|---|---|
| P03372: ESR1_HUMAN | EAGPPAFYRPNSDNR | EAGPPAFYRPNSDNR | light | 564.3 |
| P03372: ESR1_HUMAN | EAGPPAFYRPNSDNR | EAGPPAFYRPNSDNR | heavy | 567.6 |
| P03372: ESR1_HUMAN | AANLWPSPLMIK | AANLWPSPLMIK | light | 670.9 |
| P03372: ESR1_HUMAN | AANLWPSPLMIK | AANLWPSPLMIK | heavy | 674.9 |
| P03372: ESR1_HUMAN | ELVHMINWAK | ELVHMINWAK | light | 620.8 |
| P03372: ESR1_HUMAN | ELVHMINWAK | ELVHMINWAK | heavy | 624.8 |
|
Q6N021: TET2_HUMAN |
VSPDFTQESR | VSPDFTQESR | light | 583.3 |
|
Q6N021: TET2_HUMAN |
VSPDFTQESR | VSPDFTQESR | heavy | 588.3 |
|
Q6N021: TET2_HUMAN |
EGSFFGQTK | EGSFFGQTK | light | 500.7 |
|
Q6N021: TET2_HUMAN |
EGSFFGQTK | EGSFFGQTK | heavy | 504.7 |
|
Q6N021: TET2_HUMAN |
VSDVDEFGSVEAQEEK | VSDVDEFGSVEAQEEK | light | 884.4 |
|
Q6N021: TET2_HUMAN |
VSDVDEFGSVEAQEEK | VSDVDEFGSVEAQEEK | heavy | 888.4 |
|
Q6N021: TET2_HUMAN |
SGAIQVLSSFR | SGAIQVLSSFR | light | 582.8 |
|
Q6N021: TET2_HUMAN |
SGAIQVLSSFR | SGAIQVLSSFR | heavy | 587.8 |
|
Q6N021: TET2_HUMAN |
QLAELLR | QLAELLR | light | 421.8 |
|
Q6N021: TET2_HUMAN |
QLAELLR | QLAELLR | heavy | 426.8 |
|
Q6N021: TET2_HUMAN |
YPSQDPLSK | YPSQDPLSK | light | 517.8 |
|
Q6N021: TET2_HUMAN |
YPSQDPLSK | YPSQDPLSK | heavy | 521.8 |
|
Q6N021: TET2_HUMAN |
YGPDYVPQK | YGPDYVPQK | light | 533.8 |
|
Q6N021: TET2_HUMAN |
YGPDYVPQK | YGPDYVPQK | heavy | 537.8 |
|
P23771: GATA3_HUMAN mutant |
IMFATLQR | IMFATLQR | light | 490.3 |
|
P23771: GATA3_HUMAN mutant |
IMFATLQR | IMFATLQR | heavy | 495.3 |
|
P23771: GATA3_HUMAN mutant |
SSLWCLCSNH | SSLWC[+57.021464]LC[+57.021464]SNH | light | 632.3 |
|
P23771: GATA3_HUMAN mutant |
SSLWCLCSNH | SSLWC[+57.021464]LC[+57.021464]SNH | heavy | 635.8 |
|
P23771: GATA3_HUMAN |
ALGSHHTASPWNLSPFSK | ALGSHHTASPWNLSPFSK | light | 646.3 |
|
P23771: GATA3_HUMAN |
ALGSHHTASPWNLSPFSK | ALGSHHTASPWNLSPFSK | heavy | 649.0 |
|
P23771: GATA3_HUMAN |
DVSPDPSLSTPGSAGSAR | DVSPDPSLSTPGSAGSAR | light | 850.9 |
|
P23771: GATA3_HUMAN |
DVSPDPSLSTPGSAGSAR | DVSPDPSLSTPGSAGSAR | heavy | 855.9 |
|
P23771: GATA3_HUMAN |
ECVNCGATSTPLWR | EC[+57.021464]VNC[+57.021464]GATSTPLWR | light | 825.9 |
|
P23771: GATA3_HUMAN |
ECVNCGATSTPLWR | EC[+57.021464]VNC[+57.021464]GATSTPLWR | heavy | 830.9 |
|
P23771: GATA3_HUMAN |
AGTSCANCQTTTTTLWR | AGTSC[+57.021464]ANC[+57.021464]QTTTTTLWR | light | 964.9 |
|
P23771: GATA3_HUMAN |
AGTSCANCQTTTTTLWR | AGTSC[+57.021464]ANC[+57.021464]QTTTTTLWR | heavy | 969.9 |
|
P23771: GATA3_HUMAN |
NSSFNPAALSR | NSSFNPAALSR | light | 582.3 |
|
P23771: GATA3_HUMAN |
NSSFNPAALSR | NSSFNPAALSR | heavy | 587.3 |
|
P60709: ACTB_HUMAN |
VAPEEHPVLLTEAPLNPK | VAPEEHPVLLTEAPLNPK | light | 652.0 |
|
P60709: ACTB_HUMAN |
VAPEEHPVLLTEAPLNPK | VAPEEHPVLLTEAPLNPK | heavy | 654.7 |
|
P60709: ACTB_HUMAN |
EITALAPSTMK | EITALAPSTMK | light | 581.3 |
|
P60709: ACTB_HUMAN |
EITALAPSTMK | EITALAPSTMK | heavy | 585.3 |
ChIP sample preparation and sequencing
ChIP was performed as described by Papachristou et al. (2018). Briefly, cells were crosslinked at room temperature by incubating with 2 mM disuccinimidyl glutarate (DSG) for 20 min followed by 1% formaldehyde for 10 min before crosslinking was quenched with 0.1 M glycine for 10 min. Cells were then washed twice in cold PBS and harvested in cold PBS containing protease inhibitors (Roche). Crosslinked cells were incubated with lysis buffer 1 (LB1, 50 mM HEPES–KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40/Igepal CA-630, 0.25% Triton X-100) for 10 min followed by 5 min in lysis buffer 2 (LB2, 10 mM Tris–HCL, pH8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) before resuspending in lysis buffer 3 (LB3, 10 mM Tris–HCl, pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na–Deoxycholate, 0.5% N-lauroylsarcosine). Chromatin was sonicated using a Bioruptor Plus (Diagenode) for 15 min (30 s on/30 s off) to generate DNA fragments of around 100-800 bp. Beads were pre-bound with antibody overnight at 4°C, with 5 μg of the appropriate antibody (or 2.5 μg each of ERα Abcam ab3575 and ERα Millipore 06-935 where these antibodies were used in a 1:1 combination) and 50 μL of Protein A Dynabeads (Invitrogen) was used for each immunoprecipitation. After washing to remove unbound antibody, chromatin and beads were combined and samples were immunoprecipitated overnight at 4°C. The following day, beads were washed 10 times in RIPA buffer (50mM HEPES pH 7.6, 1mM EDTA, 0.7% Na-deoxycholate, 1% NP-40, 0.5M LiCl) followed by 2 washes in Tris-EDTA (10mM Tris-HCl, 1mM EDTA). Chromatin was eluted and decrosslinked by incubating samples in elution buffer (50mM TrisHCl, pH8, 10mM EDTA, 1% SDS) for 6-18 hours at 65°C. Eluted DNA was treated with RNase A (20 ng/ml) for 1 hour followed by proteinase K (200 ng/ml) for 2 hours before DNA was purified by phenol-chloroform extraction and taken forward for either qPCR or sequencing. For ChIP-qPCR analysis, ChIP DNA was used neat and input DNA diluted 1:10 for qPCR analysis. Relative enrichment was determined as % of input. For ChIP-seq library preparation and sequencing, library preparation was performed using the ThruPlex DNA-seq kit (Rubicon Genomics) or the DNA HT Dual Index Kit (Takara), and DNA was subjected to next generation sequencing on a HiSeq 4000 (Illumina) or NovaSeq 6000 (Illumina) to reach approximately 30 million reads per sample.
ChIP-seq analysis
50bp single-end (HiSeq 4000) or 50bp paired-end (NovaSeq 6000) reads were aligned to the Human Reference Genome (assembly hg38) using bowtie2 v. 2.2.6 (Langmead and Salzberg, 2012). Aligned reads with a mapping quality of less than 5 were filtered out. The read alignments from all replicates were combined into a single library and peaks were called using MACS2 v. 2.0.10.20131216 (Zhang et al., 2008) with sequences from chromatin extracts from the same cell line or PDX used as a background input control. The peaks yielded with a MACS2 q-value ≤ 1e-3 were selected for downstream analysis. Differential binding analysis was performed as described previously using DiffBind (Stark and Brown, 2011). For heatmaps, MA plots, and average plots visualizing tag density and signal distribution, consensus peak sets across the compared conditions were determined using DiffBind. Heatmaps and average plots were generated with the read coverage in a window of ± 5 kb flanking the tag midpoint using a bin size of 1/100 of the window length. MA plots were generated in R v. 3.5.1 or later, and average plots, boxplots and heatmaps were generated using MATLAB. MEME Suite (v. 4.9.1) tools were used for motif analysis. FIMO (Grant et al., 2011) was used to search all known transcription factor motifs from the JASPAR database (JASPAR CORE 2016 vertebrates) in tag-enriched sequences. Peak size-matched, randomly selected open chromatin regions based on an MCF7 MNase dataset (EBI Array Express E-MTAB-1958) were used as background controls. The motif frequencies for both tag-enriched and control sequences were calculated as the sum of motif occurrences adjusted with MEME q-value. Motif enrichment analysis was performed by calculating the odds of finding an overrepresented motif among MACS2-defined peaks by fitting Student’s t-cumulative distribution to the ratios of motif frequencies between tag-enriched and background sequences. Yielded p values were further adjusted using Benjamini-Hochberg correction. MEME (Bailey and Elkan, 1994) and DREME (Bailey, 2011) were used to perform de novo motif analysis on sequences corresponding to ChIP-seq peak regions and the resulting position weight matrix was compared to the JASPAR, Transfac and UNIPROBE databases by the TOMTOM application (Gupta et al., 2007). A p value 0.0001 was used as a threshold to define the presence of a motif.
Rapid Immunoprecipitation Mass-spectrometry of Endogenous Proteins (RIME) and whole proteome analysis
Non-quantitative RIME, qPLEX-RIME, and full proteome analysis were performed as described by Papachristou et al. (2018), with chromatin preparation and IP for RIME as described for ChIP, above, with beads undergoing an additional two washes with 100 mM ammonium hydrogen carbonate (AMBIC) after the final RIPA wash. Peptide samples were analyzed on a Dionex Ultimate LC system coupled with an LTQ Orbitrap Velos or a Fusion Lumos mass spectrometer (Thermo Scientific). For non-quantitative RIME, specific interactors were considered as those occurring in at least two out of three independent replicates. Any proteins that appeared in any one of the three IgG control RIME experiments were excluded. Raw MS files were processed with the SequestHT search engine on the Proteome Discoverer v. 1.4 or 2.1 software for peptide and protein identifications. Pre-processed quantitative datasets (peptide or protein-level TMT intensities) generated by Proteome Discoverer were imported into R and data analyzed using the qPLEXanalyzer tool (Papachristou et al., 2018).
Survival analysis
For analysis of relapse free survival, Kaplan-Meier plotter (http://kmplot.com) was used. The data and methods used for the analysis are described in Györffy et al. (2010). Briefly, patients were stratified into high or low expression groups according to the median level of the gene probe selected (TET2 JetSet probe 227624_at). ER+ and ER- cohorts were analyzed separately, with the “ER status derived from GE data” option selected. All other parameters were left as default.
Quantification and statistical analysis
All analysis additional to that described in the method-specific sections above was performed using either MS Excel, GraphPad Prism v. 8 or R v. 3.5.1. Significance was assessed using Student’s t test or Welch’s t test. Only values with a p value less than 0.05 were considered statistically significant. Error bars represent standard deviation (SD).
Acknowledgments
We thank Carlos Caldas and Alejandra Bruna for providing PDX material for the ChIP-seq experiments. We thank David Tannahill and Eun-Ang Raiber for discussions and support. We also thank the core facilities at Cancer Research UK (Genomics, Proteomics, Bioinformatics, Biorepository and Research Instrumentation). We would like to acknowledge the support of the University of Cambridge, Cancer Research UK and Hutchison Whampoa Limited. The work was supported by the NIHR Cambridge BRC. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. R.B. is supported by a CRUK PhD studentship. S.J. was funded by CRUK. J.S.C. is funded by Cancer Research UK, an ERC Consolidator Award, and a Komen Scholarship. R.S. is funded by the Novo Nordisk Foundation (NNF15OC0014136). A.M.F. is funded through the AstraZenecaPostdoctoral Programme. The Balasubramanian lab is supported by Program grant funding (C9681/A18618 and C9681/A29214) and core funding (C14303/A171970) from Cancer Research UK and a Wellcome Trust Investigator Award (209441/z/17/z).
Author contributions
Conceptualization, R.B., R.S., and J.S.C.; experimental work, R.B., A.M.F., R.S., S.J., E.K.P., S.-Q.M., V.T., and C.G.T.; data analysis, R.B., R.S., I.C., S.-Q.M., A.M., C.G.T., A.J.G., and K.K.; oversaw work, C.D., S.B., R.S., and J.S.C.; writing, R.B., A.M.F., and J.S.C.; review, all authors.
Declaration of interests
S.B. is a founder and shareholder of Cambridge Epigenetix, Ltd.
Published: February 23, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108776.
Contributor Information
Anca Madalina Farcas, Email: ancamadalina.farcas@cruk.cam.ac.uk.
Rasmus Siersbæk, Email: siersbaek@bmb.sdu.dk.
Jason S. Carroll, Email: jason.carroll@cruk.cam.ac.uk.
Supplemental information
References
- Akalin A., Kormaksson M., Li S., Garrett-Bakelman F.E., Figueroa M.E., Melnick A., Mason C.E. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzick S.L., Kononen J., Walker R.L., Azorsa D.O., Tanner M.M., Guan X.Y., Sauter G., Kallioniemi O.P., Trent J.M., Meltzer P.S. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Bachman M., Uribe-Lewis S., Yang X., Williams M., Murrell A., Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014;6:1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bird A.P., Wolffe A.P. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bruna A., Rueda O.M., Greenwood W., Batra A.S., Callari M., Batra R.N., Pogrebniak K., Sandoval J., Cassidy J.W., Tufegdzic-Vidakovic A. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell. 2016;167:260–274.e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J.S., Liu X.S., Brodsky A.S., Li W., Meyer C.A., Szary A.J., Eeckhoute J., Shao W., Hestermann E.V., Geistlinger T.R. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll J.S., Meyer C.A., Song J., Li W., Geistlinger T.R., Eeckhoute J., Brodsky A.S., Keeton E.K., Fertuck K.C., Hall G.F. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chen D., Huang S.M., Stallcup M.R. Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J. Biol. Chem. 2000;275:40810–40816. doi: 10.1074/jbc.M005459200. [DOI] [PubMed] [Google Scholar]
- Chen L.L., Lin H.P., Zhou W.J., He C.X., Zhang Z.Y., Cheng Z.L., Song J.B., Liu P., Chen X.Y., Xia Y.K. SNIP1 Recruits TET2 to Regulate c-MYC Target Genes and Cellular DNA Damage Response. Cell Rep. 2018;25:1485–1500.e4. doi: 10.1016/j.celrep.2018.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplus R., Delatte B., Schwinn M.K., Defrance M., Méndez J., Murphy N., Dawson M.A., Volkmar M., Putmans P., Calonne E. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes J., Bourdeau V., White J.H., Mader S. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J. Biol. Chem. 2007;282:17335–17339. doi: 10.1074/jbc.C700030200. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckhoute J., Keeton E.K., Lupien M., Krum S.A., Carroll J.S., Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–6483. doi: 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- Filion G.J., Zhenilo S., Salozhin S., Yamada D., Prokhortchouk E., Defossez P.A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 2006;26:169–181. doi: 10.1128/MCB.26.1.169-181.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant C.E., Bailey T.L., Noble W.S. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Hah N., Danko C.G., Core L., Waterfall J.J., Siepel A., Lis J.T., Kraus W.L. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G.C., Song C.X., Du T., Jin F., Selvaraj S., Lee A.Y., Yen C.A., Ye Z., Mao S.Q., Wang B.A. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell. 2014;56:286–297. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iurlaro M., Ficz G., Oxley D., Raiber E.A., Bachman M., Booth M.J., Andrews S., Balasubramanian S., Reik W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi: 10.1186/gb-2013-14-10-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iurlaro M., McInroy G.R., Burgess H.E., Dean W., Raiber E.A., Bachman M., Beraldi D., Balasubramanian S., Reik W. In vivo genome-wide profiling reveals a tissue-specific role for 5-formylcytosine. Genome Biol. 2016;17:141. doi: 10.1186/s13059-016-1001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.G., Zhang Z.M., Dunwell T.L., Harter M.R., Wu X., Johnson J., Li Z., Liu J., Szabó P.E., Lu Q. Tet3 Reads 5-Carboxylcytosine through Its CXXC Domain and Is a Potential Guardian against Neurodegeneration. Cell Rep. 2016;14:493–505. doi: 10.1016/j.celrep.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwik K.M., Chernukhin I., Serandour A.A., Nagarajan S., Carroll J.S. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. 2016;17:2715–2723. doi: 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose R.J., Bird A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Ko M., An J., Bandukwala H.S., Chavez L., Aijö T., Pastor W.A., Segal M.F., Li H., Koh K.P., Lähdesmäki H. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature. 2013;497:122–126. doi: 10.1038/nature12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S.L., Li G., Loh S.L., Sung W.K., Liu E.T. Cellular reprogramming by the conjoint action of ERα, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 2011;7:526. doi: 10.1038/msb.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmider O., Gelsi-Boyer V., Ciudad M., Racoeur C., Jooste V., Vey N., Quesnel B., Fenaux P., Bastie J.N., Beyne-Rauzy O. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676–1681. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H., Slorach E.M., Sternlicht M.D., Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041–1055. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.Y., Vega V.B., Thomsen J.S., Zhang T., Kong S.L., Xie M., Chiu K.P., Lipovich L., Barnett D.H., Stossi F. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin I.H., Chen Y.F., Hsu M.T. Correlated 5-Hydroxymethylcytosine (5hmC) and Gene Expression Profiles Underpin Gene and Organ-Specific Epigenetic Regulation in Adult Mouse Brain and Liver. PLoS One. 2017;12:e0170779. doi: 10.1371/journal.pone.0170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H., D’Santos C., Serandour A.A., Ali H.R., Brown G.D., Atkins A., Rueda O.M., Holmes K.A., Theodorou V., Robinson J.L. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3:342–349. doi: 10.1016/j.celrep.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papachristou E.K., Kishore K., Holding A.N., Harvey K., Roumeliotis T.I., Chilamakuri C.S.R., Omarjee S., Chia K.M., Swarbrick A., Lim E. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018;9:2311. doi: 10.1038/s41467-018-04619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik M.M., Zahid M.F., Lasho T.L., Finke C., Ketterling R.L., Gangat N., Robertson K.D., Hanson C.A., Tefferi A. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J. 2016;6:e472. doi: 10.1038/bcj.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou C.M., Sørlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Petterson A., Chung T.H., Tan D., Sun X., Jia X.Y. RRHP: a tag-based approach for 5-hydroxymethylcytosine mapping at single-site resolution. Genome Biol. 2014;15:456. doi: 10.1186/s13059-014-0456-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J., Zhang X., Zhang H.K., Yang H.M., Zhou Y.B., Han Z.G. ZBTB34, a novel human BTB/POZ zinc finger protein, is a potential transcriptional repressor. Mol. Cell. Biochem. 2006;290:159–167. doi: 10.1007/s11010-006-9183-x. [DOI] [PubMed] [Google Scholar]
- Raiber E.A., Beraldi D., Ficz G., Burgess H.E., Branco M.R., Murat P., Oxley D., Booth M.J., Reik W., Balasubramanian S. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K.D., Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K.D., Jia G., Johansen J.V., Pedersen M.T., Rapin N., Bagger F.O., Porse B.T., Bernard O.A., Christensen J., Helin K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29:910–922. doi: 10.1101/gad.260174.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K.D., Berest I., Keβler S., Nishimura K., Simón-Carrasco L., Vassiliou G.S., Pedersen M.T., Christensen J., Zaugg J.B., Helin K. TET2 binding to enhancers facilitates transcription factor recruitment in hematopoietic cells. Genome Res. 2019;29:564–575. doi: 10.1101/gr.239277.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C.S., Stark R., Holmes K.A., Schmidt D., Spyrou C., Russell R., Massie C.E., Vowler S.L., Eldridge M., Carroll J.S. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010;24:171–182. doi: 10.1101/gad.552910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scourzic L., Mouly E., Bernard O.A. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7:9. doi: 10.1186/s13073-015-0134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T., Tibshirani R., Parker J., Hastie T., Marron J.S., Nobel A., Deng S., Johnsen H., Pesich R., Geisler S. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R., Brown G.D. 2011. DiffBind: differential binding analysis of ChIP-Seq peak data.http://www.bioconductor.org/packages/release/bioc/html/DiffBind.html [Google Scholar]
- Stephens P.J., Tarpey P.S., Davies H., Van Loo P., Greenman C., Wedge D.C., Nik-Zainal S., Martin S., Varela I., Bignell G.R., Oslo Breast Cancer Consortium (OSBREAC) The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaku M., Grimm S.A., Roberts J.D., Chrysovergis K., Bennett B.D., Myers P., Perera L., Tucker C.J., Perou C.M., Wade P.A. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat. Commun. 2018;9:1059. doi: 10.1038/s41467-018-03478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou V., Stark R., Menon S., Carroll J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22. doi: 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usary J., Llaca V., Karaca G., Presswala S., Karaca M., He X., Langerød A., Kåresen R., Oh D.S., Dressler L.G. Mutation of GATA3 in human breast tumors. Oncogene. 2004;23:7669–7678. doi: 10.1038/sj.onc.1207966. [DOI] [PubMed] [Google Scholar]
- Wang Y., Xiao M., Chen X., Chen L., Xu Y., Lv L., Wang P., Yang H., Ma S., Lin H. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell. 2015;57:662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Ozark P.A., Smith E.R., Zhao Z., Marshall S.A., Rendleman E.J., Piunti A., Ryan C., Whelan A.L., Helmin K.A. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018;4:eaau6986. doi: 10.1126/sciadv.aau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K., Christensen J., Pedersen M.T., Johansen J.V., Cloos P.A., Rappsilber J., Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., D’Alessio A.C., Ito S., Wang Z., Cui K., Zhao K., Sun Y.E., Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Liu K., Lei M., Yang A., Li Y., Hughes T.R., Min J. DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Structure. 2018;26:85–95.e3. doi: 10.1016/j.str.2017.11.022. [DOI] [PubMed] [Google Scholar]
- Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356:eaaj2239. doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
hIP-seq, RNA-seq, Methyl Midi-Seq and Reduced Representation Hydroxymethylation Profiling datasets have been deposited to Gene Expression Omnibus (GEO) and are available under the accession number GSE153255. RIME, qPLEX-RIME and whole proteome data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019438. PRM datasets have been deposited to the Panorama Public database and can be accessed at http://panoramaweb.org/Panorama%20Public/2020/CRUK%20Proteomics%20Core%20-%20TET2_Project/project-begin.view? or using the dataset identifier PXD019726.