

Abstract
Following a request from the European Commission, EFSA developed updated scientific guidance to assist applicants in the preparation of applications on smoke flavouring primary products. This guidance describes the scientific data to be included in the applications for the authorisation of new smoke flavouring primary products, as well as for the renewal or for the modification of existing authorisations, submitted respectively under Articles 7, 12 and 11 of Regulation (EC) No 2065/2003. Information to be provided in all applications relates to: the characterisation of the primary product, including the description of the source materials, manufacturing process, chemical composition, specifications and stability; the proposed uses and use levels and the assessment of the dietary exposure; the safety data, including information on the genotoxic potential of the identified components and of the unidentified fraction of the primary product, toxicological data other than genotoxicity and information on the safety for the environment. For the toxicological studies a tiered approach is applied, for which the testing requirements, key issues and triggers are described. A description of the standard uncertainties relevant for the evaluation of primary products and how these are considered in the standardised risk assessment procedure is also included. The applicant should generate the data requested in each section to support the safety assessment of the smoke flavouring primary product. On the basis of the submitted data, EFSA will assess the safety of the primary product and conclude whether or not it presents risks to human health and to the environment under the proposed conditions of use.
Keywords: smoke flavourings, primary products, guidance, renewal, mixtures
Summary
The European Commission asked the European Food Safety Authority (EFSA) to develop updated consolidated guidance for submission of applications on smoke flavouring primary products under Regulations (EC) No 2065/2003 and No 1321/2013.
This document provides guidance to applicants on the scientific data to be included in applications for the authorisation of a new smoke flavouring primary product, as well as for the renewal or for the modification of existing authorisations, submitted respectively under Articles 7, 12 and 11 of Regulation (EC) No 2065/2003.
This document is also intended to outline the type and quality of information required by EFSA to carry out the evaluation of a smoke flavouring primary product and to conclude whether it is safe under the proposed conditions of use.
Sections 1, 2–3 of the guidance document reflect the structure that should be followed by applicants when preparing the dossier to support such an application:
Section 1 – Characterisation of the Primary Product, containing the information specific to the production process, compositional data, specification and stability of the smoke flavouring primary product.
Section 2 – Proposed uses and exposure assessment, including the information specific to the proposed uses and use levels and the anticipated intake of the primary product.
Section 3 – Safety data, describing the type of toxicity studies needed to demonstrate the safety of the primary product for human health and for the environment. It includes the data requirements needed to assess the genotoxic and toxicity potential of the primary product and the potential impact of its use on the environment. For the safety data requirements, a differentiation is made between applications for new primary products and applications for the renewal of already existing authorisations.
Section 4 on Uncertainty includes the characterisation of the standard uncertainties relevant to the safety assessment of a smoke flavouring primary product together with a description of how they are expected to influence the outcome of the risk assessment.
The applicant should generate the data requested in each section to support the safety assessment of the smoke flavouring primary product. Based on the submitted data, EFSA will assess the safety of the primary product and conclude whether or not it presents risks to human health and to the environment under the proposed conditions of use.
Introduction
Background as provided by the requestor
Smoke flavourings are a specific category of flavourings and are subject to the general Regulation (EC) No 1334/20081 on flavourings and certain food ingredients with flavouring properties for use in/on foods. This Regulation lays down the general requirements for safe use of flavourings, provides definitions for different types of flavourings and sets out flavouring substances for which an evaluation and approval is required.
Smoke flavourings are specifically regulated by Regulation (EC) No 2065/20032 of the European Parliament and of the Council on smoke flavourings used or intended for use in or on foods. This Regulation establishes a Community procedure for the safety assessment and the authorisation of smoke flavourings intended for use in or on foods on the basis of a high level of protection of human health and protection of consumers’ interests, as well as to ensure fair trade practices.
Regulation (EU) No 1321/20133 establishing the Union list of authorised smoke flavouring primary products for use as such in or on foods and/or for the production of derived smoke flavourings, was published on 12 December 2013. This Regulation lists the 10 authorised smoke flavouring primary products for use in or on foods and their conditions of use. This list was established on the basis of the applications submitted under Article 20 of the Regulation (EC) No 2065/20032 and after evaluation by EFSA.
As provided for under Article 7, paragraph 4 of Regulation (EC) No 2065/20032, EFSA developed the existing current guidance for the submissions of applications intended to establish the list of authorised smoke flavourings in view of their evaluation under the same Regulation.
The guidance is applicable to applications for new smoke flavouring primary products and for the renewal of the existing authorisations.
The current guidance is essentially based on a set of EFSA documents mentioned below:
-
–
Guidance on the submission of a dossier on a smoke flavouring primary product (EFSA AFC Panel, 2005)
This lays down the information required by applicants to be included in the application. It lays down requirements in terms of administrative, technical and toxicological data necessary to enable EFSA to carry out the safety assessment of a smoke flavouring primary product.
This document is supplemented by the following additional documents:
-
–
Dietary exposure assessment methods for smoke flavouring primary products (EFSA CEF Panel, 2009)
Dietary exposure for smoke flavourings is assessed using specifically developed methods, the SMK‐TAMDI and SMK‐EPIC methods.
-
–
Statement on the interpretation of the Margin of Safety for Smoke Flavouring Primary Products (EFSA CEF Panel, 2010)
This statement clarifies the use of the margin of safety for smoke flavouring primary products on the basis of the available toxicological data.
EFSA is asked to update the above‐mentioned documents and compile them in a single comprehensive document taking into account cross‐sectional guidance documents, such as:
-
–
Opinion on genotoxicity testing strategies applicable to food and feed safety assessment (EFSA Scientific Committee, 2011);
-
–
Opinion on the clarification of some aspects related to genotoxicity assessment (EFSA Scientific Committee, 2017a);
-
–
Statement on the genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019a,b,c);
-
–
Harmonised methodologies for human and animal health and ecological risk assessment of combined exposure to multiple chemicals (EFSA Scientific Committee, 2019a,b,c);
-
–
Guidance on the use of the Threshold of Toxicological Concern approach in food safety assessment (EFSA Scientific Committee, 2019a,b,c).
In addition, in the preparation of the new guidance, EFSA should also consider the latest updated version of the relevant Organisation for Economic and Co‐operation and Development Test Guidelines (OECD TG), such as:
-
–
OECD TG 488 (OECD, 2020b) Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays;
-
–
OECD TG 474 (OECD, 2016a) In vivo mammalian erythrocyte micronucleus test
-
–
OECD TG 489 (OECD, 2016b) In vivo Mammalian Alkaline Comet Assay.
As regards the exposure assessment, EFSA should take into account that the food categories used for regulatory purposes in flavourings are the food categories mentioned in Part D of Annex II of Regulation (EC) No 1333/20084 on food additives. A more refined exposure assessment could also be considered, based on actual use levels and on detailed food consumption data across different population groups and scenarios.
Besides the safety aspects derived from the general requirements for flavourings, the protection of the environment should be considered, where appropriate.
Furthermore, the relevant provisions arising from the recently published transparency Regulation5 should also be taken into account in the preparation of this updated guidance and consistency should be ensured with other sectors where similar updates will be done.
While recognising a connection with the general guidance and requirements for flavourings which may need also to be revised, the Commission considers that it is desirable, in view of the specific conditions of smoke flavourings, to consider this update of the guidance on smoke flavouring primary products separately.
Terms of Reference as provided by the requestor
The Commission requests EFSA to prepare an updated consolidated guidance for the submission of applications on smoke flavouring primary products under Regulations (EC) No 2065/20032 and No 1321/20133, taking into account the experience gained with the assessment and the regulation of the currently authorised and assessed smoke flavouring products in the EU and, notably, the numerous other relevant scientific and technical documents published by EFSA since the adoption of the current guidance related to the safety of smoke flavourings.
The guidance should be updated taking into account applications on new smoke flavourings and the renewals of the existing authorisations.
EFSA should take into account the relevant provisions of Regulation (EU) 2019/13815 of the European Parliament and of the Council on the transparency and sustainability of the EU risk assessment in the food chain in the preparation of this updated guidance and should ensure consistency with other sectors where similar updates will be done.
The Commission requests EFSA to carry out this updating within 18 months from the receipt of this letter.
Interpretation of the Terms of Reference
In accordance with the Terms of Reference as provided by the European Commission, the comparison between smoke flavouring primary products (see ‘Definitions’) and conventional methods of smoking with respect to their respective impact on human health and the environment is not considered in this guidance document, as it is outside the scope of the request.
All administrative information related to the preparation and submission of an application for a new authorisation, or for a modification, or a renewal of an existing authorisation of smoke flavouring primary products is addressed in a separate EFSA document, ‘Administrative guidance for the preparation of applications on smoke flavouring primary products’ (EFSA, 2021), which is applicable to applications submitted as of 27 March 2021.
As indicated in the Terms of Reference, this document is mainly intended to provide guidance to applicants for the preparation of applications:
-
–
for the authorisations of new smoke flavouring primary products submitted under Article 7 of Regulation (EC) No 2065/2003 and
-
–
for renewals of the existing authorisations of smoke flavouring primary products submitted under Article 12 of Regulation (EC) No 2065/2003 and Regulation (EU) No 1321/2013.
It also applies to applications for modifications of existing authorisations of smoke flavouring primary products submitted under Article 11 of Regulation (EC) No 2065/2003. Such modifications may involve changes in the conditions of use, production processes or in the specifications.
Scope of the guidance
This guidance provides information on the type and quality of the data that EFSA needs to conclude whether a smoke flavouring primary product is safe under the proposed conditions of use. Adherence to this guidance will help EFSA to carry out its evaluation and to deliver its scientific opinions in an effective and consistent way.
The main objective of applications for new smoke flavouring primary products, as well as for the renewal and modification of existing authorisations, is to demonstrate that in the light of the current knowledge, smoke flavouring primary products do not present risks to human health or to the environment, under the conditions of use, in line with Article 4 of Regulation (EC) No 2065/2003 and Article 1 of Regulation (EC) No 1334/2008.
This guidance has four main sections. Sections 1, 2–3 reflect the structure that should be followed by applicants when preparing the scientific content of a technical dossier to support an application for the authorisation of a new smoke flavouring primary product and/or for the renewal or modification of an existing authorisation.
-
–
Section 1 contains the information specific to the production process, compositional data, specification and stability of the primary product.
-
–
Section 2 contains the information specific to the proposed uses and use levels and anticipated intake of the primary product.
-
–
Section 3 contains the information related to the safety of the primary product, including data on its genotoxic potential, toxicological information and information on the safety for the environment.
-
–
Section 4 contains a characterisation of the standard uncertainties relevant to the safety assessment of the primary product together with a description of how they are expected to influence the outcome of the risk assessment.
General principles
This document should be read in conjunction with the following Regulations, which are listed in chronological order:
-
–
Regulation (EC) 178/20026, as amended by Regulation (EU) 2019/1381 of the European Parliament and of the Council of 20 June 2019 on the transparency and sustainability of the EU risk assessment in the food chain;
-
–
Commission Implementing Regulation (EU) No 1321/2013, establishing the EU list of authorised smoke flavouring primary products for use as such in or on food and/or for the production of derived smoke flavourings;
-
–
Regulation (EC) 1334/2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods;
-
–
Commission Regulation (EC) No 627/20067, implementing Regulation (EC) No 2065/2003 as regards quality criteria for validated analytical methods for sampling, identification and characterisation of primary smoke products;
-
–
Commission Regulation (EC) No 2065/2003 of the European Parliament and of the Council, on smoke flavourings used or intended for use in or on food, as amended.
In addition, the following guidance documents should be also considered:
-
–
Administrative guidance on the preparation and presentation of applications for new authorisation and for renewal of authorisation of smoke flavourings primary products (EFSA, 2021).
-
–
All the relevant cross‐sectional EFSA guidance documents cited throughout this guidance document should also be considered for the preparation of applications on smoke flavouring primary products. Applicants are advised to follow the most up‐to-date scientific knowledge, the current scientific/methodological approaches and the latest versions of EFSA guidance documents and of any other relevant guidance document, including OECD test guidelines.
In this guidance document the principles described in the Scientific Committee Guidance on Uncertainty Analysis (EFSA Scientific Committee, 2018) have been considered (see Section 4) and will be applied to the assessment of smoke flavouring primary products.
Definitions
As per Article 3 of Regulation (EC) No 2065/2003, the following definitions apply:
-
–
‘primary smoke condensate’ refers to the purified water‐based part of condensed smoke and falls within the definition of ‘smoke flavourings’;
-
–
‘primary tar fraction’ refers to the purified fraction of the water‐insoluble high‐density tar phase of condensed smoke and falls within the definition of ‘smoke flavourings’;
-
–
‘primary products’ refers to primary smoke condensates and primary tar fractions;
-
–
‘derived smoke flavourings’ refers to flavourings produced as a result of the further processing of primary products and which are used or intended to be used in or on foods in order to impart smoke flavour to those foods.
Content of the technical dossier
1. Characterisation of smoke flavouring primary products
The data requirements for the characterisation of the primary product described in the following sections apply to the assessment of new smoke flavouring primary products as well as to renewals and modifications of existing authorisations. Proposed modifications of the production process have to be assessed for a potential impact on the composition of the primary product and should be reflected in the specifications. If the existing specifications of a primary product are not met or if the detailed chemical analysis reveals changes in the chemical composition as a result of the modifications of the production process, this triggers the need for the submission of a new application.
1.1. Manufacturing process
1.1.1. Source materials for the primary product
All source materials used for the production of the primary product must be listed. They should comply with the provisions of Article 5 and Annex I of Regulation (EC) No 2065/2003. Full botanical names should be provided; in particular, the species of trees (woods) used for the production of the primary product must be specified. If more than one species of wood or other ingredients form the basis of the primary product, the proportions and ranges should be indicated. If primary products are produced from different species of trees as source materials they are considered as different primary products, triggering the need for separate applications.
1.1.2. Method of manufacture of the primary product
The process by which the raw materials are converted into the primary product should be described. The description should be detailed enough to allow the evaluators to understand the key steps involved in the production of the primary product. In particular, the fractions of the smoke condensate used to obtain the primary product, i.e. the water‐soluble phase (primary smoke condensate) and/or the water‐insoluble tar phase (primary tar fraction), and the employed purification steps should be described in detail. A flow chart diagram showing the most important steps in the process should accompany the description; it should clearly indicate the primary product. Description of the operational limits and how key parameters such as moisture of feedstock, oxygen content of pyrolysis atmosphere, residence time, condensation temperature and cooling time are controlled should be given. Measures implemented for production control and quality and safety assurance should be described (e.g. Hazard Analysis and Critical Control Points (HACCP), Good Manufacturing Practices (GMP), International Organization for Standardization (ISO)).
1.2. Identity of the primary product
1.2.1. Trade names of primary product
All trade names used for the primary product should be provided.
1.2.2. Information on existing evaluation from other regulatory bodies
Information on any existing evaluations and authorisations should be provided for the smoke flavouring primary product. This should include details of the body which carried out the evaluation and when this was undertaken. Any relevant data/studies generated/conducted in the context of other regulatory frameworks should be provided in full.
1.2.3. Description of physical state and of organoleptic properties
Physicochemical parameters, e.g. solubility characteristics, specific gravity, staining index, and pH of the primary product should be provided. A description of the organoleptic properties of the primary product as such and after incorporation into the formulations where it is intended to be used should be provided.
1.2.4. Chemical composition
1.2.4.1. General requirements
The analytical methods for sampling, identification and characterisation of the primary product should comply with the quality criteria laid down in Commission Regulation (EC) No 627/2006.
Analyses should be performed in an accredited laboratory. Quality systems in place for control and documentation should be indicated. Information on the accreditation of the facilities involved and certificates of analyses should be provided.
The proportion of solvent‐free mass (% m/m) in the primary product, as defined in Commission Regulation (EC) No 627/2006, should be provided with an explanation how it was determined.
The proportion of volatile fraction (% m/m) in the primary product, as defined in Commission Regulation (EC) No 627/2006, should be provided with an explanation how it was determined.
1.2.4.2. Chemical characterisation
Information on the primary product should be provided via chemical sum parameters, i.e. parameters determining the content (% m/m) of major classes of components with common structural aspects. In general, these structural classes (acids, carbonyls or phenols) should be determined as such, e.g. by colorimetric methods or titration, rather than by summing up the respective individual constituents.
1.2.4.3. Identification and quantification of individual components
Since the previous assessments of the currently authorised smoke flavouring primary products, as listed in Regulation (EU) No 1321/2013, there has been considerable analytical progress allowing improved qualitative and quantitative analyses of both volatile and non‐volatile target compounds. This offers applicants the opportunity and the obligation to minimise the unidentified fraction of smoke flavouring primary products. Therefore, without prejudice to the provisions in Commission Regulation (EC) No 627/2006 on the minimum proportions of the solvent‐free mass and the volatile fraction that should be identified and quantified, the components of the primary products should be characterised as fully as possible. This information is particularly required as the basis for the component‐based approach employed in the course of the genotoxicity assessment of primary products (see Section 3.2).
1.2.4.3.1. Identification and quantification of the volatile fraction
Capillary gas chromatography coupled with mass spectrometry (for identification) and with flame ionisation detection (for quantification) are state‐of‐the‐art techniques suitable for the analysis of the volatile fraction.
Unequivocal chemical identifications (names and CAS numbers) of the individual components of the volatile fraction should be provided. The criteria underlying the identifications should be clearly listed. In general, the identification of a component requires a comparison of at least two criteria, i.e. chromatographic (retention times or retention indices) and mass spectral data of the individual components with those of authentic reference substances. The identification of a component must be considered as ‘tentative’, if authentic reference substances are not available and the identification is solely based on the comparison of mass spectral data of the components to those of a fragmentation mass spectral library.
‘Tentatively’ identified components should be considered as part of the unidentified fraction (see Section 1.2.4.4). However, the information gained in the course of the tentative identification of components may assist in the assessment of the unidentified fraction, by taking into account the structural elements and possible similarities to identified constituents. To this end, the criteria underlying the tentative identifications of the components should be clearly described. For example, it should be stated if the tentative identifications are based on the comparison of the chromatographic (retention times/indices, specifying the type(s) of stationary phase(s) used) and mass spectral data of the components to the corresponding tabulated data for the reference compounds (extracted from databases) or just based on the comparison of the mass spectrometry fragmentation pattern of homologous compounds. The related analytical data supporting the tentative identifications performed should be provided.
Information on the concentrations of the individual components of the volatile fraction should be provided, as well as information on the principles underlying the quantification. For example, it should be stated whether internal standards or response factors have been used. Validation data for the limits of detection, limits of quantification, repeatability and reproducibility of the employed methods should be given.
If components of the volatile fraction remain unidentified, information on their quantitative contribution to the total volatile fraction should be provided, e.g. using peak areas determined by gas chromatography‐flame ionisation detector (GC‐FID) analysis to estimate the proportions of unidentified components.
1.2.4.3.2. Characterisation of the non‐volatile fraction
The Panel recognises the difficulties in identifying and quantifying individual components in the non‐volatile fraction of smoke flavouring primary products. However, the applicant should make use of meanwhile routinely available analytical approaches, e.g. gel permeation chromatography (GPC) or high‐performance liquid chromatography (HPLC) coupled with dedicated mass spectrometers. This should allow, for example, different classes to be characterised, e.g. lignin‐derived polymers, and to get more detailed information on the non‐volatile fraction.
1.2.4.4. Unidentified fraction
The proportion of the unidentified fraction (% m/m) in the primary product should be provided, encompassing unidentified volatile as well as non‐volatile constituents.
Any analytical information available to characterise the type and to estimate the proportions of chemical classes of components constituting the unidentified fraction should be presented.
Explanations should be provided as to why the unidentified fraction could not be reduced via manufacturing steps and why no higher proportion of the product could be identified.
1.2.4.5. Polycyclic aromatic hydrocarbons
The concentrations of the 16 polycyclic aromatic hydrocarbons (PAHs) listed in Appendix A should be provided. The method applied must fulfil the performance criteria of Commission Regulation (EC) No 627/2006. However, without prejudice to these requirements, the best analytical techniques currently available for analysis of PAHs should be applied, and thus it is expected that PAHs are now determined at lower limits of detection (LODs) and limits of quantification (LOQs) than those reported in the Annex of Regulation (EC) No 627/2006. Besides the concentrations of the 15 PAHs reported by Regulation (EC) No 627/2006, the concentration of benzo[c]fluorene should also be determined (JECFA, 2005; EFSA CONTAM Panel, 2008). The analytical data provided should be supported by adequate certificates of analysis, specifying the methodology(ies) applied for the analytical determinations along with their respective performances (i.e. reporting how the LOD and LOQ values have been established by the laboratories).
1.2.4.6. Batch‐to‐batch variability
Batch‐to‐batch variability should be investigated in at least five batches from different production runs. Information on how these batches were selected should be provided. The proportions of source materials (e.g. woods) used to produce the analysed batches should be described; the batches analysed should cover the range and the different proportions of the source materials subjected to the pyrolysis step, as described in the specifications. In addition, the range of conditions used in the pyrolysis step (such as time and temperature ranges, gas flow rates, etc.) should be represented by the tested samples.
Information on batch‐to‐batch variability for the measured chemical sum parameters (see Section 1.2.4.2) as well as for individual identified and non‐identified components of the primary product should be provided. The variability should be judged based on the relative standard deviations of the data determined on individual components in the different batches. The similarity of the different batches should be tested using appropriate statistical methods. A sole provision of GC chromatogram overlays is not sufficient to properly judge the batch‐to‐batch variability of a primary product smoke flavouring.
Analytical data should be given demonstrating that the sample(s) tested toxicologically fall within the range expected from the determined batch‐to‐batch variability and are considered to be representative of the primary product.
1.3. Specifications
Specifications of the primary product that include identity parameters (e.g. source materials used, proportions of the major classes of components and the 20 principal constituents of the volatile fraction) and purity criteria (e.g. maximum levels for PAHs and toxic elements) should be provided. Any proposed specifications of the primary product should be supported by adequate analytical data in order to demonstrate that the primary product is consistently manufactured within its proposed specifications. The proposed specifications should be submitted in line with the format presented in Appendix B.
1.4. Stability and fate in food
Information on storage stability from chemical analysis of the primary product should be provided from experimental conditions reflecting the intended shelf‐life of the product, either in real time settings or under forced, accelerated ageing. There is no fixed number of constituents which have to be assessed to demonstrate the stability of the primary product. However, the spectrum of the constituents selected should be representative of the chemical classes identified. The assessment of the stability of the primary product may also be supported by determining the areas of unidentified peaks in the chromatograms after different intervals of storage.
If available, a method for the analysis of characteristic components of the primary product in commercial formulations, derived smoke flavourings, as well as in the proposed food categories should be provided. The stability of the resulting analytical profile over time should then be followed.
2. Proposed uses and exposure assessment
2.1. Data needed for exposure assessment
As described above, this guidance deals with the authorisation of new smoke flavouring primary products and with the renewal or modification of existing authorisations of primary products. Data needed to assess the (potential) exposure to smoke flavouring primary products are described below.
2.1.1. Data to be provided for new smoke flavouring primary products
For assessing exposure to new smoke flavouring primary products, the applicant should provide proposed maximum use levels for foods within a food category for all food categories for which authorisation of the smoke flavouring primary product is requested. The food categories should be coded according to the food categories in Annex II, Part D, of Regulation (EC) No 1333/20088.
In addition, the applicant is encouraged to provide also expected typical use levels and to use the FoodEx2 nomenclature9 to identify the food categories in which the primary product may be used. Typical use levels are the common use levels of the primary products that are expected to be used in foods. FoodEx2 is a standardised food classification and description system developed by EFSA, which includes more detailed information about the foods in which the primary product may be used. Providing typical use levels and using FoodEx2 gives EFSA the possibility to obtain more refined exposure estimates; however, they are not mandatory. The link between the food categories of Annex II, Part D, to Regulation (EC) No 1333/2008 and FoodEx2 basic terms can be found at the following link: https://zenodo.org/record/4461577#.YBAaPuhKiUl. FoodEx2 basic terms are sometimes not sufficient to link eating events according to food categories of Annex II to Regulation 1333/2008 and therefore additional information (e.g. facets and original food descriptors, not shown in the above‐mentioned link) will be used in the risk assessment.
As the food categories in which smoke flavouring primary products are authorised to be used can be very broad, use levels should preferably be provided for specific foods in a food category in which the primary product may be used. For this level of detail, FoodEx2 nomenclature should preferably be used. The more detailed the information is on foods in which the primary product may be used, the less conservative the exposure estimate will be.
For compound foods, i.e. processed foods belonging to food category 18 in Annex II Part D, of Regulation (EC) No 1333/2008, with ingredients in which the use of smoke flavouring primary products is being intended, the use levels should be provided per ingredient (at food name level).10 It would be beneficial for the exposure assessment if the quantities of the ingredients in compound foods containing the primary product are also specified.
2.1.2. Data to be provided for renewals of authorisations of smoke flavouring primary products included in Regulation (EU) No 1321/2013
For already authorised smoke flavouring primary products, maximum permitted levels (MPLs)11 have been established for broad food categories, which are specified in Annex II, Part D, of Regulation (EC) No 1333/2008. However, these primary products may be used at a lower level than the MPL or only for some foods within a food category. Therefore, the levels actually used in food products available on the market could be used to perform a more realistic exposure assessment.
For assessing exposure to already authorised smoke flavouring primary products, the applicant should provide proposed maximum use levels for all food categories for which a renewal of the authorisation is requested. Food categories should be coded according to the food categories in Annex II, Part D, of Regulation (EC) No 1333/2008.
As for new smoke flavouring products, the applicant is encouraged to provide also typical use levels, and to use the FoodEx2 nomenclature to identify the food categories in which the primary product is used. Providing typical use levels and food categories based on FoodEx2 is not mandatory, but their use in the exposure assessment will result in more realistic exposure estimates. The link between the food categories of Annex II, Part D, to Regulation (EC) No 1333/2008 and FoodEx2 basic terms is provided at the following link: https://zenodo.org/record/4461577#.YBAaPuhKiUl. FoodEx2 basic terms are sometimes not sufficient to link eating events according to food categories of Annex II to Regulation 1333/2008 and therefore additional information (e.g. facets and original food descriptors, not shown in the above‐mentioned link) will be used in the risk assessment.
As the food categories authorised to contain smoke flavouring primary products can be very broad, use levels should preferably be provided for specific foods in a food category in which the primary product may be used. For this level of detail, FoodEx2 nomenclature should preferably be used. The more detailed the information is on foods in which the primary product may be used, the less conservative the exposure estimate will be.
For compound foods (see Section 2.1.1) with ingredients containing smoke flavouring primary products, the proposed maximum and typical use levels for the respective primary products should be provided per ingredient (at food name level). It would be beneficial for the exposure assessment if the quantities of the ingredients in compound foods containing the primary product are also specified.
2.1.3. Data to be provided in case of modifications of existing authorisations of smoke flavouring primary products
For assessing exposure in case of modifications of existing authorisations that would imply changes in the conditions of use of authorised smoke flavouring primary products, the applicant should provide proposed maximum use levels for foods within a food category, and is encouraged to also provide (expected) typical use levels, i.e. the most common use levels of the primary products used for foods in a food category.
Equivalent data as described in Sections 2.1.1 and 2.1.2 should be provided mutatis mutandis.
2.2. Exposure assessment
The applicant should provide dietary exposure estimates of smoke flavouring primary products by means of the Food Additive Intake Model (FAIM) model.12 This model uses consumption data from the EFSA Comprehensive European Food Consumption Database13 to estimate the exposure based on these use levels. Consumption data are categorised according to the food categories in Annex II, Part D of Regulation (EC) No 1333/2008. This FAIM tool is expected to overestimate the exposure; this overestimation will be particularly pronounced when the smoke flavouring is used only in specific foods within a food category as defined in the Annex II to Regulation No 1333/2008.
A second tool to estimate the exposure, the Dietary Exposure (DietEx) tool,14 is also available to the applicant. This tool uses the same food consumption data as FAIM, but the data are categorised according to FoodEx2. As FoodEx2 includes more information on the foods coded in the food consumption data, this tool potentially leads to less conservative estimates. The applicant is therefore encouraged to also use this tool to estimate the exposure, but this is not mandatory.
Both exposure tools calculate the exposure by combining consumed amounts of foods recorded in the EFSA Comprehensive Database with use levels inserted by the user of the tool. The applicant should perform separate calculations with the maximum and with the typical use levels, if available, using FAIM (mandatory) and DietEx (optional). The tools provide mean and 95th percentile exposure estimates and information on contribution of the food categories to the mean exposure, for different age groups and countries.
If the applicant wants to enter a use level for a food category that is not available in FAIM or DietEx, the applicant should refer to the parent food category. Furthermore, the level of detail of foods which may contain the smoke flavouring primary product will often not be specific in these tools and consequently maximum or typical use levels will be assigned to whole food categories. Due to this, exposure estimates provided by both tools are expected to overestimate the dietary exposure to smoke flavouring primary products.
Exposure results obtained from the tools should be included in the dossier submitted by the applicant.
EFSA will consider the exposure estimates submitted by the applicant and will refine them, if an insufficient margin of safety (MOS) (see Section 3.3.3) is calculated on the basis of the exposure estimated either by the FAIM or DietEx. Such a refined exposure assessment will consider all use levels (both maximum and typical levels) submitted in the dossier and aims to estimate the exposure as realistically as possible based on the provided data. The refined exposure assessment will be performed using the food category nomenclature in Annex II, Part D, of Regulation (EC) No 1333/2008 or FoodEx2, if the level of detail is sufficient. Additional information, such as from the facets within the FoodEx2 nomenclature or from Mintel's GNPD,15 may be used by EFSA to refine the exposure assessment.
If after such refinement steps, the MOS is still insufficient (see Section 3.3.3), applicants will be requested to submit proposals for use that would enable the exposure to be lowered.
Exposure will be estimated for the population groups that are considered relevant. In the EFSA Comprehensive Database, consumption data are available for infants, toddlers, children, adolescents, adults and the elderly. Consideration will also be given to the possibility that some consumers may be more highly exposed than the general population.
The Panel will consider any additional information (e.g. market share data) provided by the applicant that can be used to refine the exposure; however, the Panel does not consider it mandatory that this information is submitted.
The risk assessment will be based on the exposure estimates for high consumers (95th percentile estimated exposures) across relevant population groups and countries, based on the proposed maximum use levels either calculated with one or both exposure assessment tools described above or using a refined exposure assessment.
3. Safety data
3.1. General considerations
Toxicological studies should be carried out with the smoke flavouring primary product as intended to be marketed, i.e. (i) the test material should be manufactured according to the production process as described in Section 1.1, (ii) it should meet the compositional data as presented in Section 1.2, and (iii) it should comply with the specification proposed in Section 1.3. Since adequate human data are unlikely to be available, in vivo studies using experimental animals are needed in order to assess possible risks to humans derived from the consumption of smoke flavourings.
Toxicity studies should generally be conducted in accordance with OECD TGs. If a testing method is considered necessary or useful for which there is no OECD TG, this may be acceptable on a case‐by‐case basis under the condition that the method is based on an internationally validated experimental protocol. In any case, a statement of good laboratory practices (GLPs)16 compliance is required. Alternative validated testing methods for different toxicological endpoints may be considered on a case‐by‐case basis. Such methods must provide the same level of re‐assurance as the methods they aim to replace.
Smoke flavouring primary products are complex mixtures. Accordingly, the principles outlined in the guidance document from the EFSA Scientific Committee on harmonised methodologies for risk assessment of combined exposure to multiple chemicals (EFSA Scientific Committee, 2019a) should be applied. This EFSA guidance document differentiates between component‐based and whole mixture approaches. If a mixture is judged to be fully chemically defined, the preferred approach is generally component‐based, i.e. the risk is assessed based on data for exposure and effects of its individual components. However, smoke flavouring primary products may contain substantial portions of unidentified constituents. It is acknowledged that in many cases, toxicity data on multiple individual components of smoke flavourings will be lacking and difficult to obtain. According to the EFSA guidance document, for such insufficiently chemically defined mixtures, it may only be feasible to apply a whole mixture approach, i.e. the mixture is treated as a single entity, similar to the approach used for single chemicals. The testing of the whole mixture of components for toxicity has the advantage of not only including individual components but could also reflect interactive effects of multiple components. Toxicity testing of the whole mixture therefore would be appropriate for the derivation of a reference value (see Section 3.3.3).
For the genotoxicity assessment of mixtures containing a substantial fraction of unidentified components, the respective statement of the EFSA Scientific Committee (EFSA Scientific Committee, 2019b) requires a combination of a component‐based and a whole mixture approach, since genotoxicity of individual components may not be detected in a whole mixture testing approach, e.g. as a result of dilution. According to the EFSA Scientific Committee (EFSA Scientific Committee, 2011), clear evidence of genotoxicity in somatic cells in vivo has to be considered as an adverse effect per se.
In accordance with Directive 2010/63/EU17 on the protection of animals used for experimental and other scientific purposes, the unnecessary use of animals in toxicological studies should be avoided. The studies to be carried out should be those necessary to demonstrate the safety of a smoke flavouring primary product and planned in accordance with the principles of replacement, reduction and refinement of animal studies. Therefore, for new applications, characterisation of individual components and an assessment of their genotoxic potential, as well as the assessment of the genotoxic potential of the unidentified constituents in a primary product should be carried out before embarking on any in vivo toxicity studies, other than to test for genotoxicity. However, considering the legal deadlines applicable to renewal applications according to Article 12 of Regulation (EC) No 2065/2003 as well as the new provisions introduced by Regulation (EU) No 2019/1381, it is acknowledged that the strict application of this rule is not possible. Therefore, for renewal applications, simultaneous assessment of genotoxic potential and other toxicological properties may be acceptable.
Since the compositions of smoke flavouring primary products may differ from one product to another, and since a significant proportion of a primary product may remain unidentified, read‐across of toxicity data from one primary product to another is not considered justified.
3.2. Genotoxicity
Smoke flavouring primary products are complex mixtures that may contain a substantial fraction of unidentified components. The recommended approach for the genotoxicity assessment of such type of mixtures is described by the statement of the EFSA Scientific Committee (EFSA Scientific Committee, 2019b).
In line with this statement, an evaluation scheme describing the recommended approach for the genotoxicity assessment of smoke flavouring primary products is also reported in Appendix C. This evaluation scheme is equally applicable to renewal applications as to applications for the authorisation of new smoke flavouring primary products.
As a first step, the mixture should be chemically characterised as fully as possible. Concentrations of the identified components in the primary product should be provided (see Section 1.2).
The genotoxic potential of the chemically identified components in a smoke flavouring primary product should be assessed individually, using all available data. Genotoxicity data should be collected and evaluated based on the Scientific Committee guidance on genotoxicity (EFSA Scientific Committee, 2011, 2017a, 2020). Conclusions on genotoxicity are required for all identified components.
Structure‐activity relationship (SAR) information about the genotoxic potential of an identified component may be considered (for details, see Section 3.2.1) when no adequate information on genotoxicity from published or unpublished studies is available.
If only in silico predictions of the genotoxicity endpoints are available for an identified component and for its predicted or reported metabolites, and it is assessed as negative in a combination of independent and scientifically valid (Q)SAR models, (i.e. it is required to run more than one (Q)SAR model for each genotoxicity endpoint), the substance may be considered not to raise a concern for genotoxicity and, accordingly, no experimental genotoxicity testing may be necessary. In fact, the combination of different (Q)SAR models increases the overall sensitivity, and the occurrence of fully negative patterns of predictions reduces the probability of false negatives. However, EFSA will consider the information and in specific cases additional data may be requested.
For more details on the conditions needed to consider the results of (Q)SAR analyses as reliable for risk assessment, please refer to Section 3.2.1.
A positive in silico prediction sets the chemical as potentially genotoxic and it must be further investigated with appropriate experimental testing, to be conducted in line with the Scientific Committee guidance on genotoxicity (EFSA Scientific Committee, 2011, 2017a, 2020). Negative results in in vitro genotoxicity tests, would rule out the positive in silico prediction. On the other hand, a positive result in in vitro genotoxicity tests requires an appropriate in vivo follow‐up test to complete the assessment (steps A.1 and A.1.1 of the evaluation scheme reported in Appendix C) (EFSA Scientific Committee, 2011, 2017a, 2020). The outcome of the follow‐up testing in A.1.1 leads to the decision (step A.1.1.1) to proceed via either step A.2 or step A.3. If genotoxicity tests are required on identified components that are structurally related, read across principles for the selection of a representative chemical substance may be considered. In this case, the selected representative substance should then be tested with respect to genotoxicity and used as an indicator substance for all structurally related components that it represents. This assessment on genotoxicity should be carried out in line with the Scientific Committee guidance on genotoxicity (EFSA Scientific Committee, 2011, 2017a, 2020).
For grouping of chemicals and the selection of representative substances for testing, the criteria outlined in ECHA guideline R6 (ECHA, 2008) and practical guidance (ECHA, 2012), should be applied. Applicants should provide documentation to substantiate the applicability of the grouping and read‐across. There are several software tools available that may be used to identify structurally related substances such as the OECD QSAR Toolbox (see Section 3.2.1).
The choice of a representative substance among the structurally related substances that may be present in the primary product should be justified; for example, based on the presence of experimental data, or because it is expected to have the highest genotoxic potential based on, for example DNA or protein reactivity.
If a primary product contains one or more components that have been assessed (i.e. they are already known) to be genotoxic in vivo via a relevant route of administration (i.e. after oral exposure), then the primary product raises a concern for genotoxicity and the risk to human health related to this identified hazard needs to be taken into account in the risk assessment (step A.2 of the evaluation scheme reported in Appendix C).
If none of the identified chemical substances in a primary product raises a concern for genotoxicity (step A.3 of the evaluation scheme reported in Appendix C), as a following step, the Scientific Committee recommends evaluating the genotoxic potential of the unidentified fraction of the mixture. Experimental testing of the fraction of unidentified components should be considered as a first option (step B.1 of the evaluation scheme reported in Appendix C) or, if this is not feasible and a scientific justification can be provided, the whole mixture should be tested (step B.2 of the evaluation scheme reported in Appendix C) (EFSA Scientific Committee, 2019a). It is recognised that for primary products unidentified components may be in the volatile as well as in the non‐volatile fraction, therefore a clear separation of identified and unidentified components might be difficult. Nevertheless, attempts to fractionate the test material should be made on a case‐by‐case basis to minimise the dilution of the components of interest or to remove highly cytotoxic components from the tested sample.
The testing strategy for individual components, the whole mixture or its fraction(s) should follow the Scientific Committee's testing strategy guidance for individual chemical substances (EFSA Scientific Committee, 2011), according to which the following two in vitro tests are recommended as the first step:
-
–
A bacterial reverse mutation assay, Test No. 471 (OECD, 2020a), and
-
–
An in vitro mammalian cell micronucleus test, Test No. 487 (OECD, 2016c).
As recommended in the OECD test guidelines for in vitro genotoxicity testing, the maximum test concentration is based, amongst other criteria, on the cytotoxicity. In the Ames test, the recommended maximum test concentration for soluble non‐cytotoxic substances is 5 mg/plate. In the in vitro micronucleus test, if no precipitate or limiting cytotoxicity is observed, the highest test concentration should correspond to 10 mM or 2 mg/mL. However, if the test substance is not of defined composition, such as in the case of primary products, the recommended top concentration may need to be higher (e.g. 5 mg/mL), in the absence of sufficient cytotoxicity, to increase the concentration of each of the components (OECD, 2016c).
If testing of the whole smoke flavouring mixture or all of its fractions in an adequately performed set of in vitro assays, following the Scientific Committee testing strategy (EFSA Scientific Committee, 2011), provides clearly negative results and if no concern for genotoxicity is raised for any of the identified components, the primary product could be considered to be of no concern with respect to genotoxicity and no further testing would be required.
If testing the whole mixture or its fraction(s) or identified components in an adequately performed set of in vitro assays provides one or more positive results, in vivo follow‐up testing should be conducted to assess the relevance of these findings for risk assessment (step B.3 of the evaluation scheme reported in Appendix C). The follow‐up study should be tailored case‐by‐case based on the activity profile/mode of action observed in vitro, following the Scientific Committee genotoxicity testing strategy (EFSA Scientific Committee, 2011, 2017a, 2020), and taking into account any other relevant information (e.g. on source material, production process and available physicochemical information on the primary product).
The in vivo tests recommended by the EFSA Scientific Committee (EFSA Scientific Committee, 2011, 2017a, 2020) are:
-
–
In vivo transgenic rodent somatic and germ cell gene mutation assay, Test No. 488 (OECD, 2020b), to follow‐up in vitro positive results for gene mutations,
-
–
In vivo mammalian alkaline comet assay, Test No. 489 (OECD, 2016b) to follow‐up in vitro positive results for gene mutations and/or structural chromosomal aberrations,
-
–
In vivo mammalian erythrocyte micronucleus assay, Test No. 474 (OECD, 2016a) to follow‐up in vitro positive results for structural and numerical chromosomal aberrations.
A combination of an in vivo micronucleus and a comet assay, as recommended by the EFSA Scientific Committee (EFSA Scientific Committee, 2011), should be performed as a follow‐up to a positive in vitro micronucleus assay.
If the in vivo testing of a primary product or, a fraction thereof or of its components provides negative results, the relevance of these findings will be evaluated based on the recommendations given by the Scientific Committee in the guidance documents on genotoxicity (EFSA Scientific Committee 2011, EFSA Scientific Committee, 2017a,b,c) and in the statement on genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019b) (steps B.4 and B.4.1 of the evaluation scheme reported in Appendix C).
If positive results are observed in the in vivo test(s), the primary product raises a concern for genotoxicity (step B.5 of the evaluation scheme reported in Appendix C).
If a component of a primary product is evaluated to be genotoxic in vivo via a relevant route of administration and no relevant carcinogenicity data are available, it might be possible to apply the Threshold of Toxicological Concern (TTC) concept (EFSA Scientific Committee, 2019c). This would be applicable when the estimated exposure to the indentified genotoxic component(s) is very low, i.e. below the TTC value of 0.0025 μg/kg body weight (bw) per day (or 0.15 μg/person per day) for DNA‐reactive mutagens and/or carcinogens, and if the(se) component(s) is/are unavoidably resulting from the production process of the primary product. In such circumstances, it can be concluded that there is a low probability of adverse health effects (EFSA Scientific Committee, 2012, 2019c) (steps A.4 and A.4.1 of the evaluation scheme reported in Appendix C). On the other hand, if the estimated exposure of such component(s) is higher than the above‐mentioned TTC value for DNA‐reactive mutagens and/or carcinogens, the primary product raises a concern for genotoxicity (steps A.4 and A.4.2 of the evaluation scheme reported in Appendix C).
3.2.1. In silico methods for the prediction of genotoxicity
In silico predictive methods include: (a) structure–activity relationships (SAR) and quantitative structure–activity relationships (QSAR) models – collectively referred to as (Q)SAR – that qualitatively or quantitatively predict the toxicological endpoint from the knowledge of their chemical structure; and (b) read‐across, that uses data on one or more analogues (the ‘source’) to make a prediction about a query compound or compounds (the ‘target’) recognised to be ‘similar’ to the analogues.
These methods can only be applied to individual chemicals and not to mixtures. Therefore, when used in the context of an application for a primary product, they may be applied to the chemical structure of an identified component. Chemical identifiers such as CAS numbers and simplified molecular‐input line‐entry system (SMILES) codes should be provided by the applicant for all identified components of the primary product. Applicants should submit this information as part of the application dossier in an appropriate electronic format (either an Excel sheet or a text file) that allows for direct in silico analyses.
Whenever in silico methods are used, the general provisions outlined in ECHA Guidance R6 should be followed (ECHA, 2008) both for (Q)SAR and for read‐across analyses. Further practical guidance is provided in (ECHA, 2016) for (Q)SAR, and in (ECHA, 2012) for read‐across.
(Q)SAR models are implemented in a wide range of commercial and public software tools. Table D.1 in Appendix D provides some examples of available software tools for predicting the various genotoxicity endpoints. Many of these software platforms also support read‐across.
Table D.1.
Examples of in silico computational platforms that can be used to estimate the toxic effects or properties of individual chemicals. The table provides references to the full description of the platforms and the methods, together with their status (commercial or public)
| Commercial | |
|---|---|
| ADMET | https://www.simulations-plus.com/software/admetpredictor/ |
| ACD | https://www.acdlabs.com/ |
| Leadscope | https://www.leadscope.com/ |
| Lhasa | https://www.lhasalimited.org/ |
| CASE | http://www.multicase.com/case-ultra |
| TIMES | http://oasis-lmc.org/products/software/times.aspx |
| Public | |
| Danish QSAR DB | http://qsar.food.dtu.dk/ |
| Lazar | https://www.in-silico.de/ |
| OECD (Q)SAR Toolbox | https://www.oecd.org/chemicalsafety/risk-assessment/oecd-qsar-toolbox.htm |
| ToxTree | http://toxtree.sourceforge.net/ |
| VEGA platform | https://www.vegahub.eu/ |
The software tools usually include separate (and sometimes multiple) (Q)SAR for predicting the genotoxicity endpoints (i.e. in vitro/in vivo gene mutations, chromosomal aberrations).If (Q)SAR analyses were applied, it is required that the whole spectrum of genotoxicity endpoints is predicted by the applicant. The Panel however acknowledges that (Q)SAR predictions on aneugenicity are less advanced than those for other genotoxicity endpoints, due to e.g. limited number and structural diversity of known aneugens.
As already mentioned in Section 3.2, it is required to run more than one (Q)SAR model for each of the following aspects: DNA binding, bacterial gene mutation and chromosomal damage. The models should be independent from each other (i.e. the algorithms are based on different descriptors, structural alerts or training sets). As an example, when employing the OECD QSAR Toolbox the following combination of profilers (i.e. (Q)SAR models) may be used: (1) DNA binding by OASIS‐Laboratory of Mathematical Chemistry (OASIS‐LMC); 2) DNA binding by OECD; (3) DNA alerts for Ames, chromosomal aberrations (CA) and micronucleus test (MN) by OASIS‐LMC; (4) in vitro mutagenicity (Ames test) alerts by Istituto Superiore di Sanità (ISS); (5) in vivo mutagenicity (micronucleus) alerts by ISS; (6) protein binding alerts for chromosomal aberrations by OASIS‐LMC. The applicant has to submit the reasoning for the selection of the (Q)SAR models finally applied for the prediction.
The results of (Q)SAR methods may be considered as sufficient in the risk assessment provided that the following conditions are met:
(Q)SAR models for which scientific validity has been established are used. In particular the models should comply with the five OECD principles for (Q)SAR validation (OECD, 2007)18;
the substance falls within the applicability domain of the (Q)SAR models;
the predictions are relevant for the regulatory purpose; and
the information on the models and the predictions is well documented.
More detailed information on the above conditions is available from ECHA (2008, 2016).
3.3. Toxicity other than genotoxicity
Since smoke flavouring primary products are mixtures, the principles outlined by the EFSA Scientific Committee on the testing of combined exposures (EFSA Scientific Committee, 2019a) are to be followed for the assessment of potential toxicity. As explained above (see Section 3.1), toxicity testing of primary products should be based on the assessment of the whole mixture for derivation of the reference point. Applicants are reminded that for new applications, before conducting tests for in vivo toxicity, other than genotoxicity, any concern for genotoxicity should be ruled out. For renewal applications of authorisations as laid down in Regulation (EU) No 1321/2013, simultaneous testing of genotoxic potential and other toxicological properties is acceptable, since otherwise for these applications it would not be possible to meet the legal deadlines as foreseen by Article 12 of Regulation (EC) No 2065/2003.
Diagrams outlining the recommended tiered toxicity testing for primary products, as described in this chapter, are given in Appendix E.
3.3.1. Acute toxicity
In general, from past experience obtained from subchronic toxicity studies, there were no indications that primary products are potent acute toxicants. Therefore, there is no requirement to submit acute toxicity data. If, however, the applicants consider it appropriate, the WHO EHC 240 Section 5.2.9 (WHO/IPCS, 2009) could be consulted for derivation of an acute reference dose.
3.3.2. Toxicokinetics (absorption, distribution, metabolism, excretion)
Absorption, distribution, metabolism, excretion (ADME) studies can only address the kinetics of identified individual constituents. However, smoke flavouring primary products are complex mixtures of components belonging to many different chemical classes, for which significant differences in toxicokinetics may be anticipated. In addition, a substantial part of the primary products may be unidentified; therefore, a full prediction of their toxicokinetic behaviour is difficult. Considering these limitations, the Panel does not ask for ADME studies with primary products.
Based on the information available from previous evaluations, it can be assumed that many primary products will contain constituents that will be absorbed in the gastrointestinal tract, and given the molecular structures and molecular weights of the constituents identified up to now, the absorption in the gastrointestinal tract can be anticipated to be substantial. It can therefore be concluded that toxicity data as specified in Section 3.3.3 are needed for the safety assessment of these primary products.
3.3.3. Testing for repeated dose, reproductive and developmental toxicity
For primary products an individual evaluation should be performed, since they are complex mixtures for which read‐across to other primary products is not applicable. Further, due to the presence of a fraction of unidentified substances in these primary products, an evaluation according to the Threshold of Toxicological Concern (TTC) principles is not applicable.
From previous evaluations it has become clear that exposure levels of smoke flavouring primary products approach those observed for food additives. Consequently, toxicity data are needed in line with the principles that steer the data requirements for food additives. Comparable to food additives, the toxicity data required for smoke flavouring primary products are set following a tiered approach. The underlying rationale and detailed considerations for the toxicological requirements were set out in the guidance for submission for food additive evaluations (EFSA ANS Panel, 2012). Based on already available knowledge on smoke flavouring primary products as presented in previous EFSA's scientific opinions, it can be assumed that at least part of any orally administered primary product will be absorbed and systemically available. As a result of this anticipated absorption of constituents of a smoke flavouring primary product, data on subchronic oral toxicity and developmental and reproductive toxicity will be needed and are included as a requirement in Tier I.
It is recognised that all the data needed at Tier I can be obtained from an Extended One Generation Reproductive Toxicity study (EOGRTS), according to OECD TG 443 (OECD, 2018c). In the EOGRTS study, testing should be in both male and female animals covering a defined pre‐mating period (minimum of two weeks) and a two‐week mating period, with parental males being treated until at least the weaning of the F1, for a minimum of 10 weeks, and parental females during pregnancy and lactation until weaning of the F1. Dosing of the F1 offspring should begin at weaning and continue until scheduled necropsy in adulthood. The EOGRTS will provide information evaluating specific life stages not covered by the other toxicity studies: on fertility and reproductive function, and on short‐ to long‐term developmental effects from exposure during pregnancy, lactation and pre‐pubertal phases, as well as effects on juveniles and adult offspring. In addition, an EOGRTS will provide information on immunotoxicity and neurotoxicity. However, since the performance of an EOGRTS may require a time span of approximately two years, for renewal applications, it is considered not feasible to finalise and assess such a study within the foreseen current legal deadline. Although for these applications submission of an EORGTS would still be the preferred option, a reduced data package (see below) will also be acceptable, but will create greater uncertainty, which will have to be taken into account in the assessment. For applications for the authorisation of a new smoke flavouring primary product, the submission of an EOGRTS is mandatory at Tier I.
The toxicity studies that are to be used in the assessment should be designed in such a way that they provide reliable and useful lower confidence limit of the benchmark dose (BMDL)–upper confidence limit of the benchmark dose (BMDU) intervals19 in accordance with the EFSA Guidance on Dose Response Modelling (EFSA Scientific Committee, 2017b) or with the most recent version thereof. For all parameters studied, the data should be submitted in an appropriate electronic format (i.e. spreadsheet) that allows for direct use and evaluation of the data.
New applications
Data requirements at Tier I
In the light of the likely absorption of the constituents of a smoke flavouring primary product, an EOGRTS (OECD TG 443) is mandatory. This study should always comprise the full arms of the parental cohorts as well as cohorts 1A, 1B, 2A, 2B and 3. It is recommended to perform a dose range‐finding study, e.g. according to OECD TG 422 (Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening, Test No. 422 (OECD, 2016d), as also recommended by OECD TG 443. A scheme outlining the tiered toxicity testing for authorisations of new smoke flavouring primary products is presented in Appendix E (1).
Data requirements at Tier II
Depending on the results of the toxicity studies in Tier I, additional toxicity data may be required in Tier II.
A scheme by which it will be decided whether there is a need for additional toxicity testing in Tier II is given in Figure F.1 in Appendix F. The decision is based on the outcome of the Tier I testing for subchronic repeated dose toxicity and reproductive–developmental toxicity testing in combination with the outcome of the exposure assessment. For both aspects of toxicity, sufficiently large margins of safety (MOS) must be calculated to conclude that no additional toxicity testing is needed.
Figure F.1.

The decision scheme is based on the outcome of the Tier I testing for subchronic repeated dose toxicity and reproductive–developmental toxicity testing in combination with the outcome of the exposure assessment. It is only applicable to applications for the authorisation of new smoke flavouring primary products (for renewal applications see Figure F.2). The scheme is the conceptual representation of the considerations leading to either the identification of needs for Tier II testing or to the conclusion of no concern on the basis of the data available after Tier I. Following Tier II, a final conclusion will be reached, which could be either that there is a safety concern for the smoke flavouring primary product based on the proposed uses and use levels or that there is no safety concern for the primary product based on the proposed uses and use levels.
The decision scheme starts at the top with the derivation of the reference point from the subchronic repeated dose toxicity (subchr) resulting from the Tier I testing (yellow shading). The initial exposure estimate (yellow shading) is also input data that is needed for the calculation of the MOS for subchronic repeated dose toxicity (MOSsubchr) at Tier I. The reference point for reproductive‐developmental toxicity (repro‐dev) after Tier I (blue shading) is consecutive input data which should be combined with the exposure estimate as based on the information initially submitted by the applicant or with a lowered exposure estimate following the Tier I assessment of subchronic toxicity (blue shading). From these the MOS for reproductive/developmental toxicity can be calculated (MOSrepro‐dev). The diamonds in the decision scheme include two types of questions: a) whether the MOSsubchr or MOSrepro‐dev or the MOS for chronic toxicity and carcinogenicity study (MOSchronic/carc) are sufficient to conclude that the primary product can be considered to be of no safety concern under the proposed conditions of use. For more details on the numerical cut‐offs for the MOS, refer to Section 3.3.3; b) whether it is possible to lower the exposure estimates. This could be achieved by refining the exposure estimates (to be done by EFSA during the risk assessment). If this does not result in a sufficiently high MOS, the exposure can subsequently be lowered by lowering the (proposed) use levels and/or by reducing the uses (to be done by the applicant). If the answer to the questions in the diamonds is No (N), i.e. if the MOS are too low and if it is not possible to lower the exposure, a need for additional toxicity testing in Tier II is triggered, either for chronic toxicity and carcinogenicity testing (chronic/carc) and/or for additional testing to follow‐up toxicity effects observed in the EOGRTS (e.g. endocrine‐, neuro‐ and immuno‐toxicity effects). If after the Tier II testing the MOS are still too low, the smoke flavouring primary product is concluded to be of safety concern under the proposed conditions of use. On the other hand, if the answer to the questions in the diamonds addressing the magnitude of the MOS is Yes (Y), it can be concluded that the smoke flavouring primary product is not of safety concern under the proposed conditions of use.
For repeated dose toxicity, conventionally in the case of smoke flavourings, an MOS of at least 300 is required (EFSA CEF Panel, 2010) if the reference point originates from a 90‐day subchronic oral toxicity study. This criterion would not only apply to an MOS based on an no‐observed‐adverse‐effect level (NOAEL) as reference point, but also to an MOS which is calculated from a BMDL, provided that the benchmark response (BMR) on which this BMDL is based can be considered as a change of low biological relevance (EFSA Scientific Committee, 2017b).
An MOS of less than 300 (irrespective of whether it is based on an NOAEL or on a BMDL) would indicate that a combined chronic oral toxicity/carcinogenicity study, Test No. 453 (OECD, 2018d) would be required in Tier II testing. To avoid the requirement for Tier II testing, the applicant may lower the exposure to the primary product by limiting the number of food categories for its use and/or the maximum use levels applied. Based on updated exposure estimates the MOS can be recalculated (see Figure F.1 in Appendix F). Even if the MOS is greater than 300, a need for further testing in Tier II for chronic toxicity/carcinogenicity may also emerge from histological changes that could be indicative of potential pre‐carcinogenic lesions, considering also their biological relevance (EFSA Scientific Committee, 2017c). An MOS which is lower than 100, obtained after Tier II testing for chronic toxicity/carcinogenicity would raise a safety concern.
In addition, a need for Tier II testing may emerge from toxicity observed in the EOGRTS on reproductive (including possible endocrine effects) and developmental toxicity parameters and/or neuro‐ or immunotoxic effects in the different cohorts. In that case, the MOS criterion of 300 mentioned above may not apply. The minimal MOS requirement which is applicable for effects observed in the reproductive–developmental toxicity leg in the EOGRTS may well be less than 300, depending on the nature of the effects observed. However, no general strategy has been developed yet to give a precise cut‐off value here and a case‐by‐case assessment will be needed to decide on the need for a follow‐up in Tier II. Nevertheless, similar to what has been described above for repeated dose toxicity, the applicant may try to eliminate the need for testing in Tier II by limiting the number of food categories for use of the smoke flavouring primary product and/or the maximum use levels applied.
It is further noted that the primary product should be evaluated according to both legs of the scheme in Figure F.1 in Appendix F and that it is not enough to consider only the endpoint for which the lowest MOS is calculated. The following example may demonstrate this: assume that a MOS for subchronic toxicity of 200 were calculated and also an MOS for reproductive–developmental toxicity of 125. It may well be that in such a case the MOS for reproductive–developmental toxicity is considered sufficient. However, it would be inappropriate to conclude that the primary product is not of safety concern, since the MOS for subchronic toxicity would be too low and would indicate a need for further testing in Tier II.
Data requirements for renewal applications
For all currently authorised primary products, i.e. those already listed in Regulation (EU) No 1321/2013 for which renewal applications are submitted, subchronic studies are already available. However, these 90‐day subchronic toxicity studies have been carried out according to OECD guidance, which did not include requirements for examination of the endocrine systems. Therefore, preferably for a renewal application, the 90‐day subchronic toxicity study which is already available, is used as a dose‐range finding study before conducting an EOGRTS. The parental animals in the EOGRTS will have to be examined, and this study should further comprise the cohorts 1A, 1B, 2A, 2B and 3 as prescribed by OECD TG 443, similar to that requested for applications for new authorisations.
Alternative to the EOGRTS, however, considering the legal timeline foreseen in Article 12 of Regulation (EC) No 2065/2003, for renewal applications of authorisations as laid down in Regulation (EU) No 1321/2013, the following studies may be submitted:
-
–
a new 90‐day oral toxicity study may be submitted according to the latest version of OECD TG 408 (OECD, 2018a), including the assessment of neurotoxicity, since in the 90‐day studies already available from previous submissions neurotoxicity was either not included or inadequately addressed. This new oral 90‐day toxicity study should also include a full assessment of parameters indicative of possible effects on the endocrine system as specified in Annex B of OECD TG 408.
Furthermore, in addition to what is required for OECD 408, the following parameters indicative for effects on the immune system should be included in the same animals:
At term (at sacrifice):
Histopathology (lymphatic organs(*)), including bone marrow cellularity;
Weighing lymphoid organs(*).
In blood:
Immunoglobulin isotypes;
Complement assays: total serum haemolytic activity or individual components;
C‐reactive protein (CRP);
Total and differential white blood cell count *;
In spleen:
Phenotypic analysis of spleen cells (CD4 and CD8 T cells, regulatory T cells, B cells, natural killer (NK) cells, macrophages);
Natural killer cell functional analysis;
Phagocytic activity;
Mitogen stimulation assays for B and T cells.
(*) Standard parameters in OECD TG 408.
The proposed parameters allow the evaluation of a sufficiently wide range of immunological parameters to investigate the immunotoxic potential of a substance, providing information on innate and acquired immunity and inflammatory status. The above listed parameters are considered to provide a sufficient level of evidence to identify immunologic hazard. Many of the immune parameters are inter‐related and the weight given to any parameter should take this into account. Results should not therefore be interpreted individually but judged as a whole. Also parameters need to be evaluated in relation to other parameters on the animal's state of health, for example a reduction in body weight (overt toxicity) may result in thymus atrophy, which could be due to stress rather than a direct immunotoxic effect. A change in cellularity, without changes in functional parameters should be judged as equivocal evidence of toxicity to the immune system and should not be used to derive a reference point.
Some guidance for investgating these additional parameters may be found for example in ‘Methods in Immunotoxicology’ (Burleson et al., 1995), or in ‘Immunotoxicity testing. Methods and protocols’ (DeWitt et al., 2018) or in the WHO/IPCS Guidance for immunotoxicity risk assessment for chemicals (WHO/IPCS, 2012).
Since Tier II testing is not possible owing to the timeline constraints, indication of potential immunotoxicity in any of these parameters will have to be covered by additional uncertainty factors or will have to be flagged in the conclusions of the assessment.
-
–
For renewal applications also a developmental toxicity test in rats according to OECD TG 414 (OECD, 2018b) should be submitted when the applicant decides to submit a new 90‐day oral toxicity study, rather than the preferred EOGRTS.
Contrary to applications for new smoke flavouring primary products, for renewal applications follow‐up testing in a second tier will not be feasible due to timeline constraints. The general principles with respect to the interpretation of the magnitude of the MOS discussed above for new applications will be used. However, since the data set for renewal applications may be less comprehensive than that for new applications, any remaining uncertainty or concern will have to be covered by additional uncertainty factors (e.g. for possible chronic toxicity/carcinogenicity). Alternatively, these will have to be highlighted, if no uncertainty factors can be applied (e.g. for possible effects on reproduction). For renewal applications the evaluation will proceed following the decision scheme in Figure F.2 of Appendix F, irrespective of whether the preferred EOGRTS is submitted or whether a reduced data set (i.e. a new oral 90‐day toxicity study (OECD TG 408) plus a developmental toxicity study (OECD TG 414)) are submitted.
Figure F.2.
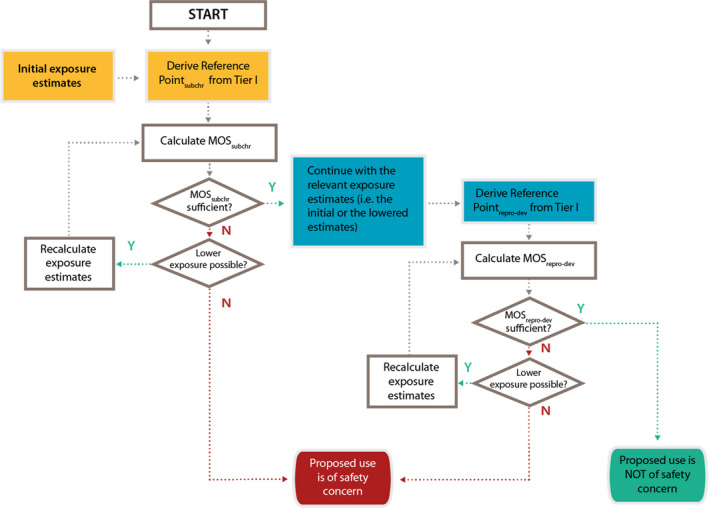
The decision scheme is based on the outcome of the Tier I testing for subchronic repeated dose toxicity and (preferably) reproductive–developmental toxicity testing (EOGRTS) or developmental toxicity (OECD TG 414) in combination with the outcome of the exposure assessment. It is applicable only to renewal applications for smoke flavouring primary products (for applications for the authorisation of new smoke flavouring primary products see Figure F.1). The scheme is the conceptual representation of the considerations leading to a final conclusion, which could be either that there is a safety concern for the smoke flavouring primary product based on the proposed uses and use levels or that there is no safety concern for the primary product based on the proposed uses and use levels. The decision scheme starts at the top with the derivation of the reference point from the subchronic repeated dose toxicity (subchr) (yellow shading). The initial exposure estimate (yellow shading) is also input data that is needed for the calculation of the MOS for subchronic repeated dose toxicity (MOSsubchr). The Reference Point for developmental or reproductive‐developmental toxicity (repro‐dev; blue shading) is consecutive input data which should be combined with the exposure estimate based on the information initially submitted by the applicant or with a lowered exposure estimate following the assessment of subchronic toxicity (blue shading). From these the MOS for reproductive/developmental toxicity can be calculated (MOSrepro‐dev). The diamonds in the decision scheme include two types of questions: (a) whether the MOSsubchr or MOSrepro‐dev are sufficient to conclude that the primary product can be considered to be of no safety concern under the proposed conditions of use. For more details on the numerical cut‐offs for the MOS, refer to Section 3.3.3; (b) whether it is possible to lower the exposure estimates. This could be achieved by refining the exposure estimates (to be done by EFSA during the risk assessment). If this does not result in a sufficiently high MOS, the exposure can subsequently be lowered by lowering the (proposed) use levels and/or by reducing the uses (to be done by the applicant). If the answer to the questions in the diamonds is No (N), i.e. if the MOS are too low and if it is not possible to lower the exposure estimate, the smoke flavouring primary product is concluded to be of safety concern under the proposed conditions of use. On the other hand, if the answer to the questions in the diamonds addressing the magnitude of the MOS is Yes (Y), it can be concluded that the smoke flavouring primary product is not of safety concern under the proposed conditions of use.
Because of the timeline constraints that are applicable for renewal applications, it is of paramount importance to provide in the submission the lowest possible recommended uses and use levels since there will be limited time for an iterative process to modify the use levels and/or food categories so as to match the MOS requirements (see Appendix F).
3.3.4. Additional information
Apart from data specifically requested in this guidance document, there may be additional toxicity studies that could be supportive for the safety assessment. For instance, toxicity studies that are not required for evaluation of the primary products, but which may have been conducted for other purposes (e.g. acute toxicity (see Section 3.3.1), irritation and sensitisation studies). If such studies are available, they should be submitted as they may provide useful background information.
3.4. Safety for the environment
Regulation (EC) No 1334/2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods lays down rules to ensure protection, where appropriate, of the environment.
Regarding the potential impact of the use of smoke flavourings on the environment, the Panel noted the following:
Smoke flavouring primary products are produced by pyrolysis of defined types of woods, i.e. naturally occurring source materials. The type of compounds generated by this step is expected to be similar to those formed upon conventional burning of wood.
Smoke flavouring primary products are produced under controlled conditions and the manufacturing process involves, in most cases, the extraction into an aqueous phase. Constituents with high lipophilicity will, therefore, be absent or present in very low concentrations.
Since primary products are added to foods, their constituents would be subject to human consumption and metabolism in the body and degradation in the sewage treatment plant before their release into the environment. It is expected that most of the constituents present in the primary products are extensively metabolised and/or readily biodegraded in a sewage treatment plant, and therefore they are of low concern for the environment.
Based on these considerations, an environmental risk assessment is not required by default.
There may, however, be primary products for which these considerations may be less applicable or not applicable at all, e.g. primary products which are obtained by manufacturing processes resulting in an increased proportion of the water‐insoluble high‐density phase of condensed smoke (primary tar fraction), see also Section 1.1. In those cases, the applicant should investigate whether the primary product contains constituents that are not extensively metabolised and/or not readily biodegradable. For these constituents, the applicant should provide evidence to demonstrate absence of concern for the environment. The testing strategy and risk assessment schemes already described for substances with a similar emission pattern and/or exposure routes such as biocides (ECHA, 2017) or medicinal products for human use (EMA, 2019) could be followed. In this respect the generation of data using non‐testing approaches, such as (Q)SAR (ECHA, 2008), could also be considered provided they are relevant, reliable and adequate for the purpose and are documented in an appropriate manner (see also ECHA, 2008). Applicants are reminded that, before conducting any testing addressing environmental safety, where applicable, any concern for genotoxicity should be ruled out.
It is noted that smoke flavourings are complex mixtures in which a fraction of components may not be fully characterised. For the fractions which have not been chemically fully characterised, it is expected that a qualitative characterisation of the main constituents is available and that the percentage of unidentified constituents is indicated and is as low as possible (see Section 1.2.4.4). In this respect, it might be relevant to assess whether the unidentified constituents might share similar properties of the constituents in the characterised fraction. On a case‐by‐case basis, further data might be needed. As already described above, in some cases (e.g. for primary products which are obtained by manufacturing processes resulting in an increased proportion of primary tar fraction) further data on the primary product as a whole, including both the identified and unidentified fraction, may need to be generated. Further guidance can be found in the OECD guidance document on aquatic toxicity testing of difficult substances and mixtures (OECD, 2019).
3.5. Other scientific data
This section is intended to provide a description of other types of scientific data that may be used to complement and to support the studies required by this guidance document, as indicated in the above sections.
Applicants should provide all the information needed to enable a conclusion on the safety assessment of a smoke flavouring primary product. This also includes the review of the published literature on both the smoke flavouring primary product and its characterised components. This may be particularly relevant for the hazard identification related to genotoxicity and environmental safety of the characterised components (see Sections 3.2 and 3.4).
The methods used to identify relevant scientific data, including the scope and criteria for literature searches, should be described in line with the principles of the systematic review methodology which aims to systematically identify, evaluate and synthesise evidence for a specific question. In particular, the search methodology (search strategy, search terms and databases searched) and the relevance and reliability assessment for any retrieved paper should be fully documented.
This would promote a more structured and transparent use of the body of evidence, reducing bias in the selection of the studies. For more detailed instruction on how to identify and select scientific literature according to the principles of the systematic literature review, applicants should refer to the EFSA guidance on application of systematic review methodology to food and feed safety assessments to support decision making (EFSA, 2010).
4. Uncertainty
4.1. Introduction to uncertainty analysis
Uncertainty analysis is part of the risk assessment performed by EFSA. In line with the principles described in the EFSA Scientific Committee's Guidance on Uncertainty Analysis (EFSA Scientific Committee, 2018), each step of the risk assessment performed by EFSA should clearly and unambiguously document what sources of uncertainty have been identified and evaluate their impact on certainty in the assessment conclusion. An uncertainty analysis is usually planned as a process in which individual sources of uncertainty are identified, characterised and combined with the aim of evaluating overall uncertainty in the output of the assessment.
4.2. Approach to treat uncertainties in the risk assessment of smoke flavouring primary products
The risk assessment of a smoke flavouring primary product aims to evaluate whether a primary product is a concern for human health and the environment. As described in detail in the previous sections of this guidance, the risk assessment is a step‐wise approach and each step requires specific considerations with respect to uncertainties.
Identification and characterisation of uncertainties in the standardised procedure for the risk assessment for smoke flavouring primary products has been described in parallel to the specification of data requirements in this guidance document. The standardised procedure covers every step of the risk assessment and is accepted by the assessors and decision‐makers as providing an appropriate basis for decision‐making (see Section 6 of the EFSA Guidance for uncertainty analysis (EFSA Scientific Committee, 2018)).
Specifying a standardised procedure and its associated (standard) uncertainties is efficient because it helps the assessors to identify and plan for any non‐standard uncertainties in each assessment. The standardised procedure ensures that all standard uncertainties are already covered by the expert judgements made when drafting the procedure. So, if no non‐standard uncertainties are present (which needs to be checked for every assessment) then EFSA can simply apply the procedure to determine a conclusion on safety in the normal way and no further uncertainty analysis is required. If non‐standard uncertainties are present then EFSA will carry out a case‐specific uncertainty analysis. This will use the same expert judgement considerations that are generally used to address sources of uncertainty in an application, but in a more structured way. In a case‐specific assessment non‐standard uncertainties may need to be reduced if they have a high impact on the conclusion of the assessment.
The list of standard uncertainties affecting the assessment of smoke flavourings and how these are treated in the standardised procedure for smoke flavouring primary products is described in Table G.1 in Appendix G. EFSA considers that the approaches taken to address these uncertainties are sufficient to meet the protection goals as specified in Regulation (EC) No 2065/2003. The presence of non‐standard uncertainties in a risk assessment is a trigger for a more detailed uncertainty analysis when a standardised procedure may not be sufficient. Table G.1 in Appendix G includes criteria to aid EFSA in determining whether non‐standard uncertainties are present, i.e. additional uncertainties that go beyond the standard uncertainties covered by the standardised procedure.
Table G.1.
List of sources of uncertainties treated by the standardised assessment of smoke flavouring primary products, how they are treated in the standard procedure, and criteria that EFSA will apply to judge whether uncertainties are standard (see Section 4.2)
| ID | Location in the assessment | Treatment in standardised procedure | Criteria to be a standard uncertainty |
|---|---|---|---|
| 1 | Manufacturing and identity of the primary product (Sections 1.1 and 1.2) | Require the applicant to provide a detailed description of the method of manufacturing and identity of the product |
Method of manufacturing is described with enough detail to ensure a consistent production of the primary product complying with the specifications. Information about the identity according to trade names (Section 1.2.1) and physical state and organoleptic properties (Section 1.2.2) is provided. |
| 2 | Chemical composition (Sections 1.2.4.1, 1.2.4.2–1.2.4.3) | Require the applicant to apply appropriate methods to sample and to analyse the volatile and non‐volatile parts of the primary product. | Methods to analyse chemical composition are appropriate and comply with the requested performance and quality criteria. A detailed description of the methods applied is included in the dossier. |
| 3 | Unidentified components (Section 1.2.4.4) | Require the applicant to demonstrate that efforts have been made to maximise the fraction of the identified components. | Fraction of unidentified components is below the limit requested in Regulation (EC) No 627/2006 and, regardless of these limits, sufficient analytical efforts to reduce the fraction of unidentified components have been demonstrated. |
| 4 | Reproducibility of the production process of the primary product (Section 1.2.4.6) |
Require the applicant to provide analytical data on at least five batches, including a description of how they were selected and ensure that the batches analysed cover the range of different proportions of source materials intended to be used. EFSA will estimate batch‐to‐batch variability with statistical methods. |
The batches are from different production runs. If applicable, for each batch, the proportions of woods are indicated and the batches analysed cover the range of source materials as well as the range of conditions used in the pyrolysis step. Batch‐to‐batch variability per investigated component is acceptably low and is evaluated as statistically reliable. |
| 5 | Stability (Section 1.4) | Require the applicant to provide compositional data from experimental conditions reflecting the intended shelf‐life of the primary product, either in real time settings or under forced, accelerated ageing conditions. |
Investigated time intervals do reflect the expected storage time. Accelerated ageing conditions reflect real time storage conditions. The constituents selected for analysis are representative for the primary product. Variability arising from storage is lower than batch‐to‐batch variability. |
| 6 | PAHs levels (Section 1.2.4.5) |
Require the applicant to show that the methods used to analyse PAHs fulfil the performance criteria of Regulation (EC) No 627/2006. EFSA will estimate average level per PAH and batch‐to‐batch variability per PAH with statistical methods. |
The selection procedure is acceptable. The average levels of individual PAHs, the respective relative standard deviations, and the applied limits of detection and quantification have been assessed with sufficient reliability. |
| 7 | Proposed use levels (Section 2.1) | EFSA will perform the risk assessment based on the information submitted by the applicant on proposed maximum and typical use levels in different food categories. No uncertainty is taken into account in these levels, other than ensuring that the definitions of maximum and typical use levels are unambiguous to avoid different interpretations. | The proposed use levels are not a concern for non‐standard uncertainties. Definitions of use levels are judged as unambiguous by EFSA. |
| 8 | Food consumption data for the foods in which the primary product is (proposed to be) used (Section 2.2) | EFSA will use food exposure tools that provide conservative estimates of exposures (for average and, if data allows, high consumers to consider variability) based on the food consumption data in the EFSA Comprehensive European Food Consumption Database and will consider indications of low reliability in the estimates. | Documentation of exposure tools and that there was no indication of low reliability in the exposure estimates due to limitations in the food consumption data. |
| 9 | Representativeness of the batches selected for toxicity or genotoxicity testing (Section 3.1, 3.2) | EFSA will evaluate whether the tested batch is representative of the material of commerce based on its description, its compositional data and the criteria for its selection. | Sufficient information that batch(es) used in testing are representative of the material of commerce. |
| 10 | Genotoxicity assessment (Section 3.2) |
Require the applicant to evaluate genotoxicity of the individual identified components. Require the applicant to perform genotoxicity testing of a fraction containing unidentified components or of the whole mixture (only if the identified components are of no concern). |
Genotoxicity testing performed in line with the criteria reported in relevant OECD TGs for genotoxicity testing (see Section 3.2). Absence of any other issues indicating non‐standard uncertainties affecting interpretation of the results (including representativeness when testing a fraction containing unidentified components or the whole product, see item 9 of this Table). |
| 11 | Lack of experimental data on genotoxicity on individual components (Section 3.2.1) | Require the applicant to perform in silico assessment with appropriate (Q)SARs on the individual components for which the experimental data on genotoxicity are missing or inadequate, when the conditions described in Section 3.2.1 of this guidance are met. | The conditions for the acceptability of the applied (Q)SAR models are met as described in Section 3.2.1. |
| 12 | Type of toxicity study (Section 3.3.3) | Add uncertainty factors to adjust the requirement for an adequate MOS according to the type of toxicity data available. |
See Section 3.3.3. for applications for new smoke flavouring primary products. Data from Tier I can trigger studies in Tier II that require case‐by‐case assessment. |
| 13 | Quality of toxicity studies (Sections 3.1 and 3.3) | Require the applicant to conduct and report toxicity studies in conformity with relevant OECD TGs. |
Compliance of the toxicity studies with the relevant OECD TGs. Absence of any issues indicating non‐standard uncertainties affecting interpretation of the results. Consistency of the results if more than one study of the same type is available. |
| 14 | Reference point for toxicity (Section 3.3.3) |
EFSA will estimate the benchmark dose (BMD), considering alternative dose‐response functions. EFSA will derive a lower bound of a probability or confidence interval on the BMD. To minimise errors in data management, the applicants are asked to provide raw data in a specific format (see Section 3.3.3). |
Data allow for a derivation of BMD. Transparency about the method and models used to estimate the BMDL. The BMDL estimate should be for the required effect size and should be statistically reliable (see criteria in the Scientific Committee Guidance on Dose Response Modelling (EFSA Scientific Committee, 2017b or later updates thereof). No indication that there is an issue with the setting of a benchmark response (BMR). |
| 15 | Data on environmental safety (Section 3.4) | No default requirement for environmental risk assessment, provided that the manufacturing process employed does not indicate that the primary product may contain constituents that are not extensively metabolised and/or readily biodegradable. | Assessment of the manufacturing process regarding the potential presence of constituents in the primary product that are not extensively metabolised and/or not readily biodegradable. |
4.3. What is required by the applicant
The applicant does not need to evaluate the uncertainties of the input to the risk assessment. The applicant should contribute to reducing uncertainties by providing comprehensive information on all aspects of the safety assessment as laid down in this guidance document and doing every efforts to fulfil these requirements using state‐of‐the‐art approaches. The applicant is encouraged to provide any information on potential sources of uncertainty which may be relevant for the risk assessment of the primary product.
After submission, the uncertainty in e.g. manufacturing, composition, exposure estimates and toxicity is characterised by EFSA. When assessing submitted applications for smoke flavouring primary products, EFSA will check whether there are any uncertainties beyond those listed as standard uncertainties. If so, EFSA will evaluate their impact on the results from the standardised procedure (see Section 3 in (EFSA Scientific Committee, 2018)) or, if non‐standard uncertainties are substantial, perform a case‐specific assessment (see Section 4 in (EFSA Scientific Committee, 2018)).
The applicant should be aware that a case‐specific assessment may require more data, expert judgement or modelling to alleviate non‐standard uncertainty.
Abbreviations
- ADME
absorption, distribution, metabolism and excretion
- BMD
benchmark dose
- BMDL
lower confidence limit of the benchmark dose
- BMDU
upper confidence limit of the benchmark dose
- BMR
benchmark response
- CRP
C‐reactive protein
- DietEx
Dietary Exposure tool
- EOGRTS
Extended One‐Generation Reproduction Toxicity
- FAIM
Food Additive Intake Model
- FID
flame ionisation detector
- GC
gas chromatography
- GLP
good laboratory practices
- GMP
good manufacturing practices
- GPC
gel permeation chromatography
- GNPD
global new products database GPC gel permeation chromatography
- HACCP
hazard analysis and critical control points
- HPLC
high‐performance liquid chromatography
- ISO
International Organization for Standardization
- ISS
Istituto Superiore di Sanità
- LOD
limit of detection
- LOQ
limit of quantification
- MOS
margin of safety
- MN
Micronucleus
- MPL
maximum permitted level
- NK
natural killer
- NOAEL
no‐observed‐adverse‐effect level
- OECD TG
Organisation for Economic Co‐operation and Development Test Guideline
- OASIS‐LMC
OASIS‐Laboratory of Mathematical Chemistry)
- PAHs
polycyclic aromatic hydrocarbons
- (Q)SAR
quantitative structure–activity relationship
- SAR
structure–activity relationship
- SMILES
simplified molecular‐input line‐entry system
- TTC
Threshold of Toxicological Concern
Appendix A – Priority group of PAHs to be analysed
1.
| Compounds | Structure | Molecular weight | |
|---|---|---|---|
| 1 | Benz[a]anthracene |

|
228 AMU |
| 2 | Benzo[b]fluoranthene |

|
252 AMU |
| 3 | Benzo[j]fluoranthene |

|
252 AMU |
| 4 | Benzo[k]fluoranthene |

|
252 AMU |
| 5 | Benzo[ghi]perylene |

|
276 AMU |
| 6 | Benzo[a]pyrene |

|
252 AMU |
| 7 | Chrysene |
|
228 AMU |
| 8 | Cyclopenta[cd]pyrene |
|
226 AMU |
| 9 | Dibenz[a,h]anthracene |

|
278 AMU |
| 10 | Dibenzo[a,e]pyrene |

|
302 AMU |
| 11 | Dibenzo[a,h]pyrene |

|
302 AMU |
| 12 | Dibenzo[a,i]pyrene |
|
302 AMU |
| 13 | Dibenzo[a,l]pyrene |

|
302 AMU |
| 14 | Indeno[1,2,3‐cd]pyrene |
|
276 AMU |
| 15 | 5‐Methylchrysene |
|
242 AMU |
| 16 | Benzo[c]fluorene |
|
216 AMU |
Appendix B – Format for the submission of the proposed specifications of a smoke flavouring primary product
1.
| Name of smoke flavouring primary product | |
|---|---|
| Source materials: | |
|
|
|
|
| Identity parameters: | |
|
|
|
|
|
|
|
|
|
|
| Chemical composition: | |
|
|
|
|
|
|
|
|
|
|
| Purity: | |
|
|
|
|
|
|
|
|
|
|
|
Appendix C – Genotoxicity assessment of primary products
1.
Figure C.1.
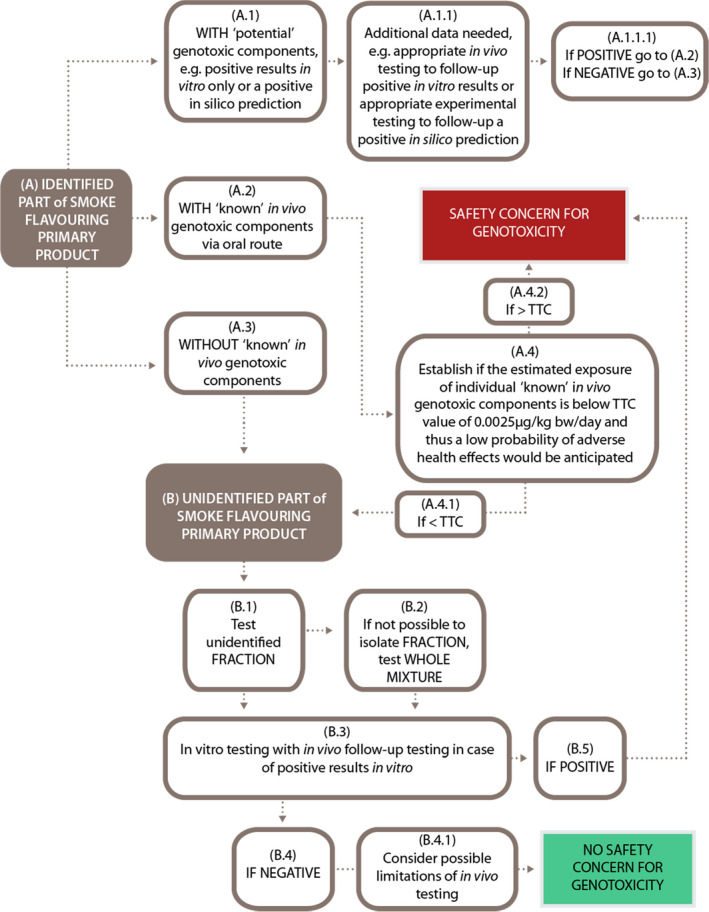
Evaluation scheme for the genotoxicity assessment of smoke flavouring primary products. The scheme is equally applicable to renewal applications as to applications for the authorisation of new smoke flavouring primary products
Appendix D – In silico computational platforms
1.
Appendix E – Tiered toxicity testing of primary products
1.
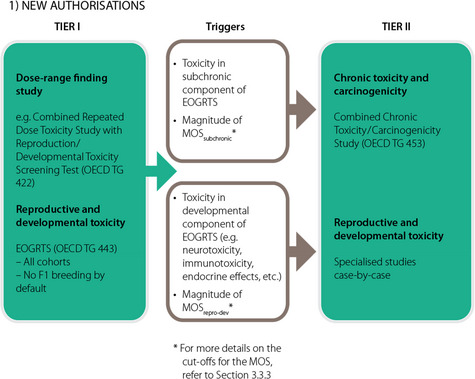

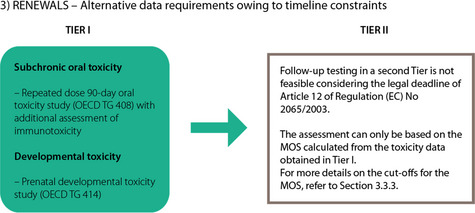
Appendix F – Decision scheme for Tier II toxicity testing
1.
1) NEW AUTHORISATIONS
2) RENEWAL APPLICATIONS
Appendix G – List of standard sources of uncertainty
1.
Appendix H – Outcome of the public consultation on the draft scientific guidance for the preparation of applications on smoke flavouring primary products
1.
Appendix H can be found in the online version of this output (‘Supporting information’ section): https://doi.org/10.2903/j.efsa.2021.6435
Supporting information
Outcome of the public consultation on the draft scientific guidance for the preparation of applications on smoke flavouring primary products
Suggested citation: EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) , Younes M, Aquilina G, Castle L, Fowler P, Frutos Fernandez MJ, Fürst P, Gundert‐Remy U, Gürtler R, Husøy T, Manco M, Mennes W, Moldeus P, Passamonti S, Shah R, Waalkens‐Berendsen I, Wölfle D, Wright M, Benigni R, Bolognesi C, Boon P, Chipman K, De Knecht J, Sahlin U, Arcella D, Barmaz S, Carfì M, Martino C, Tard A, Vianello G and Engel K‐H, 2021. Scientific Guidance for the preparation of applications on smoke flavouring primary products. EFSA Journal 2021;19(3):6435, 40 pp. 10.2903/j.efsa.2021.6435
Requestor: European Commission
Question number: EFSA‐Q‐2019‐00687
Panel members: Maged Younes, Gabriele Aquilina, Laurence Castle, Karl‐Heinz Engel, Paul Fowler, Maria Jose Frutos Fernandez, Peter Fürst, Ursula Gundert‐Remy, Rainer Gürtler, Trine Husøy, Melania Manco, Wim Mennes, Peter Moldeus, Sabina Passamonti, Romina Shah, Ine Waalkens‐Berendsen, Detlef Wölfle and Matthew Wright.
Declarations of interest: The declarations of interest of all scientific experts active in EFSA's work are available at https://ess.efsa.europa.eu/doi/doiweb/doisearch.
Acknowledgments: The FAF Panel wishes to thank the following for the support provided to this scientific output: Caroline Merten and Gianluca Rossi. The Panel wishes to thank the hearing experts: Emanuela Corsini, Andrew Hart and Henk Van Loveren.
Amendment: The following editorial corrections were carried out that do not materially affect the content or outcome of this scientific output: i) inclusion of the link to the Dietary Exposure (DietEx) tool in paragraph ‘2.2. Exposure assessment’, ii) correction of a typo in paragraph ‘3.3.3. Testing for repeated dose, reproductive and developmental toxicity’, iii) inclusion of an additional example of commercially available in silico computational platform in Appendix D. To avoid confusion, the older version of the output has been removed from the EFSA Journal, but is available on request.
Adopted: 26 January 2021
This article was originally published on the EFSA website http://www.efsa.europa.eu on 26 February 2021 as part of EFSA’s publication procedures.
The Outcome of the public consultation is available under the Supporting Information section.
Amended: 4 November 2021
Notes
Regulation (EC) No 1334/2008 of the European Parliament and of the Council of 16 December 2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods and amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC. OJ L 354, 31.12.2008, p. 34–50.
Regulation (EC) No 2065/2003 of the European Parliament and of the Council of 10 November 2003 on smoke flavourings used or intended for use in or on foods. OJ L 309, 26.11.2003, p. 1–8.
Commission Implementing Regulation (EU) No 1321/2013 of 10 December 2013 establishing the Union list of authorised smoke flavouring primary products for use as such in or on foods and/or for the production of derived smoke flavourings. OJ L 333, 12.12.2013, p. 54–67.
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives. OJ L 354, 31.12.2008, p. 16–33.
Regulation (EU) 2019/1381 of the European Parliament and of the Council on the transparency and sustainability of the EU risk assessment in the food chain. OJ L 231 of 6.9.2019, p. 1–28.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, p. 1–24.
Commission Regulation (EC) No 627/2006 of 21 April 2006 implementing Regulation (EC) No 2065/2003 of the European Parliament and of the Council as regards quality criteria for validated analytical methods for sampling, identification and characterisation of primary smoke products. OJ L 109, 22.4.2006, p. 3–6.
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32008R1333. For a description of the food categories, see the guidance document. Available here (pdf): https://ec.europa.eu/food/sites/food/files/safety/docs/fs_food-improvement-agents_guidance_1333-2008_annex-2.pdf
See Note 4 in the Annex to Commission Regulation (EU) No 1321/2013, i.e. ‘the presence of a smoke flavouring shall be permitted: (a) in a compound food other than as referred to in the Annex, where the primary product is permitted in one of the ingredients of the compound food; (b) in a food which is to be used solely in the preparation of a compound food and provided that the compound food complies with this Regulation’.
Maximum permitted levels in this document correspond to the maximum levels included in Regulation (EU) No 1321/2013.
FAIM tool is described here: http://www.efsa.europa.eu/en/applications/foodingredients/toolsand can be accessed here: https://dwh.efsa.europa.eu/bi/asp/Main.aspx?rwtrep=FAIM
The Dietary Exposure (DietEx) tool has been released by EFSA on 1 September 2021 and is available at the following link: https://www.efsa.europa.eu/en/science/tools-and-resources/dietex. A user guide and a description of the main features of the tool are available at: https://www.efsa.europa.eu/sites/default/files/2021-08/dietex-features-instructions.pdf
The Mintel's GNPD is an online database providing information available on the packaging of foods and drinks products.
Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances. OJ L 50, 20.2.2004, p. 44–59.
Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. OJ L 276, 20.10.2010, p. 33–79.
A (Q)SAR model intended to be used for regulatory purposes should be associated with the following information: (1) a defined endpoint: the model must predict the same endpoint that would be measured to fulfil the regulatory requirements; (2) an unambiguous algorithm: the algorithm underlying the model must be available and documented to ensure transparency and reproducibility of the calculation; (3) a defined domain of applicability: the applicability domain (in terms of, e.g. physicochemical parameters or molecular substructures) and the limitations of the model have to be described to allow the assessment of the applicability domain for the specific prediction; (4) appropriate measures of goodness‐of–fit, robustness and predictivity: provide appropriate measure of the internal performance of a model (as represented by goodness‐of‐fit and robustness) and the predictivity of a model (as determined by external validation); (5) a mechanistic interpretation, if possible: reasoning on, and documenting the causal link between the descriptors used in the model and the predicted endpoint add confidence in the reliability of the predictions.
The EFSA BMDL calculation tool is available at: https://shiny-efsa.openanalytics.eu/
References
- Burleson GR, Dean JH and Munson AE (eds.), 1995. Methods in Immunotoxicology. Wiley‐Liss, New York. [Google Scholar]
- DeWitt JC, Rockwell CE and Bowman CC (ed.), 2018. Immunotoxicity testing. Methods and protocols. Springer Protocols. Humana Press, New York. [Google Scholar]
- ECHA (European Chemicals Agency), 2008. Guidance for the implementation of REACH, Chapter R.6: QSARs and grouping of chemicals. ECHA, Helsinki. 134 pp. Available online: https://echa.europa.eu/documents/10162/13632/information_requirements_r6_en.pdf/77f49f81-b76d-40ab-8513-4f3a533b6ac9
- ECHA (European Chemicals Agency), 2012. Practical Guide 6. How to report read‐across and categories. Version 2.0, December 2012. Available online: https://echa.europa.eu/documents/6362380/7127661/pg_report_readacross_en.pdf/69860e5b-c669-4a0d-b868-72f5dba5b560
- ECHA (European Chemicals Agency), 2016. Practical guide. How to use and report (Q)SARs. Version 3.1, July 2016. Available online: https://echa.europa.eu/documents/10162/13655/pg_report_qsars_en.pdf
- ECHA (European Chemicals Agency), 2017. Guidance on Biocidal Products Regulation: Volume IV Environment ‐ Assessment and Evaluation (Parts B+C). Available online: https://echa.europa.eu/documents/10162/23036412/bpr_guidance_ra_vol_iv_part_b-c_en.pdf/e2622aea-0b93-493f-85a3-f9cb42be16ae
- EFSA (European Food Safety Authority), 2010. Application of systematic review methodology to food and feed safety assessments to support decision making. EFSA Journal 2010;8(6):1637, 90 pp. 10.2903/j.efsa.2010.1637 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2021. Administrative guidance for the preparation of applications on smoke flavourings primary products. EFSA supporting publication 2032:EN‐6485. 32 pp. 10.2903/sp.efsa.2021.EN-6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA AFC Panel (Panel on Food Additives, Flavourings, Processing Aids and Materials in Contact with Food), 2005. Guidance on submission of a dossier on a Smoke Flavouring Primary Product for evaluation by EFSA. EFSA Journal 2005;3(4):492, 8 pp. 10.2903/j.efsa.2005.492 [DOI] [Google Scholar]
- EFSA ANS Panel (Panel on Food Additives and Nutrient Sources added to Food), 2012. Guidance for submission for food additive evaluations. EFSA Journal 2012;10(7):2760, 60 pp. 10.2903/j.efsa.2012.2760 [DOI] [Google Scholar]
- EFSA CEF Panel (Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids), 2009. Scientific Opinion on Dietary exposure assessment methods for smoke flavouring Primary Products. EFSA Journal 2009;RN‐284, 1–30. 10.2903/j.efsa.2009.EN-248 [DOI] [Google Scholar]
- EFSA CEF Panel (Panel on Food Contact Material, Enzymes, Flavourings and Processing Aids), 2010. Statement on the Safety Evaluation of Smoke Flavourings Primary Products: interpretation of the Margin of Safety. EFSA Journal 2010;8(1):1325, 7 pp. 10.2903/j.efsa.2009.1325 [DOI] [Google Scholar]
- EFSA CONTAM Panel (Panel on Contaminants in the Food Chain), 2008. Scientific Opinion of the Panel on Contaminants in the Food Chain on a request from the European Commission on Polycyclic Aromatic Hydrocarbons in Food. EFSA Journal 2008;724, 114 pp. 10.2903/j.efsa.2008.724 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2011. Scientific Opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Scientific Opinion on Exploring options for providing advice about possible human health risks based on the concept of Threshold of Toxicological Concern (TTC). EFSA Journal 2012;10(7):2750, 103 pp. 10.2903/j.efsa.2012.2750 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017a. Scientific Opinion on the clarification of some aspects related to genotoxicity assessment. EFSA Journal 2017;15(12):5113, 25 pp. 10.2903/j.efsa.2017.5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2017b. Update: guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017; 15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017c. Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal 2017;15(8):49–70, 73 pp. 10.2903/j.efsa.2017.4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2018. Guidance on Uncertainty Analysis in Scientific Assessments. EFSA Journal 2018;16(1):5123, 39 pp. 10.2903/j.efsa.2018.5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2019a. Guidance on harmonised methodologies for human health, animal health and ecological risk assessment of combined exposure to multiple chemicals. EFSA Journal 2019;17(3):5634, 77 pp. 10.2903/j.efsa.2019.5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2019b. Statement on the genotoxicity assessment of chemical mixtures. EFSA Journal 2019;17(1):5519, 11 pp. 10.2903/j.efsa.2019.5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2019c. Guidance on the use of the Threshold of Toxicological Concern approach in food safety assessment. EFSA Journal 2019;17(6):5708, 17 pp. 10.2903/j.efsa.2019.5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2020. Draft Guidance on aneugenicity assessment. EFSA Journal 2020, 30 pp. Draft version endorsed by EFSA Scientific Committee and released for public consultation (final version under preparation). [Google Scholar]
- EMA (European Medicines Agency), 2019. Draft guideline on the environmental risk assessment of medicinal products for human use. Draft available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-environmental-risk-assessment-medicinal-products-human-use-revision-1_en.pdf
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2005. Sixty‐fourth meeting, Rome, 8–17 February 2005. Summary and Conclusions. Available online: http://www.who.int/ipcs/food/jecfa/summaries/summary_report_64_final.pdf
- OECD (Organisation for Economic Co‐operation and Development), 2007. Guidance document on the validity of (Quantitative) Structure‐activity relation ships ((Q)SAR) models. OECD Environment Health and Safety Publications Series on Testing and Assessment No.69 ENV/JM/MONO (2007), OECD Publishing, Paris. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2007)2&doclanguage=en
- OECD (Organisation for Economic Co‐operation and Development), 2016a. Test No. 474: Mammalian erythrocyte micronucleus test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264264762-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2016b. Test No. 489: In vivo mammalian alkaline comet assay, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264264885-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2016c. Test No. 487: In Vitro Mammalian Cell Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264264861-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2016d. Test No. 422: Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264264403-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2018a. Test No. 408: Repeated Dose 90‐Day Oral Toxicity Study in Rodents (OECD TG 408), in Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption, OECD Publishing, Paris, 10.1787/9789264304741-23-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2018b. Test No. 414: Prenatal Developmental Toxicity Study, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris, 10.1787/9789264070820-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2018c. Test No. 443: Extended One‐Generation Reproductive Toxicity Study, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264185371-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2018d. Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264071223-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2019. Guidance document on aquatic toxicity testing of difficult substances and mixtures, OECD Series on Testing and Assessment, OECD Publishing, Paris. 10.1787/0ed2f88e-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2020a. Test No. 471: Bacterial Reverse Mutation Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264071247-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development), 2020b. Test No. 488: Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. 10.1787/9789264203907-en [DOI]
- WHO/IPCS , 2009. Environmental Health Criteria 240. Principles and methods for the risk assessment of chemicals in food. Available online: https://apps.who.int/iris/bitstream/handle/10665/44065/WHO_EHC_240_eng.pdf
- WHO/IPCS , 2012. Guidance for Immunotoxicity Risk Assessment for Chemicals. Harmonization Project Document No. 10). Available online: https://apps.who.int/iris/bitstream/handle/10665/330098/9789241503303-eng.pdf?sequence=1&isAllowed=y
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Outcome of the public consultation on the draft scientific guidance for the preparation of applications on smoke flavouring primary products