

Abstract
Migraine is a complex disorder, involving peripheral and central brain structures, where mechanisms and site of attack initiation are an unresolved puzzle. While abnormal pontine neuronal activation during migraine attacks has been reported, exact implication of this finding is unknown. Evidence suggests an important role of glutamate in migraine, implying a possible association of pontine hyperactivity to increased glutamate levels. Migraine without aura patients were scanned during attacks after calcitonin gene-related peptide and sildenafil in a double-blind, randomized, double-dummy, cross-over design, on two separate study days, by proton magnetic resonance spectroscopy and pseudo-continuous arterial spin labeling at 3T. Headache characteristics were recorded until 24 h after drug administrations. Twenty-six patients were scanned during migraine, yielding a total of 41 attacks. Cerebral blood flow increased in dorsolateral pons, ipsilateral to pain side during attacks, compared to outside attacks (13.6%, p = 0.009). Glutamate levels in the same area remained unchanged during attacks (p = 0.873), while total creatine levels increased (3.5%, p = 0.041). In conclusion, dorsolateral pontine activation during migraine was not associated with higher glutamate levels. However, the concurrently increased total creatine levels may suggest an altered energy metabolism, which should be investigated in future studies to elucidate the role of pons in acute migraine.
Keywords: Brainstem, creatine, energy metabolism, headache, trigeminal
Introduction
Migraine is a complex, neurological disorder involving dysfunction of the central and peripheral nervous system. It manifests as recurrent attacks of headache and sensory disturbances,1,2 which can be induced by inherent and pharmacological triggers.3 The fundamental, unresolved puzzle of migraine pathophysiology is from where in the nervous system the attacks arise. An interesting clue has been provided by several neuroimaging studies pointing to the brainstem as a possible region of migraine attack initiation.4–10 Most notably, increased cerebral blood flow (CBF) in the dorsolateral pons during attacks has been reported.4–6,9 This finding indicates an abnormally increased neuronal activity in this area, suggesting involvement of pontine nuclei. However, the exact implication of this pontine activation to the pathophysiology of migraine is not understood.
Metabolic and neurochemical changes have been implied in migraine as well,11 not least abnormal regulation of the levels of glutamate, the major excitatory neurotransmitter of the brain, increasing neuronal excitability and, potentially, susceptibility to attacks through modulation of the neuronal activation threshold.2 This notion is supported by genetic and biochemical studies2,12 as well as the glutamate-modulating properties of widely used anti-migraine medications, topiramate and sumatriptan.13,14 In addition, abnormal glutamatergic activity has been demonstrated in animal models of trigeminal sensitization.15,16 Consequently, it is plausible that the observed increased pontine neuronal activity during migraine attacks is related to, and explained by, increased glutamate levels.
Brain glutamate levels can be measured non-invasively using proton (1H) magnetic resonance spectroscopy (MRS). While previous 1H-MRS migraine studies of several brain areas including thalamus, anterior paracingulate cortex and visual cortex suggest increased glutamate levels in patients outside attacks,11,17,18 investigations of the pons have not been reported, neither during or outside attacks. This is most likely due to the technical challenges related to brainstem MRS, such as time-consuming scans and noise due to magnetic field inhomogeneity.19
We applied human pharmacological migraine models3 to rigorously study changes in pontine glutamate levels, along with CBF, during migraine attacks. We used an optimized and validated method for 1H-MRS investigation of the pons,19 while pontine CBF during attacks was investigated by the MRI method of arterial spin labeling, which does not involve the use of contrast agents or ionizing radiation. Selected pharmacological headache inducers were calcitonin gene-related peptide (CGRP) and sildenafil, as they possess different modes of action. While sildenafil crosses the blood–brain barrier,20 CGRP most likely carries its effects via the peripheral division of the trigeminal pain pathway.21
We hypothesized that glutamate levels, as well as CBF, would increase in pons, ipsilateral to the most painful side, during migraine attacks.
Materials and methods
Recruitment
Episodic migraine without aura patients were recruited via advertisements on a Danish website for participants to health research (www.forsogsperson.dk), hospitals and educational institutions. Patients were included if they were diagnosed with migraine without aura according to the International Classification of Headache Disorders,22 18–50 years of age, weighed 50–100 kg and experienced ≥1 migraine attacks every other month. Exclusion criteria were inconsistent laterality of unilateral attacks, other primary headache disorders (except episodic tension-type headache not conflicting with episodic migraine diagnosis), no use of contraceptives, daily medication intake (except oral contraceptives), pregnant/breastfeeding females, daily smoking, history of serious somatic/psychiatric disease, drug abuse, hypo-/hypertension, and MRI contraindications (including braces and teeth implants, which could cause magnetic field inhomogeneities in the pontine region). Patients underwent medical examination and female patients completed pregnancy tests as well.
The study was approved by the Ethical Committee of the Capital Region of Denmark (H-15019063) and registered at www.clinicaltrials.gov (NCT03143465). Other parts of parent study have been and/or will be published elsewhere. All patients provided written informed consent in agreement with the Declaration of Helsinki of 1964 with later revisions. Enrollment period was from August 2017 to November 2018.
Experimental design
Patients received CGRP and sildenafil in a double-blinded, randomized, double-dummy, cross-over design on two separate study days. On the CGRP day, patients received 1.5 μg/min α-CGRP infusion (Tocris Bioscience, Bristol, UK) for 20 min and placebo tablet. On the sildenafil day, patients received tablet of 100 mg sildenafil (TEVA pharmaceutical industries Ltd, Petah Tikva, Israel) and isotonic saline infusion for 20 min. Venous catheter (18G Vasofix® Safety, B.Braun, Melsungen, Germany) was inserted into a cubital vein for infusion.
Randomization and preparation of study drugs were administrated by the Hospital Pharmacy of the Capital Region of Denmark and physicians not otherwise involved in the study. Investigators of the study remained blinded until after study completion.
MRI was performed immediately before and 6 h after drug administration (Figure 1). Headache characteristics and pain intensity (range 0–10; from no pain to worst imaginable pain) were obtained using a standardized headache questionnaire interview every 10 min during a 90 min monitoring period, every 30 min until 6 h after drug administration and between each scan sequence during post drug MRI. Patients completed questionnaire hourly until 24 h (waking hours) after discharge from clinic at 7 h, at which point, acute migraine medication was allowed. Headache data were used for defining migraine-like attacks (hereinafter referred to as ‘migraine attacks’) (see online supplementary material).
Figure 1.
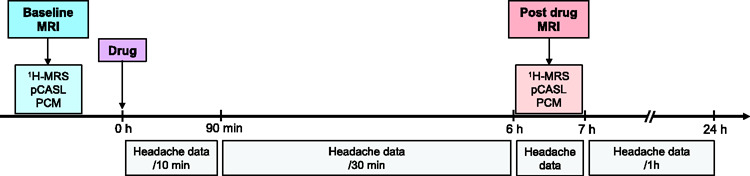
Study design was double-blinded, randomized and conducted twice on two separate study days. Headache data were obtained between scan sequences during post drug MRI. Timing of post drug MRI was selected based on previous reports of attacks after CGRP and sildenafil.3
Drug: CGRP and sildenafil; 1H-MRS: proton magnetic resonance spectroscopy; pCASL: pseudo-continuous arterial spin labeling; PCM: phase-contrast mapping; MRI: magnetic resonance imaging.
Blood pressure, heart rate, respiration rate and end-tidal CO2 tension (water trap and gas sample line, Medrad, Warrendale, PA and Veris Monitor, Medrad, Warrendale, PA) were monitored every 10 min from −10 min until 90 min after the drug administration and continuously (except blood pressure every 10 min) during the post drug scan.
Experiments were initiated in the morning on each study day. Patients were headache free for at least 48 h, fasted for 4 h, and were not allowed coffee, tea, alcohol, cocoa, and tobacco for 12 h before the start of the study. A small, standardized meal (soft bread, cheese, and banana) was provided 90 min after drug administration.
MR data acquisition and post-processing
Imaging data were obtained using a 3T Philips Achieva dStream MRI scanner (Philips Medical Systems, Best, The Netherlands) with a 32-channel phase array receiver head coil (Table 1).
Table 1.
Overview of MRI sequence parameters.
| 3D T1 | 1H-MRS | pCASLa | PCM | |
|---|---|---|---|---|
| Field of view (mm3) | 240 × 240 × 170 | – | 220 × 220 × 78.6 | 240 × 240 |
| Voxel size (mm3) | 1.00 × 1.08 × 1.10 | – | 3.44 × 3.44 × 6.6 | 0.75 × 0.75 × 8 |
| Volume-of-interest (mm3) | – | 10.5 × 12.5 × 22 | – | – |
| Repetition time (ms) | 8 | 3000 | 3000 | 12.7 |
| Echo time (ms) | 3.7 | 38.3 | 11 | 7.72 |
| Flip angle (°) | 8 | – | 40 | 10 |
| Acquisitions | 3 | 480 | – | 10 |
| Slices | 170 | – | 13 | 1 |
| Scan duration | 5 min 29 s | 24 min | 4 min 24 s | 1 min 50 s |
1H-MRS: proton magnetic resonance spectroscopy; pCASL: pseudo-continuous arterial spin labeling; PCM: phase-contrast mapping.
Multi inversion times 2D arterial spin labeling sequence with pseudo-continuous labeling duration of 1650 ms and 7 post-labeling delays (TI1/ΔTI =100 ms/300 ms) was acquired using a Look-Locker scheme with an echo planar imaging read-out.
Detailed information on MRI sequence parameters and post-processing are available in the online supplementary material.
Structural imaging
High-resolution anatomical scans were acquired by a 3D T1-weighted turbo field echo sequence and used for placement of 1H-MRS volume-of-interest (VOI) in pons as well as post-processing of pseudo-continuous arterial spin labeling (pCASL) and phase-contrast mapping (PCM) data.
Proton magnetic resonance spectroscopy
Point-resolved spectroscopy pulse sequence was used to measure pontine concentration estimate of glutamate, lactate, total creatine (creatine + phosphocreatine) and total NAA (NAA; N-acetylaspartate + NAAG; N-acetylaspartylglutamate). 1H-MRS VOI was placed ipsilaterally to the most painful side during usual unilateral migraine attacks and on the right side for patients with usual bilateral migraine attacks (Figure 2). 1H-MRS was not obtained from the thalamus due to technical issues. Metabolite quantification was performed in LCModel (Version 6.3-1 F) and was corrected for water contribution.23 Concentrations, that could not be estimated, were excluded.24 Blinded visual inspection of raw spectral data and LCModel spectra was performed to exclude poor spectral fit due to, e.g., motion.
Figure 2.

Spectroscopy region (a) with corresponding spectrum (b) and cerebral blood flow measurement in dorsolateral pons (c). Solid red line denotes spectral fit and grey line is the baseline as estimated by LCModel. The MNI-152 coordinates for the right- and left-sided pontine cerebral blood flow measurement are (x, y, z) = (±4, –32, –40).
Glu: glutamate; NAA: N-acetylaspartate.
Pseudo-continuous arterial spin labeling
Dorsolateral pontine region of interest (ROI) was selected based on a previous study reporting ipsilaterally increased CBF in that area during unilateral attacks.6 Contralateral ventroposterior medial nucleus region of thalamus (VPM), as the trigeminal somatosensory relay station,25 was delineated using Talairach atlas registered into MNI-152 space. Acquired voxel size was used for the voxel-wise analysis. CBF maps of patients, who reported predominantly right-sided pain during scanned attacks, were flipped to left side at the sagittal midline for voxel-wise analysis.
Phase-contrast mapping
Phase-contrast mapping was used as a validated method26 to assess whether quantitative global CBF was unchanged during attacks. In-house developed and validated MATLAB scripts were used.26
Statistical analysis
Sample size was calculated based on detection of at least 5% difference in glutamate levels after each drug at 5% significance, with 80% power and standard deviation of 6.9%. We estimated that at least 17 attacks in each drug group should be included. The effect size and standard deviation were based on pontine glutamate measurements in healthy volunteers.19
Primary outcomes were changes from baseline of glutamate, lactate, total NAA and total creatine in pons, regional CBF in dorsolateral pons and VPM as well as global CBF, during migraine attacks. Secondary outcomes were differences in metabolic and CBF changes between migraine attacks induced by CGRP and sildenafil, and correlation between attack-induced changes and pain intensity. Linear mixed models were used for primary (1) and secondary (2, 3) outcome analyses. Scan (baseline and post drug) and drug (CGRP and sildenafil) were fixed effects with patient identification as random effect (μ) and ε denoting the random error. Interaction term (β3) denotes significance of drug on changes from baseline. Global CBF analysis included end-tidal CO2 tension as covariate.
| (1) |
| (2) |
| (3) |
Changes from baseline of end-tidal CO2 tension and respiration rate during migraine was tested in model (1) and CO2 tension was replaced with respiration rate as covariate in the model for post hoc analyses.
Explorative, voxel-wise analyses of pCASL CBF maps was performed using in-house developed MATLAB script, applying models (1) and (2) with correction for multiple comparisons (false discovery rate, q-value of 0.05). Pontine CBF changes were explored, using model (1), on the contralateral side, in patients who developed migraine. Correlations between pontine metabolic and CBF changes to monthly attack frequency and disease duration were explored using model (3).
Data of patients, who did not develop attacks, are exploratively reported as descriptive statistics due to small sample (n = 7–8). Mean arterial pressure and heart rate are reported as descriptive statistics.
Statistical analyses were performed using R (Version 3.5.1), unless stated otherwise. P values were reported as two-tailed with a level of significance of 5%.
Results
Clinical data
Twenty-six patients were scanned during migraine (24 females; mean age 25 years, range 20–47), yielding a total of 41 scanned attacks (Figure 3), whereof 20 were scanned during an attack after CGRP and 21 after sildenafil. Fifteen patients were scanned during an attack after both CGRP and sildenafil. Median days between scans were 14 (range 6–42; one patient scanned second time after 142 days).
Figure 3.
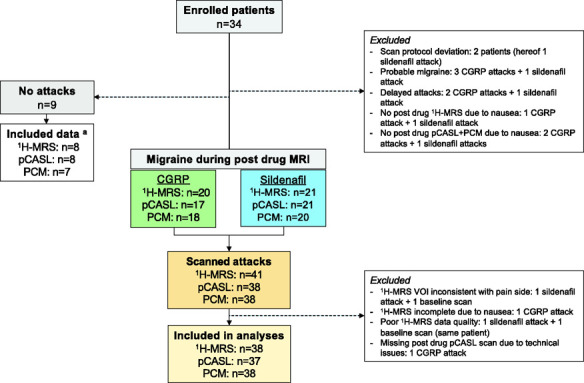
Flowchart of MRI data acquisition.
aFor no attack data: three 1H-MRS, two pCASL scans and three PCM scans were acquired after sildenafil; one 1H-MRS and one pCASL scan, from the same patient, and one baseline pCASL scan were excluded due to poor quality; two PCM scan data (incl. baseline) were excluded due to insufficient flow estimate from arteries.
CGRP: calcitonin gene-related peptide; 1H-MRS: proton magnetic resonance spectroscopy; pCASL: pseudo-continuous arterial spin labeling; PCM: phase-contrast mapping; VOI: volume-of-interest; n: number.
Headache characteristics of individual patients, who did and did not develop migraine after drugs, are available in Table 2. Median monthly attack frequency was 1.25 (range 0.5–7) and median years with migraine was 9 (range 0–25). Median time to migraine onset was 180 min (range 30–368) after CGRP and 270 min (range 80–417) after sildenafil. Five patients experienced pure unilateral attacks (three left-sided on both days, one right-sided and one left-sided after sildenafil) during scans, while most patients experienced bilateral headache with or without a predominant pain side.
Table 2.
Headache characteristics and associated symptoms.
| ID Drug | Time to peak pain | Headache characteristics | Associated symptoms | Mimics usual attacks | Migraine attack (onset time) | Treatment (time)/efficacy |
|---|---|---|---|---|---|---|
| 1 CGRP | 1 h | Bi(left)/5/T/ + | + / + / + | Yes | Yes (40 min) | NS |
| Sildenafil | 7 h | Bi(left)/5/T/ + | + / + / + | Yes | Yes (5 h) | S1 50 mg (10 h)/No; I + P (23 h)/NAa |
| 2 CGRP | 10 min | Bi/1/T/ + | –/–/– | No | No (NA) | None |
| Sildenafil | 7 h | Bi(left)/4/T/ + | –/ + / + | Yes | Yes (7 h) | P (13 h)/NSb |
| 3 CGRP | NA | No headache | NA | NA | No (NA) | None |
| Sildenafil | 8 h | Rightc/9/T + P/ + | + / + / + | Yes | Yes (4.5 h) | S1 100 mg (7 h)/No; NS (8 h)/No; NS (9 h)/Yes |
| 4 CGRP | 7 h | Bi/2/P/ + | –/–/– | No | No (NA) | None |
| Sildenafil | 7 h | Right/8/T/ + | –/ + / + | Yes | Yes (80 min) | NS |
| 5 CGRP | 80 min | Bi(left)/4/T + P/ + | –/ + / + | Yes | Yes (2.5 h) | None |
| Sildenafil | 8 h | Bi/8/T + P/ + | –/ + / + | Yes | Yes (6 h) | 2 Kodipar (9h)/Yes |
| 6 CGRP | 7 h | Bi/7/T/ + | + / + / + | Yes | Yes (3 h) | S1 50 mg (8 h, 14 h, 22 h)/No, NSb, Yes |
| 7 CGRP | 10 h | Bi(Unid)/3/T/ + | + / + / + | No | Yes (3.5 h) | None |
| Sildenafil | 50 min | Bi(left)/3/P/– | –/ + /– | No | No | None |
| 8 CGRP | 7 h | Bi(right)/5/T/ + | + / + / + | Yes | Yes (6 h) | 2 AC + P (8h)/Yes |
| Sildenafil | 7 h | Bi/3/T/ + | –/ + / + | Yes | Yes (7 h) | 2 AC (8h)/Yes |
| 9 CGRP | 70 min | Bi/4/T/ + | + / + / + | Yes | Yes (70 min) | None |
| Sildenafil | 10 h | Bi/7/T/ + | + / + / + | Yes | Yes (2.5 h) | I (11 h)/No |
| 10 CGRP | 1 h | Bi(left)/3/P/– | –/–/– | No | No | None |
| 11 CGRP | 7 h | Bi(right)/6/T/ + | + / + / + | Yes | Yes (4.5 h) | I + P (8 h)/Yes; 2 AC (12 h)/Yes |
| Sildenafil | 4.5 h | Bi(right)/7/T/ + | + / + / + | Yes | Yes (2.5 h) | Diclodan + O (7 h)/Yes; |
| 12 CGRP | 6 h | Bi(right)/7/T + P/ + | + / + / + | Yes | Yes (70 min) | S2 + O (6 h)/Yes |
| 13 CGRP | 4.5 h | Bi/4/T + P + | –/ + / + | No | Yes (6 h) | 2 AC (7h)/Yes |
| Sildenafil | 8 h | Lefte/7/T/ + | –/ + / + | Yes | Yes (5.5 h) | I + P (7h)/No; I (8h)/Yes |
| 14 CGRP | 5.5 h | Left/8/T/ + | + / + / + | Yes | Yes (3 h) | 2AC + O(7h)/No;I + P(9h)/No;2AC(12h)/Yes;AC(23)/No |
| Sildenafil | 6 h | Left/2/P / + | –/ + /– | Yes | Yes (6 h) | 1.5 AC (9h)/Yes |
| 15 Sildenafil | 5.5 h | Bi(left)/4/P/– | –/–/– | Yes | No | NA |
| 16 CGRP | 7 h | Bi(left)/7/T/ + | + / + / + | Yes | Yes (30 min) | S1 50 mg (7 h, 9 h)/No, Yes |
| Sildenafil | 7 h | Bi/9/T/ + | + / + / + | Yes | Yes (5 h) | S2 (7 h)/Yes |
| 17 Sildenafil | 6 h | Bi(left)/10/P/ + | + / + / + | Yes | Yes (5.5 h) | O + Diclodan (8h)/Yes |
| 18 CGRP | 5.5 h | Bi/8/T/ + | + / + / + | Yes | Yes (2.5 h) | S1 25 mg (7h, 10 h)/No, Yes |
| Sildenafil | 5 h | Bi/7/P/ + | + / + / + | Yes | Yes (4.5 h) | S1 25 mg (7h)/Yes |
| 19 CGRP | 50 min | Bi/1/P/ + | + /–/– | No | No | None |
| Sildenafil | NA | No headache | NA | NA | NA | NA |
| 20 CGRP | 5 h | Bi/6/T/ + | –/ + / + | Yes | Yes (5.5 h) | AC (7 h)/Yes |
| Sildenafil | 4 h | Bi/8/T + P/ + | + / + / + | Yes | Yes (3 h) | AC + O (7 h)/Yes |
| 21 CGRP | 6 h | Bi(left)/5/T/ + | + / + / + | Yes | Yes (5.5 h) | AC (8 h)/NSc; 2 AC (17h)/Yes |
| Sildenafil | 6.5 h | Left/9/T + P/ + | + / + / + | Yes | Yes (2.5 h) | S2 + O (7 h)/Yes |
| 22 CGRP | 6.5 h | Left/6/T/ + | + / + / + | Yes | Yes (3 h) | S1 50 mg (7 h, 10 h)/No, NSb |
| Sildenafil | 6.5 h | Left/8/T/ + | + / + / + | Yes | Yes (3 h) | None |
| 23 CGRP | 6 h | Bi/10/T + P/ + | + /–/– | Yes | Yes (3.5 h) | S2 (7 h)/Yes |
| 24 CGRP | 8 h | Left/5/T/ + | + / + / + | Yes | Yes (5.5 h) | None |
| Sildenafil | 6 h | Left/4/T/ + | –/ + / + | Yes | Yes (6 h) | None |
| 25 CGRP | 7 h | Bi/6/T/ + | –/ + / + | Yes | Yes (50 min) | I + P (7 h)/NA; P (8 h)/Yes |
| Sildenafil | 4 h | Bi/6/T/ + | + / + / + | Yes | Yes (3 h) | I + P (7 h)/NA; AC (10 h)/No |
| 26 Sildenafil | 9 h | Bi(left)/8/P/ + | + / + / + | Yes | Yes (3 h) | S1 50 mg (7 h)/No |
| 27 CGRP | NA | No headache | NA | NA | NA | NA |
| Sildenafil | 8 h | Bi(Unif)/4/P/ + | –/ + / + | Yes | Yes (6 h) | None |
| 28 CGRP | 7 h | Bi(right)/6/T + P/ + | –/ + / + | Yes | Yes (50 min) | S1 50 mg + I (7 h)/Yes |
| Sildenafil | 6 h | Bi(right)/5/T/ + | + / + / + | Yes | Yes (4 h) | S1 50 mg + I (8 h)/Yes |
Note: Headache characteristics: Localization/pain intensity/quality/aggravation. Associated symptoms: Nausea/photophobia/phonophobia. Migraine-like attack criteria are described in ‘Materials and Methods’ section. Treatment efficacy: ≥50% decrease in headache intensity within 2 h.
Bi: bilateral; Uni: unilateral; T: throbbing; P: pressing; Diclodan: diclofenac 100 mg (suppository); I: ibuprofen 400 mg; Kodipar: codeine 30.6 mg + paracetamol 500 mg; P: paracetamol 1 g; S1: sumatriptan (tablet); S2: sumatriptan 12 mg (injection); AC: aspirin 500 mg + caffeine 50 mg; O: ondansetron 16 mg (suppository); NS: not specified (missing data); NR: not reported.
No data from >24 h.
Went to sleep.
Bi(right) only during post drug scan.
Shifting predominant pain side; left is usual attack side and selected for analysis.
Bi(left) during scan.
Shifted from left to right at 7 h.
Regional CBF
CBF maps were acquired during 37 migraine attacks. Regional CBF in dorsolateral pons increased from baseline during attacks (13.6%, effect estimate: 0.07, 95% CI: 0.02–0.13, p = 0.009), ipsilateral to the most painful side (right side in patients with bilateral headache and no predominant pain side, except for one patient scanned during bilateral headache, who reported usual left-sided attacks; n = 12 attacks). Regional CBF did not change in the contralateral pons (5.8%, effect estimate: 0.03, 95% CI: –0.02–0.08, p = 0.278) nor in VPM contralateral to the most painful side (1.5%, effect estimate: 0.0004, 95% CI: –0.04–0.04, p = 0.982).
Regional CBF changes from baseline in dorsolateral pons, ipsilateral to pain side, did not correlate with pain intensity during attacks (p = 0.688). There was no difference in changes from baseline between CGRP and sildenafil in dorsolateral pons (6.4% and 19.7%, effect estimate: 0.09, 95% CI: –0.01–0.20, p = 0.090) or VPM (0.3% and 2.4%, effect estimate: 0.01, 95% CI: –0.07–0.09, p = 0.818). Explorative analysis revealed no correlation between pontine CBF changes and monthly attack frequency or disease duration.
Voxel-wise analysis yielded no additional regions of increased CBF from baseline in patients scanned during attacks and with no difference between drugs.
In patients, who did not develop migraine (n = 7), CBF did not increase from baseline in the dorsolateral pons on the most painful side (mean ± SD: –0.5% ± 22.0%) or contralateral VPM (–2.0% ± 9.1%).
Pontine metabolites
Spectra were obtained from 38 attacks, ipsilaterally to the most painful side (similar to regional CBF investigations). Glutamate did not increase in the pons during migraine attacks (0.9%, effect estimate: 0.03, 95% CI: –0.35–0.41, p = 0.873), compared to baseline (Table 3). There was no difference in glutamate levels between CGRP and sildenafil (2.7% and 2.7%, effect estimate: 0.18, 95% CI: –0.58–0.93, p = 0.654). Mean baseline glutamate levels were 8.30 mmol/L ± 0.19 in patients who experienced an attack and 8.32 mmol/L ± 0.44 in those who did not (n = 8).
Table 3.
Glutamate, lactate, total NAA and total creatine concentration estimates and relative changes from baseline in pons of patients who experienced migraine attacks.
| Baseline mean mmol/L ± SE |
Attack mean mmol/L ± SE |
Change from baseline (mean) |
Effect estimate and 95% CI | p | CGRP vs. sildenafil: Effect estimate and 95% CI |
CGRP vs. Sildenafil: P | |
|---|---|---|---|---|---|---|---|
| Glutamate | 8.30 ± 0.19 | 8.33 ± 0.19 | 0.9% | 0.03, –0.35–0.41 | 0.873 | 0.18, –0.58–0.93 | 0.654 |
| Total creatine | 4.57 ± 0.09 | 4.73 ± 0.08 | 3.5% | 0.16, 0.01–0.31 | 0.041 | 0.22, –0.07–0.50 | 0.145 |
| Total NAA | 10.30 ± 0.12 | 10.63 ± 0.13 | 3.5% | 0.34, 0.08–0.60 | 0.014 | 0.12, –0.41–0.63 | 0.678 |
| Lactate | 0.93 ± 0.08 | 0.78 ± 0.09 | –6.3% | –0.16, –0.34–0.30 | 0.099 | –0.05, –0.42–0.31 | 0.778 |
Note: Three baseline lactate values after CGRP and one post drug (including baseline) value after sildenafil were excluded according to protocol as described in ‘Materials and Methods’ section. Absolute mean and standard error of the mean (SE) reported from mixed model.
CI: confidence interval; NAA: N-acetylaspartate.
Total creatine (3.5%, effect estimate: 0.16, 95% CI: 0.01–0.31, p = 0.041) and total NAA levels (3.5%, effect estimate: 0.34, 95% CI: 0.08–0.60, p = 0.014) increased during attacks with no difference between drugs. Lactate levels were unchanged (–6.3, effect estimate: –0.16, 95% CI: –0.34–0.30, p = 0.099) during attacks; however, when two gross outliers of relative change from baseline (> 4 SD from mean; sildenafil days) were excluded from analysis, the lactate levels decreased (–24.5%, effect estimate: –0.22, 95% CI: –0.39 to –0.04, p = 0.016).
There was no correlation between metabolic changes and pain intensity during attacks, pontine CBF changes, monthly attack frequency or disease duration. Metabolic changes from baseline (mean ± SD) in patients, who did not develop attacks (n = 8), were 7% ± 9.8% for glutamate, –20% ± 47.4% for lactate, 3.5% ± 4.7% for total NAA and –2.6% ± 5.9% for total creatine.
Global CBF
Global CBF decreased slightly (–4.0%, effect estimate: –2.52, 95% CI: –4.70 to –0.35 p = 0.028) during attacks, with no difference between CGRP and sildenafil (–7.6% and –1%, effect estimate: 4.14, 95% CI: –0.02–8.28, p = 0.062). End-tidal CO2 tension did not change between scans (–0.9%, effect estimate: –0.05, 95% CI: –0.23–0.14, p = 0.596), while respiration rate increased during post drug scan (27.5%, effect estimate: –2.35, 95% CI: –3.92 to –0.70, p = 0.005), with no difference between drugs. When respiration rate was used as a covariate, instead of CO2 tension, the global CBF decrease was no longer significant.
Mean arterial pressure and heart rate
Mean arterial pressure and heart rate are visualized in Figure 4 for both study days of patients who were scanned during an attack.
Figure 4.
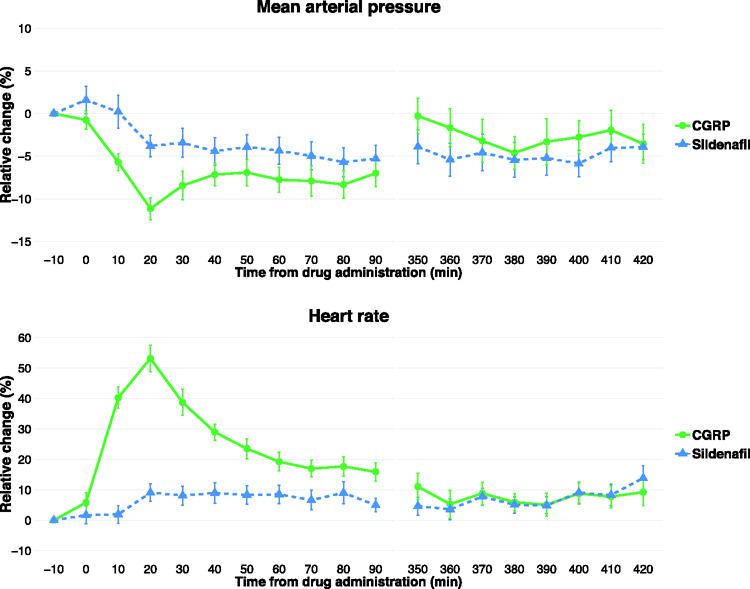
Relative percentage change of mean arterial pressure and heart rate in patients who developed attacks during scan. Error bars denote standard error of the mean.
CGRP: calcitonin gene-related peptide.
Discussion
The major novel findings of the present study were that during migraine attacks, ipsilateral to the most painful side, pontine glutamate levels did not change, while regional CBF increased in the dorsolateral pons. Interestingly, we found increase of total creatine in this region. We found no changes in CBF of the contralateral VPM or other areas during attacks.
CBF and unilateral pain
We investigated CBF in the largest sample of migraine attacks reported to date. Pontine CBF increase was higher on the most painful side, compared to the contralateral side. Previous studies have reported activation of the dorsal pons during migraine attacks as well using PET and BOLD-fMRI.4–7 Here, we measured pontine CBF changes using a novel approach by pCASL.
One PET study of right- and left-sided attacks reported activation of lower dorsal pons and rostral medulla on the pain side,6 similar to our findings, while in another study, left-sided activation was reported during right-sided attacks in rostral dorsal pons.4 Bilateral attacks produced bilateral dorsal pons activation in a previous PET study,6 which may explain the contralateral numerical CBF increase in our study. The pontine ROI in the present study was based on the previous PET study of unilateral attacks.6 While we are unable to determine exactly which nuclei are located in this area, due to spatial limitations of the method, possible areas include superior salivatory nucleus and dorsal raphe nucleus.27 Superior salivatory nucleus is part of the trigeminal autonomic reflex and may modulate spinal trigeminal nucleus activity,2 while dorsal raphe nucleus is involved in regulation of serotoninergic neurotransmission in migraine.28 Previous PET studies have reported increased activation in the locus coeruleus and suggested that it plays a role in the antinociceptive control in migraine.4,5 Locus coeruleus is likely located outside the pontine ROI of the current study.27
Increased perfusion of dorsal pons appears to be specific for migraine, as this was not detected in CBF studies of cluster headache attacks,29 or during experimental nociceptive activation of the trigeminal pain pathway using capsaicin in healthy participants.30 However, one fMRI study of experimental trigeminal pain, using olfactory stimuli, yielded brainstem activation that included the caudal pons (likely the trigeminal principal nuclei) as well.31
In the present study, there was no difference in the pontine CBF increase between CGRP and sildenafil, and patients reported similar attack characteristics after both drugs (Table 2). Consequently, pontine CBF increase is most likely directly related to the migraine attacks rather than an unspecific drug effect. Furthermore, the no difference in CBF between drugs also supports the notion that CGRP and sildenafil induce the same attacks within patients,32 despite different modes of action of the drugs.
We observed a slight decrease in global CBF (CO2 tension corrected) during migraine attacks, which was insignificant when corrected for the respiration rate increase during post drug scans. Further, voxel-wise analysis indicated that the decrease was not restricted to any specific brain regions, but rather was a true global effect. We therefore consider it possible that the initially observed CBF decrease was due to hyperventilation during migraine attacks and that nasal end-tidal CO2 tension measurements may have been inaccurate. Any decrease in global CBF during attacks would not influence our conclusions regarding regional CBF increases from baseline.
The CBF changes reported here also supports the notion that spreading depolarization is not a phenomenon of migraine without aura33 but is a more specific underlying mechanism of migraine with aura.
Glutamate levels during attacks
We sought to clarify the pathophysiological significance of the pontine activation in migraine by measuring glutamate levels in a corresponding area to the regional CBF measurement, also on the pain side. Pontine glutamate measurements in migraine patients have, to our knowledge, never been reported before, neither during nor between attacks.
We detected no changes of glutamate levels in the pons ipsilateral to pain side during attacks, corresponding to the increased CBF. Thus, the pontine neuronal activation, denoted by increased CBF, is likely not associated with abnormal glutamate levels.
The role of increased glutamate levels in migraine patients has been indicated by genetic, biochemical and pharmacological studies.2,12 Topiramate, which is widely used as a preventive migraine medication, supposedly exerts its effects, at least partially, via inhibition of glutamate receptors.13 Topiramate may also carry its effects via gamma-aminobutyric acid (GABA) receptors, calcium and sodium channels as well as carbonic anhydrase isoenzymes.13 Furthermore, the widely used acute migraine drug, triptans, are 5-hydroxytryptamine 1B/1D receptor agonists. While triptans may modulate the CGRP levels,34 it is also possible that they modulate glutamate release in the trigeminal ganglion.14 The precise mode of action is unknown. Finally, genetic studies have suggested abnormal regulation of glutamate levels in migraine.12,35 While we found no changes in the pontine glutamate levels in migraine without aura patients, such changes may be present in migraine with aura patients. More specifically, genetic studies have shown that hemiplegic migraine has a genetic mutation leading to dysfunctional regulation of the glutamate levels propelling the spreading depolarization, a phenomenon of migraine with aura as well.12,35 Moreover, studies have shown a direct effect of N-methyl-D-aspartate receptors on inhibition of spreading depolarization.36 Interestingly, one 7 T 1H-MRS study only found increased occipital glutamate levels in migraine without aura patients and not in migraine with aura patients, compared to healthy controls, however, outside attacks.17 Only one previous study investigated glutamate levels by 1H-MRS during attacks, specifically in visual cortex in migraine with aura patients, which showed no difference compared to healthy controls.37 In contrast, previous 1H-MRS studies of the interictal state demonstrated increased glutamate levels in thalamus, anterior paracingulate cortex and visual cortex, while other studies reported no changes in insula, anterior/posterior cingulate and visual cortices.11,17,18
It is possible that our MRS technique was not sensitive enough to detect potential glutamate changes of attacks, matching the pontine CBF increases. However, the sample size of the study was estimated from a power calculation, before study start, based on a methodological study measuring glutamate changes using the same 1H-MRS-sequence and hardware.19 A post hoc power analysis showed that in our sample of 38 1H-MRS scanned attacks, using a measurement variability of 6.9%,19 the minimum detectable glutamate change would be 4.1% with 95% power and a significance level of 5%. Studies of this nature and size are challenging to conduct, as they require extreme efforts from the patients to, twice on two separate days, complete a time-consuming MRI scan during their migraine attacks.
A methodological limitation was that the size of the small 1H-MRS VOI, which was already close to limits of deep brain 1H-MRS, exceeded the size of the dorsal pons ROI, which we used here for CBF analyses. The glutamate measurements consisted of the combined estimate of glutamate and glutamine due to lack of separation of their signals at clinical magnetic field strengths; however, most of the combined concentration estimate consists of glutamate (∼80%).38 Another common limitation to 1H-MRS is that only the total of the intra- and extracellular pools of glutamate is measurable by 1H-MRS. The extracellular compartments encompass 13%–22% of the brain glutamate levels,38 while ∼80% of the intracellular pool is present in the neurons and ∼20% in the astrocytes.39 Due to the technical limitations, there is an inherent uncertainty as to which compartment is mainly representative of glutamatergic changes. Several studies have suggested that during neuronal activation, the glutamate concentration, when measured by 1H-MRS, increases and correlates with the blood-oxygenation-level-dependent activation.40–43 Thus, we expected that a possible increase of the glutamate level, in our study, might reflect a proportionally higher increase in the extracellular pool (in and/or outside the synaptic cleft) of glutamate than the intracellular pool. This assumption is further supported by a study where amyotrophic lateral sclerosis patients were treated with riluzole, a drug that increases glutamate uptake in neurons of the central nervous system.44 The glutamate levels were lower in riluzole-treated patients compared to untreated patients as well to healthy controls.44 Unequivocal evidence for a measurable glutamate increase in the extracellular space has, however, only been reported for spreading depolarization and epileptic seizures.36,45 These studies reported about 2 to 4 µM glutamate in area CA1 of the rat hippocampus during epileptic seizures and extracellular glutamate increase of ∼90 µM in rat brain slices during spreading depolarization.36,45 Additionally, preclinical studies have shown a direct correlation of the extracellular glutamate level to neuronal hyperexcitability and seizure intensity.46,47 Conversely, the 7 T 1H-MRS study in migraine patients suggested that an increase in the glutamate level would reflect dynamics of the intracellular pools.17 This is likely since the glutamate levels were obtained in the interictal state, from the visual cortex, without any modalities in their study design which potentially could increase the synaptic/extracellular glutamate levels. Under normal conditions, glutamate is only released to the synaptic cleft with subsequent rapid reuptake by neurons and/or astrocytes.38
Measurement of the inhibitory neurotransmitter, GABA, using special-edited 1H-MRS sequences, may be useful to further elucidate the observed pontine activation and notion of excitability alterations in migraine.
Altered pontine energy metabolism?
Interestingly, total creatine, a component of the energy metabolism, increased by 3.5% during attacks. In patients, who did not develop attacks, the corresponding change from baseline was –2.6% during the post drug scan. Previous 1H-MRS studies reported normal cortical total creatine levels measured in the interictal state of migraine with and without aura compared to healthy controls11 and no changes of cortical total creatine during hypoxia-induced attacks in migraine with aura patients.37
Creatine kinase converts phosphocreatine to creatine in the process of ATP production, while creatine is reverted to phosphocreatine via mitochondrial oxidative phosphorylation.48 Since the total creatine, measured by 1H-MRS, consists of the combined concentration estimates of creatine and phosphocreatine, we are unable to determine exactly how the energy metabolism may be affected. The pontine total creatine level increase observed in our study may reflect neuronal activity, possibly resulting in increased supply of creatine to meet the elevated demand for energy (ATP). In support, phosphorous (31P) MRS studies, allowing for separate estimation of the energy metabolism constituents, reported that increased neuronal activation corresponded to a faster ATP-synthesis rate, with steady-state phosphocreatine levels, in healthy volunteers.49,50 The ATP-synthesis rate is likely accompanied by a corresponding increase of the total creatine level.48,51
Previous 31P-MRS studies in migraine patients reported lower phosphocreatine levels and increased ATP synthesis rate, when compared to healthy controls, during and outside attacks.11 Although these measurements were performed in cortical structures, and not in the brainstem, the findings suggest that the supply of (phospho-)creatine is not sufficient enough to meet the increased ATP synthesis rate.11 Adding to this notion, a genetic study suggested a possible component of mitochondrial dysfunction in migraine patients compared to controls.52 Release and reuptake of glutamate from synapses are important energy-consuming processes during neuronal activation, which rely on normal mitochondrial function.38 The aspect of energy metabolism alterations in the brainstem of migraine patients may be elucidated in future studies by application of 31P-MRS. Acute neuronal activation normally produces increased lactate levels.53 The possible lactate decrease in our study may reflect its conversion to pyruvate, for oxidative phosphorylation,54 to meet the increased neuronal energy demand of attacks.
Total NAA, constituted by NAA and NAAG, increased by 3.5% during attacks, similar to patients, who did not develop attacks (3.5%, n = 8). We were unable to determine the exact implication of this finding based on our study. One interesting aspect is, however, that NAAG is co-released from neurons along with glutamate upon neuronal activation and inhibits the glutamatergic transmission at high synaptic activity.55
The CBF increase of ∼14%, which was detected in the dorsolateral pontine ROI, is only accompanied by a minimal cerebral blood volume increase.56 Furthermore, the pontine CBF ROI represented a part of the larger MRS VOI (Figure 2), i.e. the CBF increase of the MRS VOI was less than 14%. Consequently, the hyperemia most likely does not influence the concentration estimates of the spectral measurements.
Conclusion
Here we report a novel study of pontine glutamate levels in migraine. While we observed increased CBF in the dorsolateral pons, on the most painful side during migraine attacks, with no change in the contralateral VPM, we did not observe corresponding changes in pontine glutamate levels. Interestingly, we detected an increase in pontine total creatine during attacks. We conclude that the headache phase of migraine encompasses activation of dorsolateral pons, which likely involves altered energy metabolism. Further investigations, including 31P MRS of brainstem during migraine attacks, could prove useful to clarify this aspect of the migraine pathophysiology.
Supplemental Material
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20906902 for Glutamate levels and perfusion in pons during migraine attacks: A 3T MRI study using proton spectroscopy and arterial spin labeling by Samaira Younis, Casper E Christensen, Mark B Vestergaard, Ulrich Lindberg, Daniel Tolnai, Olaf B Paulson, Henrik BW Larsson, Anders Hougaard and Messoud Ashina in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
The authors thank research assistants Nikolaj Malthe Toft, Thomas Søborg and Marius Lendal, and lab technicians Lene Elkjær and Winnie Grønning for their assistance with headache data collection and handling. The authors thank Drs. Anne Luise Haulund Vollesen and Nita Wienholtz for their assistance in study drug administration.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the study was supported by the Research Foundation of Rigshospitalet (E-23327-02) and Lundbeck Foundation (R155-2014-171). Funding parties had no influence on study design, patient inclusion or data interpretation.
Data availability: Data that support the findings from this study are available upon reasonable request to the corresponding author.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions: SY: Concept and design of study, acquisition, processing, analysis and interpretation of data, and drafting of article. CEC: Concept and design of study, and data acquisition. MBV: Data processing, analysis and interpretation. UL: Data processing and interpretation. DT: Data analysis and quality assurance. OBP and HBWL: Concept and design of study, and interpretation of data. AH and MA: Concept and design of study, interpretation of data, and overall supervision of study. All authors revised manuscript for intellectual content and approved the final version.
Supplemental material: Supplemental material for this article is available online.
ORCID iDs
Samaira Younis https://orcid.org/0000-0002-9750-690X
Ulrich Lindberg https://orcid.org/0000-0002-0004-6354
Olaf B Paulson https://orcid.org/0000-0001-7712-8596
References
- 1.Akerman S, Holland PR, Goadsby PJ.Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci 2011; 12: 570–584. [DOI] [PubMed] [Google Scholar]
- 2.Burstein R, Noseda R, Borsook D.Migraine : multiple processes, complex pathophysiology. J Neurosci 2015; 35: 6619–6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashina M, Hansen JM, á Dunga BO, et al. Human models of migraine – short-term pain for long-term gain. Nat Rev Neurol 2017; 13: 713–724. [DOI] [PubMed] [Google Scholar]
- 4.Weiller C, May A, Limmroth V, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med 1995; 1: 658–660. [DOI] [PubMed] [Google Scholar]
- 5.Afridi S, Giffin N, Kaube H, et al. A positron emission tomographic study in spontaneous migraine. Arch Neurol 2005; 62: 1270–1275. [DOI] [PubMed] [Google Scholar]
- 6.Afridi SK, Matharu MS, Lee L, et al. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain 2005; 128: 932–939. [DOI] [PubMed] [Google Scholar]
- 7.Stankewitz A, Aderjan D, Eippert F, et al. Trigeminal nociceptive transmission in migraineurs predicts migraine attacks. J Neurosci 2011; 31: 1937–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulte LH, May A.The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016; 139: 1987–1993. [DOI] [PubMed] [Google Scholar]
- 9.Hougaard A, Amin FM, Christensen CE, et al. Increased brainstem perfusion, but no blood-brain barrier disruption, during attacks of migraine with aura. Brain 2017; 140: 1633–1642. [DOI] [PubMed] [Google Scholar]
- 10.Chong CD, Plasencia JD, Frakes DH, et al. Structural alterations of the brainstem in migraine. NeuroImage Clin 2017; 13: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Younis S, Hougaard A, Vestergaard MB, et al. Migraine and magnetic resonance spectroscopy : a systematic review. Curr Opin Neurol 2017; 30: 246–262. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann J, Charles A.Glutamate and its receptors as therapeutic targets for migraine. Neurotherapeutics 2018; 15: 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linde M, Mulleners W, Chronicle E, et al. Topiramate for the prophylaxis of episodic migraine in adults (Review). Cochrane Database Syst Rev 2013; 6: CD010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma QP.Co-localization of 5-HTIB/ID/IF receptors and glutamate in trigeminal ganglia in rats. Neuroreport 2001; 12: 1589–1591. [DOI] [PubMed] [Google Scholar]
- 15.Greco R, Demartini C, Zanaboni AM, et al. Effects of kynurenic acid analogue 1 (KYNA-A1) in nitroglycerin-induced hyperalgesia: targets and anti-migraine mechanisms. Cephalalgia 2017; 37: 1272–1284. [DOI] [PubMed] [Google Scholar]
- 16.Laursen JC, Cairns BE, Dong XD, et al. Glutamate dysregulation in the trigeminal ganglion: a novel mechanism for peripheral sensitization of the craniofacial region. Neuroscience 2014; 256: 23–35. [DOI] [PubMed] [Google Scholar]
- 17.Zielman R, Wijnen JP, Webb A, et al. Cortical glutamate in migraine. Brain 2017; 140: 1859–1871. [DOI] [PubMed] [Google Scholar]
- 18.Bathel A, Schweizer L, Stude P, et al. Increased thalamic glutamate/glutamine levels in migraineurs. J Headache Pain 2018; 19: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Younis S, Hougaard A, Christensen CE, et al. Effects of sildenafil and calcitonin gene-related peptide on brainstem glutamate levels: a pharmacological proton magnetic resonance spectroscopy study at 3.0 T. J Headache Pain 2018; 19: 1– 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez-Vallejo V, Ugarte A, García-Barroso C, et al. Pharmacokinetic investigation of sildenafil using positron emission tomography and determination of its effect on cerebrospinal fluid cGMP levels. J Neurochem 2016; 136: 403–415. [DOI] [PubMed] [Google Scholar]
- 21.Eftekhari S, Warfvinge K, Blixt FW, et al. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 2013; 14: 1289–1303. [DOI] [PubMed] [Google Scholar]
- 22.Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013; 33: 629–808. [DOI] [PubMed] [Google Scholar]
- 23.Quadrelli S, Mountford C, Ramadan S.Hitchhiker’s guide to voxel segmentation for partial volume correction of in vivo magnetic resonance spectroscopy. Magn Reson Insights 2016; 9: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Provencher S. LCModel & LCMgui User’s Manual, http://lcmodel.ca (2018, accessed 4 February 2020).
- 25.Younis S, Hougaard A, Noseda R, et al. Current understanding of thalamic structure and function in migraine. Cephalalgia 2019; 39: 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vestergaard MB, Lindberg U, Aachmann-Andersen NJ, et al. Comparison of global cerebral blood flow measured by phase-contrast mapping MRI With 15 O-H2O positron emission tomography. J Magn Reson Imaging 2017; 45: 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haines DE.Neuroanatomy in clinical context: an atlas of structures, sections, systems, and syndromes. Philadelphia, PA: Lippincott Wolters Kluwer, 2014. [Google Scholar]
- 28.Deen M, Hansen HD, Hougaard A, et al. Low 5-HT1B receptor binding in the migraine brain: a PET study. Cephalalgia 2018; 38: 519–527. [DOI] [PubMed] [Google Scholar]
- 29.May A, Bahra A, Büchel C, et al. PET and MRA findings in cluster headache and MRA in experimental pain. Neurology 2000; 55: 1328–1335. [DOI] [PubMed] [Google Scholar]
- 30.May A, Kaube H, Buchel C, et al. Experimental cranial pain elicited by capsaicin: a PET study. Pain 1998; 74: 61–66. [DOI] [PubMed] [Google Scholar]
- 31.Stankewitz A, May A, Peschke C, et al. A new trigemino-nociceptive stimulation model for event-related fMRI. Cephalalgia 2009; 30: 475–485. [DOI] [PubMed] [Google Scholar]
- 32.Younis S, Christensen CE, Toft NM, et al. Investigation of distinct molecular pathways in migraine induction using calcitonin gene-related peptide and sildenafil. Cephalalgia 2019; 39: 1776–1788. [DOI] [PubMed] [Google Scholar]
- 33.Olesen J, Tfelt-Hansen P, Henriksen L, et al. The common migraine attack may not be initiated by cerebral ischaemia. Lancet 1981; 318: 438–440. [DOI] [PubMed] [Google Scholar]
- 34.Goadsby PJ, Edvinsson L.The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993; 33: 48–56. [DOI] [PubMed] [Google Scholar]
- 35.Gasparini CF, Smith RA, Griffiths LR.Genetic insights into migraine and glutamate: a protagonist driving the headache. J Neurol Sci 2016; 367: 258–268. [DOI] [PubMed] [Google Scholar]
- 36.Zhou N, Rungta RL, Malik A, et al. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab 2013; 33: 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arngrim N, Schytz HW, Britze J, et al. Migraine induced by hypoxia: an MRI spectroscopy and angiography study. Brain 2016; 139: 723–737. [DOI] [PubMed] [Google Scholar]
- 38.Danbolt NC.Glutamate uptake. Prog Neurobiol 2001; 65: 1–105. [DOI] [PubMed] [Google Scholar]
- 39.Erecińska M, Silver IA.Metabolism and role of glutamate in mammalian brain. Prog Neurobiol 1990; 35: 245–296. [DOI] [PubMed] [Google Scholar]
- 40.Ip IB, Berrington A, Hess AT, et al. Combined fMRI-MRS acquires simultaneous glutamate and BOLD-fMRI signals in the human brain. Neuroimage 2017; 155: 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanley JA. nad Raz N. Functional magnetic resonance spectroscopy: the ‘new’ MRS for cognitive neuroscience and psychiatry research. Front Psychiatry 2018; 9: 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cleve M, Gussew A, Wagner G, et al. Assessment of intra- and inter-regional interrelations between GABA + , Glx and BOLD during pain perception in the human brain – a combined 1H fMRS and fMRI study. Neuroscience 2017; 365: 125–136. [DOI] [PubMed] [Google Scholar]
- 43.Bednařík P, Tkáč I, Giove F, et al. Neurochemical and BOLD responses during neuronal activation measured in the human visual cortex at 7 Tesla. J Cereb Blood Flow Metab 2015; 35: 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foerster BR, Pomper MG, Callaghan BC, et al. An imbalance between excitatory and inhibitory neurotransmitters in amyotrophic lateral sclerosis revealed by use of 3-T proton magnetic resonance spectroscopy. JAMA Neurol 2013; 70: 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Song Y, Xiao G, et al. Bio-electrochemical microelectrode arrays for glutamate and electrophysiology detection in hippocampus of temporal lobe epileptic rats. Anal Biochem 2018; 550: 123–131. [DOI] [PubMed] [Google Scholar]
- 46.Hunsberger HC, Konat GW, Reed MN.Peripheral viral challenge elevates extracellular glutamate in the hippocampus leading to seizure hypersusceptibility. J Neurochem 2017; 141: 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunsberger HC, Wang D, Petrisko TJ, et al. Peripherally restricted viral challenge elevates extracellular glutamates and enhances synaptic transmission in the hippocampus. J Neurochem 2016; 138: 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyss M, Kaddurah-Daouk R.Creatine and creatinine metabolism. Physiol Rev 2017; 80: 1107–1213. [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Zhu XH, Adriany G, et al. Increase of creatine kinase activity in the visual cortex of human brain during visual stimulation: a 31P NMR magnetization transfer study. Magn Reson Med 1997; 38: 551–557. [DOI] [PubMed] [Google Scholar]
- 50.Chen C, Stephenson MC, Peters A, et al. 31P magnetization transfer magnetic resonance spectroscopy: assessing the activation induced change in cerebral ATP metabolic rates at 3 T. Magn Reson Med 2018; 79: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sartorius A, Lugenbiel P, Mahlstedt MM, et al. Proton magnetic resonance spectroscopic creatine correlates with creatine transporter protein density in rat brain. J Neurosci Methods 2008; 172: 215–219. [DOI] [PubMed] [Google Scholar]
- 52.Guo S, Esserlind A, Andersson Z, et al. Prevalence of migraine in persons with the 3243A>G mutation in mitochondrial DNA. Eur J Neurol 2016; 23: 175–181. [DOI] [PubMed] [Google Scholar]
- 53.Schaller B, Xin L, O’Brien K, et al. Are glutamate and lactate increases ubiquitous to physiological activation? A 1H functional MR spectroscopy study during motor activation in human brain at 7 Tesla. Neuroimage 2014; 93: 138–145. [DOI] [PubMed] [Google Scholar]
- 54.Lemire J, Mailloux RJ, Appanna VD.Mitochondrial lactate dehydrogenase is involved in oxidative-energy metabolism in human astrocytoma cells (CCF-STTG1). PLoS One 2008; 3: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benarroch EE.N-Acetylaspartate and N-acetylaspartylglutamate: neurobiology and clinical significance. Neurology 2008; 70: 1353–1357. [DOI] [PubMed] [Google Scholar]
- 56.Ito H, Kanno I, Ibaraki M, et al. Changes in human cerebral blood flow and cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab 2003; 23: 665–670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20906902 for Glutamate levels and perfusion in pons during migraine attacks: A 3T MRI study using proton spectroscopy and arterial spin labeling by Samaira Younis, Casper E Christensen, Mark B Vestergaard, Ulrich Lindberg, Daniel Tolnai, Olaf B Paulson, Henrik BW Larsson, Anders Hougaard and Messoud Ashina in Journal of Cerebral Blood Flow & Metabolism